

Abstract
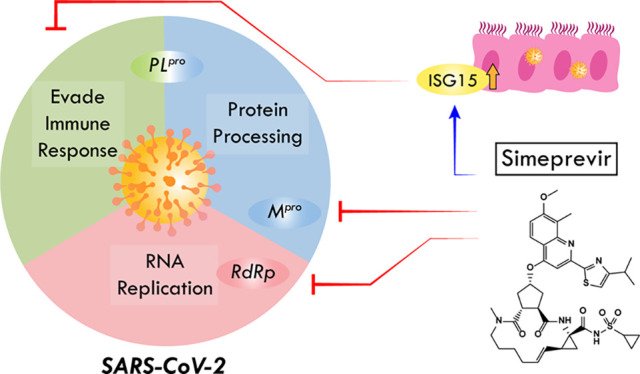
The outbreak of coronavirus disease 2019 (COVID-19), caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), is a global threat to human health. Using a multidisciplinary approach, we identified and validated the hepatitis C virus (HCV) protease inhibitor simeprevir as an especially promising repurposable drug for treating COVID-19. Simeprevir potently reduces SARS-CoV-2 viral load by multiple orders of magnitude and synergizes with remdesivir in vitro. Mechanistically, we showed that simeprevir not only inhibits the main protease (Mpro) and unexpectedly the RNA-dependent RNA polymerase (RdRp) but also modulates host immune responses. Our results thus reveal the possible anti-SARS-CoV-2 mechanism of simeprevir and highlight the translational potential of optimizing simeprevir as a therapeutic agent for managing COVID-19 and future outbreaks of CoV.
Short abstract
Simeprevir, an HCV drug, suppresses SARS-CoV-2 replication by inhibiting two viral proteins and modulating host immune responses, thus providing novel insights in anti-CoV drug design.
Introduction
The outbreak of infection by the novel betacoronavirus severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has spread to almost all countries and claimed more than 2.2 million lives worldwide.1 Alarming features of COVID-19 include a high risk of clustered outbreak both in community and nosocomial settings and a portion of up to one-fifth severe/critically ill symptomatic inpatients reported.2−5 Furthermore, a significant portion of infected individuals are asymptomatic, substantially delaying their diagnoses, hence facilitating the widespread dissemination of COVID-19.6 On the other hand, as more infectious mutants of SARS-CoV-2 emerge around the world, there is a dire need for effective therapeutics that combat the possibly altered drug sensitivities of the virus.7−9 In order to reduce both clinical severity and viral shedding, numerous antiviral candidates have been under clinical trials or in compassionate use for the treatment of SARS-CoV-2 infection.10
Several antivirals under study are hypothesized or proven to target the key mediator of a specific step in the SARS-CoV-2 viral replication cycle. For instance, lopinavir/ritonavir (LPV/r) and danoprevir have been proposed to inhibit the SARS-CoV-2 main protease (Mpro, also called 3CLPro) needed for the maturation of multiple viral proteins; chloroquine (CQ)/hydroxychloroquine (HCQ) [alone or combined with azithromycin (AZ)] may abrogate viral replication by inhibiting the endosomal acidification which is crucial for viral entry;11,12 nucleoside analogues such as remdesivir, ribavirin, favipiravir, and EIDD-2801 likely inhibit the SARS-CoV-2 nsp12 RNA-dependent RNA polymerase (RdRp) and/or induce lethal mutations during viral RNA replication.13−16 Unfortunately, from the clinical perspective, LPV/r failed to demonstrate clinical benefits in well-powered randomized controlled trials (RCTs), while HCQ and/or AZ also failed to demonstrate benefits in observational studies.17−19 Meanwhile, LPV/r, CQ/HCQ, and AZ may even increase the incidence of adverse events.19−21 Although remdesivir is widely considered as one of the most promising candidates, latest RCTs only revealed marginal shortening of disease duration in patients treated.22 Therefore, further efforts are required to search for more potent, readily repurposable therapeutic agents for SARS-CoV-2 infection, either as a sole therapy or in combination with other drugs to enhance their efficacy.
Ideally, the candidate drugs need to be readily available as intravenous and/or oral formulation(s), possess favorable pharmacokinetics properties as anti-infectives, and not cause adverse events during the treatment of SARS-CoV-2 infection (e.g., nonspecific immunosuppression, arrhythmia, or respiratory side effects). Two complementary approaches have been adopted to identify novel drugs or compounds that can suppress SARS-CoV-2 replication. One approach relies on in vitro profiling of the antiviral efficacy of up to thousands of compounds in early clinical development or drugs already approved by the U.S. Food and Drug Administration (FDA).23−28 On the other hand, as the crystal structure of the Mpro,29,30 papain-like protease (PLpro),31 and the cryo-EM structure of the nsp12 nsp7 nsp8 RdRp complex15,32 of the SARS-CoV-2 virus became available, the structure-based development of their specific inhibitors has become feasible. Structure-aided screening will enable the discovery of novel compounds as highly potent inhibitors33 as well as the repurposing of readily available drugs as anti-CoV agents for fast-track clinical trials.
Here we report our results regarding the discovery of FDA-approved drugs potentially active against the SARS-CoV-2. In vitro experiments led to the identification of simeprevir, a hepatitis C virus (HCV) NS3A/4 protease inhibitor,34 as a potent inhibitor of SARS-CoV-2 replication (Figure 1A). While simeprevir was proposed as a potential therapeutic using computational approaches,35−37 our study validated it with in-depth experimental investigation of its mechanism of action, thus providing solid evidence for its therapeutic potential. Importantly, simeprevir acts synergistically with remdesivir, whereby the effective dose of remdesivir could be lowered by multiple-fold by use of simeprevir at physiologically feasible concentrations. Interestingly, biochemical and molecular characterizations revealed that simeprevir inhibits both the Mpro protease and RdRp polymerase activities. This unexpected anti-SARS-CoV-2 mechanism of simeprevir provides hints on novel antiviral strategies.
Figure 1.

Repurposing FDA-approved drugs for SARS-CoV-2 through cellular screening. (A) Summary of methodology used in this paper. (B) Screening for FDA-approved small molecule therapeutics for activities in suppressing SARS-CoV-2 replication in Vero E6 cells. Dose–response curves in the suppression of SARS-CoV-2 replication in Vero E6 cells and cytotoxicity for simeprevir (C) and remdesivir (D) are shown. Data points in all plots represent mean ± S.E.M. For all data points, n = 3 replicates.
Results
Prioritized Screening Identifies Simeprevir As a Potent Suppressor of SARS-CoV-2 Replication in a Cellular Infection Model
Given our goal of identifying immediately usable and practical drugs against SARS-CoV-2, we prioritized a list of repurposed drug candidates for in vitro testing based on joint considerations of safety, pharmacokinetics, drug formulation availability, and feasibility of rapidly conducting clinical trials (Table S1). We focused on FDA-approved antivirals (including simeprevir, saquinavir, daclatasvir, ribavirin, sofosbuvir, and zidovudine) and drugs whose primary indication was not antiviral but had reported antiviral activity (including bromocriptine and atovaquone). Remdesivir was also tested for comparison of efficacy and as a positive control.
In a Vero E6 cellular infection model, we found the macrocyclic HCV protease inhibitor simeprevir as the only prioritized drug candidate that showed potent suppression of SARS-CoV-2 replication in the ≤10 μM range (Figure 1B). More detailed dose–response characterization found that simeprevir has a potency comparable to remdesivir (Figure 1C, D). The half-maximal effective concentration (EC50) of simeprevir was determined to be 4.08 μM, while the 50% cytotoxicity concentration (CC50) was 19.33 μM (Figure 1C, D). Interestingly, there was an atypical increase in the MTT assay readout at ∼10 μM simeprevir, which might be explained by an increase in cellular metabolic activity. In a physiologically relevant human lung epithelial cell model, ACE2-expressing A549 cells (A549-ACE2) infected with SARS-CoV-2, we also observed the strong antiviral effect of simeprevir38 (Supplementary Figure 1). The cytotoxicity data are also in line with the reported in vitro and in vivo safety pharmacological profiling using human cell lines, genotoxicity assays, and animal models.34,39 These data suggest that a desirable therapeutic window exists for the suppression of SARS-CoV-2 replication with simeprevir.
Simeprevir Potentiates the Suppression of SARS-CoV-2 Replication by Remdesivir
While simeprevir is a potential candidate for clinical use alone, we hypothesized that it may also have a synergistic effect with remdesivir, thereby mitigating its reported adverse effects, improving its efficacy, and broadening its applicability.22 Indeed, combining simeprevir and remdesivir at various concentrations apparently provided much greater suppression of SARS-CoV-2 replication than remdesivir alone, while they did not synergize to increase cytotoxicity (Figure 2A). Importantly, such effects were not merely additive, as the excess over Bliss score suggested synergism at 3.3 μM simeprevir and 1.1–10 μM remdesivir in suppressing SARS-CoV-2 replication (Figure 2B). Such a strong drug synergism was also independently reported in a recent preprint.40
Figure 2.

(A) Viral replication–suppression efficacies of different combinations of simeprevir and remdesivir concentrations. The numbers after S (simeprevir) and R (remdesivir) indicate the respective drug concentrations in μM. Data points in all plots represent mean ± S.E.M. For all data points, n = 3 replicates. (B) Bliss score analyses of synergism. (left) Diagram showing 12 combinations of simeprevir and remdesivir and their respective percentage inhibition (% inhibition, color-coded) of SARS-CoV-2 replication in Vero E6 cells compared to DMSO controls. (right) Excess over Bliss score (ΔBliss, color-coded) of different drug combinations. A positive and negative number indicates a likely synergistic and antagonistic effect, respectively, while a zero value indicates independence of action.
Simeprevir Weakly Inhibits the Mpro and RdRp but Does Not Inhibit PLpro at Physiologically Feasible Concentrations
The desirable anti-SARS-CoV-2 effect of simeprevir prompted us to determine its mechanism of action. Given that simeprevir is an HCV NS3/4A protease inhibitor, we first investigated its inhibitory activity against SARS-CoV-2 Mpro and PLpro using biochemical assays41,42 (Figure 3). We found inhibition of Mpro by simeprevir with half-maximal inhibitory concentration (IC50) of 9.6 ± 2.3 μM (Figure 3A), two times higher than the EC50 determined from our cell-based assay. The substrate cleavage was further verified with SDS-PAGE (supplementary Figure 3). Docking simeprevir against the apo protein crystal structure of SARS-CoV-2 Mpro suggested a putative binding mode with a score of −9.9 kcal mol–1 (supplementary Figure 4). This binding mode is consistent with a recent docking study using a homology model of SARS-CoV-2 Mpro.43 On the other hand, no inhibition of PLpro activity was observed at physiologically feasible concentrations of simeprevir, with either ISG15 or ubiquitin as substrate (Figure 3B and supplementary Figures 5 and 6).
Figure 3.

Simeprevir weakly inhibits Mpro and RdRp. Assay scheme and enzyme activity of main protease (Mpro), papain-like protease (PLpro), and RNA-dependent RNA polymerase (RdRp) under various concentrations of simeprevir. (A) For Mpro, a CFP-YFP conjugate with a Mpro cleavage site linker is utilized, where relative activity is determined by the residual FRET efficiency after cleavage. (B) For PLpro, rhodamine-conjugated ISG15 is used as a substrate for the enzyme, whose relative activity is determined by release of the fluorophore. (C) For RdRp, an extension assay based on the dsRNA-binding property of the intercalating agent Picogreen was established. Data points in all plots represent mean ± S.E.M. For all data points, n = 3 replicates.
We speculated that the weak inhibition of Mpro protease activity by simeprevir could not fully account for its antiviral effect toward SARS-CoV-2. To identify additional target(s), we next docked simeprevir alongside several nucleoside analogues (remdesivir, ribavirin, and favipiravir) against the motif F active site of the cryo-EM structure of the SARS-CoV-2 nsp12 RdRp (supplementary Figure 7A). Interestingly, simeprevir presented a higher binding score than the nucleoside analogues (supplementary Figure 7B). To test this experimentally, we established and performed two RdRp assays using a complex of recombinant nsp12, nsp7L8, and nsp8 of SARS-CoV.44 Intriguingly, simeprevir showed low micromolar-range inhibition toward SARS-CoV RdRp in a Picogreen fluorescence-based assay, with an IC50 value of 5.5 ± 0.2 μM (Figure 3C).45 The inhibitory effect was validated by a gel-based primer extension assay, albeit with lower efficiency (supplementary Figure 8). Collectively, the assay data suggested that simeprevir inhibits the enzymatic activities of both Mpro and RdRp but not PLpro.
RNA Sequencing Identifies Significant Modulation of Viral Defense Responses upon the Treatment of Simeprevir
While inhibition of viral targets seems to be a primary mechanism of action of simeprevir, the weak inhibitory effects observed in biochemical assays suggest the possibility of additional host-mediated antiviral response. To further elucidate the antiviral mechanism of simeprevir, we next performed RNA sequencing on Vero E6 cells to reveal the transcriptomic changes upon drug treatment (Figure 4A). In line with the literature, SARS-CoV-2 infection induced type I interferon and chemokine response (supplementary Figure 9).46,47 In mock-infected cells, simeprevir treatment (at 1.1 μM or 3.3 μM) did not induce any significant changes of differentially expressed genes (DEGs); while in SARS-CoV-2-infected cells, a small number of DEGs was observed (Figure 4B). Gene set enrichment analyses (GSEA) of infected cells using Reactome gene sets revealed significant positive enrichment of 93 gene sets in the simeprevir-treated samples, including histone lysine/arginine methylation, histone demethylation, and cell cycle control (Figure 4C and supplementary Table 2). On the other hand, gene sets with the gene ontology (GO) terms “defense response to virus” and “response to type I interferon” were negatively enriched, suggesting overall downregulation of these gene sets (Figure 4D). In the latter set, green monkey orthologs of crucial human innate immune-related genes (e.g., IFIT1–3, USP18) and interferon-stimulated genes (e.g., ISG15) were downregulated with simeprevir treatment in a dose-dependent manner (Figure 4E). Similarly, the downregulation of some of these genes as well as the proinflammatory cytokines IL-6 and interferon IFNL1 were also observed with remdesivir treatment (supplementary Figure 10).
Figure 4.

RNA-seq analysis and validation of simeprevir-mediated host response and antiviral activity. (A) Schematic representation of RNA-seq sample preparation. Treatment sequence and incubation time of simeprevir and SARS-CoV-2 was indicated with arrows and legends. (B) Venn diagrams showing differentially expressed genes (DEGs) comparing simeprevir-treated (1.1 or 3.3 μM), infected, and mock-infected samples. (C) Bubble plot of top 20 hits of positively enriched reactome gene sets under simeprevir treatment using gene set analysis (GSEA). Enriched gene sets were filtered with criteria false discovery rate (FDR) q-value < 0.25 and nominal p-value < 0.05 before ranked with their normalized enrichment scores (NES). (D) Enrichment plots of GSEA results using gene ontology (GO) gene sets. (E) Clustered heatmap showing the row-normalized expression level of genes belonging to GO term “response to type I interferon”. (F) Viral titration assay using A549-ACE2 cells treated with 4 μM simeprevir and/or 1 μM JAK inhibitor I. (G) Relative RNA level of ISG15 in uninfected or infected A549-ACE2 cells, treated with 4 μM simeprevir and/or 1 μM JAK inhibitor I. Data points in all plots represent mean ± S.D. For all data points, n = 3 replicates; * p-value < 0.05, *** p-value < 0.005.
Next, we further investigated the antiviral effects of simeprevir in human alveolar epithelial A549 cells expressing ACE2. As expected, there was a dose-dependent reduction of viral titer in culture supernatant of infected A549-ACE2 cells by using the TCID50 assay48 (supplementary Figure 11A). We also evaluated the mRNA expression profiles of antiviral genes altered by simeprevir (supplementary Figure 11B). Importantly, although simeprevir up-regulated ISG15 in infected cells at 48 h postinfection, the ISG15 level reduced to near normal levels at 72 h, coincident with a decline in viral replication (supplementary Figure 11A, B). The addition of JAK inhibitor I, which blocks signal transduction downstream of interferon receptors,48 also abolished the antiviral effects of simeprevir (Figure 4F). Concomitantly, the up-regulation of ISG15 by simeprevir in infected cells was also abolished by JAK inhibitor treatment (Figure 4G) while there were no significant differences of mRNA expression of the other tested genes (supplementary Figure 12). These results suggest that the antiviral effects of simeprevir may also stem from the induction of ISG15 during SARS-CoV-2 infection.
Discussion
The novel coronavirus SARS-CoV-2 has gone from an emerging infection to a global pandemic with its high transmissibility. As human activities are becoming more aggressive and damaging to nature, future coronavirus pandemics are bound to happen. It is therefore essential to reduce the casualties by effective pharmacological management. Our study has successfully identified the readily repurposable, clinically practical antiviral simeprevir that could target SARS-CoV-2. Specifically, we found up to fourth-order suppression of viral genome copies by simeprevir at ≤10 μM in cell-based viral replication assays—a concentration that is expected to be attainable in human lung tissues with ≥150 mg daily dosing based on available pharmacokinetic data.49,50 We observed a high Hill coefficient (>1) for simeprevir, which is in line with previous studies regarding HIV-1 protease inhibitors and is potentially important for the antiviral activity.51,52
In addition, we discovered that simeprevir can synergize with remdesivir in inhibiting SARS-CoV-2 replication in a cellular model, potentially allowing lower doses of both drugs to be used to treat COVID-19. In a global pandemic with patients having diverse clinical characteristics, providing additional therapeutic options to remdesivir will be important to treat those who are intolerant or not responding to the drug,22 which can easily amount to tens of thousands of patients. As there is only one confirmed and approved therapy for COVID-19, a potentially repurposable drug can be rapidly tested in animal models before clinical trials to prepare for supply shortages or when remdesivir-resistant mutations arise. Combination treatment, such as simeprevir–remdesivir, may also help to reduce the dose required to alleviate side effects.
We note, however, there are also several limitations of simeprevir and the proposed simeprevir–remdesivir combination. Simeprevir requires dose adjustments in patients with Child-Pugh Class B or C cirrhosis, as well as in patients with East Asian ancestry.49 In addition, simeprevir has been taken off the market since 2018 due to the emergence of next-generation HCV protease inhibitors; hence, its supply may not be ramped up easily. It is noteworthy that simeprevir is metabolized by the CYP3A4 enzyme with saturable kinetics49 while remdesivir itself is not only a substrate of CYP3A4 but also a CYP3A4 inhibitor. Whether such theoretical pharmacokinetic interaction will exacerbate liver toxicity or provide additional pharmacokinetic synergy (in addition to pharmacodynamic synergy) in vivo remains to be tested.
Mechanistically, we found that simeprevir suppresses SARS-CoV-2 replication by targeting at least two viral proteins—it weakly inhibits Mpro at ∼10 μM and unexpectedly inhibits RdRp at ∼5 μM. The potency toward Mpro is consistent with the IC50 of ∼13.7 μM as determined in a parallel study.42 Given that simeprevir is originally a non-nucleoside antiviral targeting HCV protease, its inhibition toward RdRp is largely unexpected and represents a novel mechanism of action. Simeprevir thus holds promise to be a lead compound for the future development of dual inhibitors of Mpro and RdRp. The precise molecular basis of simeprevir inhibition of the RdRp activity is an important subject of future investigation. These results also suggest possible mechanisms of drug synergy between simeprevir and remdesivir, such as cooperative binding to RdRp by targeting different sites of the enzyme, which warrants additional investigation.
The discrepancy between RdRp and Mpro inhibitory potency versus in vitro inhibitory potency of SARS-CoV-2 replication suggested additional mechanism(s) of action of simeprevir. Our RNA-seq and GSEA analysis revealed several molecular pathways that warrant future investigation. A possible direction is immune modulation via epigenetic regulations, which could mediate viral infection (via SWI/SNF chromatin remodeling complex and histone H3.3 complex),53 interferon-induced antiviral response (via H3K79 methylation),54 and host immune evasion (via alteration of DNA methylome).55 By blocking the JAK signaling pathways in human alveolar epithelial cells, we demonstrated that simeprevir exerts its antiviral effect via modulating ISG15 induction. Collectively, simeprevir inhibits two viral proteins and also modulates host immune responses to suppress SARS-CoV-2 replication. Further investigation of the mechanism of action of simeprevir may uncover new druggable targets and guide the development of broad-spectrum coronavirus antivirals.
Materials and Methods
No unexpected or unusually high safety hazards were encountered in this study.
Chemicals and Reagents
Bromocriptine mesylate (BD118791), saquinavir (BD150839), bictegravir (BD767657), atovaquone (BD114807) and asunaprevir (BD626409) were purchased from BLD Pharmatech (Shanghai, China). Entecavir (HY-13623), zidovudine (HY-17413), sofosbuvir (HY-15005), daclatasvir (HY-10466), simeprevir (HY-10241), remdesivir (HY-104077), and remdesivir triphosphate sodium (HY-126303A) were purchased from MedChemExpress (Monmouth Junction, NJ). Drug stocks were made with DMSO. JAK inhibitor I (420099) was purchased from Merck Millipore.
In Vitro SARS-CoV-2 Antiviral Tests
SARS-CoV-2 virus (BetaCoV/Hong Kong/VM20001061/2020, SCoV2) was isolated from the nasopharyngeal aspirate and throat swab of a COVID-19 patient in Hong Kong using Vero E6 cells (ATCC CRL-1586). Vero E6 or A549-ACE2 cells were infected with SCoV2 at a multiplicity of infection (MOI) of 0.05 or 0.5, respectively, in the presence of varying concentrations and/or combinations of the test drugs. DMSO as the vehicle was used as a negative control. Drugs or vehicle are present in the supernatant 1 h before infection, during infection, and 48 h after infection, while the virus was washed off after infection. Antiviral activities were evaluated by quantification of SARS-CoV-2 ORF1b copy number in the culture supernatant by using quantitative real-time RT-PCR (qPCR) at 48 h postinfection with specific primers targeting the SARS-CoV-2 ORF1b.56
Viral Titration by TCID50 Assay
Vero E6 cells were seeded on a 96-well tissue culture plate 1 day before the titration assay. The confluent cells were washed once with PBS and replenished with 2% FBS-DMEM medium supplemented with 100 units/mL penicillin and 100 mg/mL streptomycin. Virus supernatants were serially diluted from 0.5 log to 7 log in a dilution plate. Diluted supernatants were added to the cell plates in quadruplicate. The plates were observed for cytopathic effect daily. The end-point of viral dilution leading to CPE in 50% of inoculated wells was estimated using the Karber method.
JAK Pathway Blocking Study
A549-ACE2 cells were pretreated for 1 h with simeprevir, JAK inhibitor I, or combined treatment of simeprevir and JAK inhibitor I before infection. DMSO was used as the vehicle control. Cells were replenished with fresh medium in the presence of the above drugs after infection. Viral titer was determined by TCID50 assay as detailed above. Regarding the real-time PCR assay, the RNA of infected cells were extracted at 48 and 72 h post infection using a MiniBEST universal RNA extraction kit (Takara Biotechnology). RNA was reverse-transcribed by using oligo-dT primers with RT-PCR kit (Takara). mRNA expression of target genes was performed using an ABI ViiA 7 real-time PCR system (Applied Biosystems). The gene expression profiles were quantified and normalized with beta-actin as previously described.48
In Vitro Drug Cytotoxicity Assays
In vitro cytotoxicity of the tested drugs was evaluated using thiazolyl blue tetrazolium bromide (MTT, Sigma-Aldrich)-based cell viability assays. Vero E6 cells were seeded onto 48-well plates and treated with indicated concentrations of simeprevir and/or remdesivir for 48 h. Treated cells were incubated with DMEM supplemented with 0.15 mg/mL MTT for 2 h, and formazan crystal products were dissolved with DMSO. Cell viability was quantified with colorimetric absorbance at 590 nm.
Molecular Docking Simulations
Three-dimensional representations of chemical structures were extracted from the ZINC15 database (http://zinc15.docking.org),57 with the application of three selection filters—Protomers, Anodyne, and ref. ZINC15 subset DrugBank FDA (http://zinc15.docking.org./catalogs/dbfda/) were downloaded as the mol2 file format. The molecular structures were then converted to the pdbqt format (the input file format for AutoDock Vina) using MGLTools2–1.1 RC1 (subprogram “prepare_ligand”) (http://adfr.scripps.edu/versions/1.1/downloads.html). AutoDock Vina v1.1.2 was employed to perform docking experiments.58 Docking of simeprevir on SARS-CoV-2 Mpro was performed with the target structure based on an apo protein crystal structure (PDB ID: 6YB7); the A:B dimer was generated by crystallographic symmetry. Docking was run with the substrate-binding residues set to be flexible. Docking of simeprevir and other active triphosphate forms of nucleotide analogues was performed against the nsp12 portion of the SARS-CoV-2 nsp12 nsp7 nsp8 complex cryo-EM structure (PDB ID: 6M71).
Expression and Purification of Mpro and its Substrate for FRET Assay
The sequence of SARS-CoV-2 Mpro was obtained from GenBank (accession number: YP_009725301), codon-optimized, and ordered from GenScript. A C-terminal hexahistidine-maltose binding protein (His6-MBP) tag with two in-between Factor Xa digestion sites were inserted. Expression and purification of SARS-CoV-2 Mpro was then performed as described for SARS-CoV Mpro.41 The protein substrate, where the cleavage sequence “TSAVLQSGFRKM” of Mpro was inserted between a cyan fluorescent protein and a yellow fluorescent protein, was expressed and purified as described.41
In Vitro Mpro Inhibition Assay
The inhibition assay was based on fluorescence resonance energy transfer (FRET) using a fluorescent protein-based substrate previously developed for SARS-CoV Mpro.41,59 A 0.1 μM portion of purified SARS-CoV-2 Mpro was preincubated with 0–250 μM simeprevir in 20 mM HEPES pH 6.5, 120 mM NaCl, 0.4 mM EDTA, and 4 mM DTT for 30 min before the reaction was initiated by addition of 10 μM protein substrate.42 Protease activity was followed at 25 °C by FRET with excitation and emission wavelengths of 430 and 530 nm, respectively, using a multiplate reader as previously described.41,59 Reduction of fluorescence at 530 nm was fitted to a single exponential decay to obtain the observed rate constant (kobs). Relative activity of Mpro was defined as the ratio of kobs with inhibitors to that without. The relative IC50 value of simeprevir was determined by fitting the relative activity at different inhibitor concentration to a four-parameter logistics equation.
Expression and Purification of SARS-CoV nsp7L8, nsp8, nsp12
The fusion protein nsp7 nsp8 (nsp7L8) was generated by inserting a GSGSGS linker sequence between the nsp7 and nsp8 coding sequences.44 The nsp7L8, nsp8, and nsp12 were produced and purified independently as described.60 The complex, referred to as the replication/transcription complex (RTC), was reconstituted with a 1:3:3 ratio of nsp12:nsp7L8:nsp8.
In Vitro RdRp Inhibition Assay—Fluorescence-Based
The assay was performed as previously described.45 The compound concentration leading to a 50% inhibition of RTC-mediated RNA synthesis was determined as previously described. Briefly, 350 nM poly(A) template and 150 nM SARS-CoV RTC were incubated 5 min at room temperature and then added to increasing concentration of compound. Reaction was started by adding 500 μM UTP and incubated 20 min at 30 °C. Reaction assays were stopped by the addition of 20 μL 100 mM EDTA. Positive and negative controls consisted of a reaction mix with 5% DMSO (final concentration) or EDTA (100 mM) instead of compounds, respectively. Picogreen fluorescent reagent was diluted to 1/800 final in TE buffer according to the data manufacturer and aliquots were distributed into each well of the plate. The plate was incubated for 5 min in the dark at room temperature and the fluorescence signal was then read at 480 nm (excitation) and 530 nm (emission) using a TecanSafire2 microplate reader. IC50 was determined using the following equation: % of active enzyme = 100/(1 + (I)2/IC50), where I is the concentration of inhibitor and 100% of activity is the fluorescence intensity without inhibitor. IC50 was determined from curve-fitting using the GraphPad Prism 8.
In Vitro RdRp Inhibition Assay—Gel-Based
Enzyme mix (10 μM nsp12, 30 μM nsp7L8, 30 μM nsp8) in complex buffer (25 mM HEPES pH 7.5, 150 mM NaCl, 5 mM TCEP, 5 mM MgCl2) was incubated for 10 min on ice and then diluted with reaction buffer (20 mM HEPES pH 7.5, 50 mM NaCl, 5 mM MgCl2) to 2 μM nsp12 (6 μM nsp7L8 and nsp8) to a final volume of 10 μL. The resulting RTC complex was mixed with the 10 μL of 0.8 μM primer/template (P/T) carrying a Cy5 fluorescent label at the 5′ end (P: Cy5-GUC AUU CUC C, T: UAG CUU CUU AGG AGA AUG AC) in reaction buffer and incubated at 30 °C for 10 min. Inhibitor was added in 2 μL to the elongation complex, and reactions were immediately started with 18 μL of NTP mix in the reaction buffer. Final concentrations in the reactions were 0.5 μM nsp12 (1.5 μM nsp7L8 and nsp8), 0.2 μM P/T, 50 μM NTPs, and the given concentrations of inhibitors. Samples of 8 μL were taken at given time points and mixed with 40 μL of formamide containing 10 mM EDTA. Then, 10-μL samples were analyzed by denaturing PAGE (20% acrylamide, 7 M urea, TBE buffer), and product profiles were visualized by a fluorescence imager (Amersham Typhoon). Quantification of product bands and analyses were performed using ImageQuant and Excel.
In Vitro PLpro Inhibition Assay—Fluorescence-Based
The purification and assay of PLpro activity was adapted from as previously described.31 Briefly, a ISG15-C-term protein tagged with rhodamine was used as a substrate for the enzymatic assay. Because of the solubility of simeprevir, we used an optimized reaction buffer (5% DMSO, 15% PEG300, 50 mM Tris-HCl (pH 7.5), 50 mM NaCl, 5 mM DTT). A 5 μL solution containing 0–2 mM of simeprevir, and 2 μM of ISG15c-rhodamine were aliquoted into a 384-well plate. Reaction was initiated by addition of 5 μL of 40 nM PLpro to the well. Initial velocities of rhodamine release (36–240 s) were normalized against DMSO control. Reactions were conducted for 12 min with monitoring of fluorescence intensity at 485/520 nm using a microplate reader (PHERAstar FSX, BMG Labtech). The same experiment was repeated with a ubiquitin-rhodamine substrate.
In Vitro PLpro Inhibition Assay—Gel-Based
All proteins used for ubiquitination and PLpro biochemical assays were expressed in E. coli RIL cells with 0.6 mM IPTG overinduction at 16 °C. Rsp5 WW3-HECT with 2 mutations (Q808M, E809L) at the C-terminus was expressed using GST-fusion affinity tag followed by TEV protease digestion and purification by size exclusion chromatography. His-tagged PLpro, UBA1, UBCH7, ubiquitin K63R with extra cysteine at the N-terminus were also expressed and purified similarly, except hexahistidine tag used. Ubiquitin was further cross-linked with fluorescein-5-maleimide (Anaspec, Fremont, CA, US) and the poly ubiquitination sample was generated following previous protocol.61 Deubiquitination assays using PLpro (SARS-CoV-2) were carried out at 37 °C for 10 min using 1–100 μM simeprevir and 1 μM PLpro. The final DMSO concentration of each reaction is 2%. The reaction was quenched by SDS sample buffer and analyzed by 4–20% SDS-gel (GenScript, Piscataway, NJ, US). Fluorescent ubiquitin signals were imaged using Thermo iBright exposed for 750 ms.
Sample Preparation for RNA-seq
Approximately 4 × 105 Vero E6 cells were seeded onto each well of 12-well plates, in DMEM supplemented with 2% FBS, 4.5 g/L d-glucose, 4 mM l-glutamine, 25 mM HEPES, and 1% penicillin/streptomycin. Infections of SARS-CoV-2 were performed at an MOI of 1 for 24 h, followed by drug treatment or 2% DMSO in triplicates. Uninfected cells were also treated with the same concentrations of drug or DMSO in triplicates. After 24 h of drug treatment, total RNA from infected cells and uninfected cells were extracted using Qiagen RNeasy mini kit (Qiagen) following the manufacturer’s instructions. Then, we performed pair-end sequencing on a NovaSeq 6000 PE150 platform and generated 20 million reads per sample at Novogene Bioinformatics Institute (Novogene, Beijing, China).
Bioinformatic Analyses
The raw reads quality was checked by the FastQC (0.11.7) and aligned to ChlSab1.1 (Chlorocebus sabaeus) reference genome by the STAR (2.5.0a)63 with default parameters. The count matrix was generated by the featureCounts64 (as a component of Subread package 2.0.1) program. Differentially expressed genes (DEGs) were calculated by the DESeq2 package (1.26.0)65 under an R environment (3.6.1) and characterized for each sample (|L2FC| > 1, p-adjusted-value < 0.05). Gene set enrichment analysis (GSEA) was performed as previously described using normalized counts with orthology gene converting to human gene by the biomaRt package (2.42.1).62,66 Bubble plots and heatmaps were generated using the ggplot package and heatmap() function in R respectively.
Statistical Analyses
For in vitro experiments, four-parameter dose response curve fitting was performed with constraints: top = 1, IC50 > 0 using GraphPad Prism 8. Statistical significance between different drug treatment groups in A549-ACE2 cells were calculated by two-way ANOVA with Tukey’s multiple comparisons test using GraphPad Prism 8.
Acknowledgments
We thank Dr. Martin Chan for helpful discussions during the early phase of this project. We thank Diana Grewe, Etienne Decroly, and Adrien Delpal for technical assistance. We also thank Prof. Vincent Lee and Dr. Ivanhoe Leung for the critical comments on this manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscentsci.0c01186.
Supplementary Figures and Tables (PDF)
Author Contributions
■ H.S.L., K.P.Y.H., and H.-M.L. contributed equally to this work. W.L.N. and M.C.W.C. oversaw the project. H.S.L., H.-M.L., H.K., and W.L.N. wrote the manuscript with input from all authors. K.P.Y.H., K.-C.N., C.K.P.M., and J.C.W.H. contributed to the viral infection studies. H.S.L., X.H., S.K., H.-M.L., J.H., B.S., C.E., J.-C.G., B.C., B.M., and W.S.A. contributed to the development of the RdRp assays. K.S.K., H.H.-Y.C., and K.B.W. contributed to the development of the Mpro assay. D.S., M.-H.L., K.P.W., I.D., and P.H.L. contributed to the development of the PLpro assay. K.W. contributed to protein expression and purification. H.S.L., A.K.N.C., Y.W.C., and P.M.H.C. contributed to molecular modeling. Z.L. contributed to the mechanistic study. H.-M.L., Z.Z., J.H., F.K.L.C., D.S.C.H., V.C.T.M., H.K., M.C.W.C., and W.L.N. contributed to the conceptual design of the project.
W.L.N. acknowledges funding support from CUHK (“Improvement on competitiveness in hiring new faculties funding scheme”, a seed fund from the Faculty of Medicine and a PIEF grant [Ph2/COVID/19]) and the Croucher Foundation (start-up fund). M.C.W.C. acknowledges funding support by the Theme Based Research Scheme [T11-322 712/19-N], Research Grants Council, Hong Kong SAR, and the US National Institute of Allergy and Infectious Diseases (NIAID) under the Centers of Excellence for Influenza Research and Surveillance (CEIRS) [HHSN272201400006C]. W.S.A. acknowledges HKBU’s funding support through the Tier2 Start-up Grant (RC-SGT2/18-19/SCI/003). D.S. acknowledges the funding support of National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (no. 2021R1C1C1003961). The work of the BC group was supported by the Fondation pour la Recherche Médicale (Aide aux équipes), the SCORE project H2020 SC1-PHE-Coronavirus-2020 (grant #101003627) and the CARE project (grant #101005077) within IMI2 Europe’s partnership for health.
The authors declare the following competing financial interest(s): CUHK and HKU have filed a US provisional patent application based on the finding of this manuscript. W.L.N., M.C.W.C., K.P.Y.H., H.S.L., K.S.K., H.K., and H.-M.L. are inventors of the patent. F.K.L.C. has served as a consultant to Eisai, Pfizer, Takeda, and Otsuka and has been paid lecture fees by Eisai, Pfizer, AstraZeneca, and Takeda.
Notes
The raw data and data analysis codes used in this project are available from the authors upon reasonable request.
Supplementary Material
References
- Weekly Epidemiological Update–2 February 2021; World Health Organization, 2021. [Google Scholar]
- Huang C.; Wang Y.; Li X.; Ren L.; Zhao J.; Hu Y.; Zhang L.; Fan G.; Xu J.; Gu X.; et al. Clinical Features of Patients Infected with 2019 Novel Coronavirus in Wuhan, China. Lancet 2020, 395 (10223), 497–506. 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen N.; Zhou M.; Dong X.; Qu J.; Gong F.; Han Y.; Qiu Y.; Wang J.; Liu Y.; Wei Y.; et al. Epidemiological and Clinical Characteristics of 99 Cases of 2019 Novel Coronavirus Pneumonia in Wuhan, China: A Descriptive Study. Lancet 2020, 395 (10223), 507–513. 10.1016/S0140-6736(20)30211-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F.; Yu T.; Du R.; Fan G.; Liu Y.; Liu Z.; Xiang J.; Wang Y.; Song B.; Gu X.; et al. Clinical Course and Risk Factors for Mortality of Adult Inpatients with COVID-19 in Wuhan, China: A Retrospective Cohort Study. Lancet 2020, 395 (10229), 1054–1062. 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan W.-J.; Ni Z.-Y.; Hu Y.; Liang W.-H.; Ou C.-Q.; He J.-X.; Liu L.; Shan H.; Lei C.-L.; Hui D. S. C.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020, 382 (18), 1708–1720. 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.; Pei S.; Chen B.; Song Y.; Zhang T.; Yang W.; Shaman J. Substantial Undocumented Infection Facilitates the Rapid Dissemination of Novel Coronavirus (SARS-CoV-2). Science 2020, 368 (6490), 489–493. 10.1126/science.abb3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise J. Covid-19: New Coronavirus Variant Is Identified in UK. BMJ. 2020, 371, m4857. 10.1136/bmj.m4857. [DOI] [PubMed] [Google Scholar]
- Plante J. A.; Liu Y.; Liu J.; Xia H.; Johnson B. A.; Lokugamage K. G.; Zhang X.; Muruato A. E.; Zou J.; Fontes-Garfias C. R.; et al. Spike Mutation D614G Alters SARS-CoV-2 Fitness. Nature 2021, 592, 116. 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachetti M.; Marini B.; Benedetti F.; Giudici F.; Mauro E.; Storici P.; Masciovecchio C.; Angeletti S.; Ciccozzi M.; Gallo R. C.; et al. Emerging SARS-CoV-2 Mutation Hot Spots Include a Novel RNA-Dependent-RNA Polymerase Variant. J. Transl. Med. 2020, 18 (1), 179. 10.1186/s12967-020-02344-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders J. M.; Monogue M. L.; Jodlowski T. Z.; Cutrell J. B. Pharmacologic Treatments for Coronavirus Disease 2019 (COVID-19): A Review. JAMA 2020, 323 (18), 1824–1836. 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- Pastick K. A.; Okafor E. C.; Wang F.; Lofgren S. M.; Skipper C. P.; Nicol M. R.; Pullen M. F.; Rajasingham R.; McDonald E. G.; Lee T. C.; et al. Review: Hydroxychloroquine and Chloroquine for Treatment of SARS-CoV-2 (COVID-19). Open Forum Infect. Dis. 2020, 7 (4), ofaa130. 10.1093/ofid/ofaa130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A.; Chan J. F. W.; Azhar E. I.; Hui D. S. C.; Yuen K.-Y. Coronaviruses - Drug Discovery and Therapeutic Options. Nat. Rev. Drug Discovery 2016, 15 (5), 327–347. 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan T. P.; Sims A. C.; Zhou S.; Graham R. L.; Pruijssers A. J.; Agostini M. L.; Leist S. R.; Schäfer A.; Dinnon K. H.; Stevens L. J. An Orally Bioavailable Broad-Spectrum Antiviral Inhibits SARS-CoV-2 in Human Airway Epithelial Cell Cultures and Multiple Coronaviruses in Mice. Sci. Transl. Med. 2020, 12 (541), eabb5883. 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon C. J.; Tchesnokov E. P.; Woolner E.; Perry J. K.; Feng J. Y.; Porter D. P.; Götte M. Remdesivir Is a Direct-Acting Antiviral That Inhibits RNA-Dependent RNA Polymerase from Severe Acute Respiratory Syndrome Coronavirus 2 with High Potency. J. Biol. Chem. 2020, 295 (20), 6785–6797. 10.1074/jbc.RA120.013679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W.; Mao C.; Luan X.; Shen D.-D.; Shen Q.; Su H.; Wang X.; Zhou F.; Zhao W.; Gao M.; et al. Structural Basis for Inhibition of the RNA-Dependent RNA Polymerase from SARS-CoV-2 by Remdesivir. Science 2020, 368 (6498), 1499–1504. 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robson F.; Khan K. S.; Le T. K.; Paris C.; Demirbag S.; Barfuss P.; Rocchi P.; Ng W.-L. Coronavirus RNA Proofreading: Molecular Basis and Therapeutic Targeting. Mol. Cell 2020, 79 (5), 710–727. 10.1016/j.molcel.2020.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E. S.; Dufort E. M.; Udo T.; Wilberschied L. A.; Kumar J.; Tesoriero J.; Weinberg P.; Kirkwood J.; Muse A.; DeHovitz J.; et al. Association of Treatment With Hydroxychloroquine or Azithromycin With In-Hospital Mortality in Patients With COVID-19 in New York State. JAMA 2020, 323 (24), 2493–2502. 10.1001/jama.2020.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geleris J.; Sun Y.; Platt J.; Zucker J.; Baldwin M.; Hripcsak G.; Labella A.; Manson D. K.; Kubin C.; Barr R. G.; et al. Observational Study of Hydroxychloroquine in Hospitalized Patients with Covid-19. N. Engl. J. Med. 2020, 382 (25), 2411–2418. 10.1056/NEJMoa2012410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao B.; Wang Y.; Wen D.; Liu W.; Wang J.; Fan G.; Ruan L.; Song B.; Cai Y.; Wei M.; et al. A Trial of Lopinavir-Ritonavir in Adults Hospitalized with Severe Covid-19. N. Engl. J. Med. 2020, 382 (19), 1787–1799. 10.1056/NEJMoa2001282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gbinigie K.; Frie K. Should Chloroquine and Hydroxychloroquine Be Used to Treat COVID-19? A Rapid Review. Br J. Gen Pract Open 2020, 4 (2), bjgpopen20X101069. 10.3399/bjgpopen20X101069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chorin E.; Wadhwani L.; Magnani S.; Dai M.; Shulman E.; Nadeau-Routhier C.; Knotts R.; Bar-Cohen R.; Kogan E.; Barbhaiya C.; et al. QT Interval Prolongation and Torsade de Pointes in Patients with COVID-19 Treated with Hydroxychloroquine/Azithromycin. Heart Rhythm 2020, 17 (9), 1425–1433. 10.1016/j.hrthm.2020.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zhang D.; Du G.; Du R.; Zhao J.; Jin Y.; Fu S.; Gao L.; Cheng Z.; Lu Q.; et al. Remdesivir in Adults with Severe COVID-19: A Randomised, Double-Blind, Placebo-Controlled, Multicentre Trial. Lancet 2020, 395 (10236), 1569–1578. 10.1016/S0140-6736(20)31022-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M.; Cao R.; Zhang L.; Yang X.; Liu J.; Xu M.; Shi Z.; Hu Z.; Zhong W.; Xiao G. Remdesivir and Chloroquine Effectively Inhibit the Recently Emerged Novel Coronavirus (2019-NCoV) in Vitro. Cell Res. 2020, 30 (3), 269–271. 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy K.-T.; Wong A. Y.-L.; Kaewpreedee P.; Sia S. F.; Chen D.; Hui K. P. Y.; Chu D. K. W.; Chan M. C. W.; Cheung P. P.-H.; Huang X.; et al. Remdesivir, Lopinavir, Emetine, and Homoharringtonine Inhibit SARS-CoV-2 Replication in Vitro. Antiviral Res. 2020, 178, 104786. 10.1016/j.antiviral.2020.104786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva L.; Yuan S.; Yin X.; Martin-Sancho L.; Matsunaga N.; Pache L.; Burgstaller-Muehlbacher S.; De Jesus P. D.; Teriete P.; Hull M. V.; et al. Discovery of SARS-CoV-2 Antiviral Drugs through Large-Scale Compound Repurposing. Nature 2020, 586 (7827), 113–119. 10.1038/s41586-020-2577-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan J. T.; Cheng T. R.; Juang Y. P.; Ma H. H.; Wu Y. T.; Yang W. B.; Cheng C. W.; Chen X.; Chou T. H.; Shie J. J. Identification of Existing Pharmaceuticals and Herbal Medicines as Inhibitors of SARS-CoV-2 Infection. Proc. Natl. Acad. Sci. U. S. A. 2021, 118 (5), e2021579118. 10.1073/pnas.2021579118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko M.; Jeon S.; Ryu W.; Kim S. Comparative Analysis of Antiviral Efficacy of FDA-approved Drugs against SARS-CoV-2 in Human Lung Cells. J. Med. Virol. 2021, 93 (3), 1403–1408. 10.1002/jmv.26397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weston S.; Coleman C. M.; Haupt R.; Logue J.; Matthews K.; Li Y.; Reyes H. M.; Weiss S. R.; Frieman M. B. Broad Anti-Coronavirus Activity of Food and Drug Administration-Approved Drugs against SARS-CoV-2 In Vitro and SARS-CoV In Vivo. J. Virol. 2020, 10.1128/JVI.01218-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal Structure of SARS-CoV-2 Main Protease Provides a Basis for Design of Improved α-Ketoamide Inhibitors. Science 2020, 368 (6489), 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Du X.; Xu Y.; Deng Y.; Liu M.; Zhao Y.; Zhang B.; Li X.; Zhang L.; Peng C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582 (7811), 289–293. 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Shin D.; Mukherjee R.; Grewe D.; Bojkova D.; Baek K.; Bhattacharya A.; Schulz L.; Widera M.; Mehdipour A. R.; Tascher G.; et al. Papain-like Protease Regulates SARS-CoV-2 Viral Spread and Innate Immunity. Nature 2020, 587 (7835), 657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y.; Yan L.; Huang Y.; Liu F.; Zhao Y.; Cao L.; Wang T.; Sun Q.; Ming Z.; Zhang L.; et al. Structure of the RNA-Dependent RNA Polymerase from COVID-19 Virus. Science 2020, 368 (6492), 779–782. 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z.; Zhao Y.; Sun Y.; Zhang B.; Wang H.; Wu Y.; Zhu Y.; Zhu C.; Hu T.; Du X.; et al. Structural Basis for the Inhibition of SARS-CoV-2 Main Protease by Antineoplastic Drug Carmofur. Nat. Struct. Mol. Biol. 2020, 27 (6), 529–532. 10.1038/s41594-020-0440-6. [DOI] [PubMed] [Google Scholar]
- Rosenquist Å.; Samuelsson B.; Johansson P.-O.; Cummings M. D.; Lenz O.; Raboisson P.; Simmen K.; Vendeville S.; de Kock H.; Nilsson M.; et al. Discovery and Development of Simeprevir (TMC435), a HCV NS3/4A Protease Inhibitor. J. Med. Chem. 2014, 57 (5), 1673–1693. 10.1021/jm401507s. [DOI] [PubMed] [Google Scholar]
- Hosseini F. S.; Amanlou M. Anti-HCV and Anti-Malaria Agent, Potential Candidates to Repurpose for Coronavirus Infection: Virtual Screening, Molecular Docking, and Molecular Dynamics Simulation Study. Life Sci. 2020, 258, 118205. 10.1016/j.lfs.2020.118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- J A.; Francis D.; C. S S.; K. G A.; C S.; Variyar E. J. Repurposing Simeprevir, Calpain Inhibitor IV and a Cathepsin F Inhibitor against SARS-CoV-2 and Insights into Their Interactions with Mpro. J. Biomol. Struct. Dyn. 2020, 1–12. 10.1080/07391102.2020.1813200. [DOI] [PubMed] [Google Scholar]
- Trezza A.; Iovinelli D.; Santucci A.; Prischi F.; Spiga O. An Integrated Drug Repurposing Strategy for the Rapid Identification of Potential SARS-CoV-2 Viral Inhibitors. Sci. Rep. 2020, 10 (1), 13866. 10.1038/s41598-020-70863-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E. C.; Huang C.; Narayanan K.; Makino S.; Tesh R. B.; Peters C. J. Exogenous ACE2 Expression Allows Refractory Cell Lines to Support Severe Acute Respiratory Syndrome Coronavirus Replication. J. Virol. 2005, 79 (6), 3846–3850. 10.1128/JVI.79.6.3846-3850.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T.-I.; Lenz O.; Fanning G.; Verbinnen T.; Delouvroy F.; Scholliers A.; Vermeiren K.; Rosenquist A.; Edlund M.; Samuelsson B.; et al. In Vitro Activity and Preclinical Profile of TMC435350, a Potent Hepatitis C Virus Protease Inhibitor. Antimicrob. Agents Chemother. 2009, 53 (4), 1377–1385. 10.1128/AAC.01058-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bafna K.; White K.; Harish B.; Rosales R.; Ramelot T. A.; Acton T. B.; Moreno E.; Kehrer T.; Miorin L.; Royer C. A. Hepatitis C Virus Drugs Simeprevir and Grazoprevir Synergize with Remdesivir to Suppress SARS-CoV-2 Replication in Cell Culture. BioRxiv 2020, 10.1101/2020.12.13.422511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck C.-P.; Chong L.-T.; Chen C.; Chow H.-F.; Wan D. C.-C.; Wong K.-B. Profiling of Substrate Specificity of SARS-CoV 3CL. PLoS One 2010, 5 (10), e13197. 10.1371/journal.pone.0013197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C.; Sacco M. D.; Hurst B.; Townsend J. A.; Hu Y.; Szeto T.; Zhang X.; Tarbet B.; Marty M. T.; Chen Y.; et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30 (8), 678–692. 10.1038/s41422-020-0356-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calligari P.; Bobone S.; Ricci G.; Bocedi A. Molecular Investigation of SARS-CoV-2 Proteins and Their Interactions with Antiviral Drugs. Viruses 2020, 12 (4), 445. 10.3390/v12040445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subissi L.; Posthuma C. C.; Collet A.; Zevenhoven-Dobbe J. C.; Gorbalenya A. E.; Decroly E.; Snijder E. J.; Canard B.; Imbert I. One Severe Acute Respiratory Syndrome Coronavirus Protein Complex Integrates Processive RNA Polymerase and Exonuclease Activities. Proc. Natl. Acad. Sci. U. S. A. 2014, 111 (37), E3900–9. 10.1073/pnas.1323705111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eydoux C.; Fattorini V.; Shannon A.; Le T.-T.-N.; Didier B.; Canard B.; Guillemot J.-C. A Fluorescence-Based High Throughput-Screening Assay for the SARS-CoV RNA Synthesis Complex. J. Virol. Methods 2021, 288, 114013. 10.1016/j.jviromet.2020.114013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D.; Nilsson-Payant B. E.; Liu W.-C.; Uhl S.; Hoagland D.; Møller R.; Jordan T. X.; Oishi K.; Panis M.; Sachs D.; et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell 2020, 181 (5), 1036–1045. 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park A.; Iwasaki A. Type I and Type III Interferons - Induction, Signaling, Evasion, and Application to Combat COVID-19. Cell Host Microbe 2020, 27 (6), 870–878. 10.1016/j.chom.2020.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K. P. Y.; Lee S. M. Y.; Cheung C.-Y.; Mao H.; Lai A. K. W.; Chan R. W. Y.; Chan M. C. W.; Tu W.; Guan Y.; Lau Y.-L.; et al. H5N1 Influenza Virus-Induced Mediators Upregulate RIG-I in Uninfected Cells by Paracrine Effects Contributing to Amplified Cytokine Cascades. J. Infect. Dis. 2011, 204 (12), 1866–1878. 10.1093/infdis/jir665. [DOI] [PubMed] [Google Scholar]
- Janssen Pharmaceutica . Clinical Pharmacology and Biopharmaceutics Reivew(s) (Application Number 205123Orig1s000), Center for Drug Evaluation and Research. [Google Scholar]
- Snoeys J.; Beumont M.; Monshouwer M.; Ouwerkerk-Mahadevan S. Mechanistic Understanding of the Nonlinear Pharmacokinetics and Intersubject Variability of Simeprevir: A PBPK-Guided Drug Development Approach. Clin. Pharmacol. Ther. 2016, 99 (2), 224–234. 10.1002/cpt.206. [DOI] [PubMed] [Google Scholar]
- Shen L.; Peterson S.; Sedaghat A. R.; McMahon M. A.; Callender M.; Zhang H.; Zhou Y.; Pitt E.; Anderson K. S.; Acosta E. P.; et al. Dose-Response Curve Slope Sets Class-Specific Limits on Inhibitory Potential of Anti-HIV Drugs. Nat. Med. 2008, 14 (7), 762–766. 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L.; Rabi S. A.; Sedaghat A. R.; Shan L.; Lai J.; Xing S.; Siliciano R. F. A Critical Subset Model Provides a Conceptual Basis for the High Antiviral Activity of Major HIV Drugs. Sci. Transl. Med. 2011, 3 (91), 91ra63. 10.1126/scitranslmed.3002304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Alfajaro M. M.; DeWeirdt P. C.; Hanna R. E.; Lu-Culligan W. J.; Cai W. L.; Strine M. S.; Zhang S.-M.; Graziano V. R.; Schmitz C. O.; et al. Genome-Wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184 (1), 76–91. 10.1016/j.cell.2020.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcos-Villar L.; Díaz-Colunga J.; Sandoval J.; Zamarreño N.; Landeras-Bueno S.; Esteller M.; Falcón A.; Nieto A. Epigenetic Control of Influenza Virus: Role of H3K79 Methylation in Interferon-Induced Antiviral Response. Sci. Rep. 2018, 8 (1), 1230. 10.1038/s41598-018-19370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menachery V. D.; Schäfer A.; Burnum-Johnson K. E.; Mitchell H. D.; Eisfeld A. J.; Walters K. B.; Nicora C. D.; Purvine S. O.; Casey C. P.; Monroe M. E.; et al. MERS-CoV and H5N1 Influenza Virus Antagonize Antigen Presentation by Altering the Epigenetic Landscape. Proc. Natl. Acad. Sci. U. S. A. 2018, 115 (5), E1012–E1021. 10.1073/pnas.1706928115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui K. P. Y.; Cheung M.-C.; Perera R. A. P. M.; Ng K.-C.; Bui C. H. T.; Ho J. C. W.; Ng M. M. T.; Kuok D. I. T.; Shih K. C.; Tsao S.-W.; et al. Tropism, Replication Competence, and Innate Immune Responses of the Coronavirus SARS-CoV-2 in Human Respiratory Tract and Conjunctiva: An Analysis in Ex-Vivo and in-Vitro Cultures. Lancet Respir. Med. 2020, 8 (7), 687–695. 10.1016/S2213-2600(20)30193-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T.; Irwin J. J. ZINC 15--Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55 (11), 2324–2337. 10.1021/acs.jcim.5b00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trott O.; Olson A. J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31 (2), 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuck C.-P.; Chen C.; Ke Z.; Wan D. C.-C.; Chow H.-F.; Wong K.-B. Design, Synthesis and Crystallographic Analysis of Nitrile-Based Broad-Spectrum Peptidomimetic Inhibitors for Coronavirus 3C-like Proteases. Eur. J. Med. Chem. 2013, 59, 1–6. 10.1016/j.ejmech.2012.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shannon A.; Selisko B.; Le N.-T.-T.; Huchting J.; Touret F.; Piorkowski G.; Fattorini V.; Ferron F.; Decroly E.; Meier C.; et al. Rapid Incorporation of Favipiravir by the Fast and Permissive Viral RNA Polymerase Complex Results in SARS-CoV-2 Lethal Mutagenesis. Nat. Commun. 2020, 11 (1), 4682. 10.1038/s41467-020-18463-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W.; Wu K.-P.; Sartori M. A.; Kamadurai H. B.; Ordureau A.; Jiang C.; Mercredi P. Y.; Murchie R.; Hu J.; Persaud A.; et al. System-Wide Modulation of HECT E3 Ligases with Selective Ubiquitin Variant Probes. Mol. Cell 2016, 62 (1), 121–136. 10.1016/j.molcel.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A.; Tamayo P.; Mootha V. K.; Mukherjee S.; Ebert B. L.; Gillette M. A.; Paulovich A.; Pomeroy S. L.; Golub T. R.; Lander E. S.; et al. Gene Set Enrichment Analysis: A Knowledge-Based Approach for Interpreting Genome-Wide Expression Profiles. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (43), 15545–15550. 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A.; Davis C. A.; Schlesinger F.; Drenkow J.; Zaleski C.; Jha S.; Batut P.; Chaisson M.; Gingeras T. R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y.; Smyth G. K.; Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 2014, 30, 923. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Love M. I; Huber W.; Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durinck S.; Moreau Y.; Kasprzyk A.; Davis S.; De Moor B.; Brazma A.; Huber W. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439. 10.1093/bioinformatics/bti525. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.