

Abstract
COVID-19 has affected millions of people. Although many drugs are in use to combat disease, there is not any sufficient treatment yet. Having critical role in propagation of the novel coronavirus (SARS-CoV-2) works Main Protease up into a significant drug target. We have performed a molecular docking study to define possible inhibitor candidates against SARS-CoV-2 Main Protease enzyme. Besides docking Remdesivir, Ribavirin, Chloroquine and 28 other antiviral inhibitors (totally 31 inhibitors) to Main Protease enzyme, we have also performed a molecular docking study of 2177 ligands, which are used against Main Protease for the first time by using molecular docking program Autodock4. All ligands were successfully docked into Main Protease enzyme binding site. Among all ligands, EY16 coded ligand which previously used as EBNA1-DNA binding blocker candidate showed the best score for Main Protease with a binding free energy of −10.83 kcal/mol which was also lower than re-docking score of N3 ligand (−10.72 kcal/mol) contained in crystal structure of Main Protease. After analyzing the docking modes and docking scores we have found that our ligands have better binding free energy values than the inhibitors in use of treatment. We believe that further studies such as molecular dynamics or Molecular Mechanic Poisson Boltzmann Surface Area studies can make contribution that is more exhaustive to the docking results.
Keywords: COVID-19, Sars-CoV-2, Main protease, Molecular docking, Autodock4
Graphical abstract

1. Introduction
Arising COVID-19 outbreak in Hubei province, P.R. China was a milestone for humanity when it started to affect the entire world in a short span of time like a tornado. It has changed our lifestyles, education methods and whatever we got accustomed. Affecting all the world, COVID 19, enabled scientists, companies, and countries to act together on a topic for the first time. While World Health Organization (WHO) declared the COVID-19 as pandemic on March 11, 2020, vaccine and appropriate drugs for treatment of disease remain aliment yet. According to the current data of the WHO [1], there are a total of 120 383 919 infected people and 2 664 386 deaths in the world by the March 18th, 2021.
Being a member of Nidovirales, Coronaviridae includes Coronaviruses which can be investigated in four genera; alphacoronavirus, betacoronavirus, gammacoronavirus, and deltacoronavirus. From these species, alpha- and betacoronaviruses known to infect mammalian including human. Until 2019, it was reported that six types of coronavirus (HCoV-NL63, HCoV-229E, HCoV-HKU1, HCoV-OC43, severe acute respiratory syndrome (SARS-CoV), and Middle East respiratory syndrome coronavirus (MERS-CoV)) infected humans. Among these viruses, SARS-CoV killed 916 people in 37 countries between 2002 and 2003, MERS which was firstly reported in the Middle East killed 1791 people in 27 countries [2,3]. The severe acute respiratory syndrome (SARS-CoV) outbreak was started in 2003 in China followed by the second outbreak of the Middle East respiratory syndrome (MERS-CoV) in 2012 in Saudi Arabia. Again, the SARS-CoV-2 infection was started in later part of the year 2019 and was named as a novel coronavirus (COVID-19) on January 12, 2020, which attacks the immunity and the immune system, breaks down in the worst condition [[4], [5], [6]]. Among all other six coronaviruses, which are known to infect humans, SARS-CoV-2 seems to be the most pathogenic kind of these viruses. The SARS-CoV-2 is a new strain that has emerged due to which they are not familiar to our immune system causing infection in the respiratory tract. WHO has declared that fever, dry cough, and tiredness are the most common symptoms of COVID-19. Other symptoms are nasal congestion, diarrhea, sore throat, runny nose, and pains which are mainly mild and arise gradually. The less common symptoms are aches and pains, sore throat, conjunctivitis, headache, loss of taste or smell, a rash on skin, or discoloration of fingers or toes, as well as the serious symptoms: viz difficulty breathing or shortness of breath chest pain or pressure and loss of speech or movement. It should also be noted that not all infected people show these indications and thus most people recuperate without any special treatment. About 17% of infected patients have severe illness and breathing difficulties. Disease affect older people and people who have chronic diseases such as diabetes, heart problems, and high blood pressure more seriously [7].
Virus genome encodes two overlapping polyproteins, pp1a and pp1ab. These polyproteins are later subjected to proteolytic processing by viral proteases, main protease (Mpro; 3C-like protease) and accessory proteases (papain-like cysteine proteases) to produce functional polypeptides. Mpro is a 33.8 kDa enzyme, which also processes itself from polyproteins besides processing all other subunits of virus [8,9]. While Mpro cleaves subunits of virus at no less than 11 cleavage sites on 1 ab, this specific cutting makes Mpro an attractive drug target since no human proteases have a similar cleavage property. The main title is attributed to enzyme due to its dominant role in the processing replicase polyproteins and gene expression while 3C-like protease referred to enzyme due to similarity of the enzyme with picornavirus 3C proteases [8,10]. SARS-COV-2 main protease (Mpro) is one of the promising drug targets in the many possible drug targets of SARS-COV2 such as spike proteins, non-structural proteins (NSPs) and human receptor angiotensin-converting enzyme (ACE2). Mpro feature in mediating viral replication and transcription.
Recently, several Mpro crystal structures [9,[11], [12], [13]] were published on Protein Data Bank (www.rcsb.org). Jin et al. [9], designed a combination of high-throughput screening and structure-based virtual study over 10 000 compounds including drug candidates in clinical trials and approved drugs against Mpro enzyme. They have reported that six of these ligands have IC50 values ranging from 0.67 to 21.4 μM inhibition activity. In another study, Chandel et al. [14], used FDA approved antiviral compounds and a library of active phytochemicals through molecular docking. Durdagi et al. [15], performed a molecular docking study for 7922 compounds from NIH Chemical Genomics Center (NCGC) Pharmaceutical Collection (NPC) database (https://tripod.nih.gov/npc/) which then followed by molecular dynamics and MM/GBSA studies for best scored ligands. There are also many antivirals, antimalarials, anti-parasitic, and antibacterial are in clinical investigations for the treatment of COVID-19[16]. An in vitro study of such drugs (ribavirin, penciclovir, nitazoxanide, nafamostat, chloroquine, favipiravir, and Remdesivir) was performed by Wang et al. [17], and they have reported that Remdesivir and Chloroquine can inhibit 2019-nCov effectively.
Due to rapid transmission of the virus and urgency in discovering effective drugs for treatment of COVID-19, we have performed an in-silico study by using 2208 compounds (31 ligands approved or in clinical trial and 2177 used first time for SARS-CoV-2. The interactions of best-scored ligands were investigated deeply in this study.
2. Materials and methods
2.1. Receptor structure preparation
Recently reported (www.rcsb.org) [18] crystal structure of SARS-CoV-2 Mpro, 6lu7. pdb [9], was selected for molecular docking. Crystal structure of 6lu7 also contains N3 ligand, which was used for dock validation. The receptor and N3 ligand files were saved separately after removing water molecules from crystal structure.
2.2. Ligand library preparation
Ligand library was generated as follows; previously used for both HIV-1 IN inhibition and 3-Hydroxy-3-methyl-glutaryl-CoA Reductase Enzyme (HMGR) (10 ligands) [[19], [20], [21]] inhibition, 104 ligands used for HMGR (ligands coded as HMXXX) [21] enzyme inhibition, 1637 ligands used for EBV EBNA1-DNA binding blockers (ligands coded as EBXXX, EYXX, and EZXXXX) [22], and 426 ligands used as HIV-1 RT-IN dual inhibitors (ligands coded as HBXX) [23]. Designing procedure of these ligands have been described in the related publications. Since these ligands were optimized formerly, they need not to be optimized any more. Beside these ligands, we have also studied 31 ligands to compare docking scores of ligands which some of them are approved inhibitors and some of them are in clinical trials for treatment of COVID-19[24]. The 3D structures of inhibitors in use of treatment for COVID-19 and which are in clinical trials were downloaded from PubChem [25] and Drugbank [26].
2.3. Molecular docking
While Autodock Vina, Autodock4 with Lamarckian Genetic algorithm (LGA) and Local Search (LS) algorithms were used for validation studies, it has seen that LS algorithm of Autodock4 showed best docking pose for re-docking of N3 ligand to Mpro enzyme structure (Fig. 1 .) Docking site and box dimensions were selected by centering grid box on the N3 ligand, included in crystal structure. Docking score of N3 was −10.72 kcal/mol with a RMSD value of 0.81. Box was placed to −9.73 Å, 11.52 Å, 68.49 through x, y, and z coordinates, respectively, with the dimensions of 36 × 60 x 40 Å. Docking studies were performed using LS algorithm with a number of 300 individuals to result in 250 docking poses for each ligand.
Fig. 1.

Binding modes of crystal structure and re-docked structure of N3 ligand in the binding site of Mpro enzyme (re-docked structure: green, crystal structure: blue). (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
3. Result and discussion
Molecular docking is a key step in structure-based drug design. Allowing flexible ligand docking is also advantage of the method. In this study, we used Autodock4 docking program to investigate binding modes of a total 2177 ligands in binding site of Mpro enzyme of SARS-CoV-2. Researchers have been making many efforts to find or develop appropriate drug candidates against possible drug targets of SARS-CoV-2 since arising COVID-19 outbreak by using repurposing drugs or using different ligand sources [[27], [28], [29], [30], [31], [32], [33]]. For same goal, we have used different set of ligands in the current study.
Docking studies of 31 inhibitors, which are in usage of COVID-19 treatment or in clinical trials and 2177 ligands, which are subjected to SARS-CoV-2 Mpro enzyme structure for the first time were performed successfully. To validate the accuracy of docking procedure we have performed a re-docking study. According to docking scores, EY16 coded ligand (Fig. 2 .), which is previously used as EBNA1 inhibitor in vitro [34] and in silico [22,34], was the best-scored ligand with a score of −10.83 kcal/mol while re-docking of N3 ligand has a score of −10.72 kcal/mol. Other great white hope ligands are ligands coded as EZ055, EZ0239, HB393, HB099, HB110 and HMZ24 (Fig. 2.). We also want to report that newly studied ligands have better scores than approved inhibitors or inhibitors in clinical trials where their docking scores are in the range of (−3.74)-(-8.92) kcal/mol. While Danoprevir (Fig. 2.) has the best docking score (−8.93 kcal/mol), Acetylcysteine has the lowest docking score (−3.74 kcal/mol) among this set of ligands.
Fig. 2.
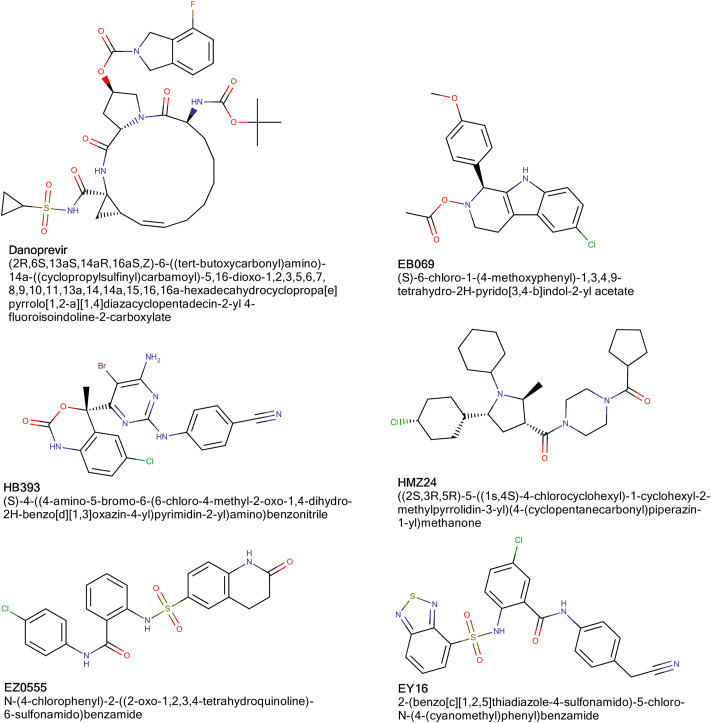
2D representations of best-scored ligands in their ligand sets.
Docking scores of 20 ligands in each set and docking scores of inhibitors in clinical trials or in use are shown in Table 1 and Table 2 , respectively. Docking scores of all ligands were added as supplementary material (S.M. Table 1).
Table 1.
Docking scores of 20 best ligands of each sets of ligands.
| EBNA1 ligand libraries |
HIV1-RT, IN ligand library |
HMGR ligand library |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Mol.a | Score | Mol. | Score | Mol. | Score | Mol. | Score | Mol. | Score |
| EB069 | −9,84 | EY16 | −10,83 | EZ0555 | −10,46 | HB393 | −10,51 | HMZ24 | −10,51 |
| EB071 | −9,55 | EY06 | −9,67 | EZ0239 | −10,25 | HB099 | −10,01 | HMZ29 | −9,89 |
| EB143 | −9,48 | EY03 | −9,66 | EZ0172 | −9,92 | HB110 | −10.00 | HMZ12 | −9,74 |
| EB045 | −9,40 | EY07 | −9,63 | EZ0271 | −9,81 | HB070 | −9,82 | HMZ34 | −9,73 |
| EB047 | −9,38 | EY04 | −9,51 | EZ0464 | −9,74 | HB051 | −9,33 | HMZ19 | −9,72 |
| EB070 | −9,27 | EY32 | −9,30 | EZ0593 | −9,72 | HB104 | −9,32 | HMZ16 | −9,58 |
| EB367 | −9,24 | EY28 | −9,27 | EZ0718 | −9,68 | HB095 | −9,31 | HMZ01 | −9,54 |
| EB183 | −9,20 | EY17 | −9,22 | EZ0171 | −9,64 | HB101 | −9,20 | HMZ26 | −9,27 |
| EB043 | −9,17 | EY01 | −9,18 | EZ0401 | −9,64 | HB061 | −9,13 | HMZ02 | −9,23 |
| EB196 | −9,13 | EY46 | −9,11 | EZ0542 | −9,60 | HB220 | −9,05 | HMZ09 | −9,05 |
| EB054 | −9,12 | EY12 | −9,10 | EZ0915 | −9,57 | HB062 | −9,01 | HMZ37 | −9,02 |
| EB190 | −9,12 | EY33 | −9,08 | EZ0763 | −9,52 | HB102 | −8,98 | HMZ10 | −8,95 |
| EB189 | −9,11 | EY45 | −8,99 | EZ0929 | −9,51 | HB105 | −8,96 | HMZ18 | −8,93 |
| EB354 | −9,08 | EY11 | −8,92 | EZ0248 | −9,49 | HB228 | −8,96 | HMM11 | −8,91 |
| EB004 | −9,03 | EY47 | −8,81 | EZ0097 | −9,48 | HB395 | −8,91 | HMZ17 | −8,85 |
| EB145 | −9,03 | EY20 | −8,73 | EZ0314 | −9,48 | HB219 | −8,84 | HMM32 | −8,84 |
| EB359 | −9,02 | EY19 | −8,59 | EZ0363 | −9,46 | HB229 | −8,83 | HMZ21 | −8,84 |
| EB122 | −8,98 | EY30 | −8,59 | EZ0296 | −9,43 | HB187 | −8,80 | HMZ25 | −8,77 |
| EB032 | −8,96 | EY29 | −8,56 | EZ0851 | −9,42 | HB071 | −8,79 | HMD04 | −8,67 |
| EB116 | −8,95 | EY14 | −8,55 | EZ0198 | −9,40 | HB107 | −8,79 | HMB04 | −8,66 |
Mol. = Molecule.
Table 2.
Docking scores of inhibitors in clinical trials or in use of COVID-19 treatment.
| Molecule | Score | Molecule | Score | Molecule | Score |
|---|---|---|---|---|---|
| Danoprevir | −8.93 | Ritonavir | −7,20 | Pirfenidone | −5,86 |
| Nelfinavir | −8,92 | Camostat | −6,91 | Triazavirin | −5,52 |
| Ebastine | −8,47 | Chloroquine | −6,89 | Azvudine | −5,38 |
| Baloxavir_marboxil | −8,23 | Cobicistat | −6,80 | Fingolimod | −5,10 |
| Losartan | −8,14 | Darunavir | −6,73 | Emtricitabine | −4,94 |
| Methylprednisolone | −8,08 | Remdesivir | −6,68 | Ribavirin | −4,48 |
| Ivermectin | −8,07 | Oseltamivir | −6,41 | Tenofovir | −4,35 |
| Ruxolitinib | −8,04 | Hydroxychloroquine | −6,37 | Favipiravir | −4,21 |
| Lopinavir | −7,90 | Dipyridamole | −6,22 | Acetylcysteine | −3,74 |
| Umifenovir | −7,65 | Tranilast | −6,17 | ||
| Thalidomide | −7,26 | Azithromycin | −6,15 |
As mentioned before, we used different sets of ligands in the study. From the three different sets of ligands formerly used for blocking DNA binding to the EBNA1, EB069, EY16, and EZ0555 were the best binding ligands with the scores of −9.84, −10.83, and −10.46 kcal/mol, respectively. HB393 coded ligand was the best-scored ligand from the set of ligands designed for dual inhibition of HIV-1 RT and IN enzymes with a binding free energy of −10.51 kcal/mol. While HMZ24, a ligand designed for inhibition of HMGR, scored −10.51 kcal/mol, Danoprevir was the best scored ligand through inhibitors in use or in clinical trials for treatment of COVID-19 with a binding free energy of −8.93 kcal/mol.
We have analyzed the interactions of receptor residues with five best-scored ligands of each sets of ligands and two best scored inhibitors, which are in use or in clinical trials for treatment of COVID-19.
We have analyzed average scores of used all ligands to define the best ligand library, which will also contribute to understanding of more appropriate structural properties of ligands. As seen from Table 3 , ligands used for EBNA1 those selected from literature [[34], [35], [36], [37], [38], [39], [40]] showed best average docking scores through all ligands with an average score of −8.05 kcal/mol.
Table 3.
Average scores of ligands (kcal/mol) according to ligand sets.
| Ligand Type | Numbers of Ligands | Average Docking Score |
|---|---|---|
| EBNA1 Designed ligand library | 409 | −7.40 |
| EBNA1 Literature ligand library | 58 | −8.05 |
| EBNA1 ZINC15 ligand library | 1170 | −7.77 |
| HIV-1 RT, IN ligand library | 426 | −6.86 |
| HMGR ligand library | 114 | −7.88 |
| COVID-19 Inhibitors | 31 | −6.56 |
| Total | 2208 | −7,42 |
Before analyzing ligand-receptor interactions, we have defined binding site residues of Mpro enzyme. Binding site of Mpro enzyme contains Thr24, Thr25, Thr26, Leu27, His41, Met49, Tyr54, Phe140, Leu141, Asn142, Gly143, Ser144, Cys145, His163, His164, Met165, Glu166, Leu167, Pro168, His172, Asp187, Arg188, Gln189, Thr190, Ala191, and Gln192 residues. The interactions of these residues with ligands’ atoms are valuable and are investigated from docking modes of ligands. Ligand N3 interacts with Mpro enzyme residues Phe140, Gly143, His163, His164, Glu166, Gln189, and Thr190 by hydrogen bonds. Other interactions are amide-π stacked interaction with Leu141 and Asn142 residues, alkyl-alkyl interactions with Met49, Met165, and Leu167 residues, π-alkyl interactions with His41, Pro168 and Ala191 residues in crystal structure 6lu7. pdb. The enzyme residues with 20 best-scored ligands and 31 drugs or inhibitors in clinical trial interacted with are summarized in Table 4 and Table 5 , respectively.
Table 4.
Mpro residues in interaction with best scored 20 ligands.
| Molecule | Score | Interacted Mpro enzyme residues |
|---|---|---|
| EY16 | −10.83 | Thr26, His41, Met49, Leu141, Ser144, Cys145, His163, His164, Met165, Glu166 |
| HB393 | −10.51 | His41, Met49, Leu141, Cys145, His164, Met165, Glu166 |
| HMZ24 | −10.51 | His41, Met49, Ser144, Cys145, Met165, Gln189, |
| EZ0555 | −10.46 | Thr24, His41, Met49, Pro52, Cys145, His164 |
| EZ0239 | −10.25 | Thr26, Leu27, His41, Met49, Gly143, Cys145, His163, Met165, His172 |
| HB099 | −10.01 | His41, Met49, Cys145, His163, His164, Glu166, Pro168, His172, Arg188, Gln189 |
| HB110 | −10.00 | His41, Cys44, Met49, Tyr54, Cys145, His163, Met165, Glu166, Pro168, Gln192 |
| EZ0172 | −9.92 | Cys145, His163, Met165, Arg188, Thr190 |
| HMZ29 | −9.89 | Leu27, Thr26, His41, Phe140, Gly143, Cys145, Met165 |
| EB069 | −9.84 | His41, Met49, Gly143, Cys145, His164, Met165, His172 |
| HB070 | −9.82 | Leu27, His41, Gly143, Cys145, Met165, Glu166, Leu167 |
| EZ0271 | −9.81 | Met49, Ser144, Cys145, His163, Met165, Glu166 |
| EZ0464 | −9.74 | His41, Met49, Cys145, His164, Met165, Arg188, Thr190, Gln192 |
| HMZ12 | −9.74 | His41, Met49, Cys145, Met165, Gln189 |
| HMZ34 | −9.73 | Glu166, Gly143, Ser144, Cys145, Met165, Pro168 |
| EZ0593 | −9.72 | His41, His163, Met165, Pro168, His172, Arg188, Gln189, Thr190, Gln192 |
| HMZ19 | −9.72 | Gly143, Ser144, Cys145, Met165, Glu166, Leu167 |
| EZ0718 | −9.68 | His41, Met49, Cys145, His164, Met165, Glu166 |
| EY06 | −9.67 | His41, Asn142, Gly143, Met49, His163, His164, Cys145, His172, Arg188 |
| EY03 | −9.66 | Leu27, His41, Cys44, Met49, Pro52, Tyr54, Asn142, Ser144, Cys145, Met165, Pro168, Arg188, Gln189 |
Table 5.
Mpro residues in interaction with drugs or inhibitors in clinical trials.
| Molecule | Score | Interactions |
|---|---|---|
| Danoprevir | −8.93 | His41, Asn142, Met165, Glu166, Thr190 |
| Nelfinavir | −8.92 | Met49, Cys145, His163, Met165, Glu166, Leu167, Pro168, Thr190, Gln192 |
| Ebastine | −8.47 | Leu27, Met49, His41, Cys145, Glu166 |
| Baloxavir_marboxil | −8.23 | Thr26, Phe140, Leu141, Gly143, Ser144, Cys145, His163, Met165, Glu166 |
| Losartan | −8.14 | Met49, Leu141, Ser144, Cys145, Glu166, Met165, Gln189 |
| Methylprednisolone | −8.08 | Cys145, Met165, Glu166, Thr190, Gln192 |
| Ivermectin | −8.07 | Thr26, His41, Met49, Gly143, Met165, Thr190, Ala191 |
| Ruxolitinib | −8.04 | His41, Met49, Cys145, Met165, Leu167, Pro168, Gln189, Thr190, Gln192 |
| Lopinavir | −7.90 | Met49, Gly143, Cys145, His164, Met165, Leu167, Pro168 |
| Umifenovir | −7.65 | His41, Met49, His164, Met165, Glu166 |
| Thalidomide |
−7.26 |
His41, Met165, Glu166, Thr190, Gln192 |
| Ritonavir | −7.20 | His41, Met49, Gly143, Cys145, Met165, Pro168 |
| Camostat | −6.91 | Phe140, Asn142, Gly143, Cys145, His163, Glu166 |
| Chloroquine | −6.89 | His41, Met49, Phe140, Cys145, His163, His164, Met165, Glu166, His172 |
| Cobicistat | −6.80 | Thr26, Ser144, Cys145, Met165, Glu166, Gln189 |
| Darunavir | −6.73 | His41, Met49, Cys145, His163, Met165, Glu166, Pro168, Gln192 |
| Remdesivir | −6.68 | Leu27, His41, Met49, Leu141, Ser144, Cys145, His164, Met165, Glu166, Thr190 |
| Oseltamivir | −6.41 | His41, Met165, Pro168, Arg188, Thr190, Gln192 |
| Hydroxychloroquine | −6.37 | Gly143, Met165, His163, Pro168, Gln189 |
| Dipyridamole | −6.22 | Met49, Cys145, Met165, Glu166 |
| Tranilast | −6.17 | Leu27, His41, Ser144, Cys145, Met165 |
| Azithromycin | −6.15 | Met49, Asn142, Cys145, His164, Met165, Glu166, Pro168, Gln189, Ala191 |
| Pirfenidone | −5.86 | His41, Met49, Met165, Gln189 |
| Triazavirin | −5.52 | Leu141, Asn142, Gly143, Cys145, Ser144 |
| Azvudine | −5.38 | Gly143, Ser144, His163, Glu166, Gln189 |
| Fingolimod | −5.10 | His41, Asn142, Cys145, Glu166 |
| Emtricitabine | −4.94 | Phe140, Asn142, Gly143, Cys145, His163, Glu166 |
| Ribavirin | −4.48 | Leu141, Asn142, Ser144, His163, Glu166 |
| Tenofovir | −4.35 | Gly143, Cys145, Glu166 |
| Favipiravir | −4.21 | Phe140, Leu141, Asn142, Ser144, Glu166 |
| Acetylcysteine | −3.74 | Leu141, Gly143, Ser144, Cys145, His164 |
Ligands coded as EB069, EY16, and EZ0555 were previously used for preventing DNA binding to EBV EBNA1 protein [22]. EY16 gave the best docking score through all ligands used in this study. As mentioned before, this ligand was studied formerly as in vitro and in silico for binding EBNA1 antigen. EB069 was designed by using some ligands as template which explained in one of our study [22] while EZ0555 (ZINC12583256) downloaded from ZINC14 database [41]. EB069 forms three hydrogen bonds with Gly143, Cys145, and His164 residues of receptor (Fig. 3 .). Other interactions seen between EB069 and receptor residues are π-sulfur interaction with the residue Met49, alkyl-alkyl interactions with residues Cys145 and Met49, π-alkyl interactions with His41, Cys145, and His172, and a halogen interaction of ligands Cl atom with Met49 as given in Fig. 3. Ligand EY16 forms four hydrogen bonds with residues Thr26, Leu141, His164, and Glu166. π-sulfur interaction with Met165, π-π T shaped interactions with His163, π-π stacked interaction with His41, alkyl-alkyl interaction with Met49 and π-alkyl interaction with Met49 and Cys145 residues are other interactions seen between EY16 and receptor residues. There is also an unfavorable H–H interaction between Ser144:HN and Y16:H15 atoms (Fig. 4 .). Ligand EZ0555 forms hydrogen bonds with Thr24, Cys145 and His164 residues, π-sulfur interaction with His41 and Cys145 residues, alkyl-alkyl interactions with Met49 and Pro52 residues and π-alkyl interaction with Met49 residue (Fig. 5 .).
Fig. 3.
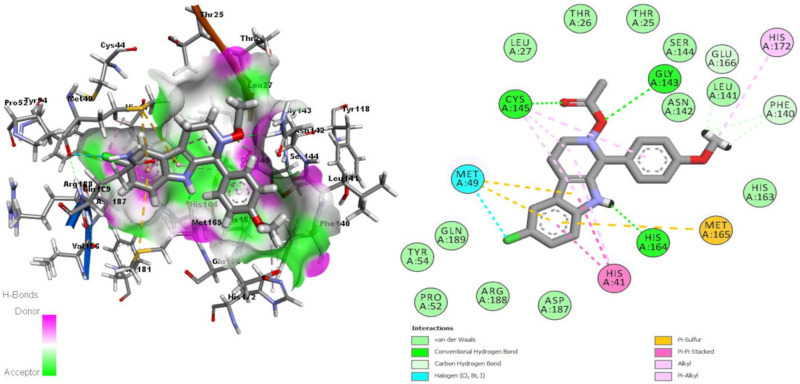
Interactions of Mpro binding site residues with EB069 (3D (right) and 2D (left)).
Fig. 4.

Interactions of Mpro binding site residues with EY16 (3D (right) and 2D (left)).
Fig. 5.
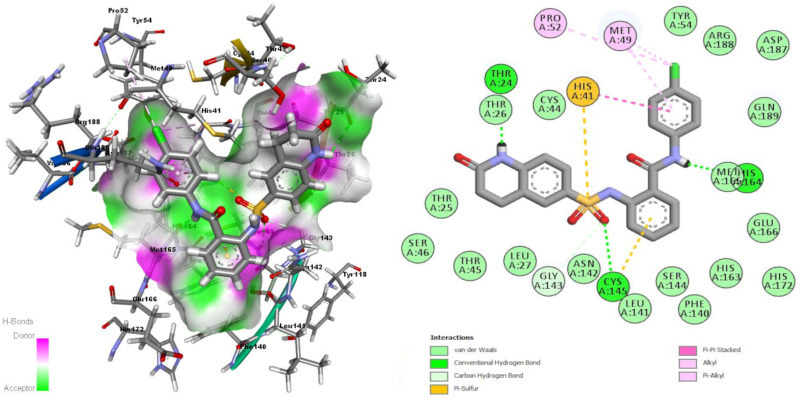
Interactions of Mpro binding site residues with EZ0555 (3D (right) and 2D (left)).
HB393 is a ligand designed for dual inhibition of HIV-1 RT and IN enzymes. Ligand showed a binding free energy of −10.51 kcal/mol for inhibition of Mpro enzyme. Ligand HB393 creates three hydrogen bonds with Leu141, His164, and Glu166 residues. HB393 also in interaction with Met49, Cys145 by forming π-sulfur interactions, with His41 by forming π-π stacked interaction and with His41 and Met165 residues by forming π-alkyl interactions (Fig. 6 .).
Fig. 6.

Interactions of Mpro binding site residues with HB393 (3D (right) and 2D (left)).
HMZ24 is the best scored ligand in the HMGR ligand set with a binding free energy of −10.51 kcal/mol. Interactions those ligand forms are; hydrogen bonds with Ser144 and Cys145 residues, alkyl-alkyl interactions with Met49, Cys145, and Met165, π-σ interaction with His41 and π-alkyl interaction with His41 residue (Fig. 7 .).
Fig. 7.

Interactions of Mpro binding site residues with HMZ24 (3D (right) and 2D (left)).
Danoprevir has the lowest binding free energy through inhibitors, which are in use for treatment of COVID-19 or in clinical trials with a binding score of −8.93 kcal/mol. Danoprevir forms two hydrogen bonds with Asn142 and Glu166 residues of enzyme. Ligand also construct π-sulfur, alkyl-alkyl and a halogen interaction with His41, Met165, and Thr190 residues, respectively (Fig. 8 .).
Fig. 8.

Interactions of Mpro binding site residues with Danoprevir (3D (right) and 2D (left)).
Nelfinavir was the second-best scored ligand through COVID-19 inhibitors with a binding free energy of −8.92 kcal/mol. Nelfinavir forms three hydrogen bonds with Gln192, Glu166, and Thr190 residues of Mpro. Alkyl-alkyl interactions with Met49, Pro168, π-sulfur interaction with Met165, and π-alkyl interactions with His41, Met49, and Pro168 are the other interactions observed between Nelfinavir and enzyme residues (Fig. 9 .).
Fig. 9.

Interactions of Mpro binding site residues with Nelfinavir (3D (right) and 2D (left)).
Among the binding site residues, His41, Met49, Gly143, Cys145, and Met165 are the most common residues, which interact with ligands’ atoms. While Met49 interacts with ligands’ atoms only by hydrophobic interactions other residues are also seen in hydrogen bond interactions with ligands’ atoms. The importance of these residues’ interactions was also discussed by Yoshino and coworkers [42]. According to binding free energy scores, we can state that the study bids fair different structural inhibitor samples, which need to be forwarded to in vitro studies.
We should mention that drugs such as Hydroxychloroquine, Remdesivir, Favipiravir and Ribavirin are currently in use of COVID 19 treatment and show high activity against SARS-Cov-2. Nguyen and coworkers [43] have performed a computational study on binding modes of Remdesivir to the RNA-Dependent RNA Polymerase and Main Protease. They have used Autodock Vina for docking studies where Remdesivir gave the binding energy of −7.9 kcal/mol for Mpro. …. .
In another study, Naik et al. [44], used LGA algorithm of Autodock4 and they have obtained the docking binding energies of −8.2 kcal/mol, −6.2 kcal/mol, −6.1 kcal/mol, −5.7 kcal/mol, −5.0 kcal/mol for Remdesivir, Ribavirin, Oseltamivir, Chloroquine, and Favipiravir, respectively. Obtaining similar results with study of Naik et al. also confirm our results since we experienced from docking tries that LGA algorithm of Autodock4 gives 2–3 kcal/mol lower scores than LS algorithm of Autodock4.
In a study performed by Chakraborti coworkers, some drugs were repurposed against main protease of SARS-CoV-2. They used Autodock Vina for docking studies of drugs and they have acquired the docking binding free energies of −8.8 kcal/mol and −8.0 kcal/mol for Danoprevir and Remdesivir, respectively which also support the better binding result of Danoprevir than Remdesivir of our study. In another docking study [45], Chloroquine and Hydroxychloroquine have been placed to different Mpro crystal structures with Autodock4 and while Chloroquine had a binding score of −4.3 kcal/mol and Hydroxychloroquine had a binding score of −4.8 kcal/mol for 6LU7 crystal Mpro structure which also have been used in our study.
Analyzing docking scores showed that newly studied two ligands have same docking score as Danoprevir, the best scored ligands through drugs in usage, while 157 ligands have a lower score than Danoprevir. This is also an indication of the studied ligands could have better inhibition effect on Mpro enzyme than drugs in usage of COVID-19 treatment. According to binding free energy scores, we can state that the study promise hope about introducing different structural inhibitor samples as inhibitor candidates for inhibition of SARS-CoV-2 Mpro enzyme.
4. Conclusion
To bring forth new type of ligands to the treatment efforts of COVID-19, we have performed a molecular docking study by using totally 2208 ligands. Our ligand library consists of ligands which are previously used for different targets as explained in ligand library preparation section.
Ligand coded EY16 was the best scored ligand through all ligands with a binding free energy of −10.83 kcal/mol. Other promising ligands are EZ055, EZ0239, HB393, HB099, HB110 and HMZ24. Although we have observed valuable results, we know that the study should be forward to molecular dynamics and MM/PB(GB)SA studies which will detail the accuracy of the molecular docking results.
Funding
There was no external support for this work.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgement
There are no acknowledgements.
Footnotes
This work was partially presented as an oral presentation at 5th International Applied Science Congress, Rize, Turkey, February 19–21,2021.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jics.2021.100041.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Who WHO coronavirus disease (COVID-19) dashboard. WHO. 2021. https://covid19.who.int/table Accessed 03.18.2021.
- 2.Fung T.S., Liu D.X. Human coronavirus: host-pathogen interaction. Annu. Rev. Microbiol. Sep 8 2019;73:529–557. doi: 10.1146/annurev-micro-020518-115759. [DOI] [PubMed] [Google Scholar]
- 3.Liu Z., Xiao X., Wei X. Composition and divergence of coronavirus spike proteins and host ACE2 receptors predict potential intermediate hosts of SARS-CoV-2. J. Med. Virol. Feb 26 2020 doi: 10.1002/jmv.25726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu H., Stratton C.W., Tang Y.W. Outbreak of pneumonia of unknown etiology in Wuhan, China: the mystery and the miracle. J Med Virol. Apr. 2020;92(4):401–402. doi: 10.1002/jmv.25678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. Oct. 2020;92(10):2249. doi: 10.1002/jmv.26234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bogoch, Watts A., Thomas-Bachli A., Huber C., Kraemer M.U.G., Khan K. Pneumonia of unknown aetiology in Wuhan, China: potential for international spread via commercial air travel. J. Trav. Med. Mar 13 2020;27(2) doi: 10.1093/jtm/taaa008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Who Q&A on coronaviruses (COVID-19). WHO. 2020. https://www.who.int/news-room/q-a-detail/q-a-coronaviruses Accessed 28.09.2020.
- 8.Yang H., Bartlam M., Rao Z. Drug design targeting the main protease, the Achilles’ heel of coronaviruses. Curr. Pharmaceut. Des. 2006;12(35):4573–4590. doi: 10.2174/138161206779010369. [DOI] [PubMed] [Google Scholar]
- 9.Jin Z., Du X., Xu Y., Deng Y., Liu M., Zhao Y., Zhang B., Li X., Zhang L., Peng C., Duan Y., Yu J., Wang L., Yang K., Liu F., Jiang R., Yang X., You T., Liu X., Yang X., Bai F., Liu H., Liu X., Guddat L., Xu W., Xiao G., Qin C., Shi Z., Jiang H., Rao Z., Yang H. Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Biorxiv. 2020 doi: 10.2210/pdb6lu7/pdb. [DOI] [PubMed] [Google Scholar]
- 10.Ziebuhr J., Snijder E.J., Gorbalenya A.E. Virus-encoded proteinases and proteolytic processing in the Nidovirales. J Gen Virol. Apr. 2000;81(Pt 4):853–879. doi: 10.1099/0022-1317-81-4-853. [DOI] [PubMed] [Google Scholar]
- 11.Fearon D., Powell A.J., Douangamath A., Owen C.D., Wild C., Krojer T., Lukacik P., Strain-Damerell C.M., Walsh M.A. 2020. von Delft, . PanDDA analysis of COVID-19 main protease against the DSI-poised Fragment Library. To be published. [DOI] [Google Scholar]
- 12.Zhang L., Lin D., Sun X. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved alpha-ketoamide inhibitors. Science. Mar 20 2020 doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mesecar A.D. 2020. A Taxonomically-Driven Approach to Development of Potent, Broad-Spectrum Inhibitors of Coronavirus Main Protease Including SARS-CoV-2 (COVID-19) [DOI] [Google Scholar]
- 14.Chandel Vr S., Rathi B., Kumar D. In silico identification of potent COVID-19 main protease inhibitors from FDA approved antiviral compounds and active phytochemicals through molecular docking: a drug repurposing approach. Preprints. 2020 doi: 10.20944/preprints202003.0349.v1. [DOI] [Google Scholar]
- 15.Durdagi S., Aksoydan Busecan, Dogan Berna, Sahin Kader, Shahraki Aida. Screening of clinically approved and investigation drugs as potential inhibitors of COVID-19 main protease: a virtual drug repurposing study. ChemRxiv Preprint. 2020 doi: 10.26434/chemrxiv.12032712.v1. [DOI] [Google Scholar]
- 16.Harrison C. Coronavirus puts drug repurposing on the fast track. Nat Biotechnol. Apr. 2020;38(4):379–381. doi: 10.1038/d41587-020-00003-1. [DOI] [PubMed] [Google Scholar]
- 17.Wang M., Cao R., Zhang L. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. Mar. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Berman H.M., Westbrook J., Feng Z. The protein Data Bank. Nucleic Acids Res. Jan 1. 2000;28(1):235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ercan S., Pirinccioglu N. Computational design of a full-length model of HIV-1 integrase: modeling of new inhibitors and comparison of their calculated binding energies with those previously studied. J. Mol. Model. 2013;19(10):4349–4368. doi: 10.1007/s00894-013-1943-4. 2013/10/01. [DOI] [PubMed] [Google Scholar]
- 20.Ercan S. Docking and molecular dynamics calculations of some previously studied and newly designed ligands to catalytic core domain of HIV-1 integrase and an investigation to effects of conformational changes of protein on docking results. JOTCSA. 2017;4(1) 270-243. [Google Scholar]
- 21.Ercan S., Çınar E., Özaydın C., Efe Ertürk N., Çakmak R. Inhibitor design for 3-hydroxy-3-methyl-glutaryl-CoA reductase enzyme; molecular docking and determination of molecular and electronic properties of ligands by density functional theory method. J. Heterocycl. Chem. 2020;57(7):2875–2888. doi: 10.1002/jhet.3996. 2020/07/01. [DOI] [Google Scholar]
- 22.Ercan S., Şenses Y. Design and molecular docking studies of new inhibitor candidates for EBNA1 DNA binding site: a computational study. Mol. Simulat. 2020;46(4):332–339. doi: 10.1080/08927022.2019.1709638. 2020/03/03. [DOI] [Google Scholar]
- 23.Ercan S., Senyigit B., Senses Y. Dual inhibitor design for HIV-1 reverse transcriptase and integrase enzymes: a molecular docking study. J Biomol Struct Dyn. Feb. 2020;38(2):573–580. doi: 10.1080/07391102.2019.1700166. [DOI] [PubMed] [Google Scholar]
- 24.Ghddi . The Global Health Drug Discovery Institute; 2020. Selected Drug Information of Current Ongoing Clinical Studies on COVID-19.https://ghddi-ailab.github.io/Targeting2019-nCoV/clinical Accessed 25.04.2020. [Google Scholar]
- 25.Kim S., Chen J., Cheng T. PubChem 2019 update: improved access to chemical data. Nucleic Acids Res. Jan 8. 2019;47(D1):D1102–D1109. doi: 10.1093/nar/gky1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wishart D.S., Feunang Y.D., Guo A.C. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucleic Acids Res. Jan 4. 2018;46(D1):D1074–D1082. doi: 10.1093/nar/gkx1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marinho E.M., Batista de Andrade Neto J., Silva J. Virtual screening based on molecular docking of possible inhibitors of Covid-19 main protease. Microb. Pathog. Jun 30 2020;148:104365. doi: 10.1016/j.micpath.2020.104365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sepay N., Sekar A., Halder U.C., Alarifi A., Afzal M. Anti-COVID-19 terpenoid from marine sources: a docking, ADMET and molecular dynamics study. J Mol Struct. Oct. 2020;10:129433. doi: 10.1016/j.molstruc.2020.129433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vardhan S., Sahoo S.K. In silico ADMET and molecular docking study on searching potential inhibitors from limonoids and triterpenoids for COVID-19. Comput. Biol. Med. Sep 2020;124:103936. doi: 10.1016/j.compbiomed.2020.103936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu J.W., Wang L., Bao L.D. Exploring the active compounds of traditional Mongolian medicine in intervention of novel coronavirus (COVID-19) based on molecular docking method. J Funct Foods. Aug 2020;71:104016. doi: 10.1016/j.jff.2020.104016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mu C., Sheng Y., Wang Q., Amin A., Li X., Xie Y. Potential compound from herbal food of rhizoma polygonati for treatment of COVID-19 analyzed by network pharmacology and molecular docking technology. J Funct Foods. Aug 14 2020:104149. doi: 10.1016/j.jff.2020.104149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abo-Zeid Y., Ismail N.S.M., McLean G.R., Hamdy N.M. A molecular docking study repurposes FDA approved iron oxide nanoparticles to treat and control COVID-19 infection. Eur. J. Pharmaceut. Sci. Oct 1 2020;153:105465. doi: 10.1016/j.ejps.2020.105465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar Y., Singh H., Patel C.N. In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing. J Infect Public Heal. Sep. 2020;13(9):1210–1223. doi: 10.1016/j.jiph.2020.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li N., Thompson S., Schultz D.C. Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening. PloS One. Apr 12 2010;5(4) doi: 10.1371/journal.pone.0010126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akihisa T., Hamasaki Y., Tokuda H., Ukiya M., Kimura Y., Nishino H. Microbial transformation of isosteviol and inhibitory effects on Epstein-Barr virus activation of the transformation products. Journal of Natural Products. Mar. 2004;67(3):407–410. doi: 10.1021/np030393q. [DOI] [PubMed] [Google Scholar]
- 36.Gianti E., Messick T.E., Lieberman P.M., Zauhar R.J. Computational analysis of EBNA1 "druggability" suggests novel insights for Epstein-Barr virus inhibitor design. J Comput Aid Mol Des. Apr. 2016;30(4):285–303. doi: 10.1007/s10822-016-9899-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hu L., Zhang Y., Zhu H. Filicinic acid based meroterpenoids with anti-epstein-barr virus activities from Hypericum japonicum. Org Lett. May 6. 2016;18(9):2272–2275. doi: 10.1021/acs.orglett.6b00906. [DOI] [PubMed] [Google Scholar]
- 38.Tikhmyanova N., Schultz D.C., Lee T., Salvino J.M., Lieberman P.M. Identification of a new class of small molecules that efficiently reactivate latent Epstein-Barr Virus. ACS Chem Biol. Mar 21. 2014;9(3):785–795. doi: 10.1021/cb4006326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ukiya M., Akihisa T., Tokuda H. Sunpollenol and five other rearranged 3,4-seco-tirucallane-type triterpenoids from sunflower pollen and their inhibitory effects on Epstein-Barr virus activation. Journal of Natural Products. Nov. 2003;66(11):1476–1479. doi: 10.1021/np030276v. [DOI] [PubMed] [Google Scholar]
- 40.Wu T., Wang Q., Jiang C. Neo-clerodane diterpenoids from Scutellaria barbata with activity against Epstein-Barr virus lytic replication. J Nat Prod. Mar 27. 2015;78(3):500–509. doi: 10.1021/np500988m. [DOI] [PubMed] [Google Scholar]
- 41.Sterling T., Irwin J.J. Zinc 15 – ligand discovery for everyone. J. Chem. Inf. Model. 2015;55(11):2324–2337. doi: 10.1021/acs.jcim.5b00559. 2015/11/23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yoshino R., Yasuo N., Sekijima M. Identification of key interactions between SARS-CoV-2 main protease and inhibitor drug candidates. Sci. Rep. Jul 27 2020;10(1):12493. doi: 10.1038/s41598-020-69337-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nguyen H.L., Thai N.Q., Truong D.T., Li M.S. Remdesivir strongly binds to both RNA-dependent RNA Polymerase and main protease of SARS-CoV-2: evidence from molecular simulations. J. Phys. Chem. B. Dec 17 2020;124(50):11337–11348. doi: 10.1021/acs.jpcb.0c07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Naik V.R., Munikumar M., Ramakrishna U. Remdesivir (GS-5734) as a therapeutic option of 2019-nCOV main protease -in silicoapproach. J. Biomol. Struct. Dynam. Jun 20 2020 doi: 10.1080/07391102.2020.1781694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nimgampalle M., Devanathan V., Saxena A. Screening of Chloroquine, Hydroxychloroquine and its derivatives for their binding affinity to multiple SARS-CoV-2 protein drug targets. J. Biomol. Struct. Dynam. Jun 23 2020 doi: 10.1080/07391102.2020.1782265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.