

Abstract
The ability to predict tandem mass (MS/MS) spectra from peptide sequences can significantly enhance our understanding of the peptide fragmentation process and could improve peptide identification in proteomics. However, current approaches for predicting high-energy collisional dissociation (HCD) spectra are limited to predict the intensities of expected ion types, i.e., the a/b/c/x/y/z ions and their neutral loss derivatives (referred to as backbone ions). In practice, backbone ions only account for < 70% of total ion intensities in HCD spectra, indicating many intense ions are ignored by current predictors. In this paper, we present a deep learning approach that can predict the complete spectra (both backbone and non-backbone ions) directly from peptide sequences. We made no assumptions or expectations on which kind of ions to predict but instead predicting the intensities for all possible m/z. Training this model needs no annotations of fragment ion nor any prior knowledge of the fragmentation rules. Our analyses show that the predicted 2+ and 3+ HCD spectra are highly similar to the experimental spectra, with average full-spectrum cosine similarities of 0.820 (±0.088) and 0.786 (±0.085), respectively, very close to the similarities between the experimental replicated spectra. In contrast, the best-performed backbone only models can only achieve an average similarity below 0.75 and 0.70 for 2+ and 3+ spectra, respectively. Furthermore, we developed a multi-task learning (MTL) approach for predicting spectra of insufficient training samples, which allows our model to make accurate predictions for electron transfer dissociation (ETD) spectra and HCD spectra of less abundant charges (1+ and 4+).
Graphical Abstract
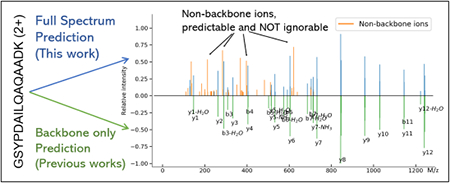
The mass spectrometry (MS) technology, in particular, the liquid chromatography coupled tandem mass spectrometry (LC-MS/MS), has evolved rapidly during the past decades. Many large-scale proteomic projects have been launched for various diseases, including cardiovascular diseases,1 diabetes2 and cancer.3 These studies often involved hundreds to thousands of clinical samples, generating massive tandem mass (MS/MS) datasets. To make the maximum use of such data, a community effort represented by the ProteomeXchange consortium4 (including the PRIDE Archive,5 Peptide Atlas,6 MassIVE7 and jPOST) was launched for public repository of proteomics data. As a result, the number of publicly accessible proteomic MS/MS datasets has grown exponentially in the past few years.8
One research that could benefit from the massive, publicly available MS/MS datasets is the prediction of peptide MS/MS spectra. The ability to predict MS/MS spectra of peptides can significantly enhance our understanding of mass spectrometry and could improve peptide identification in proteomics. Many different approaches have been proposed for the prediction of peptide MS/MS spectra. The MassAnalyzer9,10 explicitly models the chemical process of peptide fragmentation with parameters optimized using annotated MS/MS spectra. Other models like SQID11 tried to make predictions based on statistical results of peak intensities from annotated MS/MS spectra. Besides, machine learning (ML) approaches was proposed by us12,13 and others14–17 to predicting MS/MS spectra from peptide sequences. Those models are designed to be trained by annotated peptide spectra and predict the probability of observing each fragment ion (e.g., b-, y-ions and neutral loss ions) in an experimental spectrum.
Since these prediction algorithms were developed by about ten years ago, signihcant advancements have been made in mass spectrometry techniques. As shown recently, the reproducibility of peptide MS/MS spectra resulting from higher-energy collisional dissociation (HCD) is much better than the collision-induced dissociation (CID) spectra used by previous algorithms.18 On the other hand, the availability of massive identihed peptide spectra and the rapid advance of ML algorithms made it possible to train complex deep learning models, as demonstrated by recently developed predictors pDeep,19 DeepMass,20 and Prosit,21 however, these methods still followed the same framework of predicting the intensity of expected fragment ions (e.g., b/y ions) only. Hence, we refer to these approaches as the backbone-only predictors to distinguish them from our approach presented here.
About This Work
In this paper, we attempt to address, for the hrst time, the full-spectrum prediction of peptide MS/MS spectra. In contrast, all methods above are limited to predicting the intensity of expected fragment ion types (e.g., b/y ions and their neutral loss ions). We are inspired by the observation that a substantial fraction (~ 30% of total ion intensities; see Supplementary Figure S2, also reported by previous research18) in HCD spectra cannot be annotated as a/b/c/x/y/z ions or their neutral loss derivatives (referred to as the backbone ions in this paper). As a result, even for a method that can perfectly predict the intensities of all backbone ions, its predictions will still lack peaks constituting ~ 30% of total ion intensities. Our analyses show, even the hypothetical perfect predictions generated by extracting a sub-spectrum that contains only backbone ions, its average similarity with its full spectrum replicates is only ~ 0.740 for 2+ HCD spectra, still far from that between the replicated full spectra of ~ 0.837 (Supplementary Figure S1). This observation implies that the full spectrum prediction is necessary for further improving the overall similarity. Notably, the mechanic explanations of these non-backbone fragment ions are lacking, and thus it is non-trivial to provide fragmentation rules to guide machine learning algorithms to learn the intensities of these ions.
On the other hand, the recent success of deep learning models on various tasks (e.g., in the ImageNet competitions22) demonstrated that, with a sufficient amount of training samples, carefully designed deep learning models can automatically discover complex rules and patterns by itself (e.g., the patterns of natural images). This encourages us to exploit this capability of using deep learning models to discover the fragmentation rules from the massive number of training samples. In this work, we made no assumptions or expectations on which kind of ions to predict, and we provide no annotations of fragment ion or fragmentation rules to our model. Instead, we attempt to predict the intensities at all possible m/z values, which means that our model will not be limited to given ions types but preserves the ability to learn the rules to simultaneously predict the m/z values and intensities of any ions, no matter of the known or unknown ion types.
Methods
Data and Evaluation Criteria
We collected identihed HCD spectra from spectral libraries including the NIST HCD library,23 the NIST Synthetic HCD library,23 the Human HCD library from MassIVE,7 and the synthetic HCD library from ProteomeTools.24 The sizes of these datasets are summarized in Table 1. In order to guarantee the quality of testing data, the NIST HCD library and the NIST synthetic HCD library, which are relatively old and with comparably lower data quality, are used for training only. Testing samples are randomly selected from the original dataset, while we guarantee that there are no overlaps between the training and testing peptides. We further puri-hed the training and testing datasets by removing under-fragmented PSMs, over-fragmented PSMs, (less than 1%, see Supplementary section Data Selection for details) and PSMs with precursor mass difference more than 200 ppm. The complete training and testing datasets are available at the supplementary website, http://www.predfull.com/datasets.
Table 1:
The total numbers of spectra in spectra libraries used for training and testing the spectra prediction models for HCD and ETD spectra. The number in each cell means the size of training data (including about 10% of validation data, used for choosing hyper-parameters), while the numbers of testing samples are shown in the parentheses. The complete set of training and testing samples are released as the Supplemental Dataset 1 and 2.
| Type | Charge | NIST HCD | NIST Synthetic | MassIVE | ProteomeTools | Total |
|---|---|---|---|---|---|---|
| HCD | 1+ | 10,392 | 29 | 6,349 (1,262) | 0 | 16,770 (1,262) |
| 2+ | 536,701 | 320,062 | 512,105 (16,989) | 126,586 (7,620) | 1,495,454 (24,609) | |
| 3+ | 189,933 | 140,273 | 309,239 (14,342) | 59,736 (5,438) | 699,181 (19,780 | |
| 4+ | 18,190 | 15,762 | 50,428 (4,494) | 7,203 (1,046) | 91,583 (5,540) | |
| ETD | 2+ | 0 | 0 | 26,254 (4,666) | 0 | 26,254 (4,666) |
| 3+ | 0 | 0 | 129,647 (17,208) | 0 | 129,647 (17,208) | |
| 4+ | 0 | 0 | 10,274 (3,405) | 0 | 10,274 (3,405) | |
Data Pre-processing
For the learning purpose, we represent an MS/MS spectrum as a sparse one dimensional (1-D) vector by binning the m/z range between 180 and 2,000 with a given bin width. We limit the range to 0 ~ 2000 because there are very few MS/MS spectra contain peaks with m/z above 2,000. This range can be extended if a larger m/z range is needed. By default, we use a bin width of 0.1, resulting in vector representations of 20,000 dimensions.
The default bin width was chosen based on the observed m/z shifts between the corresponding peaks in replicated experimental spectra. As shown in Supplementary Figure S6, although many mass spectrum instruments often claimed a much higher mass precision, the observed m/z shifts are not ignorable when the bin width is lower than 0.05 m/z. Thus a meaningful bin width must be slightly higher, so we select the default bin width as 0.1 m/z. In fact, our experiments demonstrate that a smaller bin width (i.e., higher mass resolution) such as m/z of 0.05 will not improve the performance but requiring much longer training times.
Finally, as the absolute intensities in the MS/MS spectra are irrelevant, all spectra in training and testing sets are normalized by dividing the maximum peak intensity in each spectrum. Note that we also remove the precursor peak in each spectrum, although the precursor peak is relatively weak in most spectra.
Evaluation Criteria and Intensity Transformation
Several metrics have been proposed to measure the similarity between two MS/MS spectra in the context of spectra identihcation and spectra library search.25–28 Among those, we choose the most widely accepted metric of cosine similarity (normalized dot product) between two spectra as our evaluation standard. As pointed out by previous research,25 the similarities computed on unnormalized intensities are often misleading, because the results may be dominated by a few very intense peaks in the spectra. Our study conhrmed this observation: as shown in the hrst panel of Supplementary Figure S7, when computing using the raw intensities, although the distribution of cosine similarities between replicated spectra are high, it is largely overlapped with the distribution of the similarities between the spectra of different peptides with similar precursor masses. In practice, previous studies suggested several different transformation functions to reduce the impact of the most intense peaks when performing identihcation and comparison, such as logarithm or square root.29 In this paper, we choose the square root function for transforming peak intensities in each spectrum, because the square root function exhibited similar effects as the logarithm function while won’t introduce negative values after the transformation. As shown in the Supplementary Figure S7, after the square root transformation, the similarity distribution of replicated spectra are better separated from that of the spectra from different peptides.
Prediction of Doubly and Triply Charged HCD Spectra
We hrst focus on predicting 2+ and 3+ HCD spectra of unmodihed peptides, as a large number of identihed 2+ and 3+ HCD spectra are publicly available. We implemented a convolutional neural network (CNN) using the Keras30 framework with Tensorhow31 back-end. In total, we collected around 1.5 million 2+ spectra and 1 million 3+ spectra for training (see Table 1 for details). For testing purposes, we held out about 16 thousand 2+ and 14 thousand 3+ spectra, respectively, from the peptides that do not overlap with the remaining training samples. Finally, in this paper, we focus on the prediction of MS/MS spectra from unmodihed peptides; we plan to present the results of predicting modihed peptides in the future.
Note that when training this model, we did not distinguish the types of instruments used to acquire these HCD spectra, as we observed that the HCD spectra generated by different instruments (e.g., Orbitrap, Fusion or Q Exactive) are highly similar. Besides, as not all training data provide information about the normalized collision energy (NCE), we assume all unlabeled data having the NCE of 25%.
Architecture of the Convolutional Neural Network
We developed a generalized sequence-to-sequence (Seq2Seq) model based on the structure of the residual convolutional neural network32 for predicting the full MS/MS spectra from peptide sequences, as depicted in Figure 1. The input for our model is a 27 by 23 matrix (up to 25 amino acid residues long) that contains the peptide sequence, the amino-acid masses, and other necessary meta-information. To be specihc, the row 1 to row 22 of the matrix are the One-hot encoding of the input peptide sequence (including 20 amino acids, one ending character, and one padding character), while the last row contains the monoisotopic amino acid mass.
Figure 1:
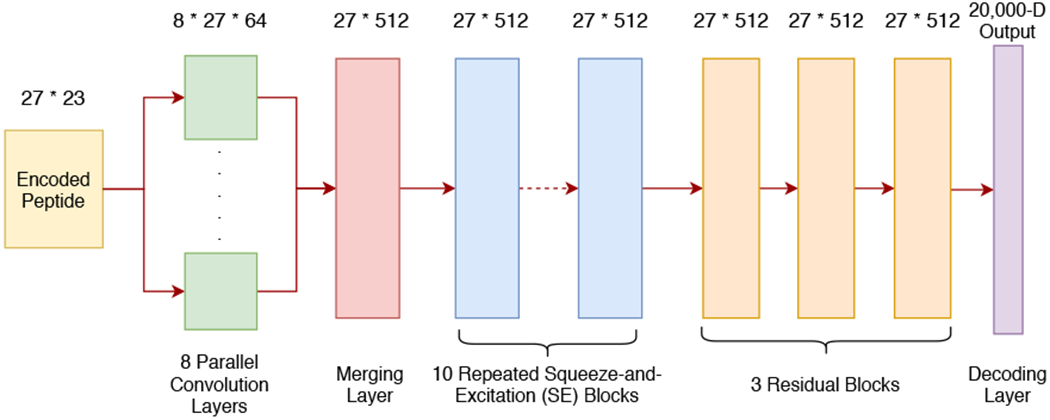
The core architecture of the residual convolutional neural network (CNN) model for spectrum prediction.
The embedded representation will hrst be fed into 8 parallel 1-dimensional convolutional layers of different kernel sizes (from 2 to 9). This step is designed to capture the correlations among subsequences of the input peptide. Afterward, the convolution results are merged into a single tensor, which is then passed through 10 consequential Squeeze-and-Excitation blocks33 (we used 10 blocks here as we cannot observe further performance improvement with more blocks in our experiments). Three subsequently residual blocks32 and the last 1-dimensional convolutional layer work as the decoder, which decodes the previous tensor into the hnal prediction vector of length 20,000 representing the hnal MS/MS spectrum. The default 20,000 length vector in our current model corresponds to the mass resolution of 0.1 m/z, as stated above.
It is worth noting that we did not incorporate any commonly used pooling layers in the architecture of our model (except the last layer). It turns out this choice along with the residual convolutional network structure, is critical for achieving a good performance according to our experiments. The entire model (Figure 1) contains about 19 million parameters and occupies a space of around 77 Mb, the details of implementation and training process can be found in the Implementation and Training section of the Supplementary Materials.
Multitask Learning Framework
Prediction of 1+ and 4+ HCD Spectra with Insufficient Training Data
As stated above, around 2.2 million training samples were used for training the model to predict 2+ and 3+ HCD spectra. Obviously, the success of 2+ and 3+ HCD spectra prediction largely depends on the abundant training datasets. As shown in Figure 2, the prediction accuracy increases significantly and steadily with more spectra are employed as training samples. Not surprisingly, we observed that the trend of performance improving gradually saturates when more than 1 million training samples were used; we estimated that the prediction accuracy of our model may not be further improved over 5% by even more training samples.
Figure 2:
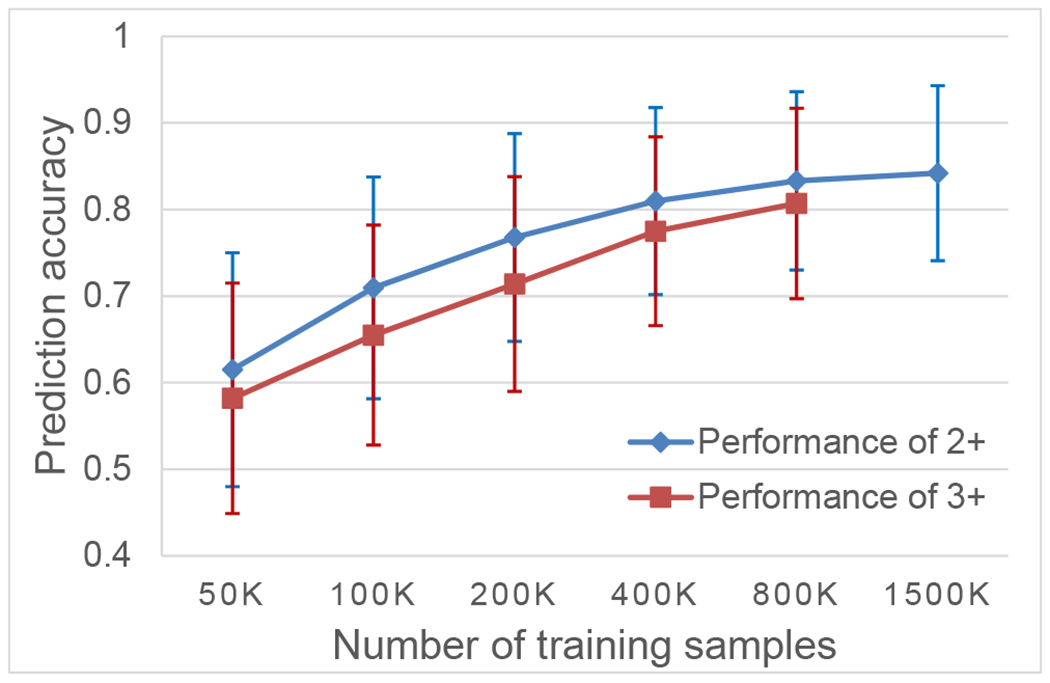
Prediction accuracy (measured by the similarity between the predicted and experimental spectra on testing data; y-axis) increases with more training data (x-axis).
However, far fewer identified HCD spectra are available for the singly (1+) and quaternarily (4+) charged peptide ions. Thus we developed a multitask learning (MTL) approach that can train our model with insufficient training samples, which significantly improves the prediction accuracy when large training sets are not available. The idea is to implement a universal model that can be trained simultaneously by HCD spectra of different charges. This approach not only saves the efforts of building many models for different charges, but also improves the prediction performance, as the fragmentation mechanisms learned from charges with abundant spectra might also guide the prediction of charges with insufficient spectra.
However, simply training a model by mixing all training samples together will not result in satisfactory performance, because the neural network can easily be overwhelmed by the most abundant 2+ and 3+ spectra in the mixed dataset (known as ‘Catastrophic Forgetting’34). As suggested by previous research on multitask learning,35,36 auxiliary tasks can be used as a focusing method. Thus we modify the original model by adding an auxiliary task branch that “predicts” the precursor charges of the HCD spectra (Figure 3). Of course, we are not interested in predicting the charge state of the precursor, which is already given in the input. However, this prediction task can inform the neural network with the importance of the desired charge state and enforce the model to balance between the training samples of different charges. Besides, we also included an auxiliary task that “predicts” the precursor mass (which is given as well). This auxiliary task works as a regulation to prevent overhtting and further stabilize the training process. With the help of those auxiliary tasks, our universal model signihcantly improved its performance on 1+ and 4+ HCD spectra (see Results section for details), conhrmed that these tasks beneht from learning spectra of different charges together.
Figure 3:
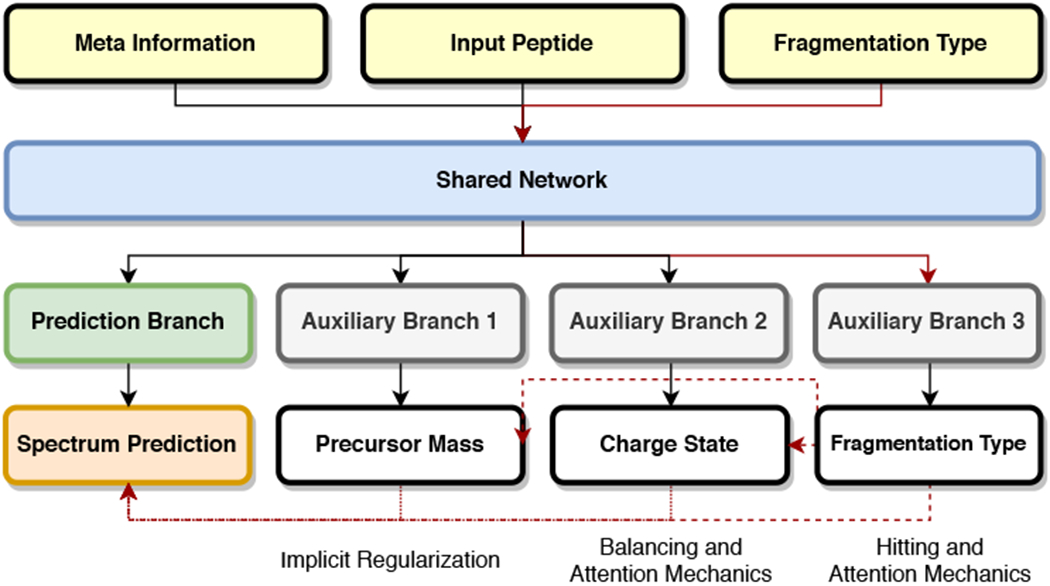
The multitask learning model for joint training of HCD and ETD Spectra with all charge states (1+, 2+, 3+ and 4+).
Prediction of ETD Spectra with Insufficient Training Data
We are also interested in predicting the MS/MS spectra resulting from Electron-Transfer Dissociation (ETD). However, we encountered the same challenge that the identihed ETD spectra we collected are much fewer: we were only able to collect around 180,000 identihed ETD spectra (i.e., < 10% of the HCD training data; Table 1). To be specihc, the ETD PSMs are obtained by MSGF+37 searching on the Kuster synthetic dataset,24 with a mass tolerance of 40 ppm and limit the QValue (similar to FDR value) up to 0.002. Furthermore, the majority (146,855 out of 191,454) of identihcation results are 3+ spectra, thus we would expect that a model trained directly on these samples will perform poorly when predicting spectra of other charges.
Again, we expect that the prediction of ETD spectra could beneht from learning HCD spectra, as they may have shared fragmentation patterns. We extend our joint model to predict both HCD and ETD spectra by adding one more auxiliary task that “predicts” the given information of the fragmentation type (Figure 3). To ensure that the given fragmentation type won’t be ignored, this auxiliary task is connected to all previous branches to allow the full network to be aware of the difference between different fragmentation types. Analysis in the Results section show that the prediction performance of ETD spectra improved signihcantly by learning HCD spectra concurrently.
Results and Discussion
Prediction Performance on 2+ and 3+ HCD Spectra of Peptides
To evaluate the accuracy of the predicted MS/MS spectra, we computed the cosine similarities between the experimental and the predicted spectra by our model on the testing data of 16 thousand 2+ and 14 thousand 3+ spectra (Table 1). For comparison, we also computed the similarities of predictions made by three best-performed models: pDeep,19 Prosit21 and DeepMass.20 Note that the similarities we report here are much lower than those reported in their original publications, because here we are computing the similarities with the complete experiment spectra but not with backbone ions solely. All these models are limited to predict backbone ions and the details of how we execute them can be found in the Running other Predictors section of Supplementary Materials. Furthermore, for each testing case, we also generated a theoretical perfect backbone spectrum consisting of only backbone ions from the experimental replicates, but removed all other ions. This represents the upper bound performance for all backbone only predictors.
As shown in Figure 4, the spectra predicted by our algorithm are highly similar with the experimental spectra, with the average full-spectrum cosine similarities of 0.820 (±0.088) and 0.786 (±0.085) for 2+ and 3+ HCD spectra, respectively, very close to the average full-spectrum cosine similarities between the replicated spectra of the same peptides are 0.837 (±0.114) and 0.806 (±0.113) for 2+ and 3+ spectra, respectively, implying that our models approach the optimal prediction accuracy. In contrast, even the generated perfect backbone spectrum (denoted as “perfect backbone” in Figure 4) can only achieve the average cosine similarities around 0.750 (±0.124) and 0.700 (±0.127) for 2+ and 3+ spectra, respectively. In practice, however, as we cannot achieve a perfect prediction, the average cosine similarities obtained by our extended implementation of pDeep19 (denoted as “full backbone” in Figure 4, see section Running other Predictors in Supplementary Materials for details) is around 0.731 (±0.126) and 0.697 (±0.107) for 2+ and 3+ spectra, respectively. The original pDeep software as well as the more recently published software tools Prosit and DeepMass, which does not consider all possible backbone ions, can only achieve an even lower average cosine similarities below 0.65 (Figure 4). Note that the similarities listed above are much lower than those reported in previous studies,19–21 because those previous results were calculated on only backbone ions but not on the full spectrum.
Figure 4:
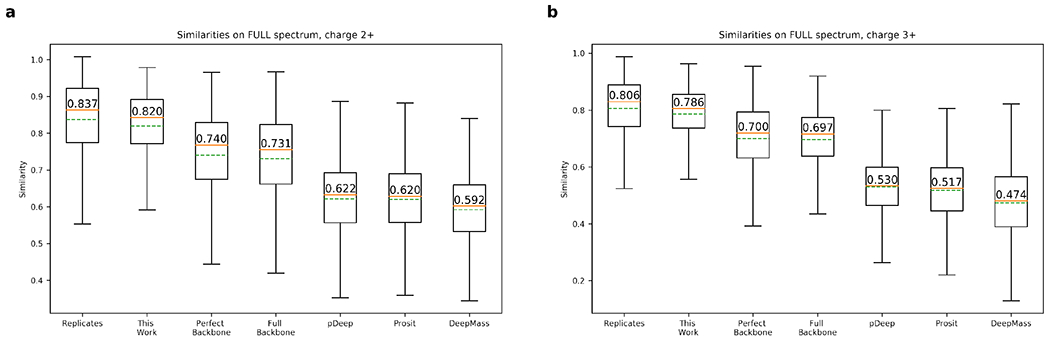
Similarities between the experimental and predicted HCD spectra for 2+ (a) and 3+ (b) precursor peptides ions, in comparison with the similarities between spectra in replicated experiments and other approaches. Evaluated on testing data.
However, even in cases we consider only backbone ions, our predictor can still outperform all previous backbone only models. In our evaluation, our model achieved highly accurate intensities prediction on b/y ions, with average cosine similarities of 0.942(±0.075) and 0.895(±0.070) for the 2+ and 3+ spectra, respectively, both approaching the similarity between replicated spectra and higher than previous models (Figure 5). It is somewhat surprising that our method performs even better than methods (pDeep, Prosit and DeepMass) that focus only on predicting backbone ions, which suggests that the full-spectrum prediction benefit from learning and predicting all ions simultaneously: knowledge learned from non-backbone ions can also guide the predicting of backbone ions.
Figure 5:
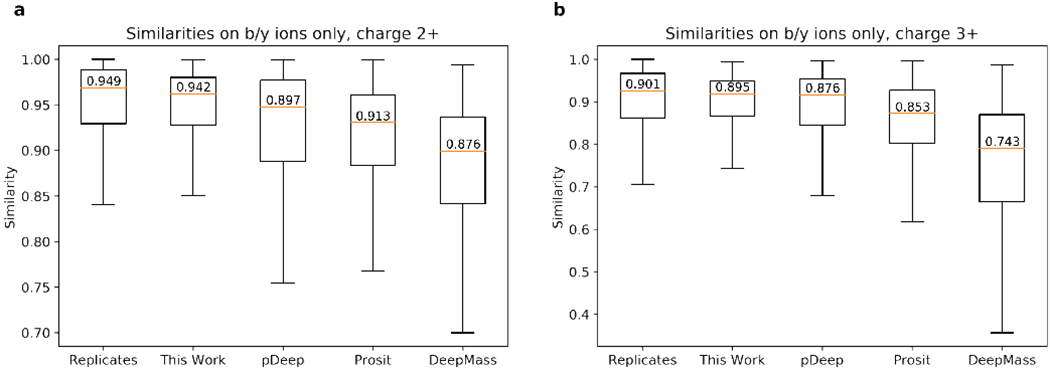
Similarities on the b/y ion intensities between the experimental and predicted HCD spectra, a, Results for charge 2+. b, Results for charge 3+.
More specihcally, as these two examples of prediction (Figure 6) show, our algorithm is capable of predicting the complete MS/MS spectra: our predictions successfully covered most intense non-backbone ion peaks observed in the experimental spectra, showing that these peaks represent fragmentation patterns that can be captured by the learning algorithm, even though the fragmentation mechanism remains unknown. Overall, our prediction demonstrates a clear improvement over previous prediction algorithms.
Figure 6:
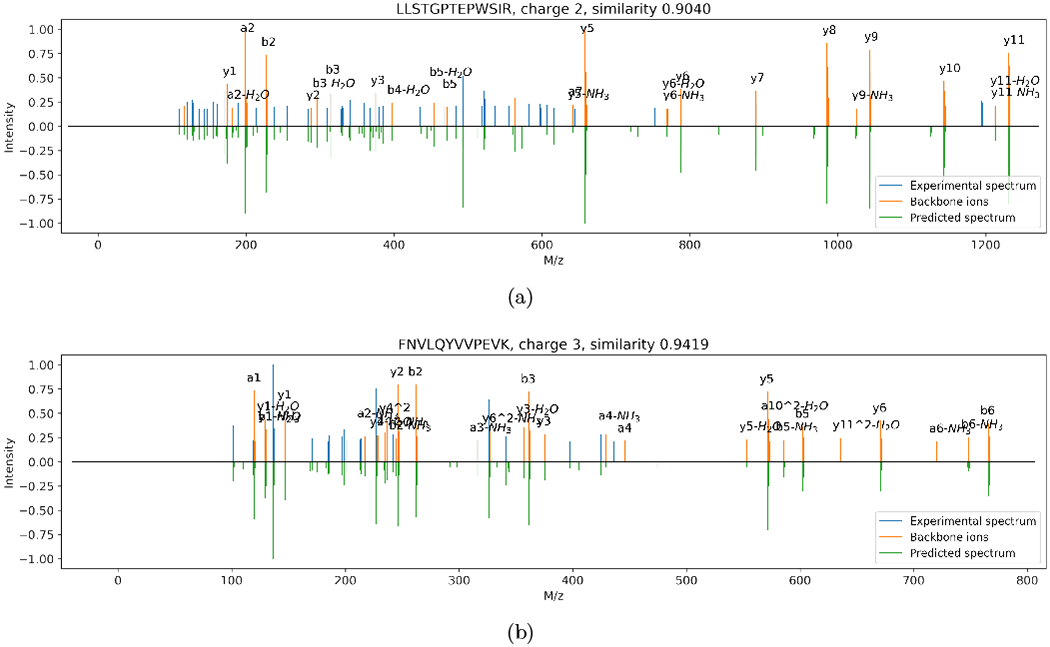
Predicted (bottom half) HCD spectra versus experimental (top half) HCD spectra of charges 2+ (a) and 3+ (b), where the backbone ions are marked in orange. Note that the intensities are transformed by the square root function.
Furthermore, we compared the composition of fragment ions in the predicted spectra versus experimental MS/MS spectra by depicting the average percentages of total intensities for different types of fragment ions (Supplementary Figure S2). The composition of fragment ions in the predicted spectra by our method is similar to that in the experimental spectra, conhrming that our prediction algorithm can reliably predict non-backbone ions. In the experimental HCD spectra, ~ 30% of total peak intensities are contributed by non-backbone ions, while for the predicted spectra it’s ~ 20%, which is smaller but still substantial. These predicted non-backbone ions significantly boosted the similarity of the predicted spectra. Note that the overall non-backbone ion intensities in the predicted spectra are slightly lower than those in the experimental spectra, probably due to the presence of non-replicable noise peaks in the experimental spectra that are not predictable.
Variation of Prediction Accuracy
We observed that the replicated spectra of some peptides exhibit relatively low similarities. We investigated if the prediction similarities of these peptides are also relatively low. As shown in the Supplementary Figure S4, the similarities between replicated HCD spectra are highly correlated with the similarities between the experimental and predicted spectra of the same peptide. This result conhrms that the prediction performance largely depends on the replicability, while most of the poor predictions are caused by those less replicable peptides.
Besides, We observed that the prediction accuracy of our model varies depending on the peptide lengths and the replicability of the MS/MS spectra. As shown in the upper panel of Supplementary Figure S5, the prediction accuracy decreases gradually with the increasing lengths of peptides, especially for peptides longer than 14 residues. Firstly, this is probably because the spectra of long peptides may exhibit more complex fragmentation patterns, and thus made the prediction of long peptides more challenging. Secondly, the training dataset contains fewer samples of longer peptides, which makes it more difficult for the model to learn the fragmentation rules and patterns for these peptides. Finally, in fact, the similarities between replicated experimental HCD spectra also decrease with the increasing peptide lengths (as shown in the lower panel of Supplementary Figure S5), indicating the signal/noise ratio decreased in spectra of relatively longer peptides.
Prediction Performance on 1+ and 4+ HCD Spectra
We evaluated the prediction performance of the MTL model using the training and testing datasets of 1+ and 4+ HCD spectra collected from the spectra libraries as described in Table 1. Because previous spectra prediction software (pDeep, DeepMass and Prosit) did not provide the option for predicting 1+ and 4+ spectra, we compared the performance of our program (i.e., the similarity between predicted and experimental spectra) with experimental replication and the prediction model trained only using the training samples with the respective charges (e.g., the model for 4+ spectra prediction trained by using only 4+ spectra in the training set).
As shown in Figure 7, the multitask learning approach yields satisfactory performance, with the similarities between the predicted and experimental spectra approaching that between the replicated spectra, much higher than those from the spectra prediction models trained directly from the subset of spectra with the specific charge (1+ or 4+).
Figure 7:
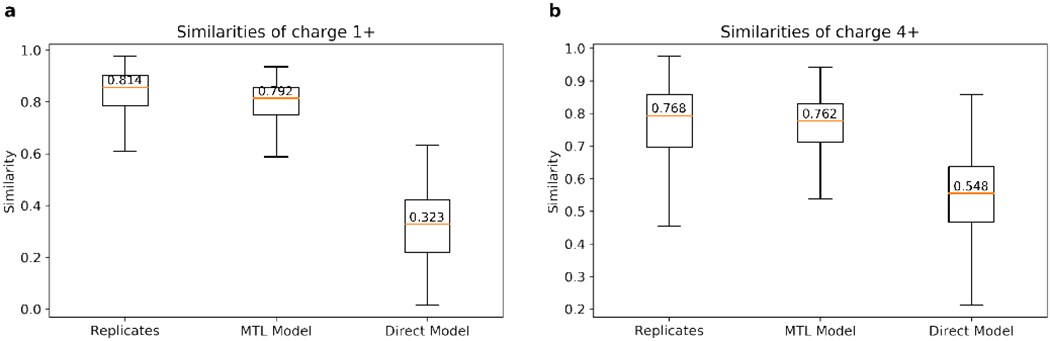
Similarities between the experimental and predicted 1+ (a) and 4+ (b) HCD spectra using MTL approach, in comparison with the similarities between spectra in replicated experiments and the direct prediction approach.
Prediction Performance on ETD Spectra of Peptides
We evaluated the prediction performance of the MTL model using the training and testing datasets of ETD spectra collected from the spectra libraries as described in Table 1. Not surprisingly, without MTL approach, the average similarity between the experimental and predicted spectra is below 0.55 (denoted as “direct training” in Figure 8), far from the average similarity between replicated ETD spectra (e.g., ~ 0.88 for 3+; Figure 8). However, with the help of our joint MTL model, we are able to achieve comparable average similarities using this relatively small ETD dataset (denoted as “multitask training” in Figure 8). An example prediction of ETD spectra is shown in Figure 9.
Figure 8:
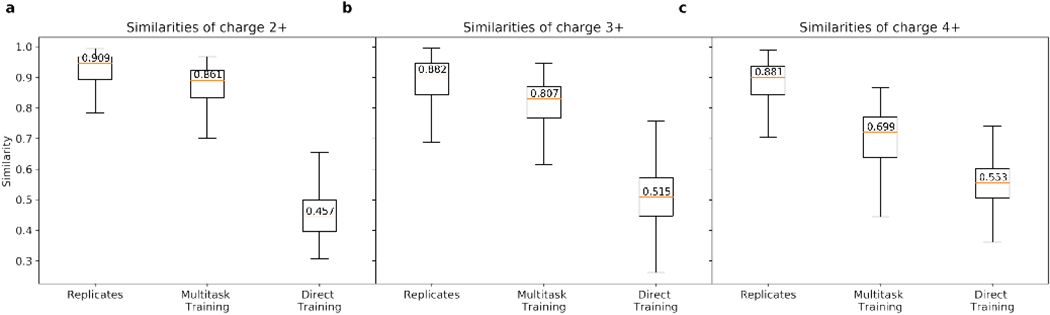
Similarities between the experimental and predicted ETD spectra using MTL approach for 2+ (a), 3+ (b) and 4+ (c) precursor peptides ions, in comparison with the similarities between spectra in replicated experiments and the direct prediction approach.
Figure 9:
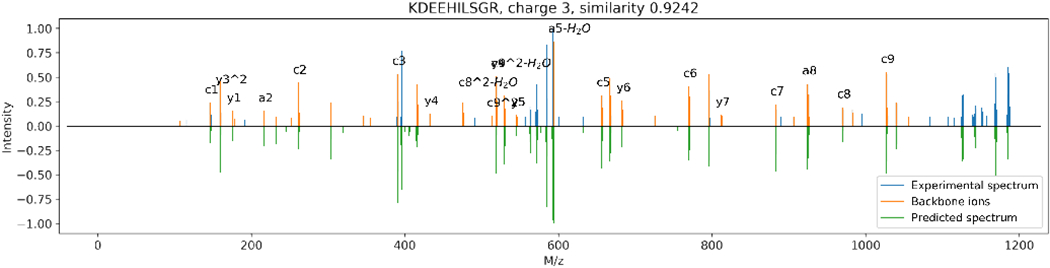
Predicted (bottom half) ETD spectra versus experimental (top half) ETD spectra of charge 3+, where the backbone ions are marked in orange. Note that the intensities are transformed by the square root function.
Interestingly, the intensity composition of the fragment ions in the predicted spectra is close to that of the experimental spectra. Like in HCD spectra where b/y ions and their neutral loss derivatives comprise more than 60% intensities (Supplementary Figure S2), c/z ions are the most intense ions in ETD spectra (Supplementary Figure S3). Notably, the fragmentation rules of these two methods (e.g., abundant b/y ions in HCD and abundant c/z ions in ETD) were not provided to the deep learning model; nonetheless, the model discovered these patterns directly from the training data.
Conclusions
In this paper, we present a deep learning approach for predicting the complete tandem mass spectra directly from peptide sequences without providing any prior knowledge. It is worth noting that this model is drastically different from existing backbone-only spectrum predictors (e.g., pDeep, Prosit and DeepMass), which are limited to predict only the intensity of an expected subset of fragment ions (i.e., backbone ions in HCD spectra). As our results showed, the non-backbone ions in HCD and ETD spectra, for which the fragmentation mechanisms may not be fully understood, can be satisfactorily predicted by our model, leading to much higher overall prediction accuracy and ion coverage (Figure 4).
We also developed a multi-task learning (MTL) approach for training a joint prediction model, which signihcantly improves the prediction accuracy for spectra with insufficient training data (e.g., 1+ and 4+ HCD spectra and ETD spectra of all charges). The testing results showed that the model trained using MTL achieved comparable performance on both types of tasks, with fewer than 200K samples were used for training.
Future works.
We note that the deep learning approaches developed here may also be extended to the prediction of MS/MS spectra using other fragmentation methods, e.g., the high energy HCD or electron transfer/ high-energy collision dissociation (EThcD), in which the fragmentation rules are more complex and less understood. Another line of research is to extend our model for predicting spectra from modihed peptides. Finally, we are interested in developing computational methods to automatically generate hypotheses about the explicit fragmentation mechanisms/rules resulting in the non-backbone ions with the help of complete spectra prediction. These applications are beyond the scope of this paper and will be presented in the future.
Supplementary Material
Supplements.pdf: Details on data selection; details on implementation and training; details of running other predictors; Figure S1: Similarities distributions of full and backbone-only spectrum with replicates; Figure S2: Intensity composition in HCD spectra; Figure S3: Intensity composition in ETD spectra; Figure S4: Relationships between similarities and replicability; Figure S5: Relationships between similarities and peptide length; Figure S6: The distribution of m/z shifts; Figure S7: The distributions of similarities by different transformation functions; Figure S8: details of training process.
Acknowledgement
The authors thank Dr. Nuno Bandeira for helpful discussions. This research was partially supported by the National Institute of Health grant 1R01AI108888 and Indiana University (IU) Precision Health Initiative (PHI).
Footnotes
Competing interests
The authors declare that they have no conflict of interest.
Supporting Information Available
The code for the prediction model Predfull is available at https://github.com/lkytal/PredFull and as a web service at http://www.predfull.com/.
References
- (1).Mokou M; Lygirou V; Vlahou A; Mischak H Proteomics in cardiovascular disease: recent progress and clinical implication and implementation. Expert review of proteomics 2017, 14, 117–136. [DOI] [PubMed] [Google Scholar]
- (2).Topf F; Schvartz D; Gaudet P; Priego-Capote F; Zufferey A; Turck N; Binz P-A; Fontana P; Wiederkehr A; Finamore F, et al. The Human Diabetes Proteome Project (HDPP): from network biology to targets for therapies and prevention. Translational proteomics 2013, 1, 3–11. [Google Scholar]
- (3).Ellis M; Gillette M; Carr S; Paulovich A; Smith R; Rodland K; Townsend R; Kinsinger C; Mesri M; Rodriguez H, et al. Clinical Proteomic Tumor Analysis Consortium (CPTAC): Connecting genomic alterations to cancer biology with proteomics: the NCI Clinical Proteomic Tumor Analysis Consortium. Cancer Discov 2013, 3, 1108–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Vizcaíno JA; Deutsch EW; Wang R; Csordas A; Reisinger F; Rios D; Dianes JA; Sun Z; Farrah T; Bandeira N, et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nature biotechnology 2014, 32, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Vizcaíno JA; Côté RG; Csordas A; Dianes JA; Fabregat A; Foster JM; Griss J; Alpi E; Birim M; Contell J, et al. The PRoteomics IDEntihcations (PRIDE) database and associated tools: status in 2013. Nucleic acids research 2012, 41, D1063–D1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Deutsch EW; Lam H; Aebersold R PeptideAtlas: a resource for target selection for emerging targeted proteomics workflows. EMBO reports 2008, 9, 429–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wang M; Wang J; Carver J; Pullman BS; Cha SW; Bandeira N Assembling the Community-Scale Discoverable Human Proteome. Cell systems 2018, 7, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Martens L; Vizcaíno JA A golden age for working with public proteomics data. Trends in biochemical sciences 2017, 42, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Zhang Z Prediction of low-energy collision-induced dissociation spectra of peptides. Analytical chemistry 2004, 76, 3908–3922. [DOI] [PubMed] [Google Scholar]
- (10).Zhang Z Prediction of low-energy collision-induced dissociation spectra of peptides with three or more charges. Analytical chemistry 2005, 77, 6364–6373. [DOI] [PubMed] [Google Scholar]
- (11).Li W; Ji L; Goya J; Tan G; Wysocki VH SQID: an intensity-incorporated protein identihcation algorithm for tandem mass spectrometry. Journal of proteome research 2011, 10, 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Arnold RJ; Jayasankar N; Aggarwal D; Tang H; Radivojac P Biocomputing 2006; World Scientihc, 2006; pp 219–230. [PubMed] [Google Scholar]
- (13).Li S; Arnold RJ; Tang H; Radivojac P On the accuracy and limits of peptide fragmentation spectrum prediction. Analytical chemistry 2011, 83, 790–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Klammer AA; Reynolds SM; Bilmes JA; MacCoss MJ; Noble WS Modeling peptide fragmentation with dynamic Bayesian networks for peptide identihcation. Bioinformatics 2008. , 24, i348–i356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Frank AM Predicting intensity ranks of peptide fragment ions. Journal of proteome research 2009, 8, 2226–2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Sun S; Yang F; Yang Q; Zhang H; Wang Y; Bu D; Ma B MS-simulator: predicting y-ion intensities for peptides with two charges based on the intensity ratio of neighboring ions. Journal of proteome research 2012, 11, 4509–4516. [DOI] [PubMed] [Google Scholar]
- (17).Degroeve S; Martens L MS2PIP: a tool for MS/MS peak intensity prediction. Bioinformatics 2013, 29, 3199–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Michalski A; Neuhauser N; Cox J; Mann M A systematic investigation into the nature of tryptic HCD spectra. Journal of proteome research 2012, 11, 5479–5491. [DOI] [PubMed] [Google Scholar]
- (19).Zhou X-X; Zeng W-F; Chi H; Luo C; Liu C; Zhan J; He S-M; Zhang Z pdeep: Predicting MS/MS spectra of peptides with deep learning. Analytical chemistry 2017, 89, 12690–12697. [DOI] [PubMed] [Google Scholar]
- (20).Tiwary S; Levy R; Gutenbrunner P; Soto FS; Palaniappan KK; Deming L; Berndl M; Brant A; Cimermancic P; Cox J High-quality MS/MS spectrum prediction for data-dependent and data-independent acquisition data analysis. Nature methods 2019, 16, 519. [DOI] [PubMed] [Google Scholar]
- (21).Gessulat S; Schmidt T; Zolg DP; Samaras P; Schnatbaum K; Zerweck J; Knaute T; Rechenberger J; Delanghe B; Huhmer A, et al. Prosit: proteome-wide prediction of peptide tandem mass spectra by deep learning. Nature methods 2019, 16, 509. [DOI] [PubMed] [Google Scholar]
- (22).Russakovsky O; Deng J; Su H; Krause J; Satheesh S; Ma S; Huang Z; Karpathy A; Khosla A; Bernstein M, et al. Imagenet large scale visual recognition challenge. International journal of computer vision 2015, 115, 211–252. [Google Scholar]
- (23).Yang X; Neta P; Stein SE Extending a Tandem Mass Spectral Library to Include MS 2 Spectra of Fragment Ions Produced In-Source and MS n Spectra. Journal of The American Society for Mass Spectrometry 2017, 28, 2280–2287. [DOI] [PubMed] [Google Scholar]
- (24).Zolg DP; Wilhelm M; Schnatbaum K; Zerweck J; Knaute T; Delanghe B; Bailey DJ; Gessulat S; Ehrlich H-C; Weininger M, et al. Building ProteomeTools based on a complete synthetic human proteome. Nature methods 2017, 14, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wan KX; Vidavsky I; Gross ML Comparing similar spectra: from similarity index to spectral contrast angle. Journal of the American Society for Mass Spectrometry 2002, 13, 85–88. [DOI] [PubMed] [Google Scholar]
- (26).Liu J; Bell AW; Bergeron JJ; Yanofsky CM; Carrillo B; Beaudrie CE; Kearney RE Methods for peptide identihcation by spectral comparison. Proteome science 2007, 5, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Shao W; Zhu K; Lam H Rehning similarity scoring to enable decoy-free validation in spectral library searching. Proteomics 2013, 13, 3273–3283. [DOI] [PubMed] [Google Scholar]
- (28).Garg N; Kapono CA; Lim YW; Koyama N; Vermeij MJ; Conrad D; Rohwer F; Dorrestein PC Mass spectral similarity for untargeted metabolomics data analysis of complex mixtures. International journal of mass spectrometry 2015. , 377, 719–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Lam H; Deutsch E; Eddes J; Eng J; King N; Yang S; Roth J; Kilpatrick L; Neta P; Stein S, et al. SpectraST: An open-source MS/MS spectra-matching library search tool for targeted proteomics. Poster at 54th ASMS Conference on Mass Spectrometry. 2006. [Google Scholar]
- (30).Chollet F, et al. Keras. https://keras.io, 2015. [Google Scholar]
- (31).Abadi M; Barham P; Chen J; Chen Z; Davis A; Dean J; Devin M; Ghemawat S; Irving G; Isard M, et al. Tensorflow: A system for large-scale machine learning. 12th {USENIX} Symposium on Operating Systems Design and Implementation ({OSDI} 16). 2016; pp 265–283. [Google Scholar]
- (32).He K; Zhang X; Ren S; Sun J Deep residual learning for image recognition. Proceedings of the IEEE conference on computer vision and pattern recognition. 2016; pp 770–778. [Google Scholar]
- (33).Hu J; Shen L; Albanie S; Sun G; Wu E Squeeze-and-Excitation Networks. IEEE transactions on pattern analysis and machine intelligence 2019, [DOI] [PubMed] [Google Scholar]
- (34).French RM Catastrophic forgetting in connectionist networks. Trends in cognitive sciences 1999, 3, 128–135. [DOI] [PubMed] [Google Scholar]
- (35).Caruana R; De Sa VR Promoting poor features to supervisors: Some inputs work better as outputs. Advances in Neural Information Processing Systems. 1997; pp 389–395. [Google Scholar]
- (36).Caruana R Neural networks: tricks of the trade; Springer, 1998; pp 165–191. [Google Scholar]
- (37).Kim S; Pevzner PA MS-GF+ makes progress towards a universal database search tool for proteomics. Nature communications 2014, 5, 5277. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplements.pdf: Details on data selection; details on implementation and training; details of running other predictors; Figure S1: Similarities distributions of full and backbone-only spectrum with replicates; Figure S2: Intensity composition in HCD spectra; Figure S3: Intensity composition in ETD spectra; Figure S4: Relationships between similarities and replicability; Figure S5: Relationships between similarities and peptide length; Figure S6: The distribution of m/z shifts; Figure S7: The distributions of similarities by different transformation functions; Figure S8: details of training process.