

Abstract
Objective
To identify an MS-specific immune cell population by deep immune phenotyping and relate it to soluble signaling molecules in CSF.
Methods
We analyzed surface expression of 22 markers in paired blood/CSF samples from 39 patients using mass cytometry (cytometry by time of flight). We also measured the concentrations of 296 signaling molecules in CSF using proximity extension assay. Results were analyzed using highly automated unsupervised algorithmic informatics.
Results
Mass cytometry objectively identified a B-cell population characterized by the expression of CD49d, CD69, CD27, CXCR3, and human leukocyte antigen (HLA)-DR as clearly associated with MS. Concentrations of the B cell–related factors, notably FCRL2, were increased in MS CSF, especially in early stages of the disease. The B-cell trophic factor B cell activating factor (BAFF) was decreased in MS. Proteins involved in neural plasticity were also reduced in MS.
Conclusion
When analyzed without a priori assumptions, both the soluble and the cellular compartments of the CSF in MS were characterized by markers related to B cells, and the strongest candidate for an MS-specific cell type has a B-cell phenotype.
The demyelinating disease MS is associated with infiltration of immune cells into the CNS and with increases in various signaling molecules in the CSF. Immune cell infiltration into the CNS is involved in causing relapses because blockade of the integrin CD49d, used by lymphocytes in crossing the blood-brain barrier, reduces relapse frequency.1 Another clinical success has been the development of monoclonal antibodies against CD20, which deplete peripheral B cells.2 These observations raise several questions regarding the pathomechanism of MS, including (1) the kinds of immune cells that enter the CNS, particularly whether the therapeutic mechanisms targeting CD49d and CD20 involve the same cell type, and (2) the particular properties of the MS brain that attract immune cell ingress. Compared with donors with noninflammatory neurologic diseases, the CSF from patients with MS contains altered proportions of T cells, B cells, monocytes, and NK cells,3 but which of these are the targets of effective therapies remains unknown. Regarding the signaling factors involved in mediating immune cell CNS ingress, techniques including ELISA4 mass spectrometry,5 and aptamer-based measurement6 have suggested several candidates, but none has been shown to be MS specific. Rather, the signaling molecules that increase in the CSF during MS resemble those involved in other inflammatory CNS diseases.
Here, we attempted to identify a pathogenic cell population in CSF using unbiased analysis of data from a large panel of markers measured by cytometry by time of flight (CyTOF). Our rationale was that conventional flow cytometry and gating would limit our examination to cell types that have been characterized largely from peripheral blood. Using a larger panel and an unsupervised clustering approach enables better identification and characterization of the cell populations actually present in CSF. We applied the same strategy to an independent, large-scale analysis of soluble signaling molecules using a proximity extension assay to measure 296 proteins and analyzed the results with bioinformatics tools developed for the analysis of genomics data sets.
Methods
Patients
Samples from 82 patients undergoing lumbar puncture for diagnosis at the University Hospital Basel were used, whereof 48 were designated controls and 34 were patients with MS. Patient characteristics are summarized in the table and detailed in table e-1, links.lww.com/NXI/A390. Thirty-nine matched blood and CSF cell samples collected between March and December of 2016 were used for CyTOF, and 74 biobanked CSF samples (without cells) were retrieved from the Basel University Hospital CSF bank for proximal extension assay (PEA). The second set of samples included aliquots frozen from the same lumbar punctures as the CyTOF samples, and some additional samples pseudo-randomly chosen from the same collection period by a technician unaware of the goals of the study.
Table.
CSF and Blood Donors

Peripheral Blood Mononuclear Cells and CSF
Peripheral blood mononuclear cells (PBMCs) were separated by density-gradient centrifugation (Lymphoprep, Axis-Shield, Oslo) according to the manufacturer's recommendations, then permeabilized and fixed for 15 minutes at room temperature in permeabilization buffer with 0.1% bovine serum albumin (BSA), and 4% paraformaldehyde in at 5 million cells per milliliter. Fixed, permeabilized cells were then washed with cell staining buffer (PBS 0.1% BSA, 0.01% NaN3, 2 mM EDTA), frozen on dry ice and stored at ‒80°C until labeling. CSF samples were centrifuged at 400g for 10 minutes at room temperature within 1 hour of lumbar puncture. Supernatants were aspirated, and pellets were resuspended, fixed, and permeabilized exactly like PBMCs.
Barcoding and Labeling for CyTOF
Frozen cells were thawed, pelleted at 420g for 5 minutes at 5°C, and resuspended in 800 μL permeabilization buffer. For postacquisition separation of CSF- and blood-derived cells, cells were labeled with unique barcodes (Cell-IDTM 20-Plex Pd Barcoding Kit; Fluidigm, CA). After 60 minutes, samples were washed twice in 2 mL of permeabilization buffer. CSF cells and PBMCs from the same patient were then pooled and incubated 10 minutes in 5% Human TruStain FcXTM Fc receptor blocking solution (BioLegend, San Diego) diluted in permeabilization buffer with 50 μL of antibody cocktail (table e-2, links.lww.com/NXI/A391) in permeabilization buffer. After 60 minutes, cells were washed with permeabilization buffer and stained with 500 μL Cell-ID Intercalator-Ir 125 μM (Fluidigm), diluted 1:500 in PBS. After 15 minutes, cells were washed with cell staining buffer followed by CHROMASOLV water. Barcoded cells were acquired over 1,000 seconds with multiple injections on the CyTOF mass cytometer (tuned and mass calibrated according to Fluidigm instructions) at 500 cells per second. During acquisition, data were dual-count calibrated and converted to.fcs files using the manufacturer's settings. Noise reduction and cell extraction parameters were as follows: cell length 10–75 and lower convolution threshold 200.
Reagents
Antibodies were preconjugated from Fluidigm. Anti-FoxP3 antibody from BioLegend (cat: 3200001) was conjugated with 146Nd using the Maxpar® Antibody Labeling Kit from Fluidigm. DNA intercalator-Ir 191/193, 2000X, was from DVS, Cat #.: 201192B, and cisplatin from Enzo Life Sciences.
Proximity Extension Assay
Proteins in CSF were characterized using a homogeneous PEA technique7 on 4 different commercial proteomics arrays (Inflammation I 96×96 #94300 lot#A70502, CVD III 96×96 #94600 lot#A70904, Immuno-Onc I 96×96 #94310 lot#A71807 and Neurology I 96×96 #94800 lot#A71301) from Olink (Uppsala, Sweden) using a 96.96 dynamic array integrated fluidic circuits (Fluidigm) running on a BioMark HD microfluidic PCR system (Fluidigm) following Olink specifications. Each array included 92 antibody pairs labeled with complementary oligonucleotides. Data were reported as normalized protein expression levels, applying Olink algorithms that standardized Ct values of samples to internal controls.
Data Analysis
CyTOF data were analyzed using R Bioconductor package CATALYST (version 1.4.2)8 for normalization of signal drift over run time.9 We used PBMC barcode channels for normalization and a fixed separation cutoff of 0.3 for identification. Trend smoothing used k equal to 10% of the total number of cells. Cells within each run were debarcoded with the automatic cutoff algorithm of CATALYST. Doublets were removed using DNA content and event length.
The flowCore (version 1.52.1) and flowStats (version 3.44.0) packages10,11 were used for normalization of asinh-transformed (cofactor 5) marker intensities across samples. The gaussNorm method was used for CD3, CD4, CD27, CD45RA, CD45RO, CXCR5, and HLA-DR. The CD49d and CXCR3 channels were centered and scaled; other channels were left unnormalized. All CSF cells were used, and 10,000 PBMCs per sample were randomly picked. One clear outlier patient on a heatmap of normalized intensities was excluded.
A self-organizing map12 was built using FlowSOM version 1.18.0,13 and cells were assigned to 100 grid points, which were then assigned to 16 metaclusters by consensus clustering using ConsensusClusterPlus version 1.50.0.14 CSF and PBMC cells were clustered using CD3, CD4, CD8, CD11b, CD11c, CD14, CD19, CD45RA, and CD45RO, and CSF cells were also clustered independently of PBMCs using all markers. Different starting seeds were used to test the stability of clustering results, and results were visualized using FIt-SNE.15 Cluster abundances across experimental conditions were compared using a generalized linear mixed model in the R package lme4 version 1.1-21,16 controlling for batch as fixed effect, and using a random effect for patients.
PEA Data Analysis
Only markers detected in at least 2 samples were retained. To better control the behavior of variance at low intensities, the undetected values for each marker were replaced with the lowest intensity value minus 10% of the range of intensities. Repeated measurements of the same marker were averaged. Three patients treated with steroids within 6 weeks preceding venipuncture were excluded. The final data set (296 markers from 74 patients) was normalized using quantile normalization. The first principal component was strongly associated with average intensity and CSF leukocyte number across patients, and further analysis used PC2 to PC10. MS and controls were compared using Bioconductor package limma version 3.42,17 and p values were adjusted using the Benjamini-Hochberg false discovery rate (FDR) procedure.
Standard Protocol Approvals, Registrations, and Patient Consents
All patients signed an informed consent and agreed to an anonymous data analysis. The study was approved by the Ethical Commission of Basel Stadt and Basel Land.
Data Availability
Anonymous data will be shared by any qualified investigator on request.
Results
Comparison of Blood and CSF Cells
We obtained CyTOF data from CSF of 25 control donors and 14 donors with MS, of which 2 had received rituximab and were analyzed separately. The resulting data were clustered with a benchmarked automated clustering routine; the clusters are displayed on the tSNE plot in figure e-1, links.lww.com/NXI/A386. First, we clustered all events together, without separating according to the anatomic compartment (blood vs CSF) or diagnosis. The clearest divisions corresponded to the 4 categories of T cells, B cells, NK cells, and myeloid cells (figure e-1, A–D), but the clustering approach enabled visualization of finer subgroups, such as the distinction within the T-cell population of CD45RA-expressing populations (clusters 13, 14, 15, and 16, figure e-1, A and C) vs CD45RO-expressing populations (clusters 1, 3, 4, 9, and 16, figure e-1, A and C). There were large differences in the abundances of these clusters between the blood and the CSF (figure e-1). In the T-cell compartment, cells in cluster 1 (expressing CD3, CD4, CD25, CD27, CD45RO, and CXCR3, i.e., memory T cells) were relatively more abundant in the CSF than in the blood. Conversely, clusters 13 and 14, also expressing CD3 and CD4 but characterized by positive CD45RA, negative CD45RO, and negative CD27, i.e., naive T cells, were more abundant in the blood. In the non–T-cell compartments, the most striking differences between CSF and blood were seen in clusters 5 and 7, of CD14-expressing monocytes, which were more abundant in blood (figure e-1E). This pattern of more naive T cells and innate cells such as monocytes in the blood than CSF matches previous reports.3 When analyzed this way, there were no significant differences between MS and control in any of the clusters in the CSF or blood, in line with previous findings.3,18 However, because the blood cells in the pooled analysis were hundreds of times more numerous than the CSF cells, the definitions of the clusters were dominated by the blood cells, and therefore unlikely to be appropriate for categorizing the CSF cells, whose phenotypes are very different (figure e-1E).
CSF From Donors With MS Contains Characteristic B Cells
To extract cell phenotypes more representative of the CSF, we repeated the clustering using only the CSF cells and plotted it in the same way (figures 1, A–C). This revealed 2 populations with significantly different relative abundance in CSF from patients with MS vs controls. First, a naive CD8+ T-cell population (cluster 4 in figures 1 and 2), expressing CXCR5, CD25, CCR7, TIGIT, and CD45RA, was reduced in the CSF of patients with MS, albeit only significantly before correction for multiple comparisons (p = 0.039, FDR = 0.31). Second, of the 3 B-cell clusters (clusters 8, 9, and 13), the subpopulation characterized by CD49d, CD45RA, CD20, high CD27, CD69, and CXCR3 expression (cluster 8 in figures 1 and 2) was significantly increased in MS (p = 0.0012, FDR = 0.0192). Two of the 3 B-cell populations, including the MS-associated cluster 8, expressed CD20, and these were reduced in rituximab-treated patients, whereas the CD20-negative cluster 9 was not; however, the small number of rituximab-treated patients precluded statistical inference.
Figure 1. Cell Types in CSF of Patients With MS or Other Diagnoses.

(A) tSNE plot of cells from CSF. Data were clustered using k-means, and the clusters colored as shown in the key on the right of the figure, in a fashion exactly analogous to figure e-1, links.lww.com/NXI/A386. The characteristics of these clusters are shown in heatmap (C). The tSNE plot on the left shows data from cells from CSF of patients with non-MS diagnoses, the figure in the middle shows data from patients with MS, and the figure on the right shows data from patients with MS treated with rituximab. The red arrow in the bottom right corner of the middle tSNE plot indicates a cluster that is more abundant in CSF from patients with MS than in controls (see below). For this analysis, cells from CSF were clustered and analyzed independently, unlike in figure 1, enabling the detection of clusters that are specific to CSF, and too small to see against the background of the much larger dataset from blood. (B) Areas of the tSNE plots in (A) have been matched to the broad cell types myeloid, NK cells, T cells, and B cells. (C) Heatmap showing, for each of the clusters generated by k-means, the average intensity of each marker across all cells in the cluster. The colored bars at the top of the heatmap match the colors used in the tSNE plots in (A), and clusters corresponding to well-established cell types are shown on the right. Higher average expression of each marker is indicated with a red-brown color, and lower expression in blue.
Figure 2. Relative Abundances of Cell Types in CSF of Patients With MS or Other Diagnoses.

For each donor, the relative abundance of the cluster (% of total cells) was plotted for each cluster described in figure 1. Cluster identity numbers are at the top of each boxplot, and the colors of the bars at the top of each boxplot match the colors used to mark the clusters in figure 1. Within each scatter column, each filled circle represents 1 donor. The diameter of the circles reflects the absolute size of the cluster in that donor. The 35 donors are separated into 3 groups: donors with other diagnoses than MS (control, light gray boxes on the left of each boxplot); donors diagnosed with MS, but not treated with CD20-depleting antibodies (dark gray box in middle); and donors diagnosed with MS and treated with rituximab (gray box on right). Data from 2 rituximab-treated patients are shown for interest without statistical inference. The asterisks mark the cluster in which there was a statistically significant difference in abundance between control and MS (generalized linear mixed model; **p = 0.0012, FDR = 0.0192). FDR = false discovery rate.
If this MS-specific B-cell population is pathogenic, preventing its access to the CNS with natalizumab ought to increase its frequency in peripheral blood. To test this, we longitudinally analyzed blood from patients before and after the start of natalizumab. The CXCR3+ HLADR+ CD45RA+ B-cell population increased within the lymphocyte population by 63% (figure e-2, links.lww.com/NXI/A387).
Cytokines, Chemokines, and B Cell–Related Molecules are Altered in MS CSF
To relate observed cellular characteristics to soluble signaling factors, we measured 296 proteins in CSF. First, we compared the concentrations of all analytes between donors with MS and controls. At an FDR cutoff of 5%, 162 analytes were differentially abundant in CSF between donors with MS or other diagnoses. The fold change levels of these differentially abundant solutes are shown as a volcano plot in figure 3 and as a heatmap in figure e-3, links.lww.com/NXI/A388. Several molecules were differentially abundant between MS and control whose binding partners were also different. These pairs of interacting proteins are compiled in table e-3, links.lww.com/NXI/A392. Most strongly increased in MS was the costimulatory receptor CD27, whose only known binding partner, CD70, was also more abundant. Other molecules differentially abundant at FDR < 10−5 can be classified into 4 categories: chemokines, cytokines, B cell–associated proteins, and neuronal signaling molecules. CCL4, CCL17, CCL28, and CXCL12 were positively associated with MS. Other chemokines including CXCL8, CXCL10, and CXCL13 also showed large fold changes in MS compared with controls, but the p values were also larger, reflecting greater heterogeneity within the MS group. Cytokines associated with MS were broadly of the Th2 type, including IL-5 (and its receptor), IL-33 (and receptor), as well as IL-13 and IL-21. We also observed higher levels of IL-4, IL-12 (and receptor), IL-18, IL-20, TNF-alpha, and TNF-beta. Three of the molecules most clearly different between MS and controls were noticeable because they are centrally involved in B-cell biology—TNFSF13B, ICOSLG, and FCRL2. TNFSF13B (tumor necrosis factor superfamily 13B, also called BAFF) is a critical survival factor for B cells,19 and was significantly less abundant in the CSF of donors with MS. Inducible T-cell costimulator (ICOS) is expressed by T cells (especially Th2 cells), and interaction with its ligand ICOSLG on B cells is costimulatory for both, leading to shedding of ICOSLG.20 ICOSLG was less abundant in the CSF of donors with MS. The Fc-receptor-like family FCRL1-5 are principally expressed on B cells, especially FCRL2,21 which was more abundant in CSF from donors with MS. Finally, neuronal signaling molecules exhibited differences in abundance between MS and control CSF rivaling those of the immune-related molecules. For example, THY1 and NBL1 were significantly increased in MS, whereas a large number of neural signaling molecules including ROBO2 and EPHB6, were reduced in MS.
Figure 3. Volcano Plot of Soluble Markers in CSF From Donors With MS or Non-MS Diagnoses.

The vertical axis is the log10 of the p value for the comparison. The horizontal axis is the log2 of the fold change. Names of analytes with FDR of less than 0.05 are printed beside the point. Red points correspond to analytes that are significantly more abundant in CSF from patients with MS, and blue points to analytes that are less abundant in MS. Analytes discussed in the text are indicated with arrows, whose directions and colors are explained on the right of the figure. FDR = false discovery rate.
To visualize the relationships between the soluble molecules and the clinical parameters, we subjected the Olink data to principal component analysis (PCA), whose major contributing factors are shown in figure 4A and plotted the individual donors on plots of the principal components PC1 and PC2 (figure 4B). Two findings emerge. First, the MS and non-MS CSF samples are displaced along a vector dominated by molecules involved in neuronal development and plasticity, such as ROBO2, EPHB6, CNTN5, UNC5C, RGMB, and MDGA1. Second, the molecules that we found to be strongly associated with MS, including soluble CD27, CXCL10, FCRL2, CHIT1, CD8A, IL-12, and TNFSF14 form a second axis, orthogonal to the neuronal plasticity vector. On this MS disease activity vector, the early stages of MS are associated with a shift toward the lower right of the plot (clinically isolated syndrome and relapsing-remitting forms), whereas samples from donors at later stages of disease are found closer to the non-MS controls (figure 4B). Samples from treated patients also appeared closer to the center of the plot, although comparison of treated vs untreated patients with MS did not reveal significantly different molecules.
Figure 4. Principal Component Analysis of Soluble Molecules Measured by PEA.
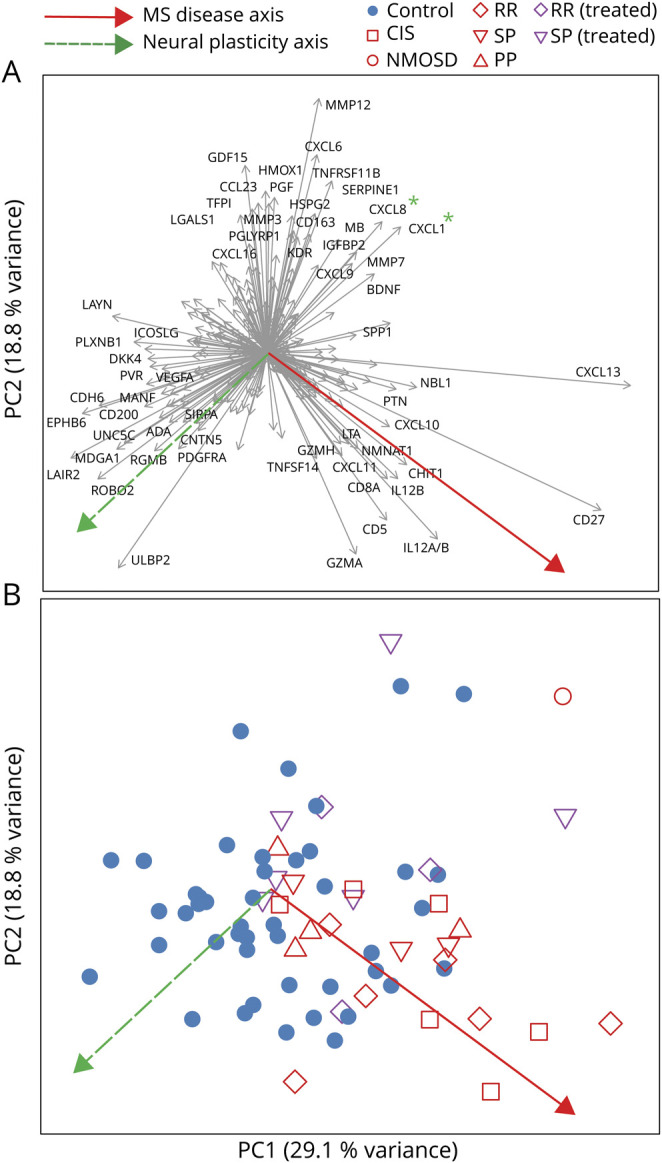
(A) The contribution of each of the soluble molecules to the first 2 principal components PC1 and PC2 is shown by plotting each as a vector. For example, CXCL13 makes a large contribution to PC1, whereas HMOX1 mostly contributes to PC2. For clarity, only the 60 most strongly discriminating molecules are labeled. The broken green arrow pointing down and left indicates a hypothetical axis of neural plasticity, inferred from the preponderance of molecules involved in neural development, axonal signaling, and so on. The red arrow pointing down and right indicates a hypothetical axis of disease activity, inferred from the preponderance of inflammatory molecules, and the presence in this quadrant of early disease stages (see plot B). Candidate MS-associated, plasticity-suppressive chemokines CXCL1 and CXCL8 (see Discussion) are marked with green asterisks. (B) PCA sample scores plot, where each point represents one donor, and the color and shape of each point indicate the diagnosis, disease stage, and treatment of the donor, as shown in the legend below the plot. Green and red arrows are identical in meaning to (A). PEA = proximal extension assay.
Soluble CD27 and FCRL2 Correlate Strongly With MS-Associated B-Cell Population
To elucidate the relationship between these soluble molecules and the cellular phenotypes characterized by CyTOF, we examined the correlation between each of the differentially expressed solutes and the abundances of the differentially abundant cell clusters (figure e-4, links.lww.com/NXI/A389). This revealed a group of genes including CD27, FCRL2, CXCL10, CXCL13 TNFSF14, and lymphotoxin-alpha, almost perfectly overlapping with the genes of the MS disease axis shown in figure 4, whose expression was strongly positively correlated with the abundance of the MS-associated B cell population (cluster 8 in figures 1 and 2). Independently from this group, BAFF (TNFSF13B) was negatively correlated with cluster 8. Also using this hierarchical clustering of the soluble proteins, based on correlation with the cell-type abundances, a clear group emerges that resembles the neural plasticity axis from figure 4. Proteins of this group do not show a clear relationship with the abundance of the MS-associated B-cell population, reinforcing the inference that these 2 axes (disease activity and neural plasticity) are independent.
Discussion
CyTOF enabled the identification of an MS-associated B-cell population using an unsupervised clustering of all CSF cells. This population does not fit perfectly into the established phylogeny of precursor, naive, memory, and plasma cells and may be a CSF niche phenotype. Other than the MS-associated subpopulation, B cells were present in similar proportions in MS and control CSF, although previous studies have found slightly elevated B-cell frequencies in relapsing remitting multiple sclerosis.3 In comparison with the other B cell types, the most striking peculiarities of the MS-associated cluster are strong expression of CXCR3 and CD27, and expression of CD69. Very high CD27 is a characteristic of plasma cells, but neither bone marrow plasma cells nor circulating plasmablasts express CD20, which was weakly expressed on the MS-associated CSF B cells. The CD27-high, CD20-positive phenotype is observed in tonsil plasma cells and can be recapitulated in vitro by stimulating memory B cells with TLR9 ligand.22 This treatment drives secretion of IgM, but the MS-associated B-cell population is IgM negative. The features of CD20 and CD49d expression make this B-cell population a target for CD20− depleting and integrin-blocking treatments, and consistent with previous reports,23 cells with this phenotype accumulate in peripheral blood during natalizumab treatment. CXCR3 expression, which enables CXCL10-driven chemotaxis, may drive migration into the CNS. CXCR3 expression is atypical for blood B cells but characteristic of transformed B cells in chronic lymphocytic leukemia,24 although an increased expression of CXCR3 on B cells in the CSF of patients with MS has been described.25 It was recently reported that CXCR3 expression on B cells in MS is driven by IFN-gamma and TLR9 stimulation.26 Although either stimulus induced the transcription factor t-bet in B cells, TLR9 stimulation was associated with switch to IgG1 memory B cells, while IFN-gamma stimulation lead to differentiation into plasmablasts. Differentiation into long-lived plasma cells can enable survival for decades in the CNS,27 whereas the phenotype of a memory cell is consistent with recent CNS ingress from the blood. The finding that MS CSF is characterized by a subtype of B cells is not so surprising, in view of the common presence of oligoclonal bands in the CSF in this disease. The MS-associated reduction in the B-cell survival factor BAFF might be expected to have a negative influence on B cell abundance in the CSF, but it has been reported that the trophic action of astrocyte secretions for B cells in vitro is BAFF-independent.28
These MS-associated B cells in the CSF have 3 possible effector mechanisms—they express HLA-DR and could stimulate T cells, and they could secrete pathogenic antibodies or inflammatory cytokines. The population is a small fraction of CSF cells, but its presence is robust to reanalysis with very stringent parameters for elimination of possible artifacts.
Several of the differentially expressed soluble factors suggest a significant involvement of B cells in MS and correlated with the MS-associated B-cell population. Most of the soluble markers could have various immune and nonsources, but FCRL2 is thought to be B cell restricted and to inhibit B-cell receptor signaling.29 Its expression in blood was reported to correlate with neurodegeneration in MS,30 and it was identified as a potential MS-associated gene,31 and the possibility of a pathogenic involvement in MS is strengthened by our findings.
Our PCA suggests that an axis of disease is reflected in high levels of soluble CD27, FCRL2, CXCL10, CXCL13, TNFSF14, and IL-12, and with advancing disease duration, this trend is reversed. Orthogonal to this MS-disease-stage axis is the vector dominated by molecules involved in neural development. Controls were distributed along this vector, but samples from patients with MS were skewed toward the upper right, i.e., showed reduced expression of these genes. This can be interpreted as suggesting that MS suppresses the ability of the CNS to counteract neurodegeneration with plasticity. Obvious candidates for plasticity-suppressive, MS-associated molecules include CXCL8 (IL-8) and CXCL1, whose vectors are diametrically opposed to the neural plasticity axis. Both are ligands of CXCR2, which is involved in the differentiation and migration of oligodendrocyte precursors, and whose disruption enhances remyelination in a mouse model.32 This interpretation has the encouraging implication that it should be possible to target this neural plasticity therapeutically, independently from immunomodulatory treatments.
CyTOF and the proximity ligation assay are well suited for assessing the complex phenotypes of cells and soluble molecules in CSF. Our integration of the 2 kinds of data identified a small population of B cells expressing CD27, CD20, and CXCR3 as the most characteristic cell population in the CSF of patients with MS and a soluble molecule phenotype characterized by CD27, CXCL10, and the B cell–specific membrane receptor FCRL2. Both the cellular and soluble signatures were observed in earlier disease stages and were diminished in secondary progressive patients. The MS-associated B-cell population that we observed, also expressing CD49d and CD20, ought to be responsive to already available treatments, albeit possibly more effectively so at earlier stages of the disease, before these cells enter the CNS. Other possible therapeutic options include targeting CXCR3 or CXCR2.
Acknowledgment
The authors are grateful to patients and staff of the University Hospital Basel for sample donation, sciCORE (scicore.unibas.ch/) scientific computing center at the University of Basel for computing resources and Aude Sylvain and Hélène Rossez for technical help.
Glossary
- BSA
bovine serum albumin
- CyTOF
cytometry by time of flight
- FDR
false discovery rate
- ICOS
inducible T-cell costimulator
- PCA
principal component analysis
- PEA
proximal extension assay
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate-buffered saline
Appendix. Authors
Contributor Information
David Johansson, Email: david.johansson@unibas.ch.
Céline Rauld, Email: celine.rauld@novartis.com.
Julien Roux, Email: julien.roux@unibas.ch.
Camille Regairaz, Email: camille.regairaz@novartis.com.
Edoardo Galli, Email: edoardo.galli@usb.ch.
Ilaria Callegari, Email: ilaria.callegari@unibas.ch.
Layla Raad, Email: layla.raad@novartis.com.
Annick Waldt, Email: annick.waldt@novartis.com.
Rachel Cuttat, Email: rachel.cuttat@novartis.com.
Guglielmo Roma, Email: guglielmo.roma@novartis.com.
Martin Diebold, Email: martin.diebold@usb.ch.
Burkhard Becher, Email: becher@immunology.uzh.ch.
Jens Kuhle, Email: jens.kuhle@usb.ch.
Tobias Derfuss, Email: tobias.derfuss@usb.ch.
José M. Carballido, Email: jose.carballido@novartis.com.
Study Funding
Swiss Multiple Sclerosis Society; Swiss National Science Foundation grant numbers 149966, 169674, 189043; Swiss Personalized Health Network (SPHN) project PRECISE; Novartis Institutes for BioMedical Research; and University Hospital Basel.
Disclosure
D. Johansson, J. Roux, E. Galli, I. Callegari, M. Diebold, B. Becher, and N.S.R. Sanderson report no disclosures. C. Rauld, C. Regairaz, L. Raad, A. Waldt, R. Cuttat, G. Roma, and J.M. Carballido are employees of Novartis Institutes for BioMedical Research. M. Diebold received speaker honoraria from Biogen Switzerland, which were used exclusively for research purposes. J. Kuhle received travel support, research support, speaker fees, and/or served on advisory boards by ECTRIMS, Swiss MS Society, Swiss National Research Foundation (320030_160221), University of Basel, Teva, Roche, Protagen AG, Novartis, Merck, Genzyme, Biogen, and Bayer. T. Derfuss serves on scientific advisory boards for Novartis, Merck, Biogen, Genzyme, GeNeuro, Mitsubishi Pharma, Actelion, Roche, Alexion, and Celgene; has received funding for travel and/or speaker honoraria from Biogen, Genzyme, Novartis, Merck, and Roche; and received research support from Biogen, Novartis, Roche, the European Union, the Swiss National Science Foundation, and the Swiss MS Society. Go to Neurology.org/NN for full disclosures.
References
- 1.Polman CH, O'Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006;354:899–910. [DOI] [PubMed] [Google Scholar]
- 2.Montalban X, Hauser SL, Kappos L, et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N Engl J Med 2017;376:209–220. [DOI] [PubMed] [Google Scholar]
- 3.Han S, Lin YC, Wu T, et al. Comprehensive immunophenotyping of cerebrospinal fluid cells in patients with neuroimmunological diseases. J Immunol 2014;192:2551–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Novakova L, Axelsson M, Khademi M, et al. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J Neurochem 2017;141:296–304. [DOI] [PubMed] [Google Scholar]
- 5.Jia Y, Wu T, Jelinek CA, et al. Development of protein biomarkers in cerebrospinal fluid for secondary progressive multiple sclerosis using selected reaction monitoring mass spectrometry (SRM-MS). Clin Proteomics 2012;9:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barbour C, Kosa P, Komori M, et al. Molecular-based diagnosis of multiple sclerosis and its progressive stage. Ann Neurol 2017;82:795–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Assarsson E, Lundberg M, Holmquist G, et al. Homogenous 96-plex PEA immunoassay exhibiting high sensitivity, specificity, and excellent scalability. PLoS One 2014;9:e95192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chevrier S, Crowell HL, Zanotelli VRT, Engler S, Robinson MD, Bodenmiller B. Compensation of signal spillover in suspension and imaging mass cytometry. Cell Syst 2018;6:612–e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Finck R, Simonds EF, Jager A, et al. Normalization of mass cytometry data with bead standards. Cytometry A 2013;83:483–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hahne F, LeMeur N, Brinkman RR, et al. flowCore: a Bioconductor package for high throughput flow cytometry. BMC Bioinformatics 2009;10:106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahne F, Khodabakhshi AH, Bashashati A, et al. Per-channel basis normalization methods for flow cytometry data. Cytometry A 2010;77:121–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nowicka M, Krieg C, Crowell HL, et al. CyTOF workflow: differential discovery in high-throughput high-dimensional cytometry datasets. F1000Res 2017;6:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Gassen S, Callebaut B, Van Helden MJ, et al. FlowSOM: using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 2015;87:636–645. [DOI] [PubMed] [Google Scholar]
- 14.Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linderman GC, Rachh M, Hoskins JG, Steinerberger S, Kluger Y. Fast interpolation-based t-SNE for improved visualization of single-cell RNA-seq data. Nat Methods 2019;16:243–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bates D, Maechler M, Bolker B, Walker S. Fitting linear mixed-effects models using lme4. JSS J Stat Softw 2015;67:1–48. [Google Scholar]
- 17.Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galli E, Hartmann FJ, Schreiner B, et al. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med 2019;25:1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smulski CR, Eibel H. BAFF and BAFF-receptor in B cell selection and survival. Front Immunol 2018;9:2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lownik JC, Luker AJ, Damle SR, et al. ADAM10-Mediated ICOS ligand shedding on B cells is necessary for proper T cell ICOS regulation and T follicular helper responses. J Immunol 2017;199:2305–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shea LK, Honjo K, Redden DT, et al. Fc receptor-like 2 (FCRL2) is a novel marker of low-risk CLL and refines prognostication based on IGHV mutation status. Blood Cancer J 2019;9:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Geffroy-Luseau A, Chiron D, Descamps G, Jego G, Amiot M, Pellat-Deceunynck C. TLR9 ligand induces the generation of CD20+ plasmablasts and plasma cells from CD27+ memory B-cells. Front Immunol 2011;2:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Saraste M, Penttilä TL, Airas L. Natalizumab treatment leads to an increase in circulating CXCR3-expressing B cells. Neurol Neuroimmunol Neuroinflamm 2016;3:e292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trentin L, Agostini C, Facco M, et al. The chemokine receptor CXCR3 is expressed on malignant B cells and mediates chemotaxis. J Clin Invest 1999;104:115–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sørensen TL, Trebst C, Kivisäkk P, et al. Multiple sclerosis: a study of CXCL10 and CXCR3 co-localization in the inflamed central nervous system. J Neuroimmunol 2002;127:59–68. [DOI] [PubMed] [Google Scholar]
- 26.van Langelaar J, Rijvers L, Janssen M, et al. Induction of brain-infiltrating T-bet-expressing B cells in multiple sclerosis. Ann Neurol 2019;86:264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Reiber H, Ungefehr S, Jacobi C. The intrathecal, polyspecific and oligoclonal immune response in multiple sclerosis. Mult Scler 1998;4:111–117. [DOI] [PubMed] [Google Scholar]
- 28.Touil H, Kobert A, Lebeurrier N, et al. Human central nervous system astrocytes support survival and activation of B cells: implications for MS pathogenesis. J Neuroinflammation 2018;15:114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jackson TA, Haga CL, Ehrhardt GR, Davis RS, Cooper MD. FcR-like 2 Inhibition of B cell receptor-mediated activation of B cells. J Immunol 2010;185:7405–7412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Comabella M, Cantó E, Nurtdinov R, et al. MRI phenotypes with high neurodegeneration are associated with peripheral blood B-cell changes. Hum Mol Genet 2016;25:308–316. [DOI] [PubMed] [Google Scholar]
- 31.Disanto G, Kjetil Sandve G, Ricigliano VA, et al. DNase hypersensitive sites and association with multiple sclerosis. Hum Mol Genet 2014;23:942–948. [DOI] [PubMed] [Google Scholar]
- 32.Marro BS, Skinner DD, Cheng Y, et al. Disrupted CXCR2 signaling in oligodendroglia lineage cells enhances myelin repair in a viral model of multiple sclerosis. J Virol 2019;93:e00240–e00319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymous data will be shared by any qualified investigator on request.