

Abstract
Oral vaccination enables pain-free and self-administrable vaccine delivery for rapid mass vaccination during pandemic outbreaks. Furthermore, it elicits systemic and mucosal immune responses. This protects against infection at mucosal surfaces, which may further enhance protection and minimize the spread of disease. The gastrointestinal (GI) tract presents a number of prospective mucosal inductive sites for vaccine targeting, including the oral cavity, stomach, and small intestine. However, currently available oral vaccines are effectively limited to live-attenuated and inactivated vaccines against enteric diseases. The GI tract poses a number of challenges, including degradative processes that digest biologics and mucosal barriers that limit their absorption. This review summarizes the approaches currently under development and future opportunities for oral vaccine delivery to established (intestinal) and relatively new (oral cavity, stomach) mucosal targets. Special consideration is given to recent advances in oral biologic delivery that offer promise as future platforms for the administration of oral vaccines.
Keywords: oral biologic delivery, oral vaccines, mucosal immunization, protein, DNA, mRNA
INTRODUCTION
Oral delivery is the preferred route for the administration of therapeutics. Compared to parenteral administrations requiring injection, oral delivery is noninvasive, convenient, and can be self-administered (1), which leads to increased compliance and greater potential for improved population coverage and health outcomes. Due to physical barriers and degradative processes in the gastrointestinal (GI) tract, however, efficient oral delivery of fragile macromolecules such as biologics has remained an almost unattainable goal (2). The key exceptions and successes are select types of vaccines (3–5) and, more recently, biologics in combination with permeation enhancers (PEs) (6, 7). The first oral vaccines were developed against poliovirus in the 1950s by Hilary Koprowski and Albert Sabin, with Sabin’s trivalent oral polio vaccine (OPV) the most utilized option for much of the twentieth century. Since then, oral vaccines have been developed against cholera, typhoid fever, gastroenteritis (rotavirus), and acute respiratory disease (adenovirus) (3, 4). Most pathogens enter the body through the GI, urogenital, and respiratory mucosal surfaces (4). Oral vaccination elicits both systemic and mucosal immune responses, providing an additional immunological benefit by protecting against infection at mucosal surfaces. While intramuscular (IM) vaccine administration elicits effective systemic immunity, it provides weak mucosal immunity (4, 5) and requires trained personnel to administer, which limits vaccination reach in pandemic outbreaks. Furthermore, mucosal infections may persist in IM-immunized individuals (8, 9), which can contribute to subsequent transmission to unimmunized or immunocompromised individuals. Despite these advantages, however, the proportion of vaccines administered orally remains small.
The GI tract provides significant challenges to the oral delivery of vaccines, including degradation, poor uptake through mucosal barriers, and a bias toward tolerability of antigens (3). A number of regions within the GI tract present prospective inductive sites for vaccine delivery, including the oral cavity (10), stomach (11), and intestines (3, 4), with each presenting unique challenges and opportunities. No comprehensive clinical solution for efficient oral delivery of a broad range of biologics or vaccines has yet been achieved, though this remains a highly active area of research (6, 7, 12–14). Approved oral vaccines are currently limited to diseases that affect the GI tract (3) and whole-virus/cell vaccines (i.e., either attenuated live or inactivated). These structurally intact pathogens are highly immunogenic; can better withstand GI degradative processes; and adhere to or replicate within mucosa, which improves their uptake. Other types of vaccines such as subunit (proteins and polysaccharides), DNA, and messenger RNA (mRNA) vaccines are sensitive to degradation, are considerably more challenging to deliver orally, elicit strong GI immune responses, and have not been successfully translated in the clinic. Furthermore, DNA and mRNA vaccines are of particular interest in response to pandemic outbreaks given their potential for rapid and cost-effective manufacture (15, 16).
Oral biologic delivery systems for both vaccination (3, 4, 17) and systemic therapies (2, 12, 13) are currently under development. Oral vaccination approaches typically exploit the body’s ability to distinguish and process threats at mucosal surfaces, resulting in pathogen-specific mucosal uptake, processing, and the initiation of immune responses. These approaches have been widely investigated for oral vaccination through intestinal delivery but are unsuitable for the oral cavity or stomach. Systemic delivery of biologics requires physical or chemical mechanisms to pass through mucosal barriers, and in the last few years, several promising platform technologies have been developed for the oral delivery of biologic therapeutics (7, 18–23), which may enable vaccination to a wider array of GI mucosa. This review summarizes the potential and the challenges of oral vaccine delivery to different GI mucosal inductive regions; key design considerations for oral delivery of subunit and nucleic acid vaccines; and the current state of research, including recent advances in oral biologic delivery more broadly, which may ultimately enable new approaches for oral vaccinations.
CHALLENGES OF ORAL VACCINE DELIVERY TO GASTROINTESTINAL MUCOSA
In their simplest form, the GI mucosae consist of a protective mucus membrane and a cellular epithelial layer (Figure 1), which protect the host from unregulated internalization of pathogens and harmful substances. The mucus lining, as the first point of contact, provides a physical barrier that limits molecular diffusion and microorganism intrusion to the epithelial layer. The structure and function of the epithelial layer vary considerably between oral, gastric, and intestinal mucosa and even within the oral cavity, but a consistent function is the prevention of unwanted uptake of pathogens and macromolecules, which is achieved through several tightly packed cellular architectures. Additional protection against microorganisms is imparted by the secretion of antimicrobial compounds within the mucus membrane, as well as by the innate and adaptive mucosal immune system. Regions of low pH (stomach), bile salts (intestines), and digestive enzymes add an extra layer of protection and challenge for oral vaccine delivery. The GI tract is also a dynamic system, with the residence time of contents limited by gastric emptying and intestinal motility, which restrict opportunities for the absorption of macromolecules. In summary, effective delivery of oral vaccines requires overcoming degradative processes, mucosal barriers, transient contact with epithelia, and a relatively tolerant immune system (discussed in the next section). The exact nature of these challenges and opportunities also depends on the target vaccination site within the diverse environment that is the GI tract.
Figure 1.
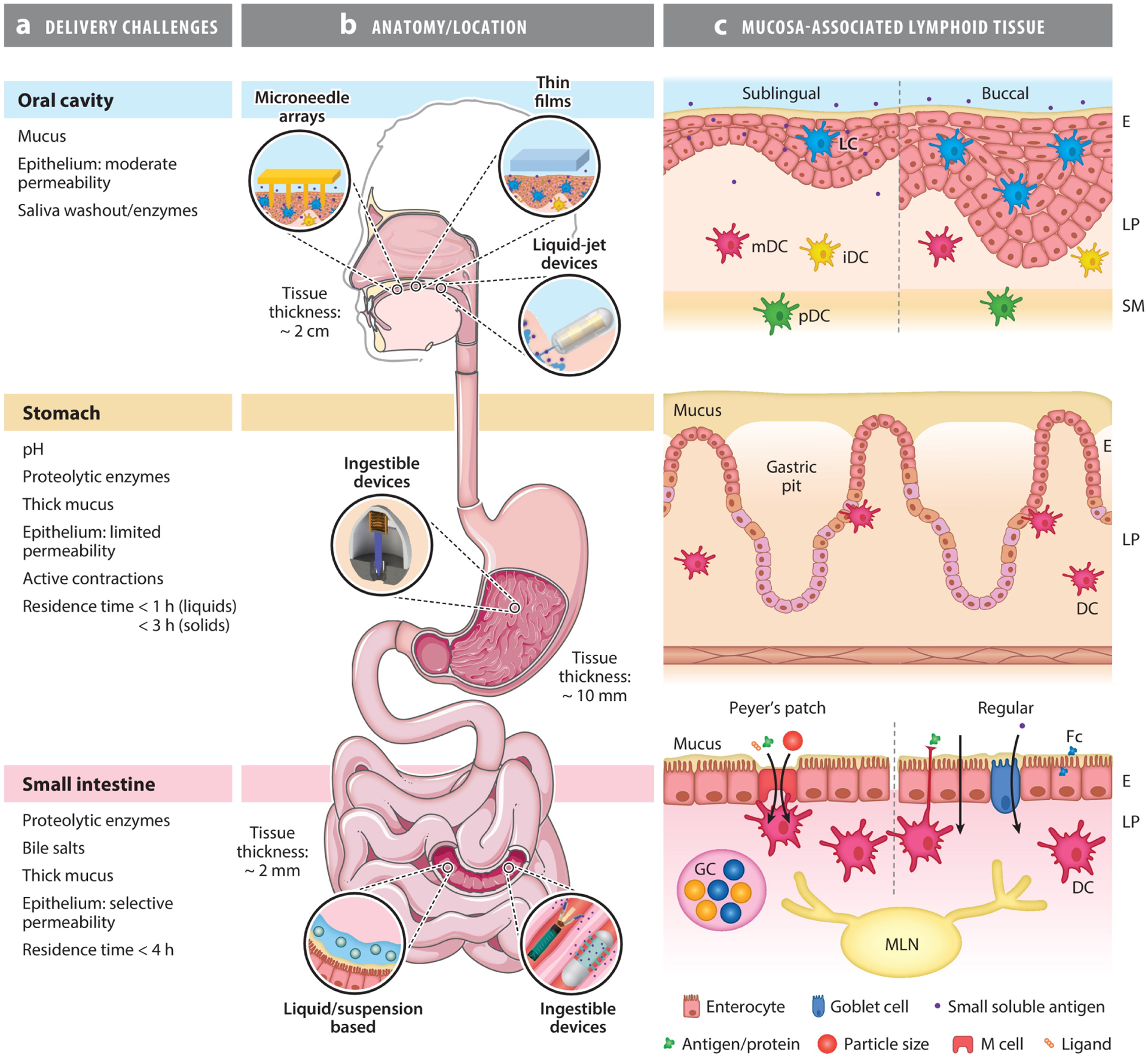
Challenges and opportunities for oral vaccine delivery to the oral cavity, stomach, and small intestine and anatomy of their respective mucosal inductor sites. Each region has unique physiochemical barriers to vaccine absorption and organization of immune inductive tissue. (a) Summary of the delivery challenges to these regions. (b) Relative location of target mucosae and examples of broad delivery approaches that have been used to deliver vaccines or biologics to each site. Panel b partially adapted from Servier Medical Art (https://smart.servier.com/smart_image/complete-digestive-apparatus/) and Reference 69. Reprinted with permission from Servier Medical Art and AAAS. (c) The structures of the mucosae and inductor sites at each location. The location of key known APCs, including DCs, and lymphoid tissue are indicated. The sublingual and buccal mucosae contain nonkeratinized stratified squamous epithelia, which differ in thickness (100–200 μm and 500–800 μm, respectively). This allows for increased diffusion of macromolecules in the sublingual mucosa compared to the thicker buccal mucosa. LCs are present in the epithelium, mDCs and iDCs in the lamina propria, and pDCs in the submucosa. The gastric mucosa is lined with gastric pits, a comparatively thick mucus lining, and a simple columnar epithelium with tight cell-cell junctions that prevent macromolecule absorption. Characterization of the gastric mucosa as an inductor site is limited; however, DCs have been observed associated with the epithelia of gastric pits. The intestinal mucosa contains a protective mucus lining and simple columnar epithelium that is capable of recognizing and uptaking pathogens/particles/antigens via several routes, depending on the properties of the pathogen/particle/antigen. These routes occur via specialist M cells (in the follicle-associated epithelium of Peyer’s patches only, which have a thinner mucus lining), interdigitated DCs, enterocytes and goblet cells, and through intercellular junctions. Note the intestinal epithelium forms high-surface-area villi, which are not shown schematically due to scale. Abbreviations: APC, antigen-presenting cell; DC, dendritic cell; E, epithelium; Fc, fragment crystallizable; GC, germinal center; iDC, interstitial dendritic cell; LC, Langerhans cell; LP, lamina propria; M cell, microfold cell; mDC, myeloid dendritic cell; MLN, mesenteric lymph node; pDC, plasmacytoid dendritic cell; SM, submucosa.
THE GASTROINTESTINAL IMMUNE SYSTEM
The basis of mucosal immunity is the migration of cells that recognize antigens [antigen-presenting cells (APCs)] from mucosal inductor sites through the lymphatics to recruit immune cells that protect against foreign pathogens to mucosal effector sites (24). The GI tract has a well-established immune system that prevents against mucosal infections (though significantly, not all organs/mucosae have been well characterized) (8, 25). Despite this, GI mucosal surfaces are colonized with an abundance of microorganisms, particularly in the intestines, some of which impart significant benefits to the host (26). Colonization is facilitated by complex mechanisms of immune tolerance, with inductor sites distinguishing between commensal and potentially harmful bacteria (27) as well as many benign environmental antigens (e.g., in food). There is thus a substantial bias toward immune tolerance to avoid unnecessary responses to these bacteria/antigens, which poses a challenge in generating immune responses to vaccination (25, 28). Inductor sites from multiple mucosae (e.g., respiratory, GI, urogenital) form a network known as mucosa-associated lymphoid tissue (MALT). Inductor sites from one mucosa may initiate immune responses at distal mucosae (8, 29–31) in what is referred to as the common mucosal immune system; however, mucosal responses are generally strongest proximal to the site of induction. For example, vaccination to the oral cavity can induce responses in the small intestine and respiratory mucosa (29), while some mucosal compartments [e.g., rectal delivery (32)] induce local rather than broad mucosal responses. Similarly, for optimum oral vaccine delivery, it may be necessary to understand a particular pathogen’s route of entry and target a specific delivery site within the GI tract that could offer the best balance of regional mucosal and systemic immune responses.
Several regions of the GI tract offer potential targets for oral vaccination, including the oral cavity, stomach, and small intestine (Figure 1). The immune system of the small intestine is the most extensively studied and the target of all currently approved oral vaccines (3, 4, 8). The oral cavity has received far less attention for vaccination (10), though it has been well studied for allergen immunotherapy (33, 34) and is the most easily accessible GI inductor site. Recently, however, there has been increased development of devices enabling efficient vaccine delivery to the oral cavity mucosa (35) (see Figure 1b). Conversely, the stomach has received little attention as a site for vaccination; however, recent demonstrations of ingestible devices for biologic delivery to the stomach (20) and of gastric tissue being an inductive site (11) suggest that the stomach has potential as a future vaccination site.
Oral Cavity
The oral cavity is the most accessible mucosal inductive site for vaccine delivery. The sublingual (SL) region (underside of the tongue and bottom surface of the mouth) and buccal mucosa (cheeks, gums, and inner surface of the lips) are the most prominent targets for vaccine delivery. Their thinner, nonkeratinized epithelia (Figure 1c) allow for both some adsorption of macromolecules and easier mechanical disruption for rapid macromolecule delivery compared with the keratinized epithelium present elsewhere in the oral cavity (e.g., the hard palate). Furthermore, the lipid lamellae between epithelial cells in nonkeratinized epithelia are comparatively less organized, which allows for increased diffusion and uptake of biomacromolecules (36). APCs that are present in these mucosal areas include Langerhans cells [LCs; suprabasal layer of the epithelium (10)] and several types of dendritic cells (DCs), including myeloid, interstitial [lamina propria (LP)], and plasmacytoid cells (in the submucosal tissue, which infiltrate the mucosa during inflammation). There are key differences between the buccal and SL mucosae, such as epithelial layer thickness and relative abundance of APCs, which play a role in site selection for vaccination. The SL mucosa has a particularly thin and permeable epithelium, which allows for absorption of macromolecules, and has been extensively investigated for sublingual immunotherapy (33, 34). In contrast, the buccal mucosal epithelium is thicker and considered to be less absorptive. Saliva flow may also dilute or wash out vaccine from the oral cavity before it is significantly absorbed, and it contains some potentially degradative enzymes. Thus, perturbing the mucosal barrier for rapid uptake is advantageous for vaccine delivery to both the buccal and SL mucosae (Figure 1c). Furthermore, interstitial CD11b+CD11c+ myeloid DCs appear to be the major DCs responsible for T cell priming in the oral mucosa, which is crucial for the adaptive immune response. These are abundant in the LP of both the buccal mucosa and, to a lesser degree, the SL mucosa. LCs in the epithelium, which are also more abundant in the buccal mucosa, are thought to have a primarily tolerogenic role (37). These mucosae are therefore particularly attractive vaccination sites; however, it is likely beneficial to deliver antigen in sufficient quantities to the depth of these DCs in preference over more superficial LCs, highlighting the need for efficient delivery technologies such as mechanical perforation.
Stomach
The stomach is the next most accessible part of the upper GI tract (Figure 1). Ingested material takes only seconds to minutes to pass through the esophagus. The gastric mucosa contains a relatively thick mucus lining and simple columnar epithelia with tight cell-cell junctions that prevent the absorption of molecules and acidic gastric fluid. Knowledge of the stomach immune system is limited and has been largely characterized in the context of Helicobacter pylori infection. The presence of DCs and inductive site capacity in healthy gastric mucosa has been doubted (38–40). However, HLAhigh/CD13low and CD13high DCs have now been definitively identified in healthy human stomach and implicated in the development of a Th1 response to H. pylori (38). Primary and secondary lymphoid follicles have also been described in healthy human gastric mucosae (41) and the mucosae and submucosae of healthy piglets (42). Recently, it was shown that subserous delivery of an H. pylori vaccine to the murine stomach wall (via surgical laparotomy) generated new perivascular lymphocyte clusters and CD4+ tissue resident memory T cells (11, 43, 44), further confirming inductive potential. Moreover, our group has developed an ingestible self-orienting device that can inject a dose of biologic directly into the gastric wall (20). Thus, while the stomach has been understudied for vaccine delivery, recent demonstrations of its inductive potential and capacity for biologic delivery support its potential as a site for vaccination.
Small Intestine
The small intestine is the target site for all currently approved oral vaccines and for the majority of investigational reports (3, 4, 17). Its mucosal immune system has been extensively characterized, and a review of its key mechanisms elucidates why it has been traditionally considered an ideal target for oral vaccines. Unlike the lining of the oral and gastric cavities, the intestinal mucosa can recognize foreign antigens and transport them through mucosal barriers for presentation to the immune system (25, 45) (Figure 1c). Multiple inductor sites have been identified in the intestinal mucosa, which are collectively referred to as gut-associated lymphoid tissue (GALT). The most prominent of these inductor sites is the Peyer’s patch (PP), consisting of small discrete areas of highly ordered and specialized immune cells beneath a follicle-associated epithelium (FAE). The FAE recognizes and aids the transport of specific pathogens and antigens into the PPs. Microfold cells (M cells) of the FAE act as the primary gateway for antigens into the PPs. These M cells express a diverse variety of receptors to foreign antigens, including pattern recognition receptors (PRRs) (46), on their apical surface through which M cells recognize and transcytose foreign pathogens to the basolateral membrane and deliver them to underlying APCs (45). Furthermore, M cells may also transcytose particles based on size ranges comparable to those of pathogens (47–51). The surface of M cells also has a thinner or absent mucus lining that lacks or contains shorter microvilli than neighboring epithelial cells, which facilitates pathogen contact with the cell (52). Beneath the FAE is the subepithelial dome, which contains DCs, the major APC of the immune system. Pathogens and antigens are transferred by M cells to these DCs, which process antigens and present fragments to downstream cells (naïve CD4+ T cells) in the immune system (45). Additionally, the PP also contains germinal centers that are rich in both B and T cells.
Other discrete inductor sites in the intestinal mucosa include mesenteric lymph nodes (MLNs) and the lesser studied lymphoid follicles. DCs may also be dispersed in smaller numbers throughout the mucosa and, unique to GI DCs, possess dendrites that extend through the epithelia into the lumen and may directly recognize and uptake antigens (53, 54). Enterocytes—the most ubiquitous cells of intestinal epithelia—are also capable of taking up and presenting some antigens via the neonatal fragment crystallizable (Fc) receptor (55). Finally, some studies suggest synthetic particles may be transported by enterocytes through non-Fc receptor–mediated pathways or by paracellular transport between enterocyte cell junctions, presenting possible opportunities for vaccine design (56). In contrast, goblet cells have been shown to transport low molecular weight–soluble antigens to DC subsets that promote tolerance (57). In summary, a range of pathogenic features that are recognized by the intestinal mucosa enable internalization and direct transfer to APCs for presentation to the immune system, including particle size, antigenic epitopes, molecular patterns, and other targeting ligands, which have the potential to be leveraged as mechanisms for vaccine uptake.
Effector Sites and the Mucosal Immune Response
Following the initial exposure to antigen in the GI tract, mucosal lymphocytes migrate through the lymphatics, priming and activating immune cells that then home to effector sites. The major effector sites of the GI tract are the LP (24); the surface of the epithelium (24); and, in the oral cavity, the secretory saliva glands (58, 59). DCs at mucosal inductor sites typically internalize and process antigens into small fragments for presentation to naïve T cells via one of two routes. A portion of DCs migrate via draining lymphatics to germinal centers in the MLNs, where they prime naïve CD4+ T cells (which are crucial in initiating an immune response). Primed CD4+ T cells then activate neighboring antigen-specific B cells in the germinal center, which class-switch to become IgA+ B cells. The IgA+ B cells then enter the bloodstream before homing to tissue effector sites. Alternatively, DCs may prime local naïve CD4+ T cells within mucosal germinal centers, e.g., within the PPs. These T cells in turn activate antigen-specific B cells, which class-switch to IgA+ B cells and then leave the PP through the lymphatics and MLN and reach the circulation for homing to effector sites.
DCs in the GALT also produce retinoic acid, which interacts with activated T cells to release homing receptors. These homing receptors direct the migration of circulating IgA+ B cells to mucosal effector sites in the LP. There, they differentiate and mature to become IgA-producing plasma cells, which produce a high-affinity version of antigen-specific IgA. Enterocytes in the epithelium express the polymeric Ig receptor (pIgR) on their basal surface, which binds to IgA (and also IgM antibodies). Thus, IgA from the LP binds to the basolateral pIgR, which is internalized and transported across the epithelial cell. The IgA is then internalized and translocated to the luminal surface where it is released as secretory IgA (sIgA), the primary molecule responsible for mucosal immunity. This sIgA can then neutralize pathogens (24), conferring the epithelial surface as a major effector site of the GI immune system. In the oral cavity, sIgA may also be produced in salivary glands (58, 59).
CLINICALLY APPROVED ORAL VACCINES
Currently, all approved oral vaccines (see Table 1) protect against diseases that affect the GI tract or against pathogens that have a crucial life cycle stage within the GI tract. These include cholera, gastroenteritis (rotavirus), polio, typhoid fever, and acute respiratory disease (adenovirus), which although not pathogenic in the GI tract, establishes an asymptomatic infection in the intestine that is exploited for vaccination. All of the approved oral vaccines for these diseases employ live-attenuated or inactivated pathogens. As with the wild-type forms of these pathogens, the attenuated and inactivated forms can endure degradation or remain sufficiently intact through transit in the gastric and intestinal environment and elicit a stronger immunogenic response in the GI mucosa than other vaccine types (e.g., subunit and nucleic acid vaccines). Importantly, adenovirus immunization demonstrates the principle that GI immunization can be effective against systemic diseases without GI pathogenicity, and this has been extensively investigated as a vector for oral delivery of plasmid DNA vaccines (covered later). Some clinical trials have investigated the GI tract for immunization against diseases that are not related to the GI tract, such as influenza (60, 61), and that use other vaccine types and/or adjuvants, e.g., DNA vaccines (61) and RNA adjuvants (60), but these have not yet resulted in approved commercial products. Thus, there is significant opportunity for advances in vaccine delivery technology that can efficiently protect vulnerable vaccine components in the gastric environment, deliver them effectively to the epithelia and APCs, and co-stimulate (i.e., adjuvant) immune responses to typically less antigenic vaccines (e.g., subunit, DNA, and mRNA vaccines).
Table 1.
Clinically approved oral vaccines
| Disease (pathogen) | Vaccine | Antigen/vaccine type | Adjuvant | Formulation |
|---|---|---|---|---|
| Cholera (Vibrio cholerae) | Dukoral | Inactivated V. cholerae recombinant CTB | NA (CTB) | Liquid |
| Vaxchora | Live-attenuated V. cholerae O1 Inaba 569B strain | NA | Liquid | |
| Gastroenteritis (rotavirus) | RotaRix | Live-attenuated | NA | Liquid |
| RotaTeq | Live-attenuated pentavalent reassortment human/bovine viruses G1, G2, G3, G4, and P1A[8] | NA | Liquid | |
| Polio | Orimune | Live-attenuated poliovirus serotypes Sabin 1, 2, and 3 | NA | Liquid |
| Typhoid fever (Salmonella typhi) | Vivotif | Live-attenuated S. typhi | NA | Enteric capsule |
| Acute respiratory syndrome (adenovirus) | Approved for military populations only | Adenovirus types 4 and 7 | NA | Enteric capsule |
Abbreviations: CTB, cholera toxin B; NA, not applicable.
STRATEGIES AND DELIVERY SYSTEMS TOWARD ORAL VACCINATION
A broad range of approaches have been investigated to overcome the challenges of oral vaccine delivery (Figure 2). These approaches typically vary depending on the characteristics of the target inductor site, i.e., the oral cavity, stomach, or intestines (see Table 2). An overview of salient GI delivery challenges and strategies for overcoming them includes the following five points. First, fragile proteins, polysaccharides, and nucleic acids must be protected from degradation during gastric and intestinal transit, but less so in the oral cavity. This may be achieved by loading vaccine into particles (3, 4, 48), enteric capsules (62), or physical devices (20). Second, enhanced uptake across mucosal barriers is required and can be achieved by mechanical disruption of the epithelial barrier (35), the use of targeting ligands (63, 64), or vaccine-carrying particles with sizes that are preferentially absorbed mucosally (small intestine only) (50). Third, non-cell-based soluble antigens lack the key immunogenic characteristics of whole pathogens that the body’s immune cells are effective at recognizing (46), which is particularly problematic in the tolerogenic environment of the GI tract. To overcome this, approaches have been developed that incorporate mucosal adjuvants (65) and/or delivery vehicles that mimic pathogenic properties and increase immunogenicity such as pathogen-sized particles as vaccine carriers (50), functionalization with molecular patterns typically presented by pathogens (46, 66), and the development of virus-like particles (VLPs)—individually expressed viral structural proteins that self-assemble into particles that resemble viruses (67). Fourth, the GI tract is a dynamic environment, giving potential vaccines limited resident times through each portion of the GI tract (Figure 1a). Contact time with the mucosa can be enhanced by the use of mucoadhesive strategies (68) or avoided altogether by physical devices that rapidly deliver a payload into the mucosa (20, 35, 69, 70). Fifth, the thermostability of vaccine formulations also needs to be considered, as it is critical in developing countries where cold-chain infrastructure is limited (71); this is particularly true for oral vaccines, given that their potential for self-administration is a key opportunity to improve coverage in remote and developing areas where infectious diseases are often most prevalent. The incorporation of stabilizing excipients and/or use of dry solid formulations has been shown to improve thermostability (72) and may need to be employed with oral vaccine formulations. This section summarizes major design considerations when developing oral vaccine approaches and the types of delivery systems that have been employed.
Figure 2.
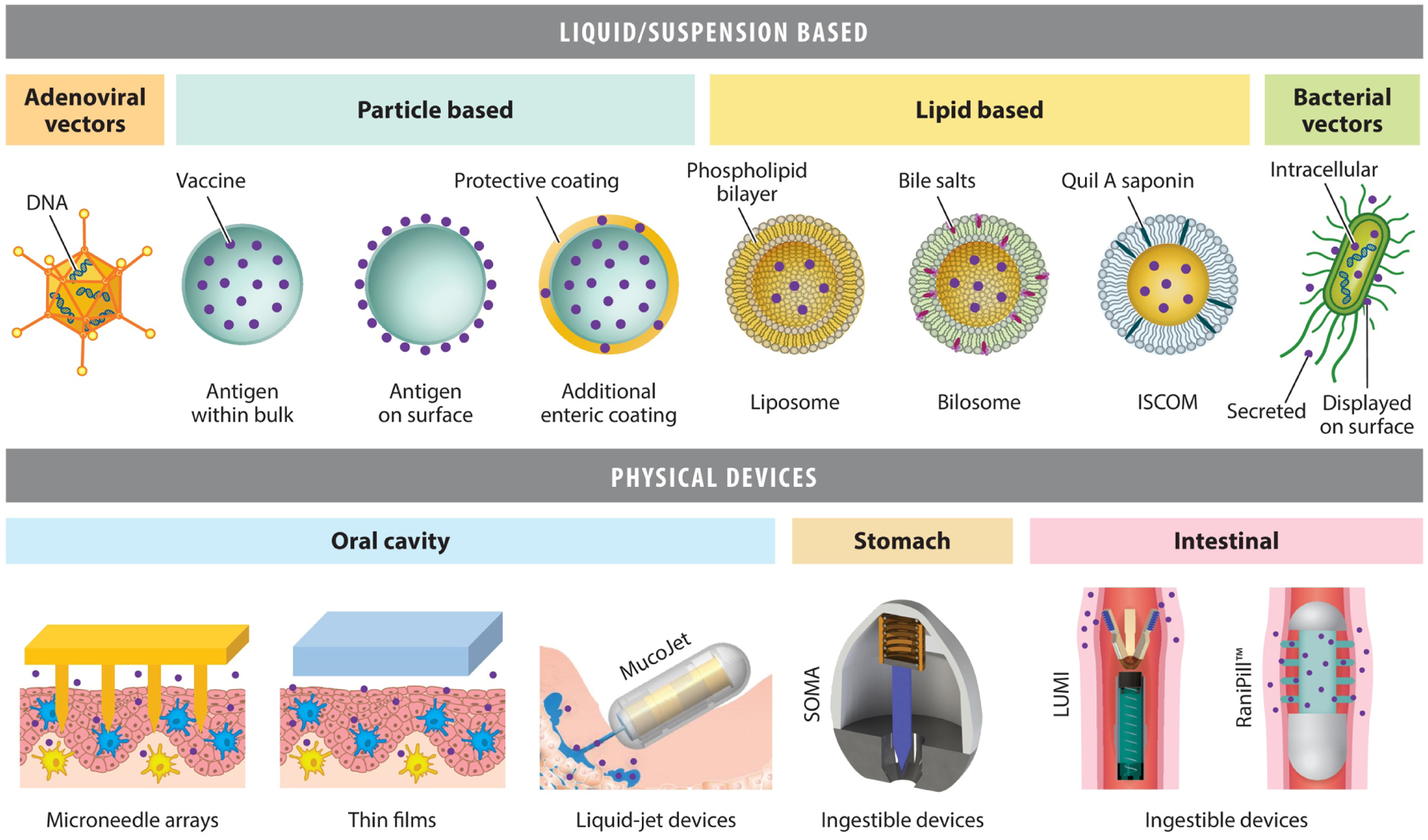
Summary of the main delivery technologies in development for oral vaccination. A variety of liquid/suspension-based approaches have been investigated, including microparticles and nanoparticles, adenoviral and bacterial vectors, and lipid-based approaches. Adenoviral vectors contain a DNA-based cargo that is delivered to and expressed as antigen by host cells in vivo. Bacterial vectors are engineered with DNA to express vaccine antigens that may be either secreted, presented on the cell surface, or contained intracellularly and released in vivo. Particle- and lipid-based approaches may potentially carry vaccine either as preformed antigens or as nucleic acids for expression in vivo. More recently, there has been an expanding range of physical devices that localize vaccine release and/or mechanically disrupt mucosa for highly efficient delivery of a broad range of biologics to target sites in the gastrointestinal tract. These include microneedle arrays, thin films/patches (e.g., mucoadhesive and/or disintegrating), liquid-jet injectors (such as the MucoJet), SOMA, LUMI, and the RaniPill™. MucoJet panel reproduced from Reference 69. Reprinted with permission from AAAS. Abbreviations: ISCOM, immune-stimulating complex; LUMI, luminal unfolding microneedle injector; SOMA, self-orientating millimeter-scale actuator.
Table 2.
Delivery technologies for oral vaccination
| Delivery system/mode of delivery | GI mucosal target | Vaccine type | Adjuvant | Targeting ligand/mucoadhesion/localization | Common materials and/or form |
|---|---|---|---|---|---|
| Microparticles/nanoparticles | Oral (85) (limited) sI (47, 48, 51, 63, 93, 158) |
Subunit (47, 48, 159) DNA (76, 77) mRNAa |
Self-adjuventing (47, 88, 159) Codelivery (160) |
Targeting ligand (63, 66, 93) Mucoadhesion (68) Localization (85) |
PLGA/PLA (47, 48, 76, 93), PA (102), PEI (104), chitosan (68), PCL (103), alginate (105) |
| Liposomes/bilosomes/ISCOMs | sI (111) | Subunit (116) DNA (111) mRNAa |
Codelivery (117) | Targeting ligand (114) Mucoadhesion (161) |
Lipids (162), lipid + salts (65, 116), lipid + adjuvant (163) |
| Bacterial and viral vectors | Oral (123) sI (125, 164) |
DNA (125) mRNAa |
Self-adjuventing | Mucoadhesion (165) | Adenovirus (125) Salmonella (166) Poxvirus (167) VLPs (66) |
| Physical devices | Oral (146) Stomacha (20) sIa (22, 150) |
Subunit (69) DNAb mRNAa |
Mechanical (147, 148) Codeliveryc |
Device localization (20, 22, 69, 70, 145, 146, 150) | Microneedles (145, 146), liquid jet (69, 70), ingestible devices (20, 22, 150), thin films (85) |
Delivery technology is highly suitable for mRNA vaccine but to our knowledge has not been demonstrated for vaccination through the oral route.
Delivery technology is highly suitable for DNA vaccine but to our knowledge has not been demonstrated for vaccination through the oral route.
Platform technology is capable of delivering biologics orally, including vaccines and/or adjuvants; however, it has not yet been demonstrated for vaccination.
Codelivery refers to incorporation within a micro- and/or nanoparticle or lipid-based vehicles.
Abbreviations: ISCOM, immune-stimulating complex; mRNA, messenger RNA; PA, polyanhydride; PCL, poly-ε-caprolactone; PEI, polyethyleneimine; PLA, polylactic acid; PLGA, poly(lactic-coglycolic) acid; sI, small intestine; VLP, virus-like particle.
Microparticles and Nanoparticles
Microparticles and/or nanoparticles (MNPs) are the most widely investigated delivery system for oral subunit (protein and polysaccharide) vaccines (3, 4, 73–75). MNPs offer considerable versatility with the ability to both tailor and combine multiple functionalities into a single particle through variation in particle design, materials properties, and surface functionalization. MNPs also have potential for nucleic acid vaccine delivery (which requires additional transfer of nucleic acids across cell membranes) and have been used to deliver plasmid DNA encoding for antigens (76–80). Oral mRNA vaccination has not to our knowledge been reported; however, MNPs have been used to transfer mRNA vaccines into cells via other delivery routes, suggesting a potential for oral delivery (15, 81, 82). MNPs may be administered via solution (4, 83) or enteric capsules (62), where they are uptaken by GALT in the small intestine. MNPs have also been delivered via topical administration to the SL mucosa; however, due to the extremely short residence time of solutions in the oral cavity, MNPs typically require mucoadhesive (84) or localization (85) strategies to improve uptake or rapid delivery through physical injection (69, 70), particularly in the comparatively less absorptive buccal mucosa.
Protection of the vaccine from the GI environment can be achieved in a number of ways, including by loading vaccines within the MNP carrier or adsorbing them to its surface (Figure 2). While surface loading provides less protection, it may render antigens more available for immune recognition. In either case, further protection can be achieved through an additional enteric surface coating on particles (86). Importantly, many MNPs also intrinsically improve uptake across epithelial barriers and stimulate the immune system (87). It is well established that certain particle sizes, ranging from several hundred nanometers in diameter (88, 89) to approximately 10 μm (50, 51, 90), are recognized and transcytosed by M cells and enterocytes, though the exact optimal size range has been debated and may be affected by other physicochemical properties of the particle, complicating comparisons between studies. Smaller particles (<5 μm) are transported by macrophages to MLNs and ultimately the spleen, clearing them from the mucosa (47, 50, 91). Larger particles that remain in mucosa for longer periods have been shown to result in higher sIgA (50), suggesting a sustained-release mucosal depot may be an effective strategy for vaccination. Furthermore, MNPs are also taken up by APCs and DCs in a size-, shape-, and charge-dependent manner (49, 92), which could be further exploited to enhance the immunogenicity of MNP vaccines. A major advantage of MNPs is that they can be imparted with multiple functionalities through the incorporation of targeting ligands for receptors on epithelial cells and with adjuvants to stimulate immune responses. Several peptides have been shown to increase both binding to M cells and mucosal uptake when attached to MNPs (64, 93). Arginylglycylaspartic acid (or RGD peptide) has also been shown to increase uptake to M cells (63), as have mucoadhesive lectins (94). In one study, multiple Toll-like receptor (TLR)-targeting ligands (macrophage-activating lipoprotein, polyinosine-polycytidylic acid, and CpG oligodeoxynucleotide) encapsulated in MNPs were shown to target the large intestine (66), which has an abundance of GALT.
Biodegradable polymers are the most frequently used material for MNPs due to their biocompatibility and ability to provide sustained vaccine release (83, 95). Sustained release has resulted in increased antibody and T cell responses compared to pulsatile delivery (50, 96). The most widely used biodegradable polymers are polylactic acid (PLA) and poly(lactic-coglycolic) acid (PLGA) (47, 48, 50, 83, 86, 97–100), owing to their biocompatibility, biodegradability, relative ease of use, capability to tailor release rate such as for sustained release, and their wide approval by the US Food and Drug Administration for drug-release applications. Additionally, coblock polymers of PLA/PLGA with other polymers, for example poly-d,l-lactide–copoly(ethylene glycol), provide additional tuning of release kinetics (97). While PLA/PLGA MNPs protect vaccines from external degradation, their biodegradation/hydrolysis with water forms acidic breakdown components, which may affect vaccine stability and needs to be thoroughly evaluated. PLGA (76, 77) and chitosan (78) microparticles have also been used to deliver antigen-encoding DNA plasmids for oral vaccination, while biodegradable particles for DNA vaccination have been widely investigated through other routes, suggesting further potential for oral DNA vaccination (101).
Polyanhydrides (PAs) (102), polystyrene (98), poly-ε-caprolactone (103), polyethyleneimine (PEI) (104), and naturally occurring polymers such as alginate (105, 106) and chitosan (73, 78) have also been used for oral vaccine delivery (95, 107). PA has been shown to be beneficial compared with PLA/PLGA, as it does not form localized acidic breakdown components (95). PEI has been used to form polyplexes suitable for DNA vaccination (104). Similar polyplex approaches may potentially be applied in the future for mRNA vaccine delivery. Naturally occurring polymers such as alginate and chitosan typically have the benefit of relatively benign gelation conditions, allowing for easy encapsulation of proteins and nucleic acids (68). Chitosan specifically possesses mucoadhesive properties and reversibly opens epithelial tight junctions, improving mucosal permeability (54, 73). However, its high solubility in gastric pH conditions necessitates additional stabilization through cross-linking (108) or protective coatings (109). Inorganic MNPs have also been used for oral vaccination, including using gold (110) and silica (88). These MNPs are highly tailorable and have a stable rigid structure that is not affected by gastric conditions (110), while their surfaces can easily be functionalized with antigens, targeting moieties, and polymers for stability enhancement.
Lipid-Based Approaches
Liposomes are enclosed vesicles of concentric self-assembling lipid bilayers composed of phospholipids and cholesterols (Figure 2). The oral delivery of liposomes has a long history in the drug delivery field, which began in the 1970s with encapsulated insulin, but has only more recently been explored for oral vaccines. The advantages of using liposomes for mucosal vaccine delivery are that they can protect against the degradation of antigens in the harsh GI environment and facilitate uptake of antigens by APCs. Liposomes are also compatible with an array of cargo: e.g., subunit proteins, DNA, RNA, carbohydrates, and peptides, which are loaded within the aqueous core. Oral delivery to preclinical animal models of liposomes containing cargo for influenza A, Salmonella enteritidis, and Mycobacterium was shown to elicit antigen-specific humoral and cellular immune responses (111–113). In addition, immunogenicity can be enhanced by incorporating lectins into liposomes, which leads to them targeting intestinal M cells in PPs and the induction of even higher antibody titers than with liposomes alone (114). Despite these initial promising results, liposomes need to be further optimized for survival within the GI tract, as intestinal bile salts have been shown to disrupt liposome stability.
To increase the stability of oral immunizations in the GI tract in the presence of bile salts, distinct lipid-based carriers known as bilosomes, which directly incorporate bile salts into their formulation, are under investigation. The increased stability allowed enhanced protection of a variety of vaccine antigens, including tetanus toxoid (115). When delivered orally, bilosomes containing hepatitis B surface antigen and cholera toxin B subunits have also been shown to generate both systemic and local immunity (65), potentially due to M cell targeting. Importantly, bilosomes were able to induce sIgA throughout the GI tract and drive Th1 and Th2 responses in animal models (116). Further evaluation in human clinical trials will be critical to fully understand bilosomes as potential oral immunization agents.
An additional innovative form of lipid-based vehicles for vaccine delivery are immune-stimulating complexes (ISCOMs). These use unique materials (saponin, cholesterol, and phospholipids) that can self-assemble to entrap bacterial and viral envelope proteins and both protect antigenic cargo and act as immune stimulants. ISCOMs have been shown to drive potent immune responses to several antigens (75), and importantly induce both cellular and humoral immunity, likely due to the engagement of materials with both innate and adaptive immune systems. Unlike bilosomes, however, ISCOMs have not been shown to increase sIgA levels, which may limit their effectiveness in preventing GI infections (117). ISCOMs will also require further innovation and rigorous clinical testing to determine their potential as an oral immunization modality in humans.
Viral and Bacterial Vectors
The use of viral and bacterial vectors expressing recombinant antigens for other pathogens is particularly attractive for oral vaccination, because pathogens that naturally infect the GI tract are efficient vectors for mucosal delivery/colonization. Bacteria such as Salmonella typhi, Escherichia coli, Listeria, Vibrio, and Shigella and viruses such as those in the poxvirus and adenovirus families have been used for intestinal delivery (118–122). S. typhi is perhaps the most studied bacterial vector in animals, but translation to humans has proved challenging (118). Recombinant adenoviruses (rAds) are the most widely used viral or bacterial vector for oral vaccination, owing to their ability to infect and replicate in the intestinal mucosa and deliver a DNA cargo (122). rAds have also been delivered to the SL mucosa (123). IM delivery of rAds has been a mainstay in the vaccinology field, and in clinical studies, both antiviral humoral and cellular immunity have been induced (124). However, the presence of naturally occurring neutralizing antibodies against a diverse number of rAds reduces their utility for systemic immunization. In contrast, oral delivery of rAds is not affected by preexisting immunity and could represent a promising route of adenoviral immunization.
Oral vaccination with rAds has been conducted with both replication-competent and replication-defective rAds. For replication-competent vectors, adenovirus serotypes 4 and 7 have been utilized, which have a natural tropism for GI tissue. rAd4 and rAd7 vectors containing a hepatitis B surface antigen transgene were delivered to chimpanzees and human volunteers, respectively (125, 126), but neither vaccine was particularly immunogenic. This was also similar to a live rAd4 vector expressing the hemagglutinin gene of the H5N1 influenza virus, which when formulated as enteric-coated tablets and delivered to healthy human volunteers did not elicit strong immunity (61). Interestingly, when these orally vaccinated individuals were subsequently boosted by a systemically delivered H5N1 subunit vaccine, the titers and avidity of virus-specific antibodies were markedly higher than in individuals only vaccinated systemically (127), suggesting that oral delivery of replication-competent rAd4 vectors could be a useful priming agent for the induction of robust, protective immunity following systemic boost.
Orally delivered, replication-defective rAd serotype 5 vectors have been shown to be substantially more immunogenic. Clinical trials testing a rAd serotype 5 vector encoding an influenza hemagglutinin, in conjunction with a TLR3 adjuvant, as an enteric-coated tablet led to the induction of transgene-specific CD8+ T cell responses and antibodies (128). Additionally, unlike systemic immunization, oral immunization was not affected by preexisting adenovirus vector immunity (60). Follow-up studies demonstrated that delivery of rAd5 vectors using radio-controlled capsules, which allowed for the release of particles within the jejunum or ileum, led to higher rates of vaccine responders in comparison to systemically immunized individuals (129). These compelling results provide the impetus for the future investigation of oral delivery of replication-deficient adenoviral vectors.
Mucosal Adjuvants
Adjuvants are immunomodulatory agents that improve the immunogenicity of vaccine vectors. Numerous adjuvants have been developed and validated for systemic immunization, but they have proven ineffective at eliciting effective mucosal immunity, in part due to their inability to withstand the harsh conditions at mucosal sites and initiate sufficient cross-talk between innate and adaptive immune cells. Studies identifying adjuvants capable of inducing immune responses at mucosal tissue sites have utilized three mechanisms: toxin-mediated immune stimulation; TLR agonism; and natural immunomodulation using cytokines, as described below. Depending on the adjuvant and vaccine delivery vehicle, these adjuvants may be coadministered in a solution or capsule with the oral vaccine formulation or directly conjugated or incorporated within a delivery particle (e.g., an MNP or liposome).
Two of the most promising toxin-mediated adjuvants studied thus far have been the ADP-ribosylating enterotoxins cholera toxin (CT), produced by Vibrio cholerae, and heat-labile toxin (LT), made by enterotoxigenic Escherichia coli (ETEC). Both CT and LT are potent enterotoxins, but they have been modified to eliminate their endogenous toxicity while still retaining their immunostimulatory properties. In the case of LT, a double-mutant form (dmLT), containing mutations of arginine to glycine at position 192 and leucine to alanine at position 211 in the active A subunit, promoted robust mucosal immune responses when codelivered within an antigen (130) in preclinical models (131) and was subsequently deemed safe, well-tolerated, and immunogenic in human clinical trials. A phase I dose-escalating safety study of 5–100 μg of orally delivered dmLT resulted in no serious adverse events (132). Coadministration of dmLT with either whole-killed or live-attenuated ETEC in two additional trials resulted in enhanced immune responses to less immunogenic antigens and protective efficacy from oral challenge 6 months after immunization (133, 134). This is the result of dmLT having no detectable effect on intestinal epithelial cells but marked effects on DCs responsible for immune stimulation (135), making dmLT a highly promising adjuvant for future oral vaccines.
TLRs belong to the family of PRRs that are expressed on DCs, macrophages, monocytes, and epithelial cells within intestinal tissue sites. Thus, TLR agonists serve as an additional class of mucosal adjuvants, with two of these, unmethylated cytosine-guanine-containing oligonucleotides (CpG DNA) and monophosphoryl lipid A (MPL), among the most promising. MPL in particular, which is a TLR4 agonist and detoxified version of lipopolysaccharide from Salmonella minnesota R595, has been demonstrated to increase IgG and IgA levels when coadministered with a number of antigens (136). Additional TLR agonists with demonstrated mucosal immunogenicity include polyinosine-polycytidylic acid [poly(I:C)], flagellin, and imiquimod, which engage TLR3, TLR5, and TLR7/8 receptors.
Cytokines are additional soluble cell-derived mediators that are capable of exerting a profound effect on APCs, allowing them to serve as potent adjuvants at mucosal sites. Orally delivered interleukin-12 (IL-12) complexed with liposomes was able to induce elevated IgG levels to coadministered tetanus toxoid (137). In addition, IL-15 was demonstrated to be an effective mucosal adjuvant when delivered with herpes simplex virus 2, with augmented humoral and cellular responses in both the mucosal and systemic compartment (138). Collectively, these studies reveal the diversity of mucosal adjuvants, which will require further investigation to identify which are most suitable for a given vaccine application.
Physical Devices
Physical devices could also be employed to achieve vaccine delivery through GI mucosa. These may take a variety of forms, including using stored electrical or chemical energy to drive processes that permeabilize epithelia (e.g., reverse iontophoresis, electroporation, thermal ablation) (12), high-energy liquids or particles that penetrate tissue (69), or physical protrusions that mechanically disrupt epithelia [e.g., microneedle arrays (MNs)] (35, 139). In the last two decades, these approaches have been pioneered primarily to permeabilize the skin barrier for both diagnostics (140–142) and drug and vaccine delivery (143, 144). Physical devices may have the added capability to transport vaccine in an internal compartment to protect it from degradation until delivery at the intended mucosa. The accessibility of the target mucosa, however, may impose challenges requiring miniaturization and triggering/targeting release.
The oral cavity has recently been of interest for the use of physical devices for vaccination (35, 69, 70). Its ease of access enables greater freedom in device design and the translation of many approaches developed for the skin. MNs have been widely investigated to mechanically penetrate buccal mucosal/epithelial layers and deliver vaccine to APCs (35). MNs have been studied for both subunit and DNA (145) vaccines against HIV (145), influenza (146), hepatitis B virus, herpes simplex virus, and human papillomavirus (35). Additionally, MNs are a versatile delivery platform that may deliver any vaccine type in combination with particulate delivery vectors (such as MNPs or liposomes), stabilizing excipients, or adjuvants. Liquid and solid formulations may be delivered through hollow needles and dissolvable projections/coatings, respectively. Solid formulations in particular have also been widely reported to offer increased vaccine thermostability (72). Furthermore, the process of mechanically disrupting epithelial barriers has been reported to stimulate antibody responses in what has been described as physical adjuvantation (147, 148).
High-pressured liquid jets have also been used to deliver vaccine to the oral cavity. A capsule-sized device, the MucoJet, delivered a liquid-jet vaccine when placed against buccal tissue (69). The minimally invasive and self-administrable device did, however, require some user assembly prior to administration. A similar approach used a larger handheld liquid-jet device to deliver a vaccine for HIV antigen in combination with a modified vaccinia Ankara vector into buccal or SL tissue (70). Furthermore, the SL mucosa allows for slow absorption of macromolecules, which can be exploited by approaches that localize release over extended periods. Both mucoadhesive patches (84) and nonadhesive orally disintegrating films (85, 149) have been shown to limit diffusion by releasing vaccine at the mucosal surface and increase antigen uptake.
RECENT ADVANCES IN BIOLOGIC DELIVERY
Ingestible Devices for Gastric and Intestinal Biologic Delivery
The use of physical devices for vaccine delivery to the gastric and intestinal mucosa requires more advanced approaches compared to the oral cavity. Such approaches require the miniaturization of components (e.g., within ingestible capsules) and/or mechanisms to trigger actuation and vaccine delivery at the site of interest. Our group has recently reported proof-of-concept delivery of biologic therapeutics to both the stomach (20) and small intestine (22) using physical devices. The self-orienting millimeter-scale applicator (SOMA; see Figure 2) is an ingestible capsule that inserts a solid millipost composed of a biologic (with or without excipient) through the mucosa (20). The soluble millipost then dissolves, releasing drug into the gastric wall, and we have demonstrated this for the systemic absorption of insulin. We also demonstrated a device for intestinal delivery of biologic therapies: the luminal unfolding microneedle injector (LUMI) (22). The LUMI consists of a three-armed device, folded into an ingestible capsule and held together by an elastomeric core. Upon reaching the intestines, the arms and elastomeric core are ejected from the capsule and unfold to contact the lumen. Arrays of soluble microneedles at the ends of the arms deliver the biologic drug into the intestinal mucosa. Perforation of the relatively thin intestinal mucosa is avoided by the use of ultra-short (<1 mm) microneedles. Both these devices incorporate actuation (i.e., triggering mechanisms) based on contact with gastric or intestinal fluid that dissolves a soluble plug to release a compressed spring. Rani Therapeutics has also proposed a capsule device with an extendable, dissolvable solid needle controlled by an expanding balloon for delivery to the intestinal mucosa called the RaniPill™ (21, 150), which was recently tested in a phase I clinical study (151; https://clinicaltrials.gov/ct2/show/NCT03798912). Collectively, these physical delivery devices have been widely regarded as disruptive technologies for biologic delivery to the stomach and intestines (18, 19).
Because these devices are intended to be self-administrable, they may have significant potential for mass oral vaccination campaigns needed during pandemic outbreaks of novel viruses [e.g., the severe acute respiratory syndrome coronavirus 2 causing COVID-19 (coronavirus disease 2019), swine flu, avian/bird flu, Zika virus] and/or for vaccines such as seasonal influenza, human papillomavirus, and common travel vaccines that are administered to adults. As platform devices, they may be loaded with potentially any type of vaccine (i.e., pure subunit/protein, particulate, lipid based, viral/bacterial vector, VLP) or adjuvant, and their compatibility with solid formulations offers potential for thermostability prior to delivery (72, 152), with or without stabilizing excipients, giving them a high degree of versatility. A key limitation of ingestible devices for vaccination, however, may be infant immunizations, where such devices may represent choking hazards.
A Role for Permeation Enhancers?
PEs increase the uptake of peptides through the intestinal epithelium for systemic delivery and have been of major interest for pharmaceutical companies for several decades, with over 250 having been investigated (7, 153). Success has been moderate, with single-digit bioavailability of biologics commonly reported. Given this, and the existence of alternative mucosal absorption strategies (e.g., via intestinal PPs), PEs have not been rigorously investigated for oral vaccination. However, recent advancements may offer renewed promise. Ionic liquids have been shown to generate high oral bioavailability for biologics (up to 50%) via intestinal absorption (23) and may have potential for delivery of vaccines. High oral bioavailability of insulin has also recently been reported using anionic nanoparticles (154). Another study identified that the PE N-[8-(2-hydroxybenzoyl)amino] caprylate (or SNAC) (155) enabled the gastric absorption of a glucagon-like peptide-1 receptor agonist. This process mimicked thin film strategies for SL delivery in that release was localized beneath a slowly eroding tablet in the stomach, which could potentially be investigated to deliver vaccines. Several surfactants have also been investigated and show some promise for peptide delivery (7, 153, 156). The efficiency of PEs, however, is typically inversely proportional to the molecular weight of the peptide/protein (156), and this limitation needs to be considered for vaccine delivery, where shorter peptides are typically only weakly antigenic (157).
CONCLUSIONS AND FUTURE OUTLOOK
To date, the only approved orally delivered vaccines are live-attenuated and inactivated vaccines. Oral delivery of subunit and nucleic acid vaccines has many potential clinical advantages; however, the GI tract poses many challenges to the successful development of such technologies. Particulate, lipid-based, bacterial, and viral vectors designed for intestinal mucosal uptake have traditionally dominated oral vaccine design. There has been success in animal studies using these strategies primarily for intestinal delivery; however, there is a remaining need to enhance immunogenicity, improve low antigen uptake, and continue the development of mucosal adjuvants. Recent breakthrough advances in physical devices and PEs for oral biologic and vaccine delivery are enabling the investigation of alternative routes for vaccination to both the intestines and previously understudied mucosal targets in oral and gastric mucosae. These offer the promise of versatile platforms for highly efficient oral vaccine delivery, which may lead to new generations of oral subunit, DNA, and mRNA vaccine development, which are of particular use in response to the growing threat of pandemic outbreaks.
ACKNOWLEDGMENTS
J.W.C. was supported by the American Australian Association Fellowship and National Health and Medical Research Council of Australia Early Career Fellowship (C.J. Martin Overseas Biomedical Fellowship APP1144167). G.D.G. was supported by the National Institutes of Health (NIH) K08 grant AI140960 and a Burroughs Wellcome Career Award for Medical Scientists. G.T. was supported in part by a grant from Novo Nordisk, NIH EB000244, and the Department of Mechanical Engineering at MIT. The authors would like to acknowledge Grace Ingalls for her assistance in manuscript preparation and Prof. R. Langer for valuable feedback and review of the manuscript.
Footnotes
DISCLOSURE STATEMENT
J.W.C. and G.T. are coinventors on multiple patent applications describing systems capable of oral delivery of biologics, including vaccines. G.D.G. has filed a provisional patent application (62/817,094) related to HIV vaccine design. Complete details of all relationships for profit and not for profit for G.T. can be found at the following link: https://www.dropbox.com/sh/szi7vnr4a2ajb56/AABs5N5i0q9AfT1IqIJAE-T5a?dl=0.
LITERATURE CITED
- 1.Eek D, Krohe M, Mazar I, Horsfield A, Pompilus F, et al. 2016. Patient-reported preferences for oral versus intravenous administration for the treatment of cancer: a review of the [DOI] [PMC free article] [PubMed]
- 2.Durán-Lobato M, Niu Z, Alonso MJ. 2019. Oral delivery of biologics for precision medicine. Adv. Mater 32(13):1901935. [DOI] [PubMed] [Google Scholar]
- 3.Vela Ramirez JE, Sharpe LA, Peppas NA. 2017. Current state and challenges in developing oral vaccines. Adv. Drug Deliv. Rev 114:116–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marasini N, Skwarczynski M, Toth I. 2014. Oral delivery of nanoparticle-based vaccines. Expert Rev. Vaccines 13(11):1361–76 [DOI] [PubMed] [Google Scholar]
- 5.Lycke N 2012. Recent progress in mucosal vaccine development: potential and limitations. Nat. Rev. Immunol 12(8):592–605 [DOI] [PubMed] [Google Scholar]
- 6.Drucker DJ. 2019. Advances in oral peptide therapeutics. Nat. Rev. Drug Discov 19:277–89 [DOI] [PubMed] [Google Scholar]
- 7.Maher S, Brayden DJ, Casettari L, Illum L. 2019. Application of permeation enhancers in oral delivery of macromolecules: an update. Pharmaceutics 11(1):41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmgren J, Czerkinsky C. 2005. Mucosal immunity and vaccines. Nat. Med 11(Suppl. 4):S45–53 [DOI] [PubMed] [Google Scholar]
- 9.Roberts L 2013. Israel’s silent polio epidemic breaks all the rules. Science 342(6159):679–80 [DOI] [PubMed] [Google Scholar]
- 10.Kraan H, Vrieling H, Czerkinsky C, Jiskoot W, Kersten G, Amorij J-P. 2014. Buccal and sublingual vaccine delivery. J. Control. Release 190:580–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu W, Zeng Z, Luo S, Hu C, Xu N, et al. 2019. Gastric subserous vaccination with Helicobacter pylori vaccine: an attempt to establish tissue-resident CD4+ memory T cells and induce prolonged protection. Front. Immunol 10:1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Anselmo AC, Gokarn Y, Mitragotri S. 2019. Non-invasive delivery strategies for biologics. Nat. Rev. Drug Discov 18(1):19–40 [DOI] [PubMed] [Google Scholar]
- 13.Vllasaliu D, Thanou M, Stolnik S, Fowler R. 2018. Recent advances in oral delivery of biologics: nanomedicine and physical modes of delivery. Expert Opin. Drug Deliv 15(8):759–70 [DOI] [PubMed] [Google Scholar]
- 14.Morales JO, Fathe KR, Brunaugh A, Ferrati S, Li S, et al. 2017. Challenges and future prospects for the delivery of biologics: oral mucosal, pulmonary, and transdermal routes. AAPS J 19(3):652–68 [DOI] [PubMed] [Google Scholar]
- 15.Zhang C, Maruggi G, Shan H, Li J. 2019. Advances in mRNA vaccines for infectious diseases. Front. Immunol 10:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lurie N, Saville M, Hatchett R, Halton J. 2020. Developing Covid-19 vaccines at pandemic speed. N. Engl. J. Med 382(21):1969–73 [DOI] [PubMed] [Google Scholar]
- 17.Kang S, Hong S, Lee Y-K, Cho S. 2018. Oral vaccine delivery for intestinal immunity—biological basis, barriers, delivery system, and M cell targeting. Polymers 10(9):948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Service RF. 2019. Pills give patients a shot inside the stomach. Science 363(6427):571. [DOI] [PubMed] [Google Scholar]
- 19.Brayden DJ, Baird AW. 2019. Stomaching drug delivery. N. Engl. J. Med 380(17):1671–73 [DOI] [PubMed] [Google Scholar]
- 20.Abramson A, Caffarel-Salvador E, Khang M, Dellal D, Silverstein D, et al. 2019. An ingestible self-orienting system for oral delivery of macromolecules. Science 363(6427):611–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hashim M, Korupolu R, Syed B, Horlen K, Beraki S, et al. 2019. Jejunal wall delivery of insulin via an ingestible capsule in anesthetized swine—a pharmacokinetic and pharmacodynamic study. Pharmacol. Res. Perspect 7(5):e00522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abramson A, Caffarel-Salvador E, Soares V, Minahan D, Tian RY, et al. 2019. A luminal unfolding microneedle injector for oral delivery of macromolecules. Nat. Med 25(10):1512–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Banerjee A, Ibsen K, Brown T, Chen R, Agatemor C, Mitragotri S. 2018. Ionic liquids for oral insulin delivery. PNAS 115(28):7296–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McGhee JR, Fujihashi K. 2012. Inside the mucosal immune system. PLOS Biol 10(9):e1001397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mowat AM, Agace WW. 2014. Regional specialization within the intestinal immune system. Nat. Rev. Immunol 14(10):667–85 [DOI] [PubMed] [Google Scholar]
- 26.Lynch SV, Pedersen O. 2016. The human intestinal microbiome in health and disease. N. Engl. J. Med 375(24):2369–79 [DOI] [PubMed] [Google Scholar]
- 27.Aychek T, Jung S. 2014. The axis of tolerance. Science 343(6178):1439–40 [DOI] [PubMed] [Google Scholar]
- 28.Knoop KA, Miller MJ, Newberry RD. 2013. Transepithelial antigen delivery in the small intestine: different paths, different outcomes. Curr. Opin. Gastroenterol 29(2):112–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Song J-H, Nguyen HH, Cuburu N, Horimoto T, Ko S-Y, et al. 2008. Sublingual vaccination with influenza virus protects mice against lethal viral infection. PNAS 105(5):1644–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quiding M, Nordström I, Kilander A, Andersson G, Hanson LA, et al. 1991. Intestinal immune responses in humans. Oral cholera vaccination induces strong intestinal antibody responses and interferon-gamma production and evokes local immunological memory. J. Clin. Investig 88(1):143–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johansson EL, Wassén L, Holmgren J, Jertborn M, Rudin A. 2001. Nasal and vaginal vaccinations have differential effects on antibody responses in vaginal and cervical secretions in humans. Infect. Immun 69(12):7481–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kozlowski PA, Cu-Uvin S, Neutra MR, Flanigan TP. 1997. Comparison of the oral, rectal, and vaginal immunization routes for induction of antibodies in rectal and genital tract secretions of women. Infect. Immun 65(4):1387–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Radulovic S, Wilson D, Calderon M, Durham S. 2011. Systematic reviews of sublingual immunotherapy (SLIT). Allergy 66(6):740–52 [DOI] [PubMed] [Google Scholar]
- 34.Calderón MA, Simons FER, Malling H-J, Lockey RF, Moingeon P, Demoly P. 2012. Sublingual allergen immunotherapy: mode of action and its relationship with the safety profile. Allergy 67(3):302–11 [DOI] [PubMed] [Google Scholar]
- 35.Creighton RL, Woodrow KA. 2019. Microneedle-mediated vaccine delivery to the oral mucosa. Adv. Healthc. Mater 8(4):1801180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Squier CA, Kremer MJ. 2001. Biology of oral mucosa and esophagus. JNCI Monogr 2001(29):7–15 [DOI] [PubMed] [Google Scholar]
- 37.Hovav A-H. 2014. Dendritic cells of the oral mucosa. Mucosal Immunol 7(1):27–37 [DOI] [PubMed] [Google Scholar]
- 38.Bimczok D, Clements RH, Waites KB, Novak L, Eckhoff DE, et al. 2010. Human primary gastric dendritic cells induce a Th1 response to H. pylori. Mucosal Immunol 3(3):260–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nagai S, Mimuro H, Yamada T, Baba Y, Moro K, et al. 2007. Role of Peyer’s patches in the induction of Helicobacter pylori–induced gastritis. PNAS 104(21):8971–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kiriya K, Watanabe N, Nishio A, Okazaki K, Kido M, et al. 2007. Essential role of Peyer’s patches in the development of Helicobacter-induced gastritis. Int. Immunol 19(4):435–46 [DOI] [PubMed] [Google Scholar]
- 41.Carney J 2010. Gastric mucosal lymphoid follicles: histology, distribution, frequency, and etiologic features. Am. J. Surg. Pathol 34(7):1019–24 [DOI] [PubMed] [Google Scholar]
- 42.Mazzoni M, Bosi P, De Sordi N, Lalatta-Costerbosa G. 2011. Distribution, organization and innervation of gastric MALT in conventional piglet. J. Anat 219(5):611–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu C, Liu W, Xu N, Huang A, Zeng Z, et al. 2020. Perivascular lymphocyte clusters induced by gastric subserous layer vaccination mediate optimal immunity against Helicobacter through facilitating immune cell infiltration and local antibody response. J. Immunol. Res 2020:1480281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu N, Ruan G, Liu W, Hu C, Huang A, et al. 2019. Vaccine-induced gastric CD4+ tissue-resident memory T cells proliferate in situ to amplify immune response against Helicobacter pylori insult. Helicobacter 24(5):e12652. [DOI] [PubMed] [Google Scholar]
- 45.Corr SC, Gahan CCGM, Hill C. 2008. M-cells: origin, morphology and role in mucosal immunity and microbial pathogenesis. FEMS Immunol. Med. Microbiol 52(1):2–12 [DOI] [PubMed] [Google Scholar]
- 46.Tyrer P, Foxwell AR, Cripps AW, Apicella MA, Kyd JM. 2006. Microbial pattern recognition receptors mediate M-cell uptake of a gram-negative bacterium. Infect. Immun 74(1):625–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eldridge JH, Gilley RM, Staas JK, Moldoveanu Z, Meulbroek JA, Tice TR. 1989. Biodegradable microspheres: vaccine delivery system for oral immunization. Curr. Top. Microbiol. Immunol 146:59–66 [DOI] [PubMed] [Google Scholar]
- 48.Eldridge JH, Staas JK, Meulbroek JA, McGhee JR, Tice TR, Gilley RM. 1991. Biodegradable microspheres as a vaccine delivery system. Mol. Immunol 28(3):287–94 [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Anselmo AC, Banerjee A, Zakrewsky M, Mitragotri S. 2015. Shape and size-dependent immune response to antigen-carrying nanoparticles. J. Control. Release 220:141–48 [DOI] [PubMed] [Google Scholar]
- 50.Tabata Y, Inoue Y, Ikada Y. 1996. Size effect on systemic and mucosal immune responses induced by oral administration of biodegradable microspheres. Vaccine 14(17):1677–85 [DOI] [PubMed] [Google Scholar]
- 51.Eldridge JH, Hammond CJ, Meulbroek JA, Staas JK, Gilley RM, Tice TR. 1990. Controlled vaccine release in the gut-associated lymphoid tissues. I. Orally administered biodegradable microspheres target the Peyer’s patches. J. Control. Release 11(1):205–14 [Google Scholar]
- 52.Zachary JF. 2017. Mechanisms of microbial infections. In Pathologic Basis of Veterinary Disease, ed. Zachary JF, pp. 132–241. e1. St. Louis, MO: Elsevier [Google Scholar]
- 53.Chieppa M, Rescigno M, Huang AYC, Germain RN. 2006. Dynamic imaging of dendritic cell extension into the small bowel lumen in response to epithelial cell TLR engagement. J. Exp. Med 203(13):2841–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeh T-H, Hsu L-W, Tseng MT, Lee P-L, Sonjae K, et al. 2011. Mechanism and consequence of chitosan-mediated reversible epithelial tight junction opening. Biomaterials 32(26):6164–73 [DOI] [PubMed] [Google Scholar]
- 55.Snoeck V, Goddeeris B, Cox E. 2005. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect 7(7):997–1004 [DOI] [PubMed] [Google Scholar]
- 56.Ménard S, Cerf-Bensussan N, Heyman M. 2010. Multiple facets of intestinal permeability and epithelial handling of dietary antigens. Mucosal Immunol 3(3):247–59 [DOI] [PubMed] [Google Scholar]
- 57.McDole JR, Wheeler LW, McDonald KG, Wang B, Konjufca V, et al. 2012. Goblet cells deliver luminal antigen to CD103+ dendritic cells in the small intestine. Nature 483(7389):345–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brandtzaeg P 2013. Secretory immunity with special reference to the oral cavity. J. Oral Microbiol 10.3402/jom.v5i0.20401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wu R-Q, Zhang D-F, Tu E, Chen Q-M, Chen W. 2014. The mucosal immune system in the oral cavity—an orchestra of T cell diversity. Int. J. Oral Sci 6(3):125–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liebowitz D, Lindbloom JD, Brandl JR, Garg SJ, Tucker SN. 2015. High titre neutralising antibodies to influenza after oral tablet immunisation: a phase 1, randomised, placebo-controlled trial. Lancet Infect. Dis 15(9):1041–48 [DOI] [PubMed] [Google Scholar]
- 61.Gurwith M, Lock M, Taylor EM, Ishioka G, Alexander J, et al. 2013. Safety and immunogenicity of an oral, replicating adenovirus serotype 4 vector vaccine for H5N1 influenza: a randomised, double-blind, placebo-controlled, phase 1 study. Lancet Infect. Dis 13(3):238–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Uddin AN, Bejugam NK, Gayakwad SG, Akther P, D’Souza MJ. 2009. Oral delivery of gastro-resistant microencapsulated typhoid vaccine. J. Drug Target 17(7):553–60 [DOI] [PubMed] [Google Scholar]
- 63.Garinot M, Fiévez V, Pourcelle V, Stoffelbach F, des Rieux A, et al. 2007. PEGylated PLGA-based nanoparticles targeting M cells for oral vaccination. J. Control. Release 120(3):195–204 [DOI] [PubMed] [Google Scholar]
- 64.Kim S-H, Seo K-W, Kim J, Lee K-Y, Jang Y-S. 2010. The M cell-targeting ligand promotes antigen delivery and induces antigen-specific immune responses in mucosal vaccination. J. Immunol 185(10):5787–95 [DOI] [PubMed] [Google Scholar]
- 65.Shukla A, Katare OP, Singh B, Vyas SP. 2010. M-cell targeted delivery of recombinant hepatitis B surface antigen using cholera toxin B subunit conjugated bilosomes. Int. J. Pharm 385(1):47–52 [DOI] [PubMed] [Google Scholar]
- 66.Zhu Q, Talton J, Zhang G, Cunningham T, Wang Z, et al. 2012. Large intestine-targeted, nanoparticle-releasing oral vaccine to control genitorectal viral infection. Nat. Med 18(8):1291–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Warzecha H, Mason HS, Lane C, Tryggvesson A, Rybicki E, et al. 2003. Oral immunogenicity of human papillomavirus-like particles expressed in potato. J. Virol 77(16):8702–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van der Lubben IM, Verhoef JC, van Aelst AC, Borchard G, Junginger HE. 2001. Chitosan microparticles for oral vaccination: preparation, characterization and preliminary in vivo uptake studies in murine Peyer’s patches. Biomaterials 22(7):687–94 [DOI] [PubMed] [Google Scholar]
- 69.Aran K, Chooljian M, Paredes J, Rafi M, Lee K, et al. 2017. An oral microjet vaccination system elicits antibody production in rabbits. Sci. Transl. Med 9(380):eaaf6413. [DOI] [PubMed] [Google Scholar]
- 70.Jones AT, Shen X, Walter KL, LaBranche CC, Wyatt LS, et al. 2019. HIV-1 vaccination by needle-free oral injection induces strong mucosal immunity and protects against SHIV challenge. Nat. Commun 10(1):798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen D, Kristensen D. 2009. Opportunities and challenges of developing thermostable vaccines. Expert Rev. Vaccines 8(5):547–57 [DOI] [PubMed] [Google Scholar]
- 72.Chen X, Fernando GJP, Crichton ML, Flaim C, Yukiko SR, et al. 2011. Improving the reach of vaccines to low-resource regions, with a needle-free vaccine delivery device and long-term thermostabilization. J. Control. Release 152(3):349–55 [DOI] [PubMed] [Google Scholar]
- 73.van der Lubben IM, Verhoef JC, Borchard G, Junginger HE. 2001. Chitosan and its derivatives in mucosal drug and vaccine delivery. Eur. J. Pharm. Sci 14(3):201–7 [DOI] [PubMed] [Google Scholar]
- 74.Rice-Ficht AC, Arenas-Gamboa AM, Kahl-McDonagh MM, Ficht TA. 2010. Polymeric particles in vaccine delivery. Curr. Opin. Microbiol 13(1):106–12 [DOI] [PubMed] [Google Scholar]
- 75.Gregory A, Williamson D, Titball R. 2013. Vaccine delivery using nanoparticles. Front. Cell Infect. Microbiol 3:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen SC, Jones DH, Fynan EF, Farrar GH, Clegg JCS, et al. 1998. Protective immunity induced by oral immunization with a rotavirus DNA vaccine encapsulated in microparticles. J. Virol 72(7):5757–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.He X-W, Wang F, Jiang L, Li J, Liu S, et al. 2005. Induction of mucosal and systemic immune response by single-dose oral immunization with biodegradable microparticles containing DNA encoding HBsAg. J. Gen. Virol 86(3):601–10 [DOI] [PubMed] [Google Scholar]
- 78.Roy K, Mao H-Q, Huang S-K, Leong KW. 1999. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat. Med 5(4):387–91 [DOI] [PubMed] [Google Scholar]
- 79.Guo L, Yin R, Liu K, Lv X, Li Y, et al. 2014. Immunological features and efficacy of a multi-epitope vaccine CTB-UE against H. pylori in BALB/c mice model. Appl. Microbiol. Biotechnol 98(8):3495–507 [DOI] [PubMed] [Google Scholar]
- 80.Kaneko H, Bednarek I, Wierzbicki A, Kiszka I, Dmochowski M, et al. 2000. Oral DNA vaccination promotes mucosal and systemic immune responses to HIV envelope glycoprotein. Virology 267(1):8–16 [DOI] [PubMed] [Google Scholar]
- 81.Lundstrom K 2018. Latest development on RNA-based drugs and vaccines. Futur. Sci. OA 4(5):FSO300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pardi N, Hogan MJ, Porter FW, Weissman D. 2018. mRNA vaccines—a new era in vaccinology. Nat. Rev. Drug Discov 17:261–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hanes J, Chiba M, Langer R. 1995. Polymer microspheres for vaccine delivery. In Vaccine Design: The Subunit and Adjuvant Approach, ed. Powell MF, Newman MJ, pp. 389–412. New York: Springer; [DOI] [PubMed] [Google Scholar]
- 84.Mašek J, Lubasová D, Lukáč R, Turánek-Knotigová P, Kulich P, et al. 2017. Multi-layered nanofibrous mucoadhesive films for buccal and sublingual administration of drug-delivery and vaccination nanoparticles—important step towards effective mucosal vaccines. J. Control. Release 249:183–95 [DOI] [PubMed] [Google Scholar]
- 85.Gala RP, Popescu C, Knipp GT, McCain RR, Ubale RV, et al. 2017. Physicochemical and preclinical evaluation of a novel buccal measles vaccine. AAPS PharmSciTech 18(2):283–92 [DOI] [PubMed] [Google Scholar]
- 86.Delgado A, Lavelle EC, Hartshorne M, Davis SS. 1999. PLG microparticles stabilised using enteric coating polymers as oral vaccine delivery systems. Vaccine 17(22):2927–38 [DOI] [PubMed] [Google Scholar]
- 87.Yeh P-Y, Ellens H, Smith PL. 1998. Physiological considerations in the design of particulate dosage forms for oral vaccine delivery. Adv. Drug Deliv. Rev 34(2):123–33 [DOI] [PubMed] [Google Scholar]
- 88.Wang T, Jiang H, Zhao Q, Wang S, Zou M, Cheng G. 2012. Enhanced mucosal and systemic immune responses obtained by porous silica nanoparticles used as an oral vaccine adjuvant: effect of silica architecture on immunological properties. Int. J. Pharm 436(1):351–58 [DOI] [PubMed] [Google Scholar]
- 89.Awaad A, Nakamura M, Ishimura K. 2012. Imaging of size-dependent uptake and identification of novel pathways in mouse Peyer’s patches using fluorescent organosilica particles. Nanomedicine 8(5):627–36 [DOI] [PubMed] [Google Scholar]
- 90.Gutierro I, Hernández RM, Igartua M, Gascón AR, Pedraz JL. 2002. Size dependent immune response after subcutaneous, oral and intranasal administration of BSA loaded nanospheres. Vaccine 21(1):67–77 [DOI] [PubMed] [Google Scholar]
- 91.Eldridge JH, Meulbroek JA, Staas JK, Tice TR, Gilley R. 1989. Vaccine-containing biodegradable microspheres specifically enter the gut-associated lymphoid tissue following oral administration and induce a disseminated mucosal immune response. In Immunobiology of Proteins and Peptides V: Vaccines Mechanisms, Design, and Applications, ed. Atassi MZ, pp. 191–202. New York: Springer; [DOI] [PubMed] [Google Scholar]
- 92.Foged C, Brodin B, Frokjaer S, Sundblad A. 2005. Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int. J. Pharm 298(2):315–22 [DOI] [PubMed] [Google Scholar]
- 93.Jiang T, Singh B, Li H-S, Kim Y-K, Kang S-K, et al. 2014. Targeted oral delivery of BmpB vaccine using porous PLGA microparticles coated with M cell homing peptide–coupled chitosan. Biomaterials 35(7):2365–73 [DOI] [PubMed] [Google Scholar]
- 94.Jepson MA, Clark MA, Hirst BH. 2004. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv. Drug Deliv. Rev 56(4):511–25 [DOI] [PubMed] [Google Scholar]
- 95.Joshi VB, Geary SM, Salem AK. 2013. Biodegradable particles as vaccine antigen delivery systems for stimulating cellular immune responses. Hum. Vaccin. Immunother 9(12):2584–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Demento SL, Cui W, Criscione JM, Stern E, Tulipan J, et al. 2012. Role of sustained antigen release from nanoparticle vaccines in shaping the T cell memory phenotype. Biomaterials 33(19):4957–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhou S, Liao X, Li X, Deng X, Li H. 2003. Poly-d,l-lactide-co-poly(ethylene glycol) microspheres as potential vaccine delivery systems. J. Control. Release 86(2):195–205 [DOI] [PubMed] [Google Scholar]
- 98.Jepson M, Simmons N, O’Hagan D, Hirst B. 1993. Comparison of poly(dl-lactide-co-glycolide) and polystyrene microsphere targeting to intestinal M cells. J. Drug Target 1(3):245–49 [DOI] [PubMed] [Google Scholar]
- 99.Men Y, Thomasin C, Merkle HP, Gander B, Corradin G. 1995. A single administration of tetanus toxoid in biodegradable microspheres elicits T cell and antibody responses similar or superior to those obtained with aluminum hydroxide. Vaccine 13(7):683–89 [DOI] [PubMed] [Google Scholar]
- 100.Carreño JM, Perez-Shibayama C, Gil-Cruz C, Printz A, Pastelin R, et al. 2016. PLGA-microencapsulation protects Salmonella typhi outer membrane proteins from acidic degradation and increases their mucosal immunogenicity. Vaccine 34(35):4263–69 [DOI] [PubMed] [Google Scholar]
- 101.Xiang SD, Selomulya C, Ho J, Apostolopoulos V, Plebanski M. 2010. Delivery of DNA vaccines: an overview on the use of biodegradable polymeric and magnetic nanoparticles. WIREs Nanomed. Nanobiotechnol 2(3):205–18 [DOI] [PubMed] [Google Scholar]
- 102.Salman HH, Irache JM, Gamazo C. 2009. Immunoadjuvant capacity of flagellin and mannosamine-coated poly(anhydride) nanoparticles in oral vaccination. Vaccine 27(35):4784–90 [DOI] [PubMed] [Google Scholar]
- 103.Benoit M-A, Baras B, Gillard J. 1999. Preparation and characterization of protein-loaded poly(ε-caprolactone) microparticles for oral vaccine delivery. Int. J. Pharm 184(1):73–84 [DOI] [PubMed] [Google Scholar]
- 104.Howard KA, Li XW, Somavarapu S, Singh J, Green N, et al. 2004. Formulation of a microparticle carrier for oral polyplex-based DNA vaccines. Biochim. Biophys. Acta Gen. Subj 1674(2):149–57 [DOI] [PubMed] [Google Scholar]
- 105.Bowersock TL, Hogenesch H, Suckow M, Porter RE, Jackson R, et al. 1996. Oral vaccination with alginate microsphere systems. J. Control. Release 39(2):209–20 [Google Scholar]
- 106.Kim B, Bowersock T, Griebel P, Kidane A, Babiuk LA, et al. 2002. Mucosal immune responses following oral immunization with rotavirus antigens encapsulated in alginate microspheres. J. Control. Release 85(1):191–202 [DOI] [PubMed] [Google Scholar]
- 107.Pavot V, Berthet M, Rességuier J, Legaz S, Handké N, et al. 2014. Poly(lactic acid) and poly(lactic-co-glycolic acid) particles as versatile carrier platforms for vaccine delivery. Nanomedicine 9(17):2703–18 [DOI] [PubMed] [Google Scholar]
- 108.Ahire VJ, Sawant KK, Doshi JB, Ravetkar SD. 2007. Chitosan microparticles as oral delivery system for tetanus toxoid. Drug Dev. Ind. Pharm 33(10):1112–24 [DOI] [PubMed] [Google Scholar]
- 109.Borges O, Tavares J, de Sousa A, Borchard G, Junginger HE, Cordeiro-da-Silva A. 2007. Evaluation of the immune response following a short oral vaccination schedule with hepatitis B antigen encapsulated into alginate-coated chitosan nanoparticles. Eur. J. Pharm. Sci 32(4):278–90 [DOI] [PubMed] [Google Scholar]
- 110.Barhate G, Gautam M, Gairola S, Jadhav S, Pokharkar V. 2013. Quillaja saponaria extract as mucosal adjuvant with chitosan functionalized gold nanoparticles for mucosal vaccine delivery: stability and immunoefficiency studies. Int. J. Pharm 441(1):636–42 [DOI] [PubMed] [Google Scholar]
- 111.Liu J, Wu J, Wang B, Zeng S, Qi F, et al. 2014. Oral vaccination with a liposome-encapsulated influenza DNA vaccine protects mice against respiratory challenge infection. J. Med. Virol 86(5):886–94 [DOI] [PubMed] [Google Scholar]
- 112.Pang Y, Zhang Y, Wang H, Jin J, Piao J, et al. 2013. Reduction of Salmonella enteritidis number after infections by immunization of liposome-associated recombinant SefA. Avian Dis 57(3):627–33 [DOI] [PubMed] [Google Scholar]
- 113.Wang D, Xu J, Feng Y, Liu Y, Mchenga SSS, et al. 2010. Liposomal oral DNA vaccine (mycobacterium DNA) elicits immune response. Vaccine 28(18):3134–42 [DOI] [PubMed] [Google Scholar]
- 114.Gupta PN, Vyas SP. 2011. Investigation of lectinized liposomes as M-cell targeted carrier-adjuvant for mucosal immunization. Colloids Surf. B Biointerfaces 82(1):118–25 [DOI] [PubMed] [Google Scholar]
- 115.Mann JFS, Scales HE, Shakir E, Alexander J, Carter KC, et al. 2006. Oral delivery of tetanus toxoid using vesicles containing bile salts (bilosomes) induces significant systemic and mucosal immunity. Methods 38(2):90–95 [DOI] [PubMed] [Google Scholar]
- 116.Conacher M, Alexander J, Brewer JM. 2001. Oral immunisation with peptide and protein antigens by formulation in lipid vesicles incorporating bile salts (bilosomes). Vaccine 19(20):2965–74 [DOI] [PubMed] [Google Scholar]
- 117.Sanders MT, Brown LE, Deliyannis G, Pearse MJ. 2005. ISCOM™-based vaccines: the second decade. Immunol. Cell Biol 83(2):119–28 [DOI] [PubMed] [Google Scholar]
- 118.Roland KL, Brenneman KE. 2013. Salmonella as a vaccine delivery vehicle. Expert Rev. Vaccines 12(9):1033–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.da Silva AJ, Zangirolami TC, Novo-Mansur MTM, de Campos Giordano R, Martins EAL. 2014. Live bacterial vaccine vectors: an overview. Braz. J. Microbiol 45(4):1117–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kotton CN, Hohmann EL. 2004. Enteric pathogens as vaccine vectors for foreign antigen delivery. Infect. Immun 72(10):5535–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Tuboly T, Nagy É, Derbyshire JB. 1993. Potential viral vectors for the stimulation of mucosal antibody responses against enteric viral antigens in pigs. Res. Vet. Sci 54(3):345–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Draper SJ, Heeney JL. 2010. Viruses as vaccine vectors for infectious diseases and cancer. Nat. Rev. Microbiol 8(1):62–73 [DOI] [PubMed] [Google Scholar]
- 123.Choi JH, Schafer SC, Zhang L, Kobinger GP, Juelich T, et al. 2012. A single sublingual dose of an adenovirus-based vaccine protects against lethal Ebola challenge in mice and guinea pigs. Mol. Pharm 9(1):156–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Rollier CS, Reyes-Sandoval A, Cottingham MG, Ewer K, Hill AVS. 2011. Viral vectors as vaccine platforms: deployment in sight. Curr. Opin. Immunol 23(3):377–82 [DOI] [PubMed] [Google Scholar]
- 125.Lubeck MD, Davis AR, Chengalvala M, Natuk RJ, Morin JE, et al. 1989. Immunogenicity and efficacy testing in chimpanzees of an oral hepatitis B vaccine based on live recombinant adenovirus. PNAS 86(17):6763–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Tacket CO, Losonsky G, Lubeck MD, Davis AR, Mizutani S, et al. 1992. Initial safety and immunogenicity studies of an oral recombinant adenohepatitis B vaccine. Vaccine 10(10):673–76 [DOI] [PubMed] [Google Scholar]
- 127.Khurana S, Coyle EM, Manischewitz J, King LR, Ishioka G, et al. 2015. Oral priming with replicating adenovirus serotype 4 followed by subunit H5N1 vaccine boost promotes antibody affinity maturation and expands H5N1 cross-clade neutralization. PLOS ONE 10(1):e0115476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Peters W, Brandl JR, Lindbloom JD, Martinez CJ, Scallan CD, et al. 2013. Oral administration of an adenovirus vector encoding both an avian influenza A hemagglutinin and a TLR3 ligand induces antigen specific granzyme B and IFN-γ T cell responses in humans. Vaccine 31(13):1752–58 [DOI] [PubMed] [Google Scholar]
- 129.Kim L, Martinez CJ, Hodgson KA, Trager GR, Brandl JR, et al. 2016. Systemic and mucosal immune responses following oral adenoviral delivery of influenza vaccine to the human intestine by radio controlled capsule. Sci. Rep 6:37295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Clements JD, Norton EB. 2018. The mucosal vaccine adjuvant LT(R192G/L211A) or dmLT. mSphere 3(4):e00215–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Summerton NA, Welch RW, Bondoc L, Yang H-H, Pleune B, et al. 2010. Toward the development of a stable, freeze-dried formulation of Helicobacter pylori killed whole cell vaccine adjuvanted with a novel mutant of Escherichia coli heat-labile toxin. Vaccine 28(5):1404–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Holmgren J, Bourgeois A, Carlin N, Clements J, Gustafsson B, et al. 2013. Development and preclinical evaluation of safety and immunogenicity of an oral ETEC vaccine containing inactivated E. coli bacteria overexpressing colonization factors CFA/I, CS3, CS5 and CS6 combined with a hybrid LT/CT B subunit antigen, administered alone and together with dmLT adjuvant. Vaccine 31:2457–64 [DOI] [PubMed] [Google Scholar]
- 133.El-Kamary SS, Cohen MB, Bourgeois AL, Van De Verg L, Bauers N, et al. 2013. Safety and immunogenicity of a single oral dose of recombinant double mutant heat-labile toxin derived from enterotoxigenic Escherichia coli. Clin. Vaccine Immunol 20(11):1764–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lundgren A, Bourgeois L, Carlin N, Clements J, Gustafsson B, et al. 2014. Safety and immunogenicity of an improved oral inactivated multivalent enterotoxigenic Escherichia coli (ETEC) vaccine administered alone and together with dmLT adjuvant in a double-blind, randomized, placebo-controlled Phase I study. Vaccine 32(52):7077–84 [DOI] [PubMed] [Google Scholar]
- 135.Norton EB, Lawson LB, Mahdi Z, Freytag LC, Clements JD. 2012. The A subunit of Escherichia coli heat-labile enterotoxin functions as a mucosal adjuvant and promotes IgG2a, IgA, and Th17 responses to vaccine antigens. Infect. Immun 80(7):2426–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.El-Kamary SS, Pasetti MF, Mendelman PM, Frey SE, Bernstein DI, et al. 2010. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J. Infect. Dis 202(11):1649–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Albu DI, Jones-Trower A, Woron AM, Stellrecht K, Broder CC, Metzger DW. 2003. Intranasal vaccination using interleukin-12 and cholera toxin subunit B as adjuvants to enhance mucosal and systemic immunity to human immunodeficiency virus type 1 glycoproteins. J. Virol 77(10):5589–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Toka FN, Rouse BT. 2005. Mucosal application of plasmid-encoded IL-15 sustains a highly protective anti-Herpes simplex virus immunity. J. Leukoc. Biol 78(1):178–86 [DOI] [PubMed] [Google Scholar]
- 139.Arora A, Prausnitz MR, Mitragotri S. 2008. Micro-scale devices for transdermal drug delivery. Int. J. Pharm 364(2):227–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Paliwal S, Hwang BH, Tsai KY, Mitragotri S. 2013. Diagnostic opportunities based on skin biomarkers. Eur. J. Pharm. Sci 50(5):546–56 [DOI] [PubMed] [Google Scholar]
- 141.Coffey JW, Corrie SR, Kendall MAF. 2013. Early circulating biomarker detection using a wearable microprojection array skin patch. Biomaterials 34(37):9572–83 [DOI] [PubMed] [Google Scholar]
- 142.Coffey JW, Corrie SR, Kendall MAF. 2018. Rapid and selective sampling of IgG from skin in less than 1 min using a high surface area wearable immunoassay patch. Biomaterials 170:49–57 [DOI] [PubMed] [Google Scholar]
- 143.Kim Y-C, Park J-H, Prausnitz MR. 2012. Microneedles for drug and vaccine delivery. Adv. Drug Deliv. Rev 64(14):1547–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Meliga SC, Coffey JW, Crichton ML, Flaim C, Veidt M, Kendall MAF. 2017. The hyperelastic and failure behaviors of skin in relation to the dynamic application of microscopic penetrators in a murine model. Acta Biomater 48:341–56 [DOI] [PubMed] [Google Scholar]
- 145.Ma Y, Tao W, Krebs SJ, Sutton WF, Haigwood NL, Gill HS. 2014. Vaccine delivery to the oral cavity using coated microneedles induces systemic and mucosal immunity. Pharm. Res 31(9):2393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.McNeilly CL, Crichton ML, Primiero CA, Frazer IH, Roberts MS, Kendall MAF. 2014. Microprojection arrays to immunise at mucosal surfaces. J. Control. Release 196:252–60 [DOI] [PubMed] [Google Scholar]
- 147.Depelsenaire ACI, Meliga SC, McNeilly CL, Pearson FE, Coffey JW, et al. 2014. Colocalization of cell death with antigen deposition in skin enhances vaccine immunogenicity. J. Investig. Dermatol 134(9):2361–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Crichton ML, Muller DA, Depelsenaire ACI, Pearson FE, Wei J, et al. 2016. The changing shape of vaccination: improving immune responses through geometrical variations of a microdevice for immunization. Sci. Rep 6:27217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Uddin MN, Allon A, Roni MA, Kouzi S. 2019. Overview and future potential of fast dissolving buccal films as drug delivery system for vaccines. J. Pharm. Pharm. Sci 22(1):388–406 [DOI] [PubMed] [Google Scholar]
- 150.Imran M 2012. Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device US Patent 9,861,683
- 151.Ther Rani. 2020. Rani therapeutics announces positive phase I study results of oral octreotide using RaniPill™. PR Newswire, January. 30 https://www.prnewswire.com/news-releases/rani-therapeutics-announces-positive-phase-i-study-results-of-oral-octreotide-using-ranipill-300992818.html?tc=eml_cleartime
- 152.Mistilis MJ, Bommarius AS, Prausnitz MR. 2015. Development of a thermostable microneedle patch for influenza vaccination. J. Pharm. Sci 104(2):740–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Maher S, Mrsny RJ, Brayden DJ. 2016. Intestinal permeation enhancers for oral peptide delivery. Adv. Drug Deliv. Rev 106:277–319 [DOI] [PubMed] [Google Scholar]
- 154.Lamson NG, Berger A, Fein KC, Whitehead KA. 2020. Anionic nanoparticles enable the oral delivery of proteins by enhancing intestinal permeability. Nat. Biomed. Eng 4(1):84–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Buckley ST, Bækdal TA, Vegge A, Maarbjerg SJ, Pyke C, et al. 2018. Transcellular stomach absorption of a derivatized glucagon-like peptide-1 receptor agonist. Sci. Transl. Med 10(467):eaar7047. [DOI] [PubMed] [Google Scholar]
- 156.Tuvia S, Pelled D, Marom K, Salama P, Levin-Arama M, et al. 2014. A novel suspension formulation enhances intestinal absorption of macromolecules via transient and reversible transport mechanisms. Pharm. Res 31(8):2010–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Li W, Joshi MD, Singhania S, Ramsey KH, Murthy AK. 2014. Peptide vaccine: progress and challenges. Vaccines 2(3):515–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Nayak B, Panda AK, Ray P, Ray AR. 2009. Formulation, characterization and evaluation of rotavirus encapsulated PLA and PLGA particles for oral vaccination. J. Microencapsul 26(2):154–65 [DOI] [PubMed] [Google Scholar]
- 159.Slütter B, Plapied L, Fievez V, Alonso Sande M, des Rieux A, et al. 2009. Mechanistic study of the adjuvant effect of biodegradable nanoparticles in mucosal vaccination. J. Control. Release 138(2):113–21 [DOI] [PubMed] [Google Scholar]
- 160.Sarti F, Perera G, Hintzen F, Kotti K, Karageorgiou V, et al. 2011. In vivo evidence of oral vaccination with PLGA nanoparticles containing the immunostimulant monophosphoryl lipid A. Biomaterials 32(16):4052–57 [DOI] [PubMed] [Google Scholar]
- 161.Filipović-Grčić J, Škalko-Basnet N, Jalšienjak I. 2001. Mucoadhesive chitosan-coated liposomes: characteristics and stability. J. Microencapsul 18(1):3–12 [DOI] [PubMed] [Google Scholar]
- 162.Perrie Y, Obrenovic M, McCarthy D, Gregoriadis G. 2002. Liposome (Lipodine™)-mediated DNA vaccination by the oral route. J. Liposome Res 12(1–2):185–97 [DOI] [PubMed] [Google Scholar]
- 163.Kazanji M, Laurent F, Péry P. 1994. Immune responses and protective effect in mice vaccinated orally with surface sporozoite protein of Eimeria falciformis in ISCOMs. Vaccine 12(9):798–804 [DOI] [PubMed] [Google Scholar]
- 164.Top FH Jr., Grossman RA, Bartelloni PJ, Segal HE, Dudding BA, et al. 1971. Immunization with live types 7 and 4 adenovirus vaccines. I. Safety, infectivity, antigenicity, and potency of adenovirus type 7 vaccine in humans. J. Infect. Dis 124(2):148–54 [DOI] [PubMed] [Google Scholar]
- 165.Lameiro MH, Malpique R, Silva AC, Alves PM, Melo E. 2006. Encapsulation of adenoviral vectors into chitosan-bile salt microparticles for mucosal vaccination. J. Biotechnol 126(2):152–62 [DOI] [PubMed] [Google Scholar]
- 166.Shata MT, Reitz MS Jr., DeVico AL, Lewis GK, Hone DM. 2001. Mucosal and systemic HIV-1 Env-specific CD8+ T-cells develop after intragastric vaccination with a Salmonella Env DNA vaccine vector. Vaccine 20(3):623–29 [DOI] [PubMed] [Google Scholar]
- 167.Naito T, Kaneko Y, Kozbor D. 2007. Oral vaccination with modified vaccinia virus Ankara attached covalently to TMPEG-modified cationic liposomes overcomes preexisting poxvirus immunity from recombinant vaccinia immunization. J. Gen. Virol 88:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]