

Abstract
Given the rapid spread of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection and the recent implementation of SARS-CoV-2 vaccination, we have much to learn about the duration of immune protection and the interface between the immune responses to infection and to vaccination. To address these questions, we monitored immune responses to SARS-CoV-2 infection in convalescent individuals over seven months and following mRNA vaccination. Spike Receptor-Binding-Domain (RBD)-specific circulating antibodies and plasma neutralizing activity generally decreased over time, whereas RBD-specific memory B cells persisted. Additionally, using antibody depletion techniques, we showed that the neutralizing activity of plasma specifically resides in the anti-RBD antibodies. More vigorous antibody and B cell responses to vaccination were observed in previously infected subjects relative to uninfected comparators, presumably due to immune priming by infection. SARS-CoV-2 infection also led to increased numbers of double negative B memory cells, which are described as a dysfunctional B cell subset. This effect was reversed by SARS-CoV-2 vaccination, providing a potential mechanistic explanation for the vaccination-induced reduction in symptoms in patients with “Long-COVID”.
Since December 2019 (1, 2), COVID-19, the disease caused by the novel severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), has infected >130 million people and caused about three million deaths worldwide (https://coronavirus.jhu.edu/map.html). The antibody response to SARS-CoV-2 infection has been a major research focus (examples are (3–5)), as it is directly relevant for diagnosis, seroprevalence studies, vaccine development, and immunotherapy (6–8). Our current knowledge of antibody longevity and function and antibody-producing cell lineages in this infection is still evolving and additional questions keep arising. Moreover, the ongoing SARS-CoV-2 vaccination campaign worldwide, including two mRNA vaccines [Moderna (mRNA-1273) or Pfizer–BioNTech (BNT162b2)] (9, 10), raises questions about the properties and duration of humoral and cellular responses to vaccination. While studies are being rapidly published (examples are (11–15)), much remains to be learned, particularly in terms of comparing responses to infection versus vaccination and determining how prior SARS-CoV-2 infection impacts the response to the vaccines.
Here we report the results of a study of antibody and memory B cell responses to SARS-CoV-2 infection of 83 Rutgers University employees who were infected with SARS-CoV-2 during the first wave of the COVID-19 pandemic in New Jersey (March-June 2020). We also characterize antibody and memory B cell responses in a cohort subset that received full (two-dose) administration of SARS-CoV-2 mRNA vaccines and compare them to the vaccine responses observed in non-infected subjects.
SARS-CoV-2 infection, which had been confirmed in 81/83 subjects by virus-specific PCR [two subjects were diagnosed by their physicians based on household exposure history, symptoms, and chest x-ray findings], induced mild to moderate symptoms in the vast majority of subjects, with only 5 (6%) reporting COVID-19-related hospitalization (demographics and clinical information are found in Supplemental Table 1). Of these 83 subjects, 22 completed between three and six study visits between April and December 2020. The demographics and clinical severity of these 22 subjects did not substantively differ from the overall study population except that this group included no participants who had been hospitalized for COVID-19 (Supplemental Table 2). We first analyzed the plasma samples of all 83 infected subjects for levels of antibodies directed to the receptor binding domain (RBD) of the SARS-CoV-2 Spike protein. The coronavirus Spike protein mediates viral entry into host cells and determines host range and tissue tropism (16). The S1 subunit of this protein interacts with the host receptor through its RBD (17). We chose to measure antibody responses to RBD because it is immunodominant and features poor sequence conservation among coronaviruses (16), which minimizes the potential detection of cross-reactive antibodies. We found that detection of RBD-specific antibodies well separated subjects that had tested positive to SARS-CoV-2 PCR (n = 83) from negative control subjects [pre-COVID-19 (n = 104) and SARS-CoV-2 PCR-negative subjects (n = 103) that remained SARS-CoV-2 PCR-negative for at least 16 weeks after the blood draw tested in the figure] (Fig. 1A). To monitor antibody responses over time, we retained 22 subjects for serial blood draws (monthly for 3 months and then bimonthly) over a period of seven months. We observed that the trajectories of RBD-specific IgG, IgM, and IgA antibodies were heterogeneous (Fig. 1B–D). In particular, the IgG response declined over time in 16 subjects (73%) while it remained stable or increased in 6 subjects (27%) (Fig. 1B). We also analyzed the virus neutralization activity of the plasma collected at the first and last study visit, using an assay with the natural SARS-CoV-2 virus. Neutralization activity decreased in most subjects (n = 15, 68%) (Fig. 1E) [the method for calculating neutralization titer 50 (NT50) is in Supplemental Fig. 1B]. When we compared neutralization titers (NT50) (Fig. 1E) with RBD-specific antibody titers (Supplemental Fig. 1A), we observed a positive correlation (r = 0.71; p<0.0001) between neutralizing titers and RBD-specific IgG titers (Fig. 1F), as also seen by others (for example, (18)). However, establishing correlations between RBD-specific IgG levels and plasma neutralizing activity only provides indirect evidence of a link between the two parameters. Moreover, demonstrating that RBD is a target of neutralizing antibodies (19–22) does not directly address the relative contribution of RBD-specific antibodies to the overall plasma neutralizing activity. To directly test the link between RBD specificity and the antibody-mediated ability of plasma to neutralize the virus, we depleted RBD-specific antibodies from seropositive plasma samples, and, as a comparator, we also depleted viral Nucleocapsid (N)-specific antibodies, and then tested the effects on neutralizing activity. We selected 10 plasma samples with high neutralizing activity and removed the antigen-specific antibodies from the plasma by incubation over multiple rounds with RBD or N (or no antigen) in solid-phase, prior to the neutralization assay. We confirmed that the pre-incubation process with either antigen resulted in depletion of the corresponding specific IgG, as demonstrated by ELISA (Fig. 1G–H). We found that the neutralizing activity of the plasma was abrogated only when the samples were preabsorbed with RBD, but not when they were preabsorbed with N (Fig. 1I). This provides direct evidence that the plasma neutralizing activity resides in the RBD-specific antibodies. Collectively, the data show that, at least in subjects that have experienced mild symptoms, circulating levels of RBD-specific IgG tend to decrease over time, with concurrent reduction of plasma neutralizing power. Decreases in circulating antibody below levels of detection will lead to an underestimation of the prevalence of SARS-CoV-2 infection using serological methods. However, this is likely mitigated by infections occurring in the population over consecutive pandemic waves, resulting in non-contemporaneous antibody trajectories in the overall population at any given time. In addition, our antibody depletion approach (that we find used only in another recent COVID-19 report (23)) directly shows that most (if not all) neutralizing activity of plasma resides in the RBD-specific antibodies. These results connect the protective properties of plasma with the molecular characterization of neutralizing monoclonal antibodies, thus providing a framework for antibody-based therapeutics and the rigorous assessment of the value of plasma therapy of COVID-19, which remains controversial (24).
Fig. 1. SARS-CoV-2 Spike S1 Receptor Binding Domain antibody levels and plasma neutralization activity.
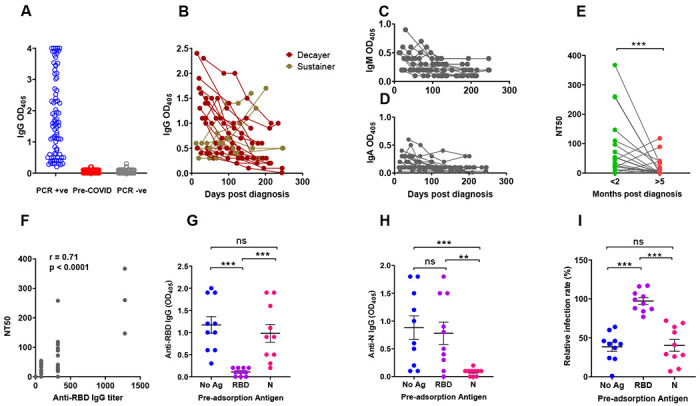
(A-D) Receptor Binding Domain (RBD)-specific antibody binding was determined by ELISA using serum/plasma samples diluted 1:80. (A) Dot plot showing anti-RBD IgG antibody levels in subjects PCR-positive for SARS-CoV-2 infection (n = 83, blue circles); pre-COVID-19 samples (n = 104; red circles); subjects SARS-CoV-2 PCR-negative for at least 16 weeks following the blood draw used in this figure (n = 103 subjects; grey circles). Each circle corresponds to one subject. (B) The line graph shows the trajectory of IgG antibody levels for 22 subjects over the indicated time post-PCR result. Symbol colors distinguish decayers (n=16) showing downward trajectories (dark red symbols) and sustainers (n=6) showing stable or upward trajectories (dark yellow symbols). (C,D) Line graphs showing IgM and IgA antibody levels, as indicated, in the same subjects as (B). (E-G) Neutralizing activity of plasma was determined utilizing ACE2-expressing HeLa cells and mNeonGreen-tagged SARS-CoV-2 virus. (E) Neutralizing titers were expressed as NT50 (reciprocal dilution of plasma yielding 50% neutralization), as obtained with samples collected <2 months (green circles) or >5 months (red circles) post-diagnosis from the same 22 subjects. Each pair of circles connected by a line represents one subject. (F) Correlation analysis between anti-RBD IgG endpoint titers (x-axis) and neutralizing titers (y-axis) (n = 44). The Spearman’s correlation coefficient r and corresponding p value were calculated. (G-H) Selected plasma samples (n = 10) exhibiting NT50 >80 were depleted of either RBD- or N-specific antibodies by pre-incubating them with either recombinant RBD or N (Nucleocapsid) proteins and then tested for ELISA reactivity. No Ag, mock pre-adsorption; RBD, pre-absorption with RBD; N, pre-absorption with N. The panels show IgG antibody binding by ELISA using RBD (panel G) or N (panel H) as solid-phase antigen and pre-adsorbed plasma (No Ag, RBD, N). (I) Neutralization activity of pre-absorbed plasma samples (No Ag, RBD, N, as in panels G-H). Relative infection rates, shown in the Y axis of the scatter plot, were obtained by dividing the fluorescence readings (derived from fluorescently tagged virus) of the sample-treated wells by the untreated control wells (infection rates above 100% result from small biological variations among untreated wells, and indicate no neutralization). In all panels, the solid black lines represent the median and Interquartile range. Statistical analyses in (G-I) were conducted by Wilcoxon matched pairs signed rank test (**, p ≤ 0.01; ***, p ≤ 0.001; ns, non-significant, p > 0.05).
The progressive decrease of protective antibodies in the circulation raises the question of whether immune protection against SARS-CoV-2 infection also wanes over time. We thus analyzed the memory B cell response in the same subjects. To do so, we developed a multicolor flow cytometry panel to measure frequencies of circulating B cells (CD19+CD20+) and B cell subsets including plasmablasts (CD27+CD38hi), naïve (CD27−IgD+), and memory B cell compartments [non-switched memory (CD27+IgD+), switched memory (CD27+IgD−), and double negative memory (CD27−IgD−)] (Fig. 2A–E). We also evaluated frequencies of RBD-specific B cells (RBD-tetramer-positive CD19+CD20+) (Fig. 2F), which we further subdivided into the memory compartments in Fig. 2E. The results of all analyses are shown in Supplemental Tables 3–4. When we compared the results obtained from our cohort of infected subjects with a group of 13 SARS-CoV-2 non-infected individuals [pre-COVID (n = 5) and SARS-CoV-2 PCR-negative samples (n = 8)], we observed that SARS-CoV-2 infection correlated with a transient increase of plasmablast frequencies (Fig. 2G) and the appearance of antigen-specific (RBD+) B cells (Fig. 2H). Plasmablast frequencies are known to increase in response to various infections, including SARS-CoV-2 (25–28). Among circulating non-plasmablasts, changes of frequencies in circulating naïve and switched memory B cells presumably reflect B cell remodeling in response to recent infection (Supplemental Table 3). Analysis of RBD+ memory B cells in the first- vs last-visit samples showed stable frequencies over time, indicating a durable memory response (Fig. 2H). Notably, the frequency of an RBD+ double negative memory (DNM) B cell subset (CD27−IgD−) increased between the two study visits (Fig. 2I, left panel). This increase was even stronger for the total pool of DNM B cells (Fig. 2I, right panel). This subset, which increases with age (29), is considered a component of the B cell memory compartment despite the absence of the CD27 memory markers, because it bears signatures of antigen experienced B cells in terms of surface phenotype, proliferation response, and patterns of somatic hypermutations (30, 31). DNM B cells likely constitute a heterogeneous B cell subset, as they have been described as exhausted / prematurely senescent B cells in HIV infection and other diseases characterized by chronic immune activation (32, 33), or as producing autoantibodies in autoimmune diseases such as systemic lupus erythematosus (SLE) (34). Interestingly, SARS-CoV-2-induced exhaustion and senescence phenotypes have been previously reported for T cells (35). Thus, our data suggest that immune exhaustion, which presumably correlates with the degree of inflammation and disease severity, occurs in both B cells and B cells during SARS-CoV-2 infection. Moreover, COVID-19 has been associated with autoantibody production in adults and children (36–38), suggesting that DNM B cells may be a source of pathogenic antibodies, particularly in the context of the enhanced immune activation and excessive cytokine production associated with severe COVID-19 (39). Further characterization of double-negative memory B cell frequencies and phenotypes across the COVID-19 severity spectrum is urgently needed.
Fig. 2. Total and SARS-CoV-2 S1 RBD-specific B cell compartments.
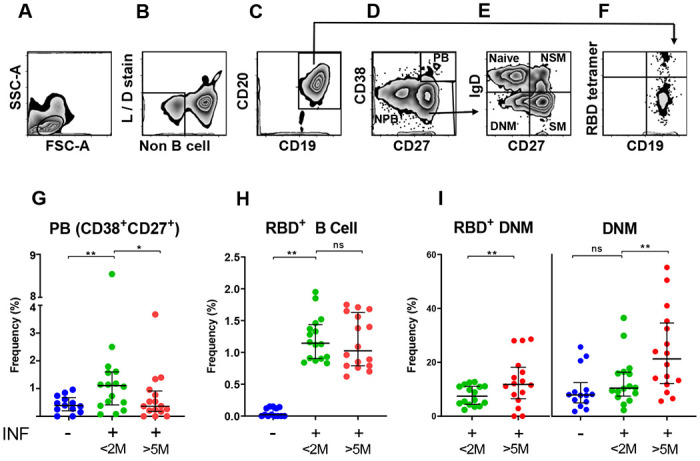
(A-F) Representative flow cytometry gating strategy for B cell responses to SARS-CoV-2 infection. (A) Physical parameters; (B) Exclusion of dead cells and non-B cells (CD14+, CD3+, CD4+, CD56+); (C) CD19+CD20+ cells (B cells) were further gated to distinguish (D) Plasmablast (CD27+CD38hl) and non-plasmablasts; (E) Non-plasmablasts were further gated on CD27 and IgD for memory phenotyping and classified as Naïve B cells (CD27−IgD+), Non-switched memory B cells (NSM; CD27+IgD+), Switched memory B cells (SM; CD27+IgD−), and Double negative memory B cell (DNM; CD27−IgD−); (F) CD19+CD20+ (B cells) were also analyzed for RBD-specificity utilizing fluorescently labeled RBD tetramers; (G-I) Frequency (%) of various B cell types, as indicated in each panel, are shown as scatter plot. Each circle represents one study subject; the solid black lines represent the median and interquartile range. <2M or >5M, months after SARS-CoV-2 diagnosis. Statistical analysis was performed either with Mann-Whitney U-test for unpaired data or Wilcoxon matched-pairs signed rank test for paired samples; * p < 0.05, ** p < 0.01.
Some members of our longitudinal cohort received COVID-19 vaccination in December 2020 - February 2021 (RNA vaccines), providing the opportunity to determine whether re-exposure to antigen via vaccination induces recall humoral and B cell responses. We measured antibody and B cell responses to full vaccination (two doses of COVID-19 RNA vaccines) in 12 cohort members (i.e., previously infected). As comparators, we also tested 8 non-infected subjects that were SARS-CoV-2 PCR-negative and seronegative prior to vaccination. We found that vaccination induced very vigorous antibody responses in both groups and that the response was much higher in infected than non-infected subjects (Fig. 3A). Moreover, IgG titers were much higher in non-infected vaccinated than infected non-vaccinated subjects at the first study visit (Fig. 3B). We also analyzed the effect of vaccination in the two groups on plasma neutralization titers and RBD-specific memory B cell frequencies. Similar to the effect on antibody response, we found that vaccination induced stronger neutralizing activity in the previously infected group (Fig. 3C). Also, vaccination of the non-infected group led to a stronger neutralizing activity than infection alone (Fig. 3D). We also found that vaccination of the infected group led to higher RBD-specific memory B cell frequencies than vaccination of the non-infected group (Fig. 3E). Collectively, these results demonstrate that administration of COVID-19 RNA vaccines induces vigorous humoral and B cell responses, and a strong recall response in subjects previously exposed to the virus. Moreover, our data suggest that vaccination elicits stronger responses than (at least mild) SARS-CoV-2 infection. Analysis of the memory B cell pool showed that, in both groups, vaccination has no effect on circulating plasmablast frequencies and is associated with remodeling and/or redistribution in the periphery of B cell memory compartments (increased naïve and decreased switched memory B cells) in response to vaccination (Supplemental Table 4). Interestingly, the frequencies of double-negative memory B cells decreases in previously infected individuals (but not in non-infected subjects) following vaccination (Fig. 3F). These declines were statistically significant despite the small sample size. Thus, our results strongly suggest that the response to vaccine counters the infection-induced increased production of potentially dysfunctional and pathogenic immune cells (Fig. 2I). This effect requires confirmation from larger studies.
Fig. 3. Humoral and B cell responses to COVID-19 RNA vaccination.
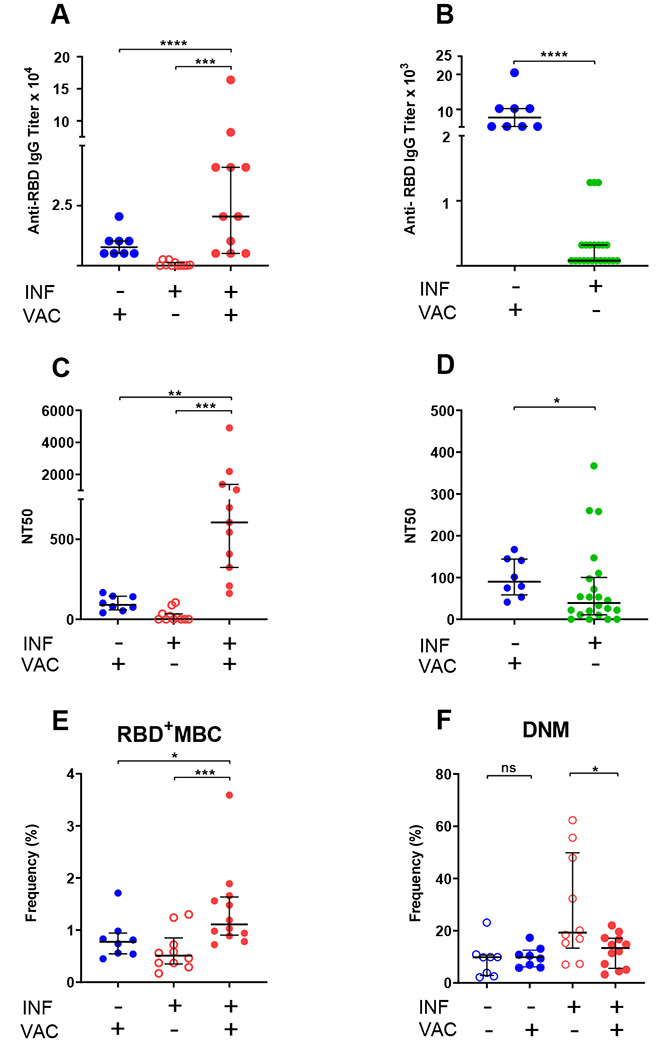
Responses to COVID-19 RNA vaccines in previously infected study subjects and non-infected comparators were determined. Receptor Binding Domain (RBD)-specific IgG antibody binding, plasma neutralizing activity, and B cell frequencies were assessed as in Fig. 1, 2. Infection (INF) and vaccination (VAC) status is indicated by (+) and (−) signs. (A,B) Titers of RBD-specific IgG by ELISA; (C,D) NT50 determinations in neutralization assays. Pre-vaccination samples of previously infected subjects in panels (A,C) were those collected last prior to vaccination to determine effect of vaccination; in panels (B,D) were those collected first (< 2 months) post SARS-CoV-2 diagnosis to compare responses to recent infection and recent vaccination. (E) Frequency of RBD-specific memory B cells (RBD+MBC). We used only one dataset for pre-vaccination infected subjects for RBD+MBC, because these cells do not vary between the <2 and >5 data points. (F) Frequency of total double-negative memory B cells (DNM, CD27−IgD−). RBD-specific DNM frequency showed the same downward trend post-vaccination (p = 0.05). In all panels, data are shown as scatter plots; each circle represents one study subject; the solid black lines represent the median and interquartile range. Statistical analysis was performed either with Mann-Whitney U-test for unpaired data or Wilcoxon matched-pairs signed rank test for paired samples; * p < 0.05, ** p < 0.01.
Conclusions.
In subjects who have experienced mild SARS-CoV-2 infection, we show that the decrease of circulating protective antibodies over time is accompanied by durable memory responses that are competent to induce potent recall responses upon re-exposure to antigen. Our works confirms studies showing potent immune responses induced by anti-SARS-CoV-2 vaccines (for example, (15, 40)). In particular, we show that the humoral and B cell response to vaccination is more vigorous than that induced by infection alone, supporting the current practice in the USA of offering SARS-CoV-2 vaccination regardless of infection history. The direct demonstration that the neutralizing activity of plasma resides mostly if not fully in the RBD antibody specificities provides a molecular foundation to the prior correlations between antibody binding and antibody-mediated neutralizing activity and to the evaluation of antibody-based COVID-19 therapies. Furthermore, our novel finding that SARS-CoV-2 infection is associated with increased numbers of double negative memory B cells, even in subjects who experienced relatively mild symptoms, suggests a hitherto unknown potential mechanism of SARS-CoV-2-induced immune dysfunction. This may explain the observed production of autoantibodies in COVID-19 patients and/or reveal the accumulation of exhausted/prematurely senescent B cells, which complements the known SARS-CoV-2-induced T cell exhaustion/senescence. It is intriguing that vaccination counters this aspect of immune dysregulation. The still anecdotal reports of symptomatic relief after vaccination in persons with post-acute sequelae of SARS-CoV-2 infection (41, 42) may find mechanistic grounds in these findings.
Materials and Methods
Study participants.
Eighty-three convalescent individuals with a history of SARS-CoV-2 infection were enrolled between April 1 and June 17, 2020 from among Rutgers University employees to monitor virus-specific immune responses. Participants showed proof of positive results of SARS-CoV-2 PCR assays that had received Emergency Use Authorization from the Food and Drug Administration with the exception of two subjects that received a clinical diagnosis only. All participants self-reported the date of symptom onset and consented to blood draws as well as the collection of demographics and clinical symptoms. Of 83 participants, 22 were retained for 3–6 study visits over a period of 7 months (once a month for the first 3 months, and then once every two months). As negative control for SARS-CoV-2 antibody testing, we used 104 stored serum/plasma samples collected prior to the COVID-19 pandemic (Institutional review board of the Rutgers New Jersey Medical School, Pro0119980237 and Pro20150001314) and 103 serum samples obtained during the pandemic from subjects that remained SARS-CoV-2 PCR-negative and seronegative for at least 16 weeks following the test blood draw (43) (ClinicalTrials.gov registration number NCT04336215). Among study participants that were vaccinated with either Moderna or Pfizer mRNA vaccines between mid-December 2020 and mid-February 2021, 12 donated blood at least two weeks after the second vaccine dose between February 22 and March 1, 2021. Between December 22, 2020 and February 11,2021, blood pre- and post-vaccination was also obtained from 8 subjects that were SARS-CoV-2 PCR-negative and seronegative prior to vaccination.
Antibody binding by enzyme-linked immunosorbent assay (ELISA)
96-well ELISA plates (Nunc MaxiSorp, ThermoFisher, Rochester NY) were coated with 2 μg/ml recombinant SARS-CoV-2 RBD overnight at 4°C. Plates were washed with washing buffer (1 X PBS containing 0.05% Tween 20) (Sigma-Aldrich, St. Louis MO) and incubated with 2% BLOTTO (Nestle Carnation, US) in PBS for 30 min at 37°C. Plasma was heat-inactivated at 56°C for 1 hour prior to use. After blocking, diluted plasma was added in blocking buffer and incubated for 1 hour at 37°C. Antigen-specific IgG was detected by adding alkaline phosphatase-conjugated goat anti-human IgG (Jackson ImmunoResearch, West Grove PA). ELISA plates were developed using phosphate substrate (Sigma-Aldrich) and the reaction was stopped with 1M NaOH. The ELISA protocol was performed using a BioTek EL406 combination washer dispenser, and absorbance (OD405nm) was measured using a BioTek Synergy Neo2 microplate reader (BioTek, Winooski VT). Each ELISA plate contained positive and negative serum/plasma controls and background control wells without primary antibody. Each sample was tested in duplicate. End-point titers were plotted for each sample using background-subtracted data. All work involving blood products from SARS-CoV-2-infected subjects were performed in a biosafety level 2+ (BSL-2+) laboratory utilizing protocols approved by the Rutgers Institutional Biosafety Committee.
Expression and purification of Recombinant SARS-COV-2 S1 RBD Protein
A DNA fragment encoding RBD (Spike residues aa. 319 to aa. 537) was amplified and cloned into the eukaryotic expression vector pcDNA3.1 (Addgene, Watertown MA). Purified plasmid was transfected into 293F cells using the Expi293 Expression system (Thermo Fisher Scientific, Waltham MA), according to the manufacturer’s protocol. Supernatants were collected on day 3 post-transfection, purified by HisPur™ Cobalt Resin (Thermo Fisher Scientific) and eluted with 200mM imidazole. Purified protein was subsequently dialyzed against PBS at 4°C. Absorbance (OD280nm) was determined by Nanodrop reading and concentrations were calculated using ExPASy Proteomics calculator. Molecular weights were adjusted to account for the number of N-linked glycosylation sites to determine the final concentration.
Absorption of convalescent plasma with SARS-CoV-2 antigens
ELISA 96-well microtiter plates were coated with 500 ng/well of SARS-CoV-2 RBD or N proteins at 4°C overnight. Coated plates were washed three times with washing buffer and blocked with PBS containing 1% BSA (Sigma-Aldrich) for 30 min at 37°C. After washing, plasma samples were diluted 1:10 in PBS containing 1% BSA (Sigma-Aldrich) and incubated up to overnight at 4°C for each cycle. Absorption was repeated at least four times utilizing fresh antigen-coated plates at each cycle. To monitor depletion of antigen-specific antibodies, RBD- and N-specific IgG titers of untreated and absorbed samples were determined as described above, prior to use in neutralization assays.
Cell lines
Vero E6 were obtained from the American Type Culture Collection (ATCC; Manassas VA) and HeLa cells stably expressing ACE2 (HeLa-ACE2) were obtained from Dennis Burton at the Scripps Research Institute (21). All cell lines were maintained in high-glucose Dulbecco’s modified Eagle’s medium (DMEM; Corning, Manassas VA) supplemented with 10% fetal bovine serum (FBS; Seradigm, Radnor PA), 2mM L-glutamine (Corning), 1% penicillin/streptomycin (Corning), and incubated in humidified atmospheric air containing 5% CO2 at 37°C.
SARS-CoV-2 virus
The virus stock of mNeonGreen (mNG) SARS-CoV-2 was obtained from Pei-Yong Shi at the University of Texas Medical Branch at Galveston. The virus stock was produced using the virus isolate of the first patient diagnosed in the US, in which the ORF7 of the viral genome was replaced with the reported mNG gene (44). Propagation of viral stocks was performed with Vero E6 cells using 2% FBS. The virus titers were determined by standard plaque assay utilizing Vero E6 cells (45) and recorded as plaque forming units per milliliter (PFU/mL). All work involving live SARS-CoV-2 were performed in a biosafety level 3 (BSL-3) laboratory utilizing protocols approved by the Rutgers Institutional Biosafety Committee.
SARS-CoV-2 neutralization assay
HeLa-Ace2 cells were seeded in 96-well black optical-bottom plates at a density of 1 × 104 cells/well in FluoroBrite DMEM (Thermo Fisher Scientific) containing 4% FBS (Seradigm), 2mM L-glutamine (Corning), 1% penicillin/streptomycin (Corning) and incubated overnight at 37°C with 5% CO2. On the following day, each sample was subjected to two-fold serial dilution in DMEM without FBS, and incubated with mNG SARS-CoV-2 at 37°C for 1.5 h. The virus-plasma mixture was transferred to 96-well plates containing Hela-Ace2 cells at a final multiplicity of infection (MOI) of 0.25 (viral PFU:cell). For each sample, the starting dilution was 1:20 and the final dilution of 1:10,240. After incubating infected cells at 37°C for 20 h, mNG SARS-CoV-2 fluorescence was measured using a Cytation™ 5 reader (BioTek). Relative fluorescent units were converted to percent neutralization by normalizing the sample-treatment to non-sample-treatment controls and plotted with a nonlinear regression curve fit to determine the titer neutralizing 50% of SARS-CoV-2 fluorescence (NT50). Each patient sample was tested in duplicate. All plasma samples were heat-inactivated at 56°C for 60 min before testing.
PBMC isolation and storage
Peripheral blood mononuclear cells (PBMC) were isolated by Ficoll density gradient centrifugation (Ficoll-Paque, GE Healthcare, Uppsala, Sweden), as described (46). Briefly, whole blood was diluted with equal volume of Roswell Park Memorial Institute Medium (RPMI; Corning) and layered on Ficoll-Paque (GE healthcare, USA). The gradient was centrifuged at 500 × g for 30 min at room temperature. Plasma was carefully removed, aliquoted, and stored at −80°C. The PBMC interface was collected, washed once, and counted using a hemocytometer. PBMCs were cryopreserved in liquid nitrogen in FBS containing 10% dimethyl sulfoxide (DMSO; Thermo Fischer Scientific) and stored until use.
B cell immunophenotyping
Biotinylated RBD (Biolegend, San Diego CA) was mixed with streptavidin PerCP-Cy5.5. (Thermo Fischer Scientific) at 4:1 molar ratio for 1 hour at 4°C to form the RBD tetramer (RBD4-PercpCy5.5). PBMCs were incubated with the fixable viability stain 780 (BD Biosciences, Franklin Lakes NJ), followed by 10 min incubation with human Fc receptor blocking reagent (BD Biosciences). For the detection of antigen-specific B cells, PBMCs were incubated with the RBD tetramer and antibodies to CD19-BV700 (HIB19, BioLegend), CD20-PE-CF594 (2H7, BD Biosciences), CD27-PE (L128, BD Biosciences), CD38-BB515 (HIT2, BD Biosciences), IgM-BV605 (MHM-88, BioLegend), IgG-PE-Cy7 (G18–145, BD Biosciences). Additionally, cells were stained with APC-labelled antibodies against CD3 (UCHT1, Thermo Fisher Scientific), CD4 (OKT4, Thermo Fisher Scientific), CD14 (C1D3, Thermo Fisher Scientific), and CD16 (CB16, Thermo Fisher Scientific) to eliminate non-B cells. Antibody binding was allowed by incubating samples for 30 minutes on ice in the dark. Cells were washed twice with FACS buffer (0.1% BSA in PBS), fixed with 4% paraformaldehyde (PFA) for 20 min, and stored at 4°C overnight. For each sample, at least 250,000 events were acquired on a BD Fortessa X-20 flow cytometer (BD Biosciences).
Statistical Analysis
All flow cytometry data were analyzed with FlowJo v12 software (FlowJo LLC, Ashland OR). Statistical analysis was performed with GraphPad Prism 8.4 (Graph Pad Software Inc., La Jolla CA). Data values are presented as median with interquartile range (IQR). Correlation analysis was performed using the non-parametric Spearman’s rank correlation. Statistical analysis was performed utilizing either Mann-Whitney U test for unpaired samples or Wilcoxon matched pairs signed rank test for paired samples. With all tests, p < 0.05 was considered significant.
Supplementary Material
Acknowledgements.
We thank the PHRI biosafety officers and RBHS Institutional Biosafety committee for fast-track review and approval of laboratory protocols and practices related to handling of SARS-CoV-2 and infected biospecimens; Daniel Fine and Steven Libutti for supporting the start of our COVID-19 work; Nancy Reilly and the entire Rutgers Corona Cohort team that established a cohort from which non-infected samples were obtained during the pandemic; Tracy Andrews of RUBIES for advice on statistical methods; Dennis Burton and Pei-Yong Shi for providing biological reagents; and Life Science Editors for professional editorial services. This work was funded by NIH grants R01 HL149450, R01 HL149450-S1, U01 AI122285-S1, and UL1 TR003017.
References
- 1.Zhou P. et al. , A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579, 270–273 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu F. et al. , A new coronavirus associated with human respiratory disease in China. Nature 579, 265–269 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Long Q. X. et al. , Antibody responses to SARS-CoV-2 in patients with COVID-19. Nat Med 26, 845–848 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Robbiani D. F. et al. , Convergent antibody responses to SARS-CoV-2 in convalescent individuals. Nature 584, 437–442 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Amanat F. et al. , A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat Med 26, 1033–1036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rostami A. et al. , SARS-CoV-2 seroprevalence worldwide: a systematic review and meta-analysis. Clin Microbiol Infect 27, 331–340 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen L., Xiong J., Bao L., Shi Y., Convalescent plasma as a potential therapy for COVID-19. Lancet Infect Dis 20, 398–400 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee C. Y., Lin R. T. P., Renia L., Ng L. F. P., Serological Approaches for COVID-19: Epidemiologic Perspective on Surveillance and Control. Front Immunol 11, 879 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson L. A. et al. , An mRNA Vaccine against SARS-CoV-2 - Preliminary Report. N Engl J Med 383, 1920–1931 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Polack F. P. et al. , Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 383, 2603–2615 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saadat S. et al. , Binding and Neutralization Antibody Titers After a Single Vaccine Dose in Health Care Workers Previously Infected With SARS-CoV-2. Jama, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Manisty C. et al. , Antibody response to first BNT162b2 dose in previously SARS-CoV-2-infected individuals. Lancet 397, 1057–1058 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Krammer F. et al. , Antibody Responses in Seropositive Persons after a Single Dose of SARS-CoV-2 mRNA Vaccine. N Engl J Med 384, 1372–1374 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goel R. R. et al. , Longitudinal Analysis Reveals Distinct Antibody and Memory B Cell Responses in SARS-CoV2 Naïve and Recovered Individuals Following mRNA Vaccination. medRxiv, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Z. et al. , mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li F., Structure, Function, and Evolution of Coronavirus Spike Proteins. Annu Rev Virol 3, 237–261 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wrapp D. et al. , Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 367, 1260–1263 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wajnberg A. et al. , Robust neutralizing antibodies to SARS-CoV-2 infection persist for months. Science 370, 1227–1230 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shi R. et al. , A human neutralizing antibody targets the receptor binding site of SARS-CoV-2. Nature, (2020). [DOI] [PubMed] [Google Scholar]
- 20.Pinto D. et al. , Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody. Nature, (2020). [DOI] [PubMed] [Google Scholar]
- 21.Rogers T. F. et al. , Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model. Science 369, 956–963 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andreano E. et al. , Extremely potent human monoclonal antibodies from COVID-19 convalescent patients. Cell 184, 1821–1835.e1816 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Greaney A. J. et al. , Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host Microbe 29, 463–476.e466 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Katz L. M., (A Little) Clarity on Convalescent Plasma for Covid-19. N Engl J Med 384, 666–668 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Garcia-Bates T. M. et al. , Association between magnitude of the virus-specific plasmablast response and disease severity in dengue patients. J Immunol 190, 80–87 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.García M. et al. , Massive plasmablast response elicited in the acute phase of hantavirus pulmonary syndrome. Immunology 151, 122–135 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vijay R. et al. , Infection-induced plasmablasts are a nutrient sink that impairs humoral immunity to malaria. Nat Immunol 21, 790–801 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.De Biasi S. et al. , Expansion of plasmablasts and loss of memory B cells in peripheral blood from COVID-19 patients with pneumonia. Eur J Immunol 50, 1283–1294 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Buffa S. et al. , B cell immunosenescence: different features of naive and memory B cells in elderly. Biogerontology 12, 473–483 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Wei C. et al. , A new population of cells lacking expression of CD27 represents a notable component of the B cell memory compartment in systemic lupus erythematosus. J Immunol 178, 6624–6633 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Ruschil C. et al. , Specific induction of double negative B cells during protective and pathogenic immune responses. bioRxiv, 2020.2009.2008.285148 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moir S. et al. , Evidence for HIV-associated B cell exhaustion in a dysfunctional memory B cell compartment in HIV-infected viremic individuals. J Exp Med 205, 1797–1805 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernard N. J., Double-negative B cells. Nat Rev Rheumatol 14, 684 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Jenks S. A. et al. , Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 49, 725–739.e726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.De Biasi S. et al. , Marked T cell activation, senescence, exhaustion and skewing towards TH17 in patients with COVID-19 pneumonia. Nat Commun 11, 3434 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Consiglio C. R. et al. , The Immunology of Multisystem Inflammatory Syndrome in Children with COVID-19. Cell 183, 968–981.e967 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bastard P. et al. , Autoantibodies against type I IFNs in patients with life-threatening COVID-19. Science 370, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y. et al. , Coagulopathy and Antiphospholipid Antibodies in Patients with Covid-19. N Engl J Med 382, e38 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu B., Huang S., Yin L., The cytokine storm and COVID-19. J Med Virol 93, 250–256 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lederer K. et al. , SARS-CoV-2 mRNA Vaccines Foster Potent Antigen-Specific Germinal Center Responses Associated with Neutralizing Antibody Generation. Immunity 53, 1281–1295.e1285 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.“https://www.theatlantic.com/health/archive/2021/03/vaccines-long-covid/618406/”.
- 42. https://www.nytimes.com/2021/03/17/health/coronavirus-patients-and-vaccine-effects.html.
- 43.Barrett E. S. et al. , Prevalence of SARS-CoV-2 infection in previously undiagnosed health care workers in New Jersey, at the onset of the U.S. COVID-19 pandemic. BMC Infect Dis 20, 853 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie X. et al. , An Infectious cDNA Clone of SARS-CoV-2. Cell Host Microbe 27, 841–848.e843 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Case J. B., Bailey A. L., Kim A. S., Chen R. E., Diamond M. S., Growth, detection, quantification, and inactivation of SARS-CoV-2. Virology 548, 39–48 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arrigucci R. et al. , FISH-Flow, a protocol for the concurrent detection of mRNA and protein in single cells using fluorescence in situ hybridization and flow cytometry. Nat Protoc 12, 1245–1260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.