

Abstract
Respiratory syncytial virus (RSV) bronchiolitis is a leading cause of infant hospitalization and mortality. We previously identified that prostaglandin D2 (PGD2), released following RSV infection of primary human airway epithelial cells or pneumonia virus of mice (PVM) infection of neonatal mice, elicits pro- or antiviral innate immune responses as a consequence of D-type prostanoid receptor 2 (DP2) or DP1 activation, respectively. Here, we sought to determine whether treatment with the DP1 agonist BW245c decreases the severity of bronchiolitis in PVM-infected neonatal mice. Consistent with previous findings, BW245c treatment increased IFN-λ production and decreased viral load in week 1 of the infection. However, unexpectedly, BW245c treatment increased mortality in week 2 of the infection. This increased morbidity was associated with viral spread to the parenchyma, an increased cellular infiltrate of TNF-α-producing cells (neutrophils, monocytes, and CD4+ T cells), and the heightened production of the pro-inflammatory cytokines TNF-α, IL-6, and IL-1β. These phenotypes, as well as the increased mortality, were significantly attenuated following the administration of anti-TNF-α to PVM-infected, BW245c-treated mice. In summary, pharmacological activation of the DP1 receptor in PVM-infected neonatal mice boosts antiviral innate and adaptive immunity, however, this is ultimately detrimental, as a consequence of increased TNF-α-induced morbidity and mortality.
Introduction
Viral bronchiolitis is one of the major causes of hospitalization and premature death in children under 5 years of age.1 An infection with respiratory syncytial virus (RSV) is the most common cause of bronchiolitis in infants. The vast majority of infant deaths occur in developing countries where access to supportive care, such as supplemental oxygen, is limited.2 Pathologically, severe bronchiolitis is characterized by an overexuberant inflammatory response, leading to oedema, epithelial sloughing, and mucus hypersecretion.2,3 The cellular inflammation is dominated by neutrophils, monocytes, CD4+ T cells, and CD8+ T cells and a pro-inflammatory cytokines milieu typified by elevated levels of TNF-α, IL-1β, and IL-6. Susceptibility to severe bronchiolitis is associated with many factors, including genetic vulnerability, poor maternal diet, smoking in pregnancy, premature birth, malnutrition, and age-related immaturity of the immune system.4–7 The latter may affect the production of antiviral cytokines, such as type I, II, and III interferons (IFNs), which promote viral clearance8 and prevent viral spread to the lung parenchyma.9,10
Prostaglandin D2 (PGD2) is a lipid mediator derived from arachidonic acid metabolism and signals via two G-protein coupled receptors, D-type prostanoid receptor 1 (DP1) and 2 (DP2).11 The prevailing view is that PGD2 is released by allergen-activated mast cells or virus-infected airway epithelial cells (AECs), whereupon it causes DP2-mediated bronchoconstriction and type 2 inflammation through the activation of multiple effector cells including CD4+ T-helper 2 (TH2) cells and type 2 innate lymphoid cells (ILC2s) in asthma.12–14 By contrast, activation of PGD2-DP1 inhibits dendritic cell (DC) migration to the lung draining lymph nodes, promotes regulatory T-cell function and induces eosinophil apoptosis, and thus confers asthma resistance.15,16 Additionally, DP1 agonism elicits TNF-α secretion from lung macrophages, leading to the recruitment and activation of neutrophils.17 More recently, PGD2-mediated activation of DP1 was found to increase type 1 IFN expression by microglia cells during neurotropic coronavirus infection, minimizing brain damage.18
We recently discovered that PGD2 levels are elevated in nasopharyngeal aspirates of young infants hospitalized with severe RSV bronchiolitis.19 RSV infection of primary human AECs from healthy pediatric donors, as well as infection of neonatal mice with pneumonia virus of mice (PVM; a mouse orthologue of RSV), increased the release of PGD2. In both the human AECs and the neonatal mice, the virus-induced PGD2 suppressed the production of antiviral cytokines, an effect that was blocked via antagonism of DP2. Intriguingly, the beneficial effect of DP2 antagonism was ablated by dual DP2 and DP1 antagonism, and could be replicated via treatment with a DP1 agonist.19 These findings suggested that DP1 agonists might have utility as a novel, adjunct therapy for the treatment of viral bronchiolitis. Here, we sought to explore this possibility and to further identify the cellular and molecular mechanisms by which DP1 agonism promotes antiviral immunity.
Results
Treatment with a DP1 agonist increases mortality in PVM-infected mice
In a neonatal mouse model of acute virus-induced lower respiratory infection (LRI), using the natural rodent pathogen PVM, we previously observed that PGD2-mediated activation of DP2 is detrimental, whereas DP1 receptor activation increases the production of IFNs and decreases viral load at 7 days post infection (dpi).19 This protective phenotype was induced via daily treatment from 3 to 9 dpi with the DP1 agonist BW245c. Here, we questioned whether decreasing the number of treatment days to 2 (days 3 and 4) or 4 (days 3, 4, 5, and 6; Fig. 1a, b) would elicit a similar protective effect. Mice were monitored until 15 dpi to assess for improvements in the clinical score. DP1 agonist treatment of PVM-infected pups significantly increased mortality (primarily between 10 and 12 dpi) regardless of the number of treatments, compared to pups infected with PVM alone (Fig. 1c). This was unexpected, however in our previous study, the mice were euthanised at 10 dpi.19 In a separate model of bronchiolitis, elicited through co-exposure to PVM and cockroach allergen (CRE),20 treatment with the DP1 agonist again led to increased mortality (Fig. S1A, B). In all three scenarios, treatment with the DP1 agonist exacerbated PVM-associated stunting of body weight gain (Figs. 1d and S1C), indicating a heightened inflammatory response.
Fig. 1. Treatment with a DP1 agonist increases mortality in PVM-infected mice.
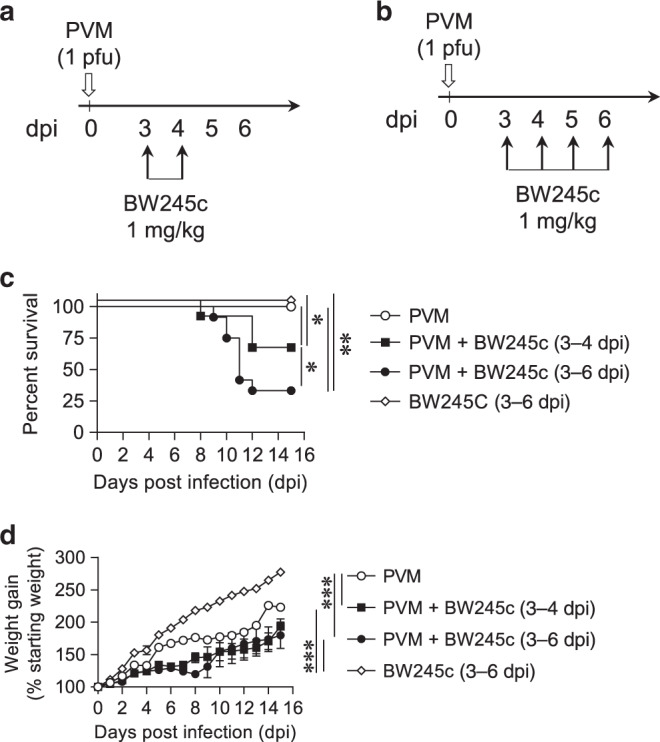
a, b WT BALB/c mice were inoculated with PVM (1 pfu) at postnatal day (PND) 7 (0 dpi). Mice were treated (i.n. route) daily with DP1 agonist (BW245c; 1 mg/kg) or vehicle from 3 dpi onwards. c, d Survival and body weight were monitored daily. Survival curves are plotted using Kaplan–Meier methods and the difference between the subgroups calculated using the Log-rank (Mental–Cox) test (pooled data from two independent experiments with n = 6–17 mice/group). Weight gain data are presented as mean ± SEM and pooled from two independent experiments with n = 6–16 mice/group. Differences between the groups were analyzed using two-way ANOVA. *p < 0.05; **p < 0.01; ***p < 0.001.
DP1 agonism-induced mortality is associated with exaggerated lung inflammation
To explore the underlying cause of the increased mortality, we focused on the mice treated with the DP1 agonist between 3 and 6 dpi. As reported previously,19 DP1 agonism increased the production of IFN-λ and IFN-γ in the lung and bronchoalveolar lavage (BAL) fluid (BALF), respectively, at 7 dpi (Fig. 2a). At 10 dpi, the virus was absent in the airway epithelium as expected,21 however, in the lung parenchyma, the viral load was significantly higher in BW245c-treated mice (Figs. 2b and S1D). This was associated with the presence of dead cells and increased levels of extracellular double-stranded DNA (dsDNA), a marker of cell death,22 in the BW245c-treated mice (Fig. 2c). PVM immunoreactivity was absent in the spleen, heart, kidney, liver, small intestine, colon, thymus, and salivary gland (Fig. S2), suggesting that the adverse response was related to a pathophysiological event in the lungs only. Although epithelial sloughing, mucus production or oedema were not apparent (data not shown), DP1 agonism led to persistent peribronchiolar and perivascular inflammation at 10 dpi (Fig. 2d), and increased production of the pro-inflammatory cytokines, TNF-α, IL-1β, and IL-6 (Fig. 2e). DP1 agonist treatment did not increase serum TNF-α expression (Fig. S1E). In the presence of the soluble IL-6 receptor (sIL-6R), IL-6 can trigger IL-6 trans-signaling23 which typically promotes an inflammatory response.24 Consistent with the elevated cellular inflammation, DP1 agonism induced a marked increase in airway sIL-6R expression at 5, 7, and 10 dpi (Fig. 2e). This exaggerated immunopathology was not associated with a change in PGD2 levels (Fig. 2f). Taken together, we show that DP1 agonism enhances antiviral cytokine production initially, but then paradoxically, delays viral clearance at 10 dpi in the parenchyma and causes increased immunopathology.
Fig. 2. DP1 agonism-induced mortality is associated with exaggerated lung inflammation.

a–e WT BALB/c mice were intranasally inoculated with PVM at PND 7 (0 dpi) and treated daily with DP1 agonist (BW245c) or vehicle from 3 to 6 dpi (intranasal). Mice were euthanized at 5, 7, and 10 dpi. a Concentrations of IFN-λ and IFN-γ in lung homogenates and BALF, respectively. b Representative micrographs (x20) of PVM immunoreactivity in the lung airway and parenchyma (scale bar = 100 µm) and quantification of PVM-positive cells in the lung parenchyma. Arrows indicate the presence of dead cells. c Concentration of dsDNA in BALF. d Representative micrographs (x20) of haematoxylin and eosin stained-lung tissue (scale bar = 100 µm) and lung inflammation score. e Concentrations of pro-inflammatory cytokines TNF-α, IL-1β, and IL-6 in the lung homogenates and sIL-6R in the BALF. f Level of PGD2 in the lung homogenate. Data are presented as box-and-whisker plots with boxes representing quartiles and whiskers indicating the range and are pooled data from two independent experiments (n = 5–12 mice per group). Two-way ANOVA with Bonferroni post hoc test was used for comparisons (*p < 0.05, **p < 0.01, ***p < 0.001).
DP1 agonism causes enhanced accumulation of neutrophils and monocytes in the lung
To characterize the cellular inflammation, we analyzed the lungs using multicolor flow cytometry (Fig. 3a). At 5 and 7 dpi, there was a significant increase in lung neutrophil (Ly6G+CD11b+) numbers in the infected BW245c-treated mice (Fig. 3b). Treatment with BW245c also increased the numbers of monocytes (CD11b+Ly6C+SiglecF−) but not interstitial macrophages (IM; SiglecF−Ly6C−CD11b+MHCII+CD64+), alveolar macrophages (AMs; SiglecF+CD11c+Ly6C−CD11b+CD64+), monocyte-derived dendritic cells (Mo-DCs; CD11c+MHCII+CD11b+CD103−CD64+), conventional type 2 dendritic cells (cDC2s; CD11c+MHCII+CD11b+CD103−CD64−) or conventional type 1 dendritic cells (cDC1s; CD11c+MHCII+CD11b−CD103+) in the lungs (Figs. 3b and S3A). This was associated with increased gene expression of the neutrophil- and monocyte-active chemokines G-CSF, CCL2, CCL7, CCL20, and M-CSF in the lungs of BW245c-treated mice (Fig. 3c). We confirmed G-CSF protein levels were significantly higher in BW245c-treated mice (Fig. 3d). CD4+ T cells and CD8+ T cells in the lung were also significantly higher in BW245c-treated mice at 5 dpi but not 7 dpi (Fig. 3e). In contrast, treatment with the DP1 agonist did not affect the numbers of FoxP3+CD4+ regulatory T cells, NK cells, ILC1s, or eosinophils (Fig. 3e and data not shown).
Fig. 3. DP1 agonism causes enhanced accumulation of neutrophils and monocytes in the lung.

a–d WT BALB/c mice were intranasally inoculated with PVM at PND 7 (0 dpi) and were treated daily with DP1 agonist (BW245c) or vehicle from 3 to 6 dpi (intranasal). Mice were euthanized at 5 and 7 dpi. a Gating strategy of lung myeloid cells. b Numbers of neutrophils [Ly6G+CD11b+] and monocytes [Ly6C+CD11b+SiglecF−] in lung tissues. c mRNA expression of G-CSF, CCL2, CCL7, CCL20, and M-CSF in whole lungs. d Concentrations of G-CSF in lung homogenates. e Numbers of CD4+T (CD3ε+CD4+), CD8+T (CD3ε+CD8+), and FoxP3+CD4+ regulatory T (CD3ε+CD4+FoxP3+) cells in lung tissues. Data are presented as box-and-whisker plots with boxes representing quartiles and whiskers indicating the range and are representative from two independent experiments (n = 6–9 mice per group). Two-way ANOVA with Bonferroni post hoc test was used for comparisons (*p < 0.05, **p < 0.01, ***p < 0.001).
DP1 agonism increases TNF-α production by neutrophils, monocytes, and CD4+ T cells
Next, we examined whether the activation status of the monocytes or neutrophils was altered in the BW245c-treated mice. Notably, the numbers of CD80+ and CD86+ neutrophil and monocytes were significantly higher at 7 dpi in BW245c-treated mice (Fig. 4a), whereas CD80 or CD86 expressing IMs, AMs, and Mo-DCs were not numerically different (Fig. S3B). Intracellular cytokine staining revealed that myeloid and T cells represented >97% of the TNF-α producing cells in both the BW245c-treated and untreated groups (data not shown). BW245c treatment increased the numbers of TNF-α producing neutrophils, monocytes, and CD4+ T cells in the lungs (Fig. 4b, c). In contrast, TNF-α-producing CD8+ T cells or other myeloid cells were not increased following BW245c treatment (Figs. 4c and S3C). These results suggest that DP1 agonism promotes the accumulation of TNF-α-producing neutrophils, monocytes, and CD4+ T in the lungs. To investigate whether AECs, neutrophils, monocytes, and CD4+ T cells were directly activated by the DP1 agonist, we FACS sorted the four cell types from the lungs of PVM-infected mice then stimulated the cells with the DP1 agonist ex vivo. DP1 agonism induced the release of TNF-α from all four cell types (Fig. 4d); however, the vast majority was produced by neutrophils and monocytes.
Fig. 4. DP1 agonism increases TNF-α production by neutrophils, monocytes, and CD4+ T cells.

a–c WT BALB/c mice were intranasally inoculated with PVM at PND 7 (0 dpi). A separate group of mice was also treated daily with DP1 agonist (BW245c) or vehicle from 3 to 6 dpi (intranasal). Mice were euthanized at 5 and 7 dpi. a Representative flow cytometry plots showing CD80 and CD86 expression on neutrophils and monocytes and the numbers of CD80 and CD86 expressing neutrophils and monocytes in lung tissues. b Representative flow cytometry plots showing TNF-α expression in different myeloid cells and T cells. c Numbers of TNF-α expressing cells in lung tissues. d TNF-α production by AECs, neutrophils (Neut), monocytes (Mono), and CD4+ T cells in response to BW245c stimulation for 4 h. Data are presented as box-and-whisker plots with boxes representing quartiles and whiskers indicating the range and are representative from two independent experiments. a–c n = 6–9 mice per group were used. d Lung tissue from three mice was pooled as one sample and n = 4 samples were used. Two-way ANOVA with Bonferroni post hoc test was used for comparisons. *p < 0.05, **p < 0.01, ***p < 0.001, comparing the groups as indicated. δδp < 0.01 and δδδp < 0.001, compared to the monocyte response.
Anti-TNF-α decreases DP1 agonist-induced cellular inflammation during an acute LRI
To investigate whether the increased inflammation, immunopathology, and mortality observed in BW245c-exposed mice is underpinned by the increased TNF-α production, we treated PVM-infected mice with BW245c and anti-TNF-α (Enbrel; i.p. route, daily from 3 dpi) or an isotype-matched control. In PVM-infected, BW245c-treated mice, anti-TNF decreased the number and activation of neutrophils and monocytes (Fig. 5b, c), as well as macrophages and DCs (Fig. S4A, B). In addition, TNF-α neutralization decreased the numbers of CD4+ and CD8+ T cells (Fig. 5d).
Fig. 5. Neutralization of TNF-α reduces the infiltration of DP1 agonism induced neutrophils, monocytes, and T cells in the lung.

a WT BALB/c mice were intranasally inoculated with PVM at PND 7 (0 dpi) and then treated with DP1 agonist (BW245c) from 3 to 6 dpi (intranasal). Mice also received TNF-α neutralizing antibody (Enbrel) or isotype control antibody once daily. Mice were euthanized 7 and 10 days later for analysis of lung inflammation. b Numbers of neutrophils [Ly6G+CD11b+] and monocytes [Ly6C+CD11b+SiglecF−] in lung tissues. c Numbers of CD80 and CD86 expressing neutrophils and monocytes in lung tissues. d Total number of CD4+T, CD8+T and FoxP3+CD4+ regulatory T cells in lung tissues. Data are presented as box-and-whisker plots with boxes representing quartiles and whiskers indicating the range and are representative from two independent experiments (n = 7–12 mice per group). Two-way ANOVA with Bonferroni post hoc test was used for comparisons (*p < 0.05, **p < 0.01, ***p < 0.001).
Neutralization of TNF-α protects against DP1 agonist-induced lethality
Lastly, we investigated the effect of anti-TNF-α on immunopathology and mortality in PVM-infected, BW245c-treated mice. Neutralization of TNF-α significantly improved weight gain and survival compared to the isotype-matched control group (Fig. 6a, b). Notably, the elevated viral load in the lung parenchyma in DP1 agonist-treated mice was reversed by anti-TNF-α (Figs. 6c and S4C), and associated with an increase in IFN-λ expression at 10 dpi (Fig. 6d). Anti-TNF also lowered the production of the pro-inflammatory cytokines IL-1β and IL-6 (Fig. 6e). However, sIL-6R levels were unaffected by anti-TNF-α treatment (Fig. 6e). Anti-TNF-α attenuated peribronchiolar and perivascular inflammation, and decreased dsDNA levels in the airways (Figs. 6f, g and S4D). Taken together, these data demonstrate that the exaggerated lung inflammation and mortality induced in PVM-infected BW245c-treated mice is in part mediated by increased levels of TNF-α.
Fig. 6. Neutralization of TNF-α protects against DP1 agonism induced lethality.

a–g WT BALB/c mice were intranasally inoculated with PVM at PND 7 (0 dpi) and then treated with DP1 agonist (BW245c) from 3 to 6 dpi (intranasal). Separate groups of mice also received TNF-α neutralizing antibody (Enbrel) once daily. Mice were monitored daily for weight gain and survival or euthanized 7 and 10 days later for analysis of lung inflammation. a Weight gain and b survival were monitored daily. Weight gain data are presented as mean ± SEM and representative of two independent experiments with n = 8 mice per group. Differences between the groups were analyzed using two-way ANOVA. Survival curves are plotted using Kaplan–Meier methods and the difference between the subgroups were calculated using the Log-rank (Mental–Cox) test (pooled data from two independent experiments with n = 16 mice per group). c Quantification of PVM-positive cells in the lung parenchyma. d Concentrations of IFN-λ in lung homogenates. e Concentrations of pro-inflammatory cytokines IL-1β and IL-6 in lung homogenates and sIL-6R in BALF. f Lung inflammation score. g Concentrations of dsDNA in BALF. Data are presented as box-and-whisker plots with boxes representing quartiles and whiskers indicating the range and are representative from two independent experiments (n = 7–12 mice per group). Two-way ANOVA with Bonferroni post hoc test was used for comparisons (*p < 0.05, **p < 0.01, ***p < 0.001).
Discussion
To preserve lung function, the inflammatory response to a respiratory virus infection must be tailored so that the invading pathogen is cleared without causing excessive collateral damage to the tissue. Previously, we identified that DP1 agonism promotes antiviral immunity in RSV-infected HBECs as well as PVM-infected mice by increasing antiviral cytokine production and decreasing viral load.19 However, here, by extending the duration of the study, we identified that pharmacological activation of DP1, even for a short period of time (i.e., 2 days), disrupts the fine balance between viral clearance and tissue damage, and ultimately leads to a severe LRI and increased mortality. Consistent with our previous findings, in the early stage of the infection, DP1 receptor activation decreased the viral load, however, at later time points, when the virus is typically cleared from the airway epithelium, there was increased spread of the virus to the lung parenchyma in the DP1 agonist-treated mice. Examination of the lungs revealed that as a consequence of increased chemokine expression, DP1 agonism increased the infiltration of immune cells, particularly hyper-activated TNF-α-producing neutrophils and monocytes, and that this led to an exaggerated pro-inflammatory cytokine response characterized by increased production of TNF-α, IL-1β, and IL-6. In the DP1 agonist-treated mice, neutralization of TNF-α attenuated the hyper-inflammatory response, reversed the viral spread to the parenchyma, and improved survival, suggesting that the deleterious effects of DP1 receptor activation are partly mediated by TNF-α.
We have previously shown that the activation of DP2 by endogenous PGD2 released in response to RSV or PVM infection, suppresses IFN-λ production, and increases viral load.19 Thus, DP2 antagonism de-represses IFN-λ production.19 Interestingly, this phenotype is attenuated by dual DP1 and DP2 antagonism, implicating DP1 activation by endogenous PGD2, and supported by the observation that DP1 agonism promotes IFN-λ and viral clearance.19 Here, we sought to further explore the cellular and molecular mechanisms by which DP1 agonism confers protection. However, by extending the model to 15 dpi to encapsulate the onset of the adaptive immune response, we discovered that DP1 agonism causes exaggerated lung inflammation and significant mortality between 10 and 12 dpi in PVM-infected mice. This deleterious response was associated with enhanced recruitment of activated neutrophils and monocytes to the lung, the predominant cell types found in the airways of infants experiencing severe bronchiolitis and in lung tissues obtained from fatal RSV bronchiolitis cases.9,25,26 AECs, neutrophils, monocytes, and CD4+ T cells express the DP1 receptor,17,19,27 However, the majority of TNF-α was produced by neutrophils and monocytes when the cells were activated directly ex vivo. Although we did not observe any oedema in DP1 agonist-treated mice, we cannot rule out a possible effect of DP1 agonism on vascular structures.
In response to Sendai virus infection, Tumala et al. found that PGD2 levels in BALF are significantly higher in neonatal mice compared to adult mice, and that this is associated with less DC activation and lower T-cell responses.28 Using adult and elderly mice, and studying the response to SARS-CoV and Influenza A, Zhao et al. found that PGD2 levels in BALF increases with age, and hence, the two studies would appear to report contradictory findings.29 However, Zhao et al. also found that the increase in PGD2 in older mice is associated with decreased DC migration and impaired T-cell activation. Others have also shown that PGD2 activation of DP1 on DCs attenuates their migration.30 In contrast to these studies, we employed a small molecule DP1 agonist based on our hypothesis that DP1 agonism would serve as a novel antiviral approach to ameliorate infant viral bronchiolitis. In our model, the DP1 agonism-induced phenotype appears to stem from a hyper-active innate immune response mediated by neutrophils and monocytes, and therefore highlights a novel immune axis separate to the DC-related phenotypes described previously.
As a consequence of the persistent inflammatory cell response induced by DP1 agonist treatment, the expression of several pro-inflammatory cytokines in the lung was heightened. A TNF-αhighIL-6highIL-1βhigh “cytokine storm” milieu during a respiratory viral infection often leads to severe respiratory failure and sudden clinical deterioration as observed in severe clinical cases of RSV bronchiolitis, COVID-19, and influenza virus infection.31–34 Severe respiratory failure is believed to occur as a consequence of cytokine-induced lung injury and fatal pneumonia rather than inadequate viral control.35 In the current study, we noticed that in PVM-infected mice, DP1 receptor activation by BW245c initiates three phases: in the first phase (3–5 dpi), DP1 agonism promotes innate antiviral immunity, lowering viral load; in the second phase (5–10 dpi), DP1 agonism amplifies the neutrophilic/monocytic response and pro-inflammatory cytokine expression, and in the third phase (10 dpi onwards), DP1 agonism promotes viral spread to the parenchyma and a cytokine storm (TNF-α, IL-6, IL-1β), leading to severe morbidity and death. The cytokines TNF-α, IL-6, and IL-1β can recruit and activate multiple different immune and stromal cells to amplify inflammatory signals.36 Left unchecked, this type 1 module of immunity (e.g., elevated neutrophilic inflammation, increased TNF-α expression by neutrophils, monocytes, and CD4+ T cells)37 can induce significant damage to alveolar epithelium, impairing gas exchange. Whether SARS-CoV-2 increases PGD2 remains to be determined, however, it is noteworthy that SARS-CoV infected mice show increased PGD2 levels in the airways and that virus-induced increases in PGD2 levels increase with age, a risk factor for COVID-19 mortality.29 It has been hypothesized that DP2 antagonists be repurposed as a novel immunotherapy for the treatment of COVID-19,38,39 and this may be appropriate in mild to moderate cases where Th1 immunity is impaired. However, in patients with severe disease, our findings suggest that a DP1 antagonism would be a more logical approach to limit the excessive immune response. Indeed, if a DP1 antagonist shunted PGD2 to promote DP2 activation, similar to our findings with DP2 antagonism, this might elicit an anti-inflammatory Th2 response, and accompanied tissue repair.
Paradoxically, TNF-α can activate both cell survival and cell death depending on the particular intracellular signaling pathway that is activated.40 The more pro-inflammatory cell death pathways, necrosis or necroptosis, cause the release of dsDNA, which can act as a pro-inflammatory damage-associated molecular pattern. Via NETosis, activated neutrophils can also release dsDNA during respiratory viral infections.41,42 We observed that DP1 agonism caused increased neutrophilia as well as enhanced release of dsDNA in the BALF, suggesting that both cell death dependent and independent pathways may have been activated. We have recently shown that inhibition of necroptosis increases antiviral immunity and decreases viral load in PVM-infected mice and RSV-infected AECs.22 Thus, it is possible that anti-TNF-α diminished the inflammatory response and viral spread by attenuating NETosis and/or necroptosis. Furthermore, we noted that anti-TNF-α did not rescue PVM-infected, BW245c-treated mice completely. This may be due to the incomplete clearance of PVM from the parenchyma with anti-TNF-α treatment. Anti-TNF-α neither lower the expression of IL-6 to the baseline levels, nor affect the expression of sIL-6R, and thus one possibility is that the residual IL-6 and sIL-6R were sufficient to maintain pro-inflammatory IL-6 trans-signaling.
We conclude that treatment with a DP1 agonist during viral infection initially promotes innate immunity to facilitate viral clearance, but eventually leads to an overexuberant inflammatory response, tissue damage, and severe morbidity. We show that DP1 agonism is a potent inducer of TNF-α-induced inflammation, suggesting that DP1 agonists may offer clinical benefit in disease settings where immunity is suppressed, e.g., cancer. In mild cases of viral bronchiolitis, DP2 antagonism is beneficial as it reverses the PGD2-dependent suppression of antiviral IFN-λ expression facilitating viral clearance.19 However, in severe LRI, where PGD2 levels are likely to be significantly elevated, treatment with a DP1 antagonist or anti-TNF-α would be more suitable to limit the excessive inflammation and collateral tissue damage.
Materials and methods
Study design
Specific pathogen-free wild-type BALB/c mice were purchased from Animal Resource Centre (Perth, Australia) and bred at QIMR Berghofer Medical Research Institute (Brisbane, Australia). Mice were time mated and neonatal mice were inoculated via intranasal route with vehicle [Dulbecco’s modified Eagle’s medium + 10% fetal calf serum (FCS) in 10 µl volume] or 1 plaque forming unit (PFU) of PVM (strain J3666; diluted in 10 µl of vehicle) at postnatal day 7 as previously described.43 In some experiments, mice were exposed (i.n. route) to 1 µg of CRE (GREER Laboratories, USA) dissolved in 10 µl of phosphate-buffered saline (PBS) at 3 dpi with PVM. To activate DP1 receptor signaling, the DP1 agonist, BW245c (1 mg/kg of body weight; diluted in 10 µl of PBS) was administered daily (i.n. route) from 3 to 6 dpi. To neutralize TNF-α, mice were treated (i.p. route) with anti-TNF-α antibody, Enbrel (2.5 mg/kg of body weight; Pfizer Australia; diluted in 50 µl PBS) or an isotype-matched control daily from 3 dpi onwards. Mice were monitored daily for clinical symptoms including weight loss, activity, fur texture, and posture (hunching). Mice with more than 10% weight loss compared to the previous day or severe clinical symptoms were euthanized in accordance with institutional animal ethics guidelines, and the date of death recorded as the next day. All animal experiments were approved by the Animal Ethics Committee of QIMR Berghofer Medical Research Institute.
Tissue harvesting and processing
Lungs were flushed with 400 µl of ice-cold PBS to harvest BAL. BAL samples were centrifuged and the cell-free supernatant stored at −80 °C prior to cytokine and dsDNA analysis. Prior to cytokine measurement by ELISA, lung lobes were placed in radioimmunoprecipitation assay buffer (150 mM sodium chloride, 1% IGEPAC, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, and 50 mM Tris buffer in water), homogenized with a tissue-tearor (Biospec, OK, USA) and the cell-debris free supernatants collected after centrifugation.
Flow cytometry
Immune cell infiltration into the lungs was assessed by preparing single-cell suspensions from the large left lung lobe by mechanical disruption against a 70 μM cell strainer (BD Biosciences, USA). Red blood cells were lysed with Gey’s lysis buffer. Cells were re-suspended in FACS buffer (2% FCS in PBS) and incubated with Fc block antibody (2.4G2) for 30 min at 4 °C to block non-specific binding. After washing, cells were incubated with the following fluorochrome-conjugated antibodies as appropriate for 30 min at 4 °C: Ly6G-FITC (1A8), CD11b-PerCp-Cy5.5 (M1/70), CD200R1-AF647 (OX-110), CD4-V500 (RM4-5), CD90.2-APC-Cy7 (53-2.1), Siglec F-PE (E50-2440) from BD Biosciences (CA, USA); CD103-AF700 (2E7), CD11c-BV785 (N418), CD19-FITC (1D3/CD19), CD3ε-FITC (17A2), CD31-FITC (MEC13.3), CD326-APC (G8.8), CD4-FITC (RM4-5), CD45-BV785 (30-F11), CD64-BV421 (X54-5/7.1), CD8-PerCp-Cy5.5 (53-6.7), CD80-APC/Fire 750 (16-10A1), CD86-PE (GL-1), F4/80-FITC (BM8), Gr-1-FITC (RB6-8C5), Ly6C-BV570 (HK1.4), MHCII-BV510 (M5/114.15.2), NKp46-BV421 (29A1.4), TCR-β-BV605 (H57-597) from Biolegend (CA, USA). For intracellular cytokine staining, cells were stimulated with phorbol 12-myristate 13-acetate and ionomycin in the presence of Brefeldin A for 3 h. Cells were harvested and washed with FACS buffer. Cells were then incubated with Fc block and surface antibodies as appropriate. The cells were processed using the FoxP3/transcription factor fixation/permeabilization kit (eBiosciences, CA, USA) according to the manufacturer instructions. After fixing and permeabilization, cells were stained with fluorochrome-conjugated antibodies (TNF-α-FITC, clone MP6-XT22, BD Biosciences). For FoxP3 staining, unstimulated cells were stained with AF647 FoxP3 antibody (MF23, BD Biosciences) as per the intracellular cytokine staining protocol described above. Cells were acquired on a BD Fortessa IV flow cytometer (BD Biosciences, USA) and data were analyzed using FACSDiva software (version 8; BD Biosciences) and FlowJo software (version 10.6; TreeStar, USA).
Cell sorting and ex vivo stimulation
At 7 dpi, the lungs of PVM-infected BALB/c neonatal mice were excised and digested using the Lung dissociation kit (Miltenyl Biotech, Germany). Following RBC lysis, the lung cells were stained with specific antibodies as above. Neutrophils [Ly6G+CD11b+], monocytes [Ly6C+CD11b+SiglecF−], and CD4+ T (CD3ε+CD4+) were sorted from the 7-AAD−CD45+ fraction and AECs were sorted as [CD326+CD31−] from the 7-AAD−CD45− fraction with >98% purity. The cells were seeded at 400,000 cells per well in 200 μl RPMI-1640 culture medium supplemented with 10% of fetal bovine serum (GE Healthcare), 1 mM of sodium pyruvate (Gibco, MA, USA), 2 mM of L-Glutamine (Gibco, MA, USA), 20 mM of HEPES (Gibco, MA, USA), 100 U/ml of Penicillin-Streptomycin (Gibco, MA, USA), and 50 μM of 2-Mercaptoethanol (Sigma-Aldrich, Mo, USA), then stimulated with BW245c (300 nM) or diluent. Culture supernatants were harvested 4 h later and stored at −80 °C prior to cytokine assay.
Histology and immunohistochemistry
Paraffin-embedded lung biopsies were processed, and histological staining and immunohistochemistry performed as described previously.44 Lung sections were stained with haematoxylin and eosin to assess lung pathology. The degree of inflammation was scored by a person who was blinded to the experimental plan and was assessed based on the peribronchiolar and perivascular cellular infiltration using the grading system described previously.45 Lung biopsies were stained immunohistochemically for PVM antigen as previously described46 and digitally scanned sections were analyzed using Aperio Image Scope Software (Leica Biosystems; Wetzlar, Germany). The number of positively PVM-stained AECs was quantified and expressed as a percentage of AECs of five airways.46 PVM-positive staining in the whole tissue was assessed using scattered point grids (>300 points per tissue) and expressed as percentage of PVM-positive points in the tissue.47
Cytokine measurement in lung and BALF
Commercially available kits were used to measure cytokine levels of INF-λ, sIL-6R (ELISA, R&D Systems, USA), IL-6, IL-1β, IFN-γ (ELISA, Biolegend, USA), and TNF-α (CBA, BD Biosciences, USA) in BALF or lung homogenate. PGD2 was measured in lung homogenates using a PGD2-MOX Express ELISA kit (Cayman Chemicals, Michigan, USA).
Quantitative real-time PCR (qRT-PCR) analysis
The superior right lobe was excised and total RNA was extracted using Tri Reagent solution (Invitrogen, CA, USA) from tissue homogenates. Following DNase treatment (Invitrogen, CA, USA), reverse transcription was performed using M-MLV reverse transcription enzyme (Invitrogen, CA, USA) and random primers (Invitrogen, CA, USA). Quantitative real-time PCR was performed with SYBR green (Applied Biosystem) using the following primers:
Mouse CCL2: forward 5′-CCAACTCTCACTGAAGCCAGCTCT-3′
reverse 5′-TCAGCACAGACCTCTCTCTTGAGC-3′
Mouse CCL7: forward 5′-GCTTCTGTGCCTGCTGCTCATA-3′
reverse 5′-CATAGCAGCATGTGGATGCATTG-3′
Mouse CCL20: forward 5′-CGACTGTTGCCTCTCGTACA-3′
reverse 5′-AGGAGGTTCACAGCCCTTTT-3′
Mouse G-CSF: forward 5′-ACTCAGGGAAGCCTTCGG-3′
reverse 5′-GCAAGTGAGGAAGATCCAGG-3′
Mouse M-CSF: forward 5′-GGTAGTGGTGGATGTTCCCA-3′
reverse 5′-CCAGGATGAGGACAGACAGG-3′
Mouse HPRT: forward 5′-AGGCCAGACTTTGTTGGATTTGAA-3′
reverse 5′-CAACTTGCGCTCATCTTAGGCTTT-3′
Gene expression was normalized relative to the housekeeping gene HPRT and expressed as a fold change relative to uninfected mice using the 2−ΔΔCt formula.
Statistical analysis
The data are displayed as Box-and-Whisker plots; boxes represent quartiles and whiskers represent range. A two-way ANOVA with Bonferroni post hoc was used for comparisons. Survival curves are estimated and plotted using Kaplan–Meier methods and the difference between the subgroups are calculated using the Log-rank (Mental–Cox) test. Statistical analyses were performed with GraphPad Prism (Version 8.0, GraphPad, USA), while statistically significant results were annotated with *p < 0.05; **p < 0.01; ***p < 0.001 and ns for not significant.
Supplementary information
Acknowledgements
This work was supported by an Australian Infectious Disease Research Centre grant and an NHMRC grant awarded to S.P. S.R. was funded by the doctorate program MOLIN (FWF, W1241) and an EMBO Short-Term Fellowship (8109). We thank the staff from the QIMR Berghofer flow cytometry laboratory, animal facility, and histology facility for assistance.
Author contributions
M.A.U. and S.P. conceived the project, designed the experiments, interpreted the data, and wrote the manuscript with input from all co-authors. M.A.U., S.R., S.Z.H., and J.L. performed the experiments and analyzed the data.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41385-021-00405-7.
References
- 1.Global Burden of Disease Pediatrics Collaboration. Kyu HH, et al. Global and national burden of diseases and injuries among children and adolescents between 1990 and 2013: findings from the global burden of disease 2013 study. JAMA Pediatr. 2016;170:267–287. doi: 10.1001/jamapediatrics.2015.4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Florin TA, Plint AC, Zorc JJ. Viral bronchiolitis. Lancet. 2017;389:211–224. doi: 10.1016/S0140-6736(16)30951-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Everard ML, et al. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch. Dis. Child. 1994;71:428–432. doi: 10.1136/adc.71.5.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levy O. Innate immunity of the newborn: basic mechanisms and clinical correlates. Nat. Rev. Immunol. 2007;7:379–390. doi: 10.1038/nri2075. [DOI] [PubMed] [Google Scholar]
- 5.Holt PG, Sly PD. Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat. Med. 2012;18:726–735. doi: 10.1038/nm.2768. [DOI] [PubMed] [Google Scholar]
- 6.Lynch JP, et al. The influence of the microbiome on early-life severe viral lower respiratory infections and asthma-food for thought? Front. Immunol. 2017;8:156. doi: 10.3389/fimmu.2017.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carroll KN, et al. Increasing burden and risk factors for bronchiolitis-related medical visits in infants enrolled in a state health care insurance plan. Pediatrics. 2008;122:58–64. doi: 10.1542/peds.2007-2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makris S, Paulsen M, Johansson C. Type I interferons as regulators of lung inflammation. Front. Immunol. 2017;8:259. doi: 10.3389/fimmu.2017.00259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 2007;20:108–119. doi: 10.1038/modpathol.3800725. [DOI] [PubMed] [Google Scholar]
- 10.Pickles RJ, DeVincenzo JP. Respiratory syncytial virus (RSV) and its propensity for causing bronchiolitis. J. Pathol. 2015;235:266–276. doi: 10.1002/path.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pettipher R, Hansel TT, Armer R. Antagonism of the prostaglandin D2 receptors DP1 and CRTH2 as an approach to treat allergic diseases. Nat. Rev. Drug Disco. 2007;6:313–325. doi: 10.1038/nrd2266. [DOI] [PubMed] [Google Scholar]
- 12.Rittchen S. & Heinemann, A. Therapeutic potential of hematopoietic prostaglandin D2 synthase in allergic inflammation. Cells8, 619 (2019). [DOI] [PMC free article] [PubMed]
- 13.Hardy CC, Robinson C, Tattersfield AE, Holgate ST. The bronchoconstrictor effect of inhaled prostaglandin D2 in normal and asthmatic men. N. Engl. J. Med. 1984;311:209–213. doi: 10.1056/NEJM198407263110401. [DOI] [PubMed] [Google Scholar]
- 14.Van Ly D, et al. Characterising the mechanism of airway smooth muscle β2 adrenoceptor desensitization by rhinovirus infected bronchial epithelial cells. PLoS ONE. 2013;8:e56058. doi: 10.1371/journal.pone.0056058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hammad H, et al. Activation of the D prostanoid 1 receptor suppresses asthma by modulation of lung dendritic cell function and induction of regulatory T cells. J. Exp. Med. 2007;204:357–367. doi: 10.1084/jem.20061196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kupczyk M, Kuna P. Targeting the PGD2/CRTH2/DP1 signaling pathway in asthma and allergic disease: current status and future perspectives. Drugs. 2017;77:1281–1294. doi: 10.1007/s40265-017-0777-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jandl K, et al. Activated prostaglandin D2 receptors on macrophages enhance neutrophil recruitment into the lung. J. Allergy Clin. Immunol. 2016;137:833–843. doi: 10.1016/j.jaci.2015.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vijay R, et al. Virus-induced inflammasome activation is suppressed by prostaglandin D2/DP1 signaling. Proc. Natl Acad. Sci. USA. 2017;114:E5444–e5453. doi: 10.1073/pnas.1704099114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Werder, R. B. et al. PGD2/DP2 receptor activation promotes severe viral bronchiolitis by suppressing IFN-lambda production. Sci. Transl. Med.10, eaao0052 (2018). [DOI] [PubMed]
- 20.Lynch JP, et al. Aeroallergen-induced IL-33 predisposes to respiratory virus-induced asthma by dampening antiviral immunity. J. Allergy Clin. Immunol. 2016;138:1326–1337. doi: 10.1016/j.jaci.2016.02.039. [DOI] [PubMed] [Google Scholar]
- 21.Simpson J, et al. The absence of interferon-beta promotor stimulator-1 (IPS-1) predisposes to bronchiolitis and asthma-like pathology in response to pneumoviral infection in mice. Sci. Rep. 2017;7:2353. doi: 10.1038/s41598-017-02564-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Simpson, J. et al. Respiratory Syncytial Virus infection promotes necroptosis and HMGB1 release by airway epithelial cells. Am. J. Respir. Crit. Care Med. 201, 1358–1371 (2020). [DOI] [PubMed]
- 23.Peters M, Müller AM, Rose-John S. Interleukin-6 and soluble interleukin-6 receptor: direct stimulation of gp130 and hematopoiesis. Blood. 1998;92:3495–3504. doi: 10.1182/blood.V92.10.3495.422k47_3495_3504. [DOI] [PubMed] [Google Scholar]
- 24.Ullah MA, et al. Allergen-induced IL-6 trans-signaling activates gammadelta T cells to promote type 2 and type 17 airway inflammation. J. Allergy Clin. Immunol. 2015;136:1065–1073. doi: 10.1016/j.jaci.2015.02.032. [DOI] [PubMed] [Google Scholar]
- 25.Heinonen S, et al. Immune profiles provide insights into respiratory syncytial virus disease severity in young children. Sci. Transl. Med. 2020;12:eaaw0268. doi: 10.1126/scitranslmed.aaw0268. [DOI] [PubMed] [Google Scholar]
- 26.McNamara PS, Ritson P, Selby A, Hart CA, Smyth RL. Bronchoalveolar lavage cellularity in infants with severe respiratory syncytial virus bronchiolitis. Arch. Dis. Child. 2003;88:922–926. doi: 10.1136/adc.88.10.922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Murata T, et al. Anti-inflammatory role of PGD2 in acute lung inflammation and therapeutic application of its signal enhancement. Proc. Natl Acad. Sci. USA. 2013;110:5205. doi: 10.1073/pnas.1218091110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tumala B, Phelps KR, Zhang S, Bhattacharya S, Shornick LP. Prostaglandin D2 levels regulate CD103(+) conventional dendritic cell activation in neonates during respiratory viral infection. Viral Immunol. 2018;31:658–667. doi: 10.1089/vim.2018.0090. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Zhao J, Legge K, Perlman S. Age-related increases in PGD(2) expression impair respiratory DC migration, resulting in diminished T cell responses upon respiratory virus infection in mice. J. Clin. Invest. 2011;121:4921–4930. doi: 10.1172/JCI59777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Angeli V, et al. Activation of the D prostanoid receptor 1 regulates immune and skin allergic responses. J. Immunol. 2004;172:3822–3829. doi: 10.4049/jimmunol.172.6.3822. [DOI] [PubMed] [Google Scholar]
- 31.Giamarellos-Bourboulis EJ, et al. Complex immune dysregulation in COVID-19 patients with severe respiratory failure. Cell Host Microbe. 2020;27:992–1000.e1003. doi: 10.1016/j.chom.2020.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zarogoulidis P, et al. Macrolides: from in vitro anti-inflammatory and immunomodulatory properties to clinical practice in respiratory diseases. Eur. J. Clin. Pharm. 2012;68:479–503. doi: 10.1007/s00228-011-1161-x. [DOI] [PubMed] [Google Scholar]
- 33.Chua, R. L. et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Biotechnol. 38, 970–979 (2020). [DOI] [PubMed]
- 34.Del Valle, D. M. et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med.26, 1636–1643 (2020). [DOI] [PMC free article] [PubMed]
- 35.Mehta P, et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395:1033–1034. doi: 10.1016/S0140-6736(20)30628-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bohmwald K, et al. Contribution of cytokines to tissue damage during human respiratory syncytial virus infection. Front. Immunol. 2019;10:452. doi: 10.3389/fimmu.2019.00452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- 38.Gupta A, Chander Chiang K. Prostaglandin D2 as a mediator of lymphopenia and a therapeutic target in COVID-19 disease. Med. Hypotheses. 2020;143:110122. doi: 10.1016/j.mehy.2020.110122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rizk, J. G. et al. Pharmaco-immunomodulatory therapy in COVID-19. Drugs 80, 1267–1292 (2020). [DOI] [PMC free article] [PubMed]
- 40.Dannappel M, et al. RIPK1 maintains epithelial homeostasis by inhibiting apoptosis and necroptosis. Nature. 2014;513:90–94. doi: 10.1038/nature13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Muraro SP, et al. Respiratory syncytial virus induces the classical ROS-dependent NETosis through PAD-4 and necroptosis pathways activation. Sci. Rep. 2018;8:14166. doi: 10.1038/s41598-018-32576-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sebina, I. & Phipps, S. The contribution of neutrophils to the pathogenesis of RSV bronchiolitis. Viruses12, 808 (2020). [DOI] [PMC free article] [PubMed]
- 43.Loh Z, et al. HMGB1 amplifies ILC2-induced type-2 inflammation and airway smooth muscle remodelling. PLoS Pathog. 2020;16:e1008651. doi: 10.1371/journal.ppat.1008651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ullah, M. A. et al. PAG1 limits allergen-induced type 2 inflammation in the murine lung. Allergy 75, 336–345 (2020). [DOI] [PubMed]
- 45.Chong L, et al. Protective effect of curcumin on acute airway inflammation of allergic asthma in mice through Notch1-GATA3 signaling pathway. Inflammation. 2014;37:1476–1485. doi: 10.1007/s10753-014-9873-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lynch JP, et al. Plasmacytoid dendritic cells protect from viral bronchiolitis and asthma through semaphorin 4a-mediated T reg expansion. J. Exp. Med. 2018;215:537–557. doi: 10.1084/jem.20170298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spann KM, et al. IRF-3, IRF-7, and IPS-1 promote host defense against acute human metapneumovirus infection in neonatal mice. Am. J. Pathol. 2014;184:1795–1806. doi: 10.1016/j.ajpath.2014.02.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.