

Abstract
Stapled α-helical RIR (Rev1-interacting region) peptides of DNA POL κ bind more effectively to the RIR-interface of the C-terminal recruitment domain of the translesion synthesis DNA polymerase Rev1 than unstapled peptide. The tightest-binding stapled peptide translocates into cells and enhances the cytotoxicity of DNA damaging agents while reducing mutagenesis. Drugs with these characteristics could potentially serve as adjuvants to improve chemotherapy and reduce acquired resistance by inhibiting Rev1-dependent mutagenic translesion synthesis.
Keywords: Translesion synthesis (TLS), Staple peptide, chemoresistance, cytotoxicity, mutagenesis
Introduction
Intrinsic and acquired resistance to DNA damaging cancer drugs such as cisplatin poses a persistent clinical challenge, one that ultimately limits successful treatment of patients [Lippert et al. , 2008]. Recent evidence from mouse models has suggested that a possible strategy to both sensitize intrinsically resistant tumors to treatment and also reduce the acquisition of drug resistance by susceptible tumors during treatment would be to inhibit the major branch of mutagenic translesion synthesis (TLS) during chemotherapy [Doles et al. , 2010; Xie et al. , 2010; Xu et al. , 2013; Sail et al. , 2017; Wojtaszek et al. , 2019]. Translesion synthesis is a DNA damage bypass process where a group of low fidelity polymerases come together to tolerate a DNA lesion [Yamanaka et al. , 2017]. This pathway is critically dependent on two specialized TLS DNA polymerases: i) Rev1, a Y family DNA polymerase that has only a limited ability to insert nucleotides opposite lesions but possesses a critically important C-terminal domain (CTD) that recruits other TLS DNA polymerases and ii) POL ζ4, a B family DNA polymerase that consists of the catalytic subunit REV3, the accessory subunit REV7, and two subunits shared with the POL δ replicative DNA polymerase, POLD2 and POLD3 [Korzhnev and Hadden, 2016].
Structural studies have suggested that an Achilles heel of the REV1/POL ζ4-dependent pathway of mutagenic TLS could be Rev1’s CTD. This 100 amino acid domain consists of an atypical four-helix bundle consisting of mixed parallel and antiparallel helices that has two distinct interfaces. One interacts with the RIR (Rev1-interacting region) peptides of POL κ POL η, POL ι, POLD3, and XRCC1, while the other interacts with REV7. Drugs that bind to either of these interfaces and thereby inhibit mutagenic TLS could potentially be used as adjuvants to improve the effectiveness of DNA damaging chemotherapy [Wojtaszek et al. , 2012a; Wojtaszek et al. , 2012b; Gabel et al. , 2013; Pustovalova et al. , 2016].
A rational strategy for obtaining such an adjuvant—which lacks small molecule-specific toxicity—is to develop a derivative of an RIR peptide that could bind to the RIR interface of REV1 CTD and interfere with the recruitment of other TLS DNA polymerases. These short α-helical RIR peptides interact with REV1 by inserting two conserved adjacent phenylalanines into preformed hydrophobic pockets in the RIR interface of CTD and make additional predominantly backbone contacts, a step important for TLS function [Ohashi et al. , 2009; Wojtaszek et al. , 2012b]. Normally only a small fraction of free RIR peptides would be in the α-helical conformation, but they can be stably locked into an α-helix by stapling as previously described [Verdine and Hilinski, 2012].
Materials and Methods
Stapled peptide synthesis
The POL κ RIR stapled peptide was synthesized on the 4-methylbenzhydrylamine (MBHA) rink amide resin (100-200 mesh, Novabiochem) using the standard Fmoc solid-phase peptide synthesis technique as described before [Kim et al. , 2011; Grossmann et al. , 2012]. Briefly, amino acids were coupled using COMU (1-Cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino—morpholino—carbenium hexafluorophosphate) and diisopropylethylamine (DIEA) in N-Methyl-2-pyrrolidone (NMP) for both standard and non-natural amino-acids in a custom-made 25 mL glass reaction vessel that is outfitted with a medium porosity frit and a “T”-bore for N2 gas bubbling. The metathesis step was performed at a 0.15 mmol scale under constant nitrogen degassing, using 10mM of 1st generation Grubbs’ catalyst in dichloroethane (DCE) in a 2 ml total solution volume, twice, for 2 hours at room temperature. Following olefin-metathesis, final Fmoc deprotection was carried out with piperidine in N,N-dimethylformamide and the peptide cleaved from the resin with a cleavage cocktail comprising TFA: TIS: H20 (95/2.5/2.5) within 3 hours. The peptide was consequently capped at its N-terminus with FITC (Fluorescein-5-isothiocyanate; 'Isomer I', [Molecular Probes] 9:1 isomer mixture) with an intervening β-alanine spacer that separated the label from the 1st amino acid of the peptide. FITC labeling enables detection of stapled peptide in cells, which appears as green foci.
The resultant crude peptide was purified and analyzed by reverse-phase HPLC using an Agilent 1200 HPLC system equipped with a DAD detector. Samples were loaded onto a Clipeus C18, 5 μm, 300 Å (150 × 4.6 mm) HPLC column Higgins Analytical, Inc.), which was equilibrated with 10% Eluent A (0.1% trifluoroacetic acid (TFA) in water). The peptides were eluted with Eluent B (0.1% TFA in acetonitrile) for 30 minutes at a 1.0 mL/min flow rate at 25 °C. The resultant elute was monitored for absorbance at 220 and 280 nm. The purified staple peptide fractions were combined, concentrated in a speed vacuum and stored as a lyophilized powder or as aqueous stock solution at −20 °C.
Stapled peptide detection was performed by electrospray ionization-mass spectrometry (ESI-MS) on an LC-MS system comprised of an Agilent 1260 LC, which is outfitted with a Poroshell 120 EC and C-18 column (2.7 μm pore, 3.0 × 50 mm, Agilent Technologies, Inc.). Agilent 6230 TOF system which houses an Agilent Jetstream ESI source is also connected to it. For peptide detection, Samples were run at a flow rate of 0.4 mL/min using a gradient of 0 to 95% of acetonitrile over 5 minute-window. High resolution mass spectroscopy data and HPLC traces of stapled peptide were obtained. Peptide quantification was carried out via UV absorbance.
Protein purification
The pET28b His-TEV hRev1 CTD plasmid containing 100 amino acids of the REV1 CTD was overexpressed in BL21RIL Escherchia coli, induced with 0.1 mM isopropyl 1-thio-β-D-galactopyranoside (IPTG) at 18 °C for 18 hours until the A600 reached 0.4. Cells were pelleted at 4000g, 20 minutes at 4 °C and resuspended in 120 μl sodium hydrogen phosphate buffer (50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 20 mM BME and protease inhibitor cocktail). Cells were lysed using French press at 30 KPsi and incubated on ice for 15 minutes following the addition of 6 μl Benzonase. Cells were then spun at 10,000g for 25 minute at 4 °C and the supernatant was incubated with 5 ml of NiNTA resin for 1 hour at 4 °C before running it through size exclusion nickel affinity chromatography. A final protein purification step was carried out by applying the elute through an equilibrated (with 1X PBS and 1 mM DTT) Sephadex 75 column.
FLAG tagged purified POL κ protein was purchased from OriGene (Catalog # TP319806).
Circular Dichroism (CD)
The stapled peptide was dissolved at a 50 μM concentration in pure water, sonicated and centrifuged to remove insoluble peptides. Peptide concentration was determined by measuring UV absorbance at 280 nm. CD measurements were collected in a quartz cuvette with a 1 nm bandwidth and 0.1 cm path length at 20 °C using CD spectrometer (Jasco J-710). Other standard spectral-measurement parameters were 190-260 nm wavelengths, 0.5 nm step resolution and 10 accumulations at 20 nm/sec speed. Spectra units were normalized to the mean residue ellipticity, [θ] (deg cm2 dmol−1) using concentration determined by UV absorbance to account for minor variations.
Fluorescence polarization assay
Binding of the stapled peptide to the REV1 CTD was carried out in triplicate as described before [Grossmann et al. , 2012]. Briefly, the stapled peptide in a concentration range of 10 nM to 50 nM was incubated with two-fold dilution of purified REV1 CTD in 300 mM NaCl, 25 mM Tris (pH 8.8), 2 mM DTT, and 2% glycerol in the 384-well black flat-bottom plates for 45 minutes. Fluorescence polarization was measured on Spectramax-M5 plate reader, with excitation wavelength of 490 and emission wavelength of 519. KD (dissociation constant) values were calculated by non-linear regression analysis using Prism software (GraphPad).
ELISA
To test whether the stapled peptide was able to inhibit REV1’s interaction with POL K, 10 picomoles of His tagged-REV1 was incubated in 200 μL of PBS/BSA (50 mM potassium phosphate, 150 mM sodium chloride, pH 7.2 with 0.2% bovine serum albumin) in each well of a 96 well NiNTA HisSorb plate, which was covered with a cover film for one hour at room temperature on a gentle rocker. Negative control wells received no His tagged-REV1. Next, the stapled peptide in a concentration range from 0 to 15 μM was added to the NiNTA HisSorb plate pre-incubated with REV1 and was incubated at room temperature with gentle shaking for 20 minutes. After this, 18 picomoles of FLAG tagged POL κ in 225 μL of PBS was added to the wells, covered with a cover film and incubated for 30 minutes at room temperature. Positive control wells did not receive the stapled peptide. Then, the cover film from the NiNTA plate is removed and the plate was washed four times with PBS-Tween (PBS with 0.05% Tween 20) by filling each well with 250 μL of wash solution, letting it sit for 10-60 seconds, dumping out the wash solution, and tapping the plate upside down on some paper towels to dry. Finally, 200 μL of anti-FLAG M2-HRP antibody was diluted to 1:20,000 in PBS/BSA and added to each well of the plate, which was sealed with a cover film and incubated for one hour at room temperature on a gentle rocker. The NiNTA plate was then washed four times with PBS-Tween as before and 200 μL of SureBlue TMB substrate was added to each well and incubated for 20 minutes. The reaction was stopped by adding 50 μL 1 M hydrochloric acid and the absorbance of each well is read within ten minutes in a Tecan 10M plate reader, 25°C, at an OD of 450 nm.
Cell Culturing
HT1080 cells (purchased from ATCC) were grown at 37 °C with 5% CO2 in RPMI 1640 (Gibco), 10% (vol/vol) FBS (HyClone), and 1% Penicillin-Streptomycin antibiotic (Corning). MEFs (wild type and Rev1 knockout) [Jansen et al. , 2006] and KP (KrasG12D;p53−/−) cells (kindly obtained from Jackson Lab, MIT) were also grown at 37 °C with 5% CO2, but in DMEM (Gibco), 10% (vol/vol) FBS (HyClone), and 1% Penicillin-Streptomycin antibiotic (Corning). All cells were trypsinized using 0.25% Trypsin-EDTA (Corning) for passaging or experimentation.
Immunofluorescence microscopy
HT1080 cells were grown on poly-L-lysine coated coverslips in 24-well plates in complete media until they reached at least 60% confluence. Plates were washed twice with PBS and then stapled peptide at 6 μM concentration in OptiMEM media was added to the wells for 18 hours. Next, cells on the coverslip were first fixed with methanol for 20 minutes at −20 °C, followed by their rehydration, by washing twice them with PBS. Then, cells were incubated in 70% ethanol for 10 minutes at 4 °C and washed twice with PBS. Finally, Prolong Gold antifade reagent with DAPI (4′, 6-diamino-2-phenylindole, Molecular Probes) was mounted on cells and the coverslips were sealed with nail polish. Foci were examined in a Nikon H600L microscope. Randomly selected fields from 3 independent coverslips were examined.
Flow cytometry
HT1080 cells were grown in complete RPMI 1640 media, washed twice with PBS and then exposed to 6 μM stapled peptide for 18 hours in OptiMEM media. Cells were trypsinized, washed twice with PBS and resuspended in 1ml paraformaldehyde in PBS overnight at 4 °C. Next, cells were pelleted and resuspended in 1ml, 1✕ PBS and were run through BD BioScience Flow cytometer. Percent stapled-peptide positive cells were calculated by dividing the number of green cells by the total number of cells that were run through the cytometer.
Cytotoxicity assay
Cytotoxicity was assessed by using the Cell-titer glo luminescence stain that monitors cell viability by quantifying the amount of ATP in the media, which is directly proportional to the number of metabolically active (viable) cells. Briefly, 5,000 cells were plated in complete media in each well of a 96-well plate for 24 hours, followed by the addition of increasing doses of cisplatin. The next day, serum-free OptiMEM media containing 6 μM stapled peptide replaces complete media and incubates over cells for 18 hours. Complete media inhibits translocation of the stapled peptide into cells, as the serum proteins tend to bind to the stapled peptide. Cell-titer glo Luminescence stain was then added to the equilibrated plates per the manufacturer’s instructions and luminescence was measured on the Tecan Spark 10M plate reader. Relative luminescence, which indicates relative survival of metabolically active cells, was calculated by dividing the luminescence of treated samples with untreated controls.
HPRT mutagenesis assay
For the hypoxanthine-guanine phosphoribosyl transferase (HPRT) mutagenesis assay, HT1080 cells were first grown in HAT (complete media with 100 μM Hypoxanthine, 0.4 μM Aminopterin and 16 μM Thymidine) media for 14 days to remove spontaneous Hprt mutants. After HAT selection, cells were exposed to cisplatin at 0.6 μM concentration for 24 hours. Then, in OptiMEM media, stapled compound was added to cells. After 18 hours hours of stapled peptide treatment, cells were trypsinized and washed with PBS. 600 cells were plated in triplicate in 6-well plates to determine clonal efficiency from the cisplatin and stapled peptide treatment in a colony survival assay (supplementary figure 10). Rest of the cells were plated in complete media to recover for 8 days. Then, 500,000 recovered cells per treatment were plated in six 10 cm dishes in 6-TG media to allow proliferation of HPRT+ cells. After 15 days, colonies were fixed (50% methanol and 10% glacial acetic acid) and stained (0.02% Coomassie brilliant blue R-250 stain in methanol: acetic acid: water in a ratio of 46.5:7:46.5 (v/v/v)) counted after 14-20 days. The HPRT mutation frequency was calculated as a ratio of the number of HPRT mutants to the number of surviving colonies plated at the time of selection [Silva et al. , 2005].
Results and Discussion
We synthesized two stapled versions of the 10 amino acid POL κ RIR peptide (WT) by introducing pairs of μ-methyl,μ-pentenylglycine residues at two different i and i+4 positions [Walensky et al. , 2004], followed by ruthenium-catalyzed ring-closing olefin metathesis to form the staple (Staple A and B) (Fig. 1a). Additional stapled variants were constructed in parallel that contained alanines instead of the conserved phenylalanines (Staple AFF to AA and Staple BFF to AA). FITC was added at the N-termini through a β-alanine linker (Supplementary Fig. 1).
Figure 1:
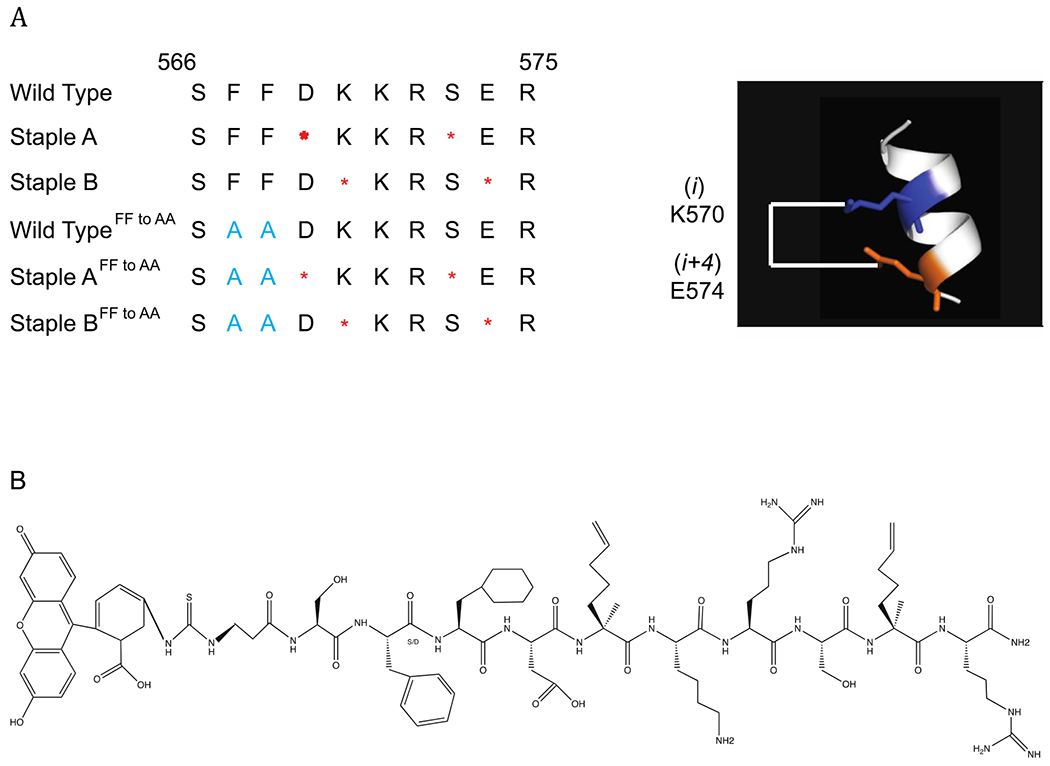
Construction and characterization of POL κ RIR Stapled peptides. (a) Sequences of wild type, Stapled A, Stapled B peptides and their corresponding Phe to Ala mutants (in blue). Red periods indicate location of staple, which confers helicity shown on right. (b) Unstapled structure of stapled peptide K, showing location of cyclohexylalanine.
Circular dichroism (CD) spectroscopy measurements of molecular ellipticity showed that Staple A and B, as well as their stapled variants, have minima at 208 and 222 nm consistent with an α-helical conformation, while the unstapled WT and WTFF to AA peptides exhibit minima around 200 nm, indicative of an unstructured peptide (Fig. 2a). Fluorescence polarization studies showed that Staple B peptide binds most efficiently to the REV1 CTD, followed by Staple A, and WT. The FF→AA derivatives of the stapled peptides interact much more weakly, indicating that Staple A and B are binding at the intended interface, while the WTFF to AA peptide does not bind detectably (Supplementary Fig. 2).
Figure 2:
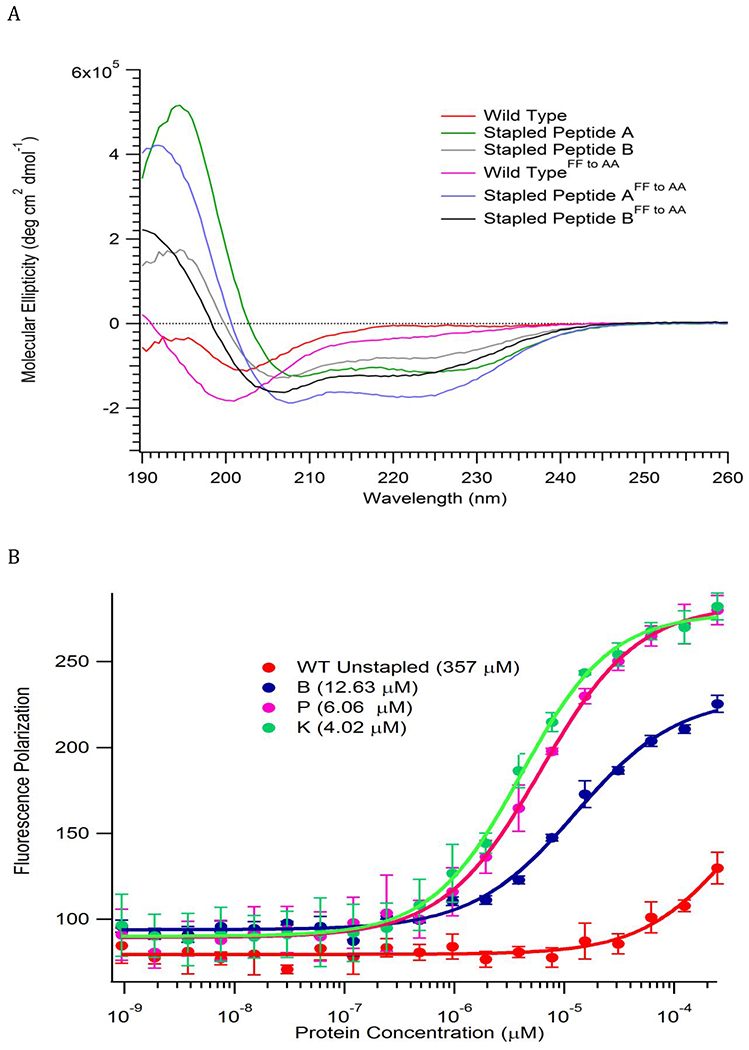
Characterization of POL κ RIR Stapled peptides. (A) CD analysis reveals enhanced α-helicity of stapled versus the unstapled peptides (B) Stapled peptides K and P bind REV1 CTD with highest affinity in a fluorescence polarization assay.
Noting that the preformed pocket in the Rev1 CTD might be able to bind a larger hydrophobic group, we replaced the conserved Phe578 residue with naphthylalanine (Staple P) and cyclohexylalanine (Staple K) (Fig. 1b). These second-generation stapled peptides showed a significantly enhanced binding to the REV1 CTD (Fig. 2b). In an ELISA assay, Staple K at IC50 (6.6 μM) successfully inhibited the REV1 CTD from interacting with POL κ protein, while the other peptides did not inhibit the interaction at the same concentration (Supplementary Fig. 3–6). This observation suggested that the Staple K could potentially inhibit the interaction of RIR-containing polymerases with REV1 in living cells.
We next tested the ability of the unstapled and stapled peptides to translocate into cells. At 6 μM concentration and 18-hour incubation in optimum media, Staple K formed discrete green foci in nuclei of cells, Staple P formed some foci and the unstapled and Staple B, failed to form visible foci (Fig. 3a, Supplementary Fig. 7). The translocation of the stapled peptide was further confirmed by FACS analysis, with ca. 85% of cells exhibiting FITC fluorescence under the same conditions (Fig. 3b).
Figure 3:
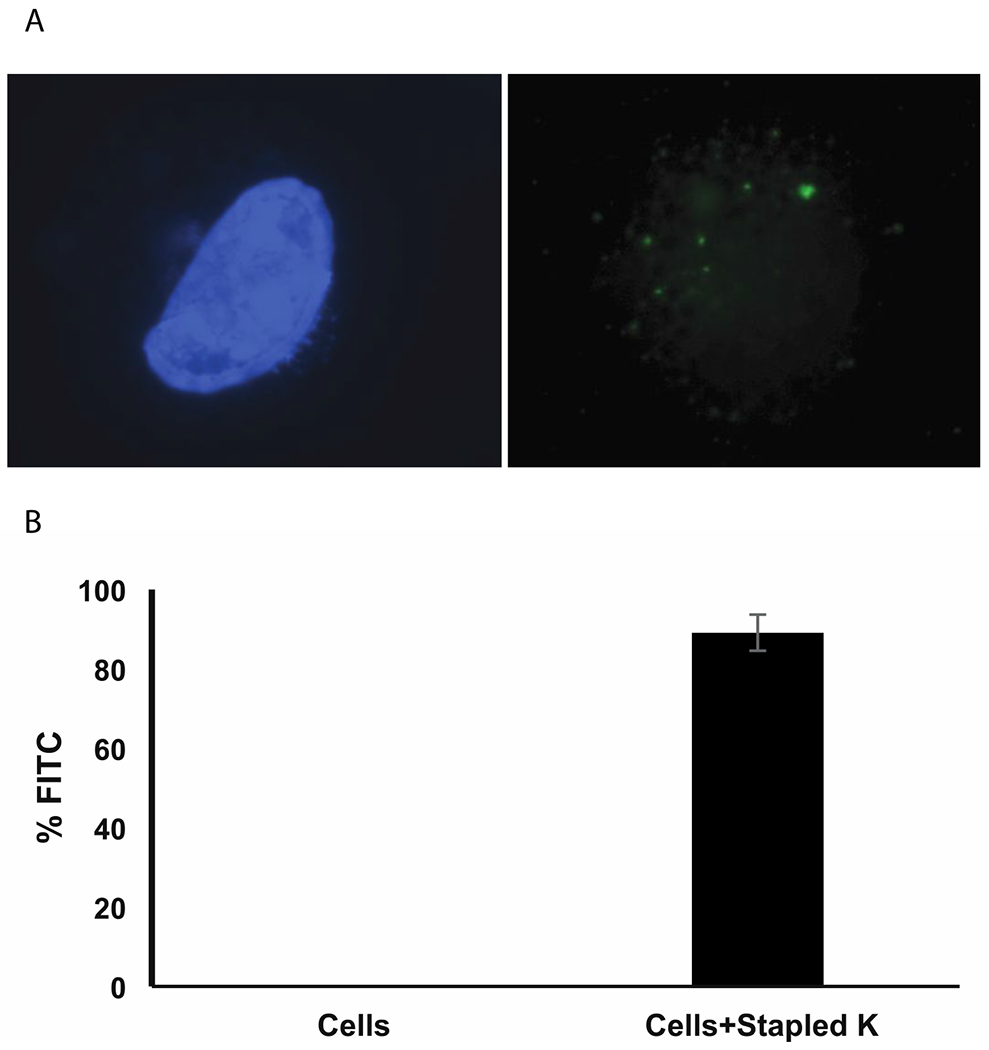
Stapled peptide K translocates into human cells in serum free media (A) Discrete foci formation of Stapled peptide K in HT1080 cells. Left panel shows blue DAPI (4’,6-diamidino-2-phenylindole) stained nuclei. Right panel shows green FITC (Fluorescein-5-isothiocyanate)-labeled stapled peptides as green foci. (B) Percentage of FITC positive HT1080 cells quantified in a flow cytometer.
Staple K had only a minimal effect on the viability of Rev1+/+ and Rev1−/− MEFs (Mouse Embryonic Fibroblasts) (Fig. 4a and 4b). In contrast, Staple K sensitized Rev1+/+ MEFs to killing by ultraviolet (UV) light but had no effect on the sensitivity of Rev1−/− MEFs to UV radiation (Fig. 4a and 4b), an important observation indicating that Staple K increases cell death in response to DNA damage by interfering with Rev1 function rather than by an off-target effect. Consistent with this conclusion, Staple K also sensitizes MEFs to killing by three additional DNA damaging agents—cisplatin, BPDE and MMS—that are known to cause greater killing of REV1-deficient cells (Supplementary Fig. 8). The Staple K peptide also sensitizes two additional cancer cell lines to killing by the widely used chemotherapeutic agent cisplatin (Supplementary Fig. 9).
Figure 4:
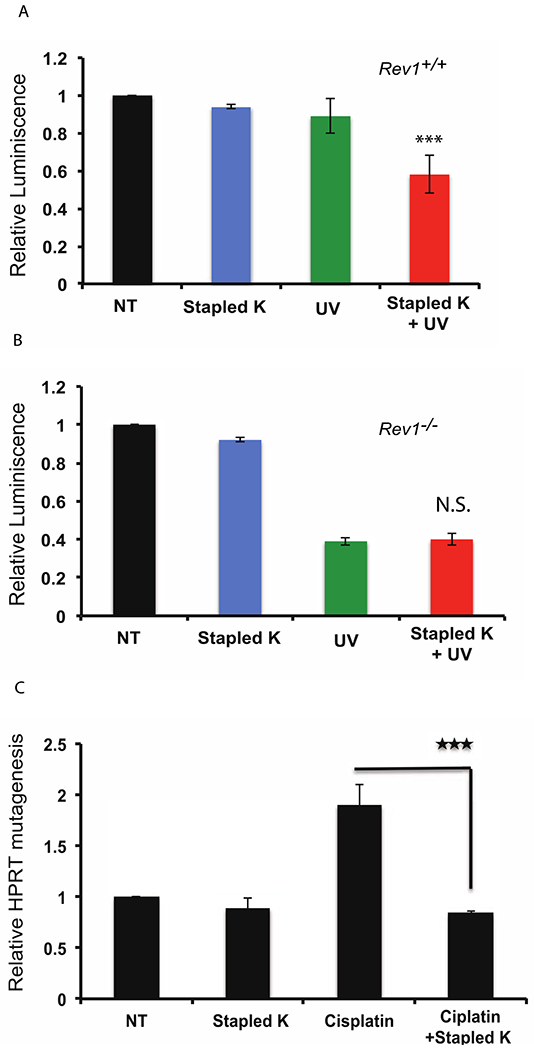
Stapled peptide K peptide enhances cytotoxicity in human cells (4A-4B). Stapled peptide K has minimal effect on Rev1+/+ (Fig. 4 A) and Rev1−/− (Fig. 4B) MEF cells, but increases UV-induced cytotoxicity only in Rev1+/+ cells (Fig. 4A) in a Cell Titer Glo (CTG) assay. 4C shows relative increase in HPRT mutagenesis in HT1080 cells. n=6; error bars represent SD; statistical significance was determined by Student’s two-tailed t test: ***P<0.001.
Importantly, our observation that Staple K additionally decreases the frequency of HPRT mutagenesis in response to cisplatin treatment indicates that Staple K is acting at least in part by interfering with the REV1/POL ζ4-dependent pathway of mutagenic TLS (Fig. 4c). It seems most likely that the effect is due to the stapled POL κ RIR peptide preventing both the RIR containing polymerases, including POLD3, a component of POL ζ4, from interacting with the RIR interface of the Rev1 CTD. Our study describes an example of a class of drug to inhibit Rev1-dependent mutagenic translesion synthesis that could potentially be used as an adjuvant to improve DNA damaging chemotherapy by increasing cell killing while reducing acquired resistance.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by NIEHS grants R01 ES015818 and R35 ES028303 to G.C.W., P30 ES002109 to MIT, and A.D.M. Research Awards Program and B.B.A., Harvard Office of Technology Development to G.L.V. G.C.W. is an American Cancer Society Professor.
Financial Interests: G.L.V. has equity stake in Hydrocarbon-stapled peptide technology licensed to Aileron Therapeutics.
REFERENCES
- Doles J, Oliver TG, Cameron ER, Hsu G, Jacks T, Walker GC, Hemann MT. 2010. Suppression of Rev3, the catalytic subunit of Pol{zeta}, sensitizes drug-resistant lung tumors to chemotherapy. Proc Natl Acad Sci U S A 107(48):20786–20791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabel SA, DeRose EF, London RE. 2013. XRCC1 interaction with the REV1 C-terminal domain suggests a role in post replication repair. DNA Repair (Amst) 12(12):1105–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann TN, Yeh JT, Bowman BR, Chu Q, Moellering RE, Verdine GL. 2012. Inhibition of oncogenic Wnt signaling through direct targeting of beta-catenin. Proc Natl Acad Sci U S A 109(44):17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JG, Langerak P, Tsaalbi-Shtylik A, van den Berk P, Jacobs H, de Wind N. 2006. Strand-biased defect in C/G transversions in hypermutating immunoglobulin genes in Rev1-deficient mice. J Exp Med 203(2):319–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YW, Grossmann TN, Verdine GL. 2011. Synthesis of all-hydrocarbon stapled alpha-helical peptides by ring-closing olefin metathesis. Nat Protoc 6(6):761–771. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Hadden MK. 2016. Targeting the translesion synthesis pathway for the development of anti-cancer chemotherapeutics. J Med Chem 59(20):9321–9336. [DOI] [PubMed] [Google Scholar]
- Lippert TH, Ruoff HJ, Volm M. 2008. Intrinsic and acquired drug resistance in malignant tumors. The main reason for therapeutic failure. Arzneimittelforschung 58(6):261–264. [DOI] [PubMed] [Google Scholar]
- Ohashi E, Hanafusa T, Kamei K, Song I, Tomida J, Hashimoto H, Vaziri C, Ohmori H. 2009. Identification of a novel REV1-interacting motif necessary for DNA polymerase kappa function. Genes Cells 14(2):101–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pustovalova Y, Magalhaes MT, D’Souza S, Rizzo AA, Korza G, Walker GC, Korzhnev DM. 2016. Interaction between the Rev1 C-Terminal Domain and the PolD3 Subunit of Polzeta Suggests a Mechanism of Polymerase Exchange upon Rev1/Polzeta-Dependent Translesion Synthesis. Biochemistry 55(13):2043–2053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sail V, Rizzo AA, Chatterjee N, Dash RC, Ozen Z, Walker GC, Korzhnev DM, Hadden MK. 2017. Identification of small molecule translesion synthesis inhibitors that target the Rev1-CT/RIR protein-protein interaction. ACS Chem Biol 12(7):1903–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MJ, Costa P, Dias A, Valente M, Louro H, Boavida MG. 2005. Comparative analysis of the mutagenic activity of oxaliplatin and cisplatin in the Hprt gene of CHO cells. Environ Mol Mutagen 46(2):104–115. [DOI] [PubMed] [Google Scholar]
- Verdine GL, Hilinski GJ. 2012. Stapled peptides for intracellular drug targets. Methods Enzymol 503:3–33. [DOI] [PubMed] [Google Scholar]
- Walensky LD, Kung AL, Escher I, Malia TJ, Barbuto S, Wright RD, Wagner G, Verdine GL, Korsmeyer SJ. 2004. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science 305(5689):1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek J, Lee CJ, D’Souza S, Minesinger B, Kim H, D’Andrea AD, Walker GC, Zhou P. 2012a. Structural basis of Rev1-mediated assembly of a quaternary vertebrate translesion polymerase complex consisting of Rev1, heterodimeric polymerase (Pol) zeta, and Pol kappa. J Biol Chem 287(40):33836–33846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek J, Liu J, D’Souza S, Wang S, Xue Y, Walker GC, Zhou P. 2012b. Multifaceted recognition of vertebrate Rev1 by translesion polymerases zeta and kappa. J Biol Chem 287(31):26400–26408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszek JL, Chatterjee N, Najeeb J, Ramos A, Lee M, Bian K, Xue JY, Fenton BA, Park H, Li D, Hemann MT, Hong J, Walker GC, Zhou P. 2019. A Small molecule targeting mutagenic translesion synthesis improves chemotherapy. Cell 178(1):152–159 e111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie K, Doles J, Hemann MT, Walker GC. 2010. Error-prone translesion synthesis mediates acquired chemoresistance. Proc Natl Acad Sci U S A 107(48):20792–20797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Xie K, Zhang XQ, Pridgen EM, Park GY, Cui DS, Shi J, Wu J, Kantoff PW, Lippard SJ, Langer R, Walker GC, Farokhzad OC. 2013. Enhancing tumor cell response to chemotherapy through nanoparticle-mediated codelivery of siRNA and cisplatin prodrug. Proc Natl Acad Sci U S A 110(46):18638–18643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka K, Chatterjee N, Hemann MT, Walker GC. 2017. Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy? PLoS Genet 13(8):e1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.