

Abstract
The extent of irreversible neuroaxonal damage is the key determinant of permanent disability in traumatic and inflammatory conditions of the central nervous system (CNS). Structural damage is nevertheless in part compensated by neuroplastic events. However, it is unknown whether the same kinetics and mechanisms of neuroaxonal de‐ and regeneration take place in inflammatory and traumatic conditions. We analyzed neuroaxonal degeneration and plasticity in early multiple sclerosis (MS) lesions and traumatic brain injury (TBI). Neuroaxonal degeneration identified by the presence of SMI31+ chromatolytic neurons and SMI32+ axonal profiles were characteristic features of leukocortical TBI lesions. Axonal transport disturbances as determined by amyloid precursor protein (APP)+ spheroids were present in both TBI and MS lesions to a similar degree. Neurons expressing growth‐associated protein 43 (GAP43) and synaptophysin (Syn) were found under both pathological conditions. However, axonal swellings immunopositive for GAP43 and Syn clearly prevailed in subcortical MS lesions, suggesting a higher regenerative potential in MS. In this context, GAP43+/APP+ axonal spheroid ratios correlated with macrophage infiltration in TBI and MS lesions, supporting the idea that phagocyte activation might promote neuroplastic events. Furthermore, axonal GAP43+ and Syn+ swellings correlated with prolonged survival after TBI, indicating a sustained regenerative response.
Keywords: axonal regeneration, GAP43, multiple sclerosis (MS), neuronal plasticity, traumatic brain injury (TBI)
INTRODUCTION
Axonal damage and loss are important pathological hallmarks in both inflammatory (40) and traumatic conditions of the central nervous system (CNS) (4). In addition, damage to and demise of neurons have lately been described as accompanying features in multiple sclerosis (MS) brain and spinal cord 22, 31, 35, 46, 47. Axonal pathology as reflected by transport disturbances and spheroid formations was mainly described as an acute and transient event occurring in active demyelinating lesions 11, 19, 28. Similarly, trauma to the CNS exhibits a variable picture of axonal damage and retrograde neuronal reactions 3, 25, 30, 39.
So far, little is known about the mechanisms and kinetics of neuroaxonal damage in MS and traumatic brain injury (TBI). In MS, acute axonal damage in early lesions is generally visualized using markers of transport disturbances (19). Nevertheless, axonal loss seems to continue also in chronic lesion stages (36). Mechanistically, mediators of the adaptive and innate immune system, as well as a lack of trophic support from oligodendrocytes, are discussed (1). In TBI, diffuse and focal axonal injuries occur early and are characteristic of the condition, whereas only a few studies have addressed the question of protracted neuroaxonal degeneration after TBI (2). Usually, a mechanical disruption of axonal membranes and the cytoskeleton followed by Ca2+ influx and protease activation are considered the earliest pathogenic events (4).
CNS lesions, be they traumatic or inflammatory in nature, are followed by plastic events leading to improved functionality. And although axonal regeneration is not as pronounced as in the peripheral nervous system, substantial reorganization can take place as evidenced in functional magnetic resonance imaging (MRI) studies 34, 37. In general, neuronal plasticity and axonal regeneration were mainly studied in the context of spinal cord trauma and cerebral ischemia 20, 27, 33. In experimental autoimmune encephalomyelitis (EAE), substantial reorganization on the cortical and spinal levels following a focal inflammatory demyelinating lesion in the spinal cord was reported (17). There, the question arose whether the inflammatory microenvironment, as opposed to a purely traumatic environment, may enhance and stimulate neuroaxonal repair. Of note, a plethora of inflammatory mediators and growth factors are secreted in response to CNS inflammation (16).
In this study, we asked whether there were differences in the extent of neuroaxonal pathology and regeneration in acute and chronic MS vs. TBI lesions. As both lesion types differ regarding their pathophysiology, we wondered whether white matter (WM) axons and adjacent gray matter (GM) neurons might share common pathological features with regard to degeneration and repair.
Under physiological conditions, large neuronal cell bodies contain nonphosporylated neurofilament proteins detected by SMI32 antibodies, whereas phosphorylated high molecular weight neurofilaments (labeled by SMI31 antibodies) can be detected in WM axons (43). Anterogradely transported amyloid precursor protein (APP) is below the immunohistochemical detection limit in healthy CNS axons. Under various pathological conditions, however, APP and nonphosphorylated neurofilaments accumulate within the axon and are considered markers of acute axonal injury. Furthermore, as a consequence of retrograde degeneration, morphologically characterized by chromatolysis, SMI31+ filaments accumulate within the soma of injured neurons. In contrast, synaptic proteins that are associated with axonal sprouting such as growth‐associated protein 43 (GAP43) and synaptophysin (Syn) may also accumulate in regenerating neurons (14).
In the present study, as expected, we found more chromatolytic SMI31+ neurons in GM TBI lesions than cortical MS lesions. However, the extent of acute axonal injury as evidenced by the presence of APP+ and SMI32+ axonal spheroids was similar in both acute WM MS and TBI lesions. In contrast and surprisingly, the number of GAP43+ and Syn+ axonal swellings was higher in MS. In a proportion of MS lesions, small GAP43+ axonal sprouts branching from established axons and neuronal cell bodies could be found. Our findings suggest that the events following axonal degeneration may be similar in both conditions, but that plastic responses are enhanced in MS as compared with TBI.
SUBJECTS AND METHODS
MS, TBI, and control tissue
The study was performed on formalin‐fixed and paraffin‐embedded biopsy and autopsy CNS tissue collected at the Institute of Neuropathology at the University Medical Center Göttingen. Acute cortico‐subcortical (= leukocortical) MS tissue from 10 MS patients was obtained upon stereotactic brain biopsy (Table 1). Additionally, tissue with chronic brainstem lesions from three MS patients obtained upon autopsy was included (Table 1). MS biopsies were from patients who had been diagnosed with MS prior to biopsy or were diagnosed upon bioptic examination. Stereotactic biopsies were carried out due to the uncertainty of diagnosis, for example, tumefactive lesions or untypical clinical symptoms. Material from eight female and four male patients diagnosed with MS was included; in one autopsy case, the sex could not be determined. In the biopsied patients, the age was between 20 years and 54 years (median 32 ± 11 years; 50 ± 21 years in the autopsied cases), and the disease duration varied in six biopsied patients between 21 days and 8 years (median 12.5 ± 7.8 years); in four biopsied patients and two autopsied patients, the disease duration could not be obtained. In eight biopsied patients, the disease course could be determined retrospectively on follow‐up: four patients with relapsing‐remitting MS (RR‐MS), one patient with primary progressive MS (PP‐MS) and three patients with a clinically isolated syndrome (CIS) were studied. In two cases, the disease course was unknown. In the autopsy samples, one chronic progressive case was included; in the remaining two patients, the disease course was unknown.
Table 1.
Clinical and pathological characteristics of MS patients. Abbreviations: CIS = clinically isolated syndrome; f = female; GM = gray matter demyelination; m = male; MS = multiple sclerosis; n/a = not available; PP = primary progressive MS; RR = relapsing‐remitting MS; WM = white matter demyelination.
| MS # | Age | Sex | Disease duration/symptoms before biopsy | Disease course | Lesion staging (21) | Lesion localization (31) |
|---|---|---|---|---|---|---|
| 1 | 32 | f | 17 days | RR | Early active, type I | Leukocortical, type I |
| 2 | 28 | f | 23 days | RR | Early active, type I | Leukocortical, type I |
| 3 | 39 | f | 8 years | PP | Late active | Leukocortical, type I |
| 4 | 31 | f | 45 days | CIS | Late active | Subcortical |
| 5 | 52 | f | n/a | RR | Late active | Leukocortical, type I |
| 6 | 44 | f | n/a | n/a | Late active | Subcortical |
| 7 | 25 | m | n/a | n/a | Late active | Leukocortical, type I |
| 8 | 54 | f | n/a | CIS | Early active | Leukocortical, type I |
| 9 | 20 | m | 45 days | CIS | Early active, type I | Leukocortical, type I |
| 10 | 32 | f | 21 days | RR | Early active, type II | Subcortical |
| 11 | n/a | n/a | n/a | n/a | Inactive | WM‐GM, medulla |
| 12 | n/a | n/a | n/a | n/a | Early inactive | WM‐GM, medulla |
| 13 | 35 | m | 21 years | Chronic progressive | Inactive | WM, medulla |
TBI autopsy tissue was obtained from eight patients (one female/seven males) with leukocortical lesions of variable age (Table 2). In three cases, brainstem trauma tissue could be included. The age of the patients was between 16 years and 60 years (median 56 ± 19 years). The survival time after TBI varied from death within the first day to 4 months survival time (median 4 ± 38 days). Of note, one patient surviving for 4 months suffered from two consecutive TBIs.
Table 2.
Clinical and pathological characteristics of TBI and control cases. Abbreviations: m = male; f = female; TBI = traumatic brain injury.
| TBI # | Age | Sex | Survival time (days) | Location | Clinical data |
|---|---|---|---|---|---|
| 1 | 44 | m | 2 | Frontobasal | Subacute contusion |
| 2 | 55 | m | 0 | Frontobasal | Acute contusion |
| 3 | 54 | m | 30 | Frontal, medulla | Subacute and chronic contusions |
| 4 | 59 | m | 1 | Frontobasal | Acute contusions (contre‐coup) |
| 5 | 18 | m | 30 | Frontal, medulla | Subacute and chronic contusions |
| 6 | 60 | m | 0 | Frontobasal | Acute contusion |
| 7 | 60 | f | 5 | Frontobasal, pons | Subacute contusions frontobasal cortex and brainstem |
| 8 | 16 | m | 120 | Frontal, mesencephalon, medulla | Consecutive contusions (acute and chronic) frontal cortex and brainstem |
| Control # | Age | Sex | Location | Cause of death |
|---|---|---|---|---|
| 1 | 51 | m | Frontal, medulla | Multiorgan failure, Hodgkin lymphoma |
| 2 | 36 | m | Frontal, pons | Aortic dissection |
| 3 | 65 | m | Frontal, mesencephalon | Acute pulmonary embolism |
| 4 | 71 | m | Frontal, medulla | Aortic dissection |
| 5 | 81 | f | Frontal | Multiorgan failure upon coronary surgery |
| 6 | 58 | f | Frontal | Pneumonia |
Leukocortical and brainstem tissue from six control patients (two females, four males; age range 36–81 years, median 68 ± 15 years) obtained at autopsy without neuropathological findings was available (Table 2). Control CNS tissue from areas matching the MS and trauma lesion locations was examined.
The study was carried out according to the national ethical guidelines and legal regulations regarding the use of archival post‐mortem material. For diagnostic purposes, hematoxylin and eosin (HE) and LFB/PAS (Luxol Fast Blue/Periodic acid Schiff) stained, and Bielschowsky silver impregnated sections were used.
Immunohistochemistry
Immunohistochemistry was carried out on 2‐µm to 3‐µm thick sections that were deparaffinized, hydrated and pretreated as described previously (35). Incubations with primary antibodies diluted in 10% fetal calf serum (FCS) in phosphate‐buffered saline (PBS) were carried out overnight at 4°C (Table 3). Antibody detection was achieved using biotinylated secondary antisera followed by avidin‐peroxidase. Diaminobenzidine (DAB) and Fast Red (Sigma‐Aldrich Inc., Saint Louis, MI, USA) were used as chromogenic substrates. Negative control sections without primary antibody or with isotype control antibody were processed in parallel. Sections were then counterstained with either hematoxylin or 4′‐6‐diamidino‐2‐phenylindole (DAPI). Double fluorescence labeling with two mouse monoclonal primary antibodies was carried out according to a previously described protocol (35). Images were taken using an Olympus BX51 microscope equipped with a DP71 CCD camera (Olympus Optical Co., Ltd, Hamburg, Germany) and a Zeiss Cell Observer microscope with an AxioCam ICc 3 CCD camera (Carl Zeiss MicroImaging, Ltd, Göttingen, Germany).
Table 3.
Antibodies used for immunohistochemistry. Abbreviations: APP = amyloid precursor protein; GAP43 = growth‐associated protein 43; H = high molecular weight; MW = microwave pretreatment; NF = neurofilament; nonP = nonphosphorylated; P = phosphorylated; pc = polyclonal.
| Antigen | Clone | Dilution | Pretreat‐ment | Source |
|---|---|---|---|---|
| MBP | Rabbit pc | 1:500 | MW | DakoCytomation, Glostrup, Denmark |
| CNPase | SMI91 | 1:200 | MW | Covance Inc., Princeton, NJ, USA |
| Macrophages/microglia | KiM1P | 1:5000 | MW | (32) |
| β‐tubulin | TUB 2.1 | 1:400 | MW | Sigma‐Aldrich Inc., Saint Louis, MI, USA |
| GAP‐43 | 9‐1E12 | 1:4000 | MW | Chemicon International, Temecula, CA, USA |
| Synaptophysin | SY38 | 1:10 | MW | DakoCytomation |
| APP | 22C11 | 1:3000 | MW | Chemicon International |
| nonP‐NF‐H | SMI32 | 1:7000 | MW | Covance Inc. |
| P‐NF‐H | SMI31 | 1:1000 | MW | Covance Inc. |
Morphometry
In WM lesions, numbers of APP+, SMI32+, GAP43+ and Syn+ axonal swellings were determined in five standardized microscopic fields of 90 000 µm2 each. In the adjacent GM, numbers of SMI31+, GAP43+ and Syn+ neuronal cell bodies were counted in five standardized microscopic fields of 390 625 µm2 each defined by an ocular morphometric grid. In WM trauma lesions, GAP43+ axonal sprouts were counted in five standardized fields of 36 100 µm2 each. All numbers are given as immunopositive profiles/mm2 ± standard deviation (SD).
Statistics
The nonparametric Kruskal–Wallis test with Dunn's multiple comparison post test was applied to compare neuronal and axonal numbers between acute and chronic MS, trauma and control tissues. Spearman's rank correlation was used to identify interdependence of confounding variables. All tests were classified as significant if P was <0.05. The GraphPad PRISM™ software was used for statistical calculations and graph generation (GraphPad Software, Inc., San Diego, CA, USA).
RESULTS
Characterization of MS and CNS trauma tissue
MS sections stained with LFB/PAS and antibodies against 2′, 3′‐cyclic nucleotide 3′‐phosphodiesterase (CNPase) and myelin basic protein (MBP) were used for lesion classification. Lesions were classified according to their localization in pure cortical or subcortical and combined leukocortical lesions (Table 1). Lesion staging was done according to previously defined criteria (36), taking into consideration the presence of myelin degradation products in macrophages (early active: CNPase+/MBP+/LFB+; late active: CNPase‐/MBP+/LFB+; inactive: CNPase‐/MBP‐/LFB‐). In general, specimens obtained from biopsies consisted of acute lesions with extensive macrophage infiltration (402 ± 143 KiM1P+ macrophages/activated microglia/mm2, mean ± SD) comprising early and late active demyelinating lesions. We characterized five leukocortical lesions as early active and five as late active (Table 1). Additionally, three brainstem autopsy samples comprising two medullary lesions and one pontine lesion displayed two chronic inactive plaques (no foamy microglia/macrophages) and one early inactive (foam cells without myelin degradation products) plaque (36) (Table 1).
TBI lesions where defined by areas of subarachnoid hemorrhage with petechial cortical bleedings in HE stain (Table 2; 1, 2). In addition, TBI lesions were characterized by strong macrophage infiltration, and abundant axonal spheroids were found in the affected adjacent subcortical WM (Figure 2C,E). The resorptive response in trauma lesions was assessed by the density of KiM1P+ macrophages. The degree of macrophage infiltration increased with prolonged survival of the TBI patients (236 ± 200 phagocytes/mm2 in subcortical WM lesions, n = 8, P = 0.015, r = 0.82). Subcortical control WM showed little or no phagocytic infiltration (Table 2).
Figure 1.
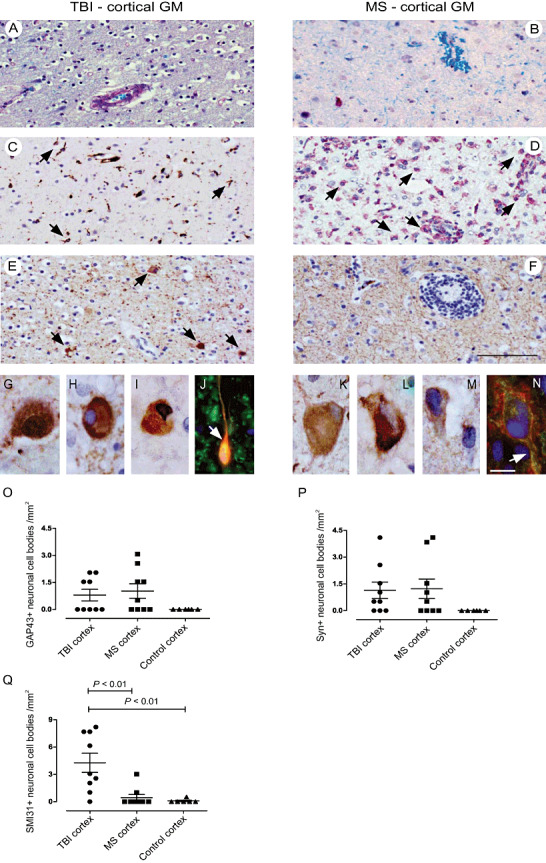
Neuronal de‐ and regeneration in cortical TBI and MS lesions. LFB‐PAS staining of cortical TBI (A) and MS (B) lesion areas with diffuse microglia activation in TBI (C) and enhanced macrophage infiltration in MS (D) cortex (KiM1P staining). Abundant SMI31+ chromatolytic neurons were found in cortical TBI (E) as compared with cortical MS lesions (F). (G–J; TBI) SMI31+ neuron in cortical TBI lesion (G); cortical neuronal cell bodies immunopositive for Syn (H) and GAP43 (I); GAP43+ (red) and β‐tubulin+ (green, arrow) neuron in mesencephalic TBI lesion (J). (K–N; MS) SMI31+ (K), Syn+ (L) and GAP43+ (M) neuronal cell bodies in cortical MS lesions; GAP43 (red) neuronal surface IR extending along a β‐tubulin+ axon (green; arrow; N). Numbers of cortical neurons with cytosolic IR for GAP43 (Ο) and Syn (P) do not differ between TBI and MS tissue. Numbers of SMI31+ cortical neurons prevail in TBI tissue (Q). Scale bars: 100 µm (A–F), 10 µm (G–N); original magnifications: ×200 (A–F), ×630 (G–I, K–M), ×1000 (J,N). GAP43 = growth‐associated protein 43; GM = gray matter; IR = immunoreactivity; MS = multiple sclerosis; Syn = synaptophysin; TBI = traumatic brain injury.
Figure 2.
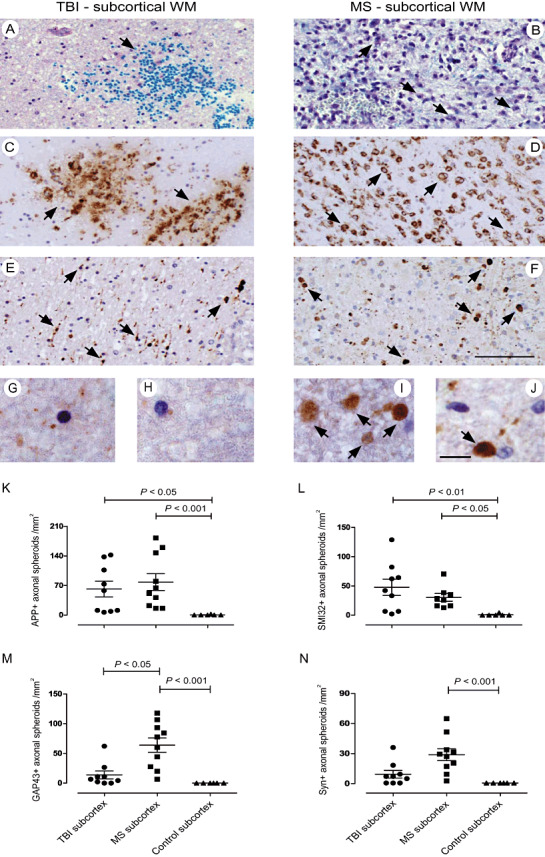
Axonal swellings immunoreactive for de‐ and regeneration markers in subcortical TBI and MS lesions. LFB‐PAS staining of subcortical TBI (A) and MS (B) lesion areas revealing myelin loss (and axon loss; not shown) and microhemorrhage (arrow) in TBI and demyelination in MS. Focal macrophage infiltrates in TBI (C; arrows) and more diffuse, extensive parenchymal macrophage infiltration in MS (B,D; arrows) subcortical lesions (KiM1P staining). APP IR showing axonal swellings and spheroid formations in TBI (E) and MS (F) white matter lesions. (G,H; TBI) Near‐absent IR for GAP43 (G) and Syn (H) in subcortical TBI tissue. (I,J; MS) Axonal spheroids with IR for GAP43 (I) and Syn (J) in a subcortical MS lesion. APP+ (K) and SMI32+ (L) axonal spheroids occur to a similar extent in subcortical MS and TBI lesions. GAP43+ (M) and Syn+ (N) axonal swellings prevail in subcortical MS lesions (I,J,M,N). Scale bars: 100 µm (A–F), 10 µm (G–J); original magnifications: ×200 (A–F), ×630 (G–J). APP = amyloid precursor protein; GAP43 = growth‐associated protein 43; IR = immunoreactivity; MS = multiple sclerosis; Syn = synaptophysin; TBI = traumatic brain injury; WM = white matter.
Cortical neuronal degeneration is much more pronounced in TBI than MS
Neuronal pathology was studied by assessing frequencies of neuronal cell bodies immunoreactive for SMI31, GAP43 and Syn in cortical TBI and MS lesions compared with control cortical tissue (Figure 1E–N). Neuronal SMI31 immunoreactivity (IR) was generally accompanied by morphological features of retrograde chromatolysis displaying swollen cell bodies with an eccentrically located nucleus (Figure 1E,G) 6, 35. GAP43+ and Syn+ neuronal somata in part showed a regular phenotype; however, chromatolytic GAP43+ or Syn+ neurons were also detected (Figure 1H–J). SMI31+ neuronal cell bodies were a common feature of cortical TBI lesions and only rarely seen in cortical MS and control tissue (TBI vs. MS: 4 ± 3 neurons/mm2 vs. 0.5 ± 1, mean ± SD, P < 0.01; TBI vs. control, P < 0.01; Figure 1E–G,K,Q). Regarding neuronal cytosolic GAP43 and Syn IR, there was no difference between cortical MS and TBI tissue (TBI: 0.8 neurons/mm2; MS: 1 neuron/mm2; Figure 1H–J,L–N,O,P). Of note, neither SMI31+ nor GAP43+ or Syn+ cell bodies were detected in cortical control tissue (Figure 1O–Q). Numbers of SMI31+ neuronal somata did neither correlate with the survival times of TBI patients nor with the disease durations of MS patients (data not shown). Albeit numbers of both GAP43+ and Syn+ neurons were correlated with increasing survival times in trauma patients (GAP43: n = 8, P = 0.005, r = 0.89; Syn: n = 8, P = 0.007, r = 0.87), no correlations could be found for GAP43 and Syn with disease duration and lesion staging in MS patients (GAP43: n = 5, P = 0.52, r = −0.34; Syn: n = 5, P = 0.78, r = 0.22). However, information on the disease duration was only available for 5 out of 10 MS patients.
Axonal transport disturbances prevail in early MS lesions, whereas alterations in neurofilament phosphorylation are more pronounced in TBI lesions
Acute axonal pathology is usually studied by measuring the density of APP+ and SMI32+ axonal spheroids (Figure 2E–J). APP+ axonal swellings are assumed to reflect disturbances of antero‐ (and retro‐)grade axonal transport (25), whereas SMI32+ axonal spheroids are indicators of changes in neurofilament phosphorylation due to axonal damage, transport disturbance and transection (38). In subcortical WM lesions, there was no statistical difference in the numbers of APP+ axonal spheroids comparing TBI with MS lesions (MS: 78 ± 63 spheroids/mm2, TBI: 62 ± 56 spheroids/mm2; mean ± SD). APP+ spheroids were only rarely seen in control WM (0 ± 1 spheroids/mm2; TBI vs. control, P < 0.05; MS vs. control, P < 0.001; Figure 2K). However, densities of SMI32+ axonal spheroids were highest in subcortical TBI tissue (48 ± 42 spheroids/mm2) as compared with MS WM lesions (30 ± 19 spheroids/mm2) and only rarely found in control WM (1 ± 2 spheroids/mm2; TBI vs. control, P < 0.01; MS vs. control, P < 0.05; Figure 2L). Regarding the temporal evolution of APP+ and SMI32+ spheroid densities, there was no correlation with both TBI survival time (APP: n = 8, P = 0.84, r = 0.098; SMI32: n = 8, P = 0.93, r = 0.04) and MS disease duration (APP: n = 6, P = 0.56, r = −0.31; SMI32: n = 5, P = 0.13, r = −0.82).
Markers of axonal regeneration and synaptic plasticity prevail in acute MS lesions and correlate with survival times in TBI patients
If axonal transport is interrupted, Syn and GAP43 accumulate along the axon similar to APP as part of axonal swellings and spheroid formations (Figure 2G–J) 3, 25. In subcortical WM MS lesions, we detected more GAP43+ and Syn+ axonal swellings (GAP43: 28 ± 19 spheroids/mm2, Syn: 64 ± 38 spheroids/mm2) in comparison with TBI (GAP43: 9 ± 12 spheroids/mm2, Syn: 14 ± 20 spheroids/mm2) and control WM lesions (GAP43: 0 ± 0 spheroids/mm2, Syn: 0 ± 0 spheroids/mm2; GAP43: MS vs. control, P < 0.001; Syn: MS vs. control, P < 0.001; MS vs. TBI, P < 0.05; Figure 2M,N). To normalize the densities of GAP43 and Syn positive axons to the total density of axons with transport impairments, we compared ratios of GAP43+/APP+ spheroids and Syn+/APP+ spheroids between MS and TBI. Syn/APP spheroid ratios were higher in MS compared with TBI subcortical WM lesions (MS: 103.4 ± 61.89%; TBI: 32.14 ± 38%; P = 0.0037). Of note, Syn+ spheroids outbalanced the number of APP+ spheroids in MS lesions (see above). In MS, there was a trend for higher GAP43/APP ratios compared with TBI subcortical WM (MS: 57.60 ± 59.41%; TBI: 19.96 ± 21.78%; P = 0.0657) (Figure 3F,G). Interestingly, axonal GAP43 IR but not Syn IR (data not shown) increased with macrophage numbers in both TBI and MS WM lesions as reflected by a positive correlation between GAP43+/APP+ spheroid ratios and KiM1P+ macrophage densities (TBI: n = 8; GAP43: P = 0.005, r = 0.9; MS: n = 10; GAP43: P = 0.04, r = 0.66; Figure 3H,I). Furthermore, the densities of GAP43+ and Syn+ axonal swellings in subcortical TBI lesions—again normalized to APP+ axonal profiles—correlated with increasing patient survival (n = 8; GAP43: P = 0.0004, r = 0.97; Syn: P = 0.045, r = 0.72; Figure 3J). In MS, no correlations for GAP43+ and Syn+ swellings with disease duration were detected (n = 6; GAP43: P = 0.7, r = −0.26; Syn: P = 0.3, r = 0.52). Increased GAP43+/APP+ ratios also correlated with younger age in the TBI (n = 8, P = 0.02, r = −0.83) but not MS group (n = 6, P = 0.18, r = 0.46), taking into account that there was a strong association with younger age and prolonged survival in TBI (n = 8, P = 0.06, r = −0.7).
Figure 3.
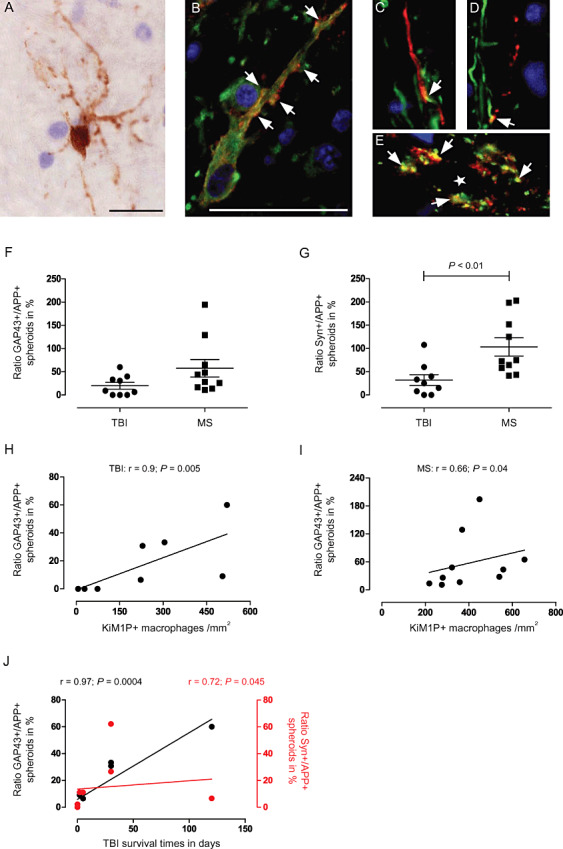
Sprouting of GAP43+ axonal filaments in TBI and MS lesions. (A) Small arborizing GAP43+ neuron in an inactive medullary MS lesion; (B) β‐tubulin (green) immunopositive neuron with GAP43+ (red) synaptic boutons (arrows) along its axon in a cortical MS lesion; (C,D) GAP43+ (red) axonal sprouts deriving (arrows) from mature SMI31+ (green) axons in a TBI WM lesion. (E) Synaptic colocalization (arrows) of GAP43 (red) and Syn (green) surrounding a cortical neuron (star) in MS. (F,G) Ratios of GAP43+/APP+ and Syn+/APP+ spheroids compared between TBI and MS subcortical WM lesions. GAP43+/APP+ spheroid ratios correlate with KiM1P+ macrophage infiltration in both TBI (H) and MS (I) lesions. In TBI, GAP43+/APP+ and Syn+/APP+ spheroid ratios correlate with survival times (J). Scale bars: 20 µm; original magnifications: ×630 (A), ×1000 (B–D). APP = amyloid precursor protein; GAP43 = growth‐associated protein 43; MS = multiple sclerosis; Syn = synaptophysin; TBI = traumatic brain injury; WM = white matter.
Consecutively, we examined the subcortical WM for GAP43+ axonal sprouting. Small‐caliber fibers that were GAP43+, and GAP43+ synaptic boutons were detected along WM axons and surrounding cortical neurons (1, 3). In double stainings, these fibers were immunonegative for established axonal markers like SMI31 (Figure 3C,D), but positive for β‐tubulin (1, 3), reflecting the immature character of those fibers. In subcortical TBI lesions, we found a correlation between GAP43+ axonal sprouts and increasing survival times of the patients (n = 8; P = 0.03, r = 0.77). A quantitative analysis of GAP43+ sprouting in early subcortical MS lesions was not feasible due to the extensive inflammation and GAP43 IR of reactive astrocytes (26). Regarding brainstem tissue, there was no difference in the axonal GAP43 IR between control and chronic MS tissue. In addition, however, single neurons with cytosolic GAP43 IR (Figure 3A) were found in pontine and medullary sections with trauma (n = 3) and MS (n = 3) lesions (P > 0.05) apart from neuronal areas with constitutive expression of GAP43 (eg, dorsal cranial nerve nuclei) as seen in corresponding control sections (n = 4).
DISCUSSION
Regenerations of axons and neurons are considered rare events in the human CNS and were only rarely described in the context of inflammatory demyelination (7). Traditionally, axonal sprouting and plasticity have been studied in experimental trauma models, and classically in spinal cord injury (33). In respect to MS, functional axonal regeneration was found to occur after the induction of focal inflammatory lesions in the spinal cord of EAE animals (17). Furthermore, the effects of regeneration‐associated proteins on axonal sprouting and functional outcome were described in animals overexpressing GAP43 (12). Albeit signs of axonal de‐ and regeneration in MS had been described more than a century ago 5, 9, only recent years brought firm evidence that neuroaxonal damage and loss take place in MS and correlate indeed with clinical disability 8, 36. In the present study, we compared the extent of neuroaxonal damage and neuroplastic responses in TBI and MS lesions of different ages applying established immunohistochemical markers for axonal regeneration and synaptogenesis as well as for axonal damage and neuronal degeneration.
Neuronal chromatolysis and axonal damage are typical features of trauma lesions
Neuronal pathology was studied by the presence of SMI31 immunoreactive cell bodies in cortical sections (35). Chromatolytic neurons, whose axons have been severed, turn SMI31+ and axonal spheroids become SMI32+. We found the highest numbers of SMI31+ neurons in cortical trauma lesions. In MS and control sections, neuronal SMI31 IR was only a rare event. In contrast to MS lesions that mainly comprise the subcortical WM, contusional lesions of the brain predominantly affect the cortical layers and subsequently lead to diffuse and widespread axonal damage in the subcortical WM (10). Neuronal SMI31 IR itself reflects alterations in neurofilament phosphorylation that might be primarily due to cytoskeletal disruptions following axonal transections, even though energy failure leading to chromatolysis and apoptosis cannot be ruled out 14, 23, 35, 36. Hence, cortical trauma lesions as compared with MS were characterized by abundant neuronal degeneration as evidenced by SMI31+ neuronal cell bodies.
In both subcortical TBI and MS lesions, we found similar numbers of axonal spheroids immunopositive for APP and SMI32. Albeit the pathogenesis differs between the two lesion types, they share a common final pathway resulting in axonal transport disturbances and transections 3, 19, 45. Of note, axonal APP accumulations reflect disturbances of the fast anterograde axonal transport and not necessarily fiber transections (18). Conversely, SMI32+ spheroids rather reflect axon transections, resulting in terminal axon ovoids with a loss of neurofilament phosphorylation (38). We observed a trend toward more SMI32+ axonal spheroids in TBI, and found slightly more APP+ spheroids in MS lesions. Thus, we reckon that trauma might favor structural damage and axon transections, whereas inflammatory demyelination might predominantly promote axonal transport impairments. Recently Nikićet al (28) have shown that indeed transient axonal swellings due to transport impairments take place in EAE and MS lesions persisting for several days and resolving spontaneously. In this context, axonal spheroids in trauma lesions might reflect substantial damage and profound cytoskeletal alterations as reflected in the higher numbers of SMI32+ spheroids. Conversely, in MS WM lesions, axonal accumulations of APP might rather reflect transient transport disturbances, and not in all cases axon transections necessarily.
Inflammatory demyelination might promote neuroaxonal regeneration
Subcortical WM MS lesions were characterized by a higher density of axonal swellings immunoreactive for both GAP43 and Syn as compared with human trauma lesions. Moreover, the ratio of Syn+ to APP+ spheroids reflecting the balance of re‐ and degeneration was increased in MS lesions (Figure 3F,G). In this context, increased GAP43+/APP+ ratios were accompanied by augmented macrophage infiltration under both pathological conditions (Figure 3H,I), supporting the idea that the presence of macrophages involved in the clearance of tissue or myelin debris may rather support than impede neuroaxonal regeneration. APP+ spheroids are known to occur in early, highly inflammatory lesion stages (19) and resolve over time. An increase of the GAP43+/APP+ spheroid ratio accompanied by ongoing inflammation and macrophage infiltration might thus indicate a delayed subacute regenerative response similar to that observed with regard to GAP43 expression in spinal neurons in MS patients (35). As both GAP43 and Syn are transported along the axon in an anterograde manner comparable with APP 18, 29, 44, transport disturbances and axon transections result in accumulation of those proteins and spheroid formations (2, 3). Nonetheless, increased accumulations of GAP43 and Syn might also reflect the neuronal expression level and amount transported to the synapses. Hence, inflammation per se and particularly the inflammatory environment in early MS lesions might promote the expression and up‐regulation of plasticity‐associated marker proteins. In line, it has been shown that macrophages and other inflammatory cells are capable of secreting neurotrophic factors promoting neuronal up‐regulation of regeneration‐associated proteins 16, 41. Furthermore, experimental models revealed that antibodies against myelin proteins (eg, Nogo‐A) can promote axonal sprouting and functional recovery after spinal cord injury (27). As demyelination, and thus a loss of presumably inhibitory myelin, takes place in MS, axons per se might exhibit a higher capacity toward regeneration and sprouting.
The number of neuronal cell bodies with strong cytosolic IR for GAP43 and Syn did not differ between trauma and MS. Thus, we assume that up‐regulation of plasticity and stress proteins is induced under both pathological conditions 13, 35. The fact that axonal GAP43 and Syn IR prevailed in MS lesions (Figure 3) despite comparable numbers of intensely immunoreactive neurons in TBI and MS might either be due to stronger axonal transport impairments leading to their accumulation, or an increased axonal transport rate reflecting a more pronounced regenerative response. In EAE, it was shown that GAP43+ axonal sprouts contribute to circuit pathways bypassing a focal lesion in the spinal cord (17). Also, it is known that under inflammatory conditions, the axonal transport of Syn can be disturbed by microglial nitric oxide (42). Moreover, cytosolic expression of GAP43 does not necessarily indicate axonal regeneration as the expression has been described in the context of general neuronal stress reactions leading to apoptosis or survival of the neuron (Figure 1I,J,M,N) 15, 35. We recently reported an early nonprogressive loss of neurons in MS spinal cord followed by a subacute enhanced neuronal IR for GAP43 (35). Furthermore, we could detect a progressive axonal loss in spinal MS WM lesions (36). Taking these previous findings into account, we assume that the increased presence of axonal swellings with GAP43 and Syn IR in subcortical MS lesions might be a transient phenomenon occurring in early highly inflammatory demyelinating lesions. Upon time, after resolution of inflammation and persistence of chronic demyelination, we then expect a diminished regenerative capacity with a shift toward a rather unfavorable tissue microenvironment 24, 36. Interestingly, in trauma WM lesions, we found both an increase of macrophage infiltration and an increase of GAP43+ and Syn+ axonal swellings with protracted survival times (Figure 3J). Thus, neurotrophic mediators secreted by phagocytes might possibly contribute to axonal regeneration over time.
Prolonged survival after TBI enhances neuroaxonal regeneration
We studied TBI lesions of different age reflecting survival times of up to 4 months, and could thus examine neuroaxonal changes over time 30, 48. Indeed, we noted a correlation in the number of GAP43+ and Syn+ neuronal cell bodies with increased survival times of the patients, whereas no temporal association was found for neuronal SMI31 IR. Furthermore, densities of axonal GAP43 and Syn IR were correlated with longer survival in the trauma patients. In this context, younger age might contribute to an enhanced regenerative capacity in TBI as reflected by higher GAP43+/APP+ ratios in younger patients. In the MS samples, no correlations were detectable, probably due to the small sample size and the rather homogenous distribution, with active demyelinating lesions being most prevalent. Of note, no correlations were found for APP+ and SMI32+ spheroids in TBI and MS lesions. Hence, prolonged survival after TBI might induce up‐regulation of plasticity‐associated proteins as a delayed response to the initial neuroaxonal damage. In this context, numbers of GAP43+ axonal sprouts also correlated with increasing survival times in trauma, most likely reflecting ongoing axonal sprouting.
CONCLUSION
Neuroaxonal damage, as assessed by the presence of SMI31+ neurons and SMI32+ axonal spheroids, is a characteristic feature of cortical and subcortical TBI lesions, whereas axonal transport impairments studied by APP+ spheroids are found in both trauma and MS lesions. Comparable numbers of neuronal cell bodies expressing GAP43 and Syn were found under both pathological conditions. Conversely, axonal swellings immunoreactive for GAP43 and Syn prevailed in subcortical MS lesions, suggesting an enhanced regenerative potential in early MS lesions. Moreover, GAP43+/APP+ axonal spheroid ratios correlated with macrophage infiltration in both TBI and MS, highlighting the possible neurotrophic effect of inflammation and phagocyte infiltration. Additionally, axonal IR for GAP43 and Syn increased with prolonged survival after TBI, indicating a sustained regenerative response.
FUNDING
This work was supported by the DFG transregional collaborative research center 43 “The brain as a target of inflammatory processes.” LS was supported by the DFG (GRK 632 “Neuroplasticity”).
STATEMENT OF CONFLICT OF INTEREST
The authors do not report any conflict of interest.
ACKNOWLEDGMENTS
The authors wish to thank Brigitte Maruschak, Jasmin Held and Katja Schulz for their expert technical assistance.
REFERENCES
- 1. Bar‐Or A, Rieckmann P, Traboulsee A, Yong VW (2011) Targeting progressive neuroaxonal injury: lessons from multiple sclerosis. CNS Drugs 25:783–799. [DOI] [PubMed] [Google Scholar]
- 2. Blaylock RL, Maroon J (2011) Immunoexcitotoxicity as a central mechanism in chronic traumatic encephalopathy‐A unifying hypothesis. Surg Neurol Int 2:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Blumbergs PC, Scott G, Manavis J, Wainwright H, Simpson DA, McLean AJ (1994) Staining of amyloid precursor protein to study axonal damage in mild head injury. Lancet 344:1055–1056. [DOI] [PubMed] [Google Scholar]
- 4. Buki A, Povlishock JT (2006) All roads lead to disconnection?—Traumatic axonal injury revisited. Acta Neurochir (Wien) 148:181–193. [DOI] [PubMed] [Google Scholar]
- 5. Charcot JM (1868) Histologie de la sclérose en plaque. Gaz Hop Civils Militaires 41:554–555, 557–558, 566. [Google Scholar]
- 6. Cragg BG (1970) What is the signal for chromatolysis? Brain Res 23:1–21. [DOI] [PubMed] [Google Scholar]
- 7. Dahl D, Perides G, Bignami A (1989) Axonal regeneration in old multiple sclerosis plaques. Immunohistochemical study with monoclonal antibodies to phosphorylated and non‐phosphorylated neurofilament proteins. Acta Neuropathol 79:154–159. [DOI] [PubMed] [Google Scholar]
- 8. De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS et al (1998) Axonal damage correlates with disability in patients with relapsing‐remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain 121:1469–1477. [DOI] [PubMed] [Google Scholar]
- 9. Doinikow B (1915) Über De‐Regenerationserscheinungen an Achsenzylindern bei der multiplen Sklerose. Z Ges Neurol Psychiat 27:151–178. [Google Scholar]
- 10. Farkas O, Povlishock JT (2007) Cellular and subcellular change evoked by diffuse traumatic brain injury: a complex web of change extending far beyond focal damage. Prog Brain Res 161:43–59. [DOI] [PubMed] [Google Scholar]
- 11. Ferguson B, Matyszak MK, Esiri MM, Perry VH (1997) Axonal damage in acute multiple sclerosis lesions. Brain 120:393–399. [DOI] [PubMed] [Google Scholar]
- 12. Frey D, Laux T, Xu L, Schneider C, Caroni P (2000) Shared and unique roles of CAP23 and GAP43 in actin regulation, neurite outgrowth, and anatomical plasticity. J Cell Biol 149:1443–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Greer JE, McGinn MJ, Povlishock JT (2011) Diffuse traumatic axonal injury in the mouse induces atrophy, c‐Jun activation, and axonal outgrowth in the axotomized neuronal population. J Neurosci 31:5089–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G et al (2011) Oxidative damage in multiple sclerosis lesions. Brain 134:1914–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Herdegen T, Skene P, Bahr M (1997) The c‐Jun transcription factor—bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci 20:227–231. [DOI] [PubMed] [Google Scholar]
- 16. Kerschensteiner M, Stadelmann C, Dechant G, Wekerle H, Hohlfeld R (2003) Neurotrophic cross‐talk between the nervous and immune systems: implications for neurological diseases. Ann Neurol 53:292–304. [DOI] [PubMed] [Google Scholar]
- 17. Kerschensteiner M, Bareyre FM, Buddeberg BS, Merkler D, Stadelmann C, Bruck W et al (2004) Remodeling of axonal connections contributes to recovery in an animal model of multiple sclerosis. J Exp Med 200:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koo EH, Sisodia SS, Archer DR, Martin LJ, Weidemann A, Beyreuther K et al (1990) Precursor of amyloid protein in Alzheimer disease undergoes fast anterograde axonal transport. Proc Natl Acad Sci USA 87:1561–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kuhlmann T, Lingfeld G, Bitsch A, Schuchardt J, Bruck W (2002) Acute axonal damage in multiple sclerosis is most extensive in early disease stages and decreases over time. Brain 125:2202–2212. [DOI] [PubMed] [Google Scholar]
- 20. Li Y, Jiang N, Powers C, Chopp M (1998) Neuronal damage and plasticity identified by microtubule‐associated protein 2, growth‐associated protein 43, and cyclin D1 immunoreactivity after focal cerebral ischemia in rats. Stroke 29:1972–1980. [DOI] [PubMed] [Google Scholar]
- 21. Lucchinetti C, Bruck W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H (2000) Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann Neurol 47:707–717. [DOI] [PubMed] [Google Scholar]
- 22. Lucchinetti CF, Popescu BF, Bunyan RF, Moll NM, Roemer SF, Lassmann H et al (2011) Inflammatory cortical demyelination in early multiple sclerosis. N Engl J Med 365:2188–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mahad D, Ziabreva I, Lassmann H, Turnbull D (2008) Mitochondrial defects in acute multiple sclerosis lesions. Brain 131:1722–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mahad DJ, Ziabreva I, Campbell G, Lax N, White K, Hanson PS et al (2009) Mitochondrial changes within axons in multiple sclerosis. Brain 132:1161–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Medana IM, Esiri MM (2003) Axonal damage: a key predictor of outcome in human CNS diseases. Brain 126:515–530. [DOI] [PubMed] [Google Scholar]
- 26. Meeuwsen S, Persoon‐Deen C, Bsibsi M, Ravid R, van Noort JM (2003) Cytokine, chemokine and growth factor gene profiling of cultured human astrocytes after exposure to proinflammatory stimuli. Glia 43:243–253. [DOI] [PubMed] [Google Scholar]
- 27. Merkler D, Metz GA, Raineteau O, Dietz V, Schwab ME, Fouad K (2001) Locomotor recovery in spinal cord‐injured rats treated with an antibody neutralizing the myelin‐associated neurite growth inhibitor Nogo‐A. J Neurosci 21:3665–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Nikić I, Merkler D, Sorbara C, Brinkoetter M, Kreutzfeldt M, Bareyre FM et al (2011) A reversible form of axon damage in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med 17:495–499. [DOI] [PubMed] [Google Scholar]
- 29. Okada Y, Yamazaki H, Sekine‐Aizawa Y, Hirokawa N (1995) The neuron‐specific kinesin superfamily protein KIF1A is a unique monomeric motor for anterograde axonal transport of synaptic vesicle precursors. Cell 81:769–780. [DOI] [PubMed] [Google Scholar]
- 30. Oppenheimer DR (1968) Microscopic lesions in the brain following head injury. J Neurol Neurosurg Psychiatry 31:299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Peterson JW, Bo L, Mork S, Chang A, Trapp BD (2001) Transected neurites, apoptotic neurons, and reduced inflammation in cortical multiple sclerosis lesions. Ann Neurol 50:389–400. [DOI] [PubMed] [Google Scholar]
- 32. Radzun HJ, Hansmann ML, Heidebrecht HJ, Bodewadt‐Radzun S, Wacker HH, Kreipe H et al (1991) Detection of a monocyte/macrophage differentiation antigen in routinely processed paraffin‐embedded tissues by monoclonal antibody Ki‐M1P. Lab Invest 65:306–315. [PubMed] [Google Scholar]
- 33. Raineteau O, Schwab ME (2001) Plasticity of motor systems after incomplete spinal cord injury. Nat Rev Neurosci 2:263–273. [DOI] [PubMed] [Google Scholar]
- 34. Rocca MA, Ceccarelli A, Rodegher M, Misci P, Riccitelli G, Falini A et al (2010) Preserved brain adaptive properties in patients with benign multiple sclerosis. Neurology 74:142–149. [DOI] [PubMed] [Google Scholar]
- 35. Schirmer L, Albert M, Buss A, Schulz‐Schaeffer WJ, Antel JP, Bruck W, Stadelmann C (2009) Substantial early, but nonprogressive neuronal loss in multiple sclerosis (MS) spinal cord. Ann Neurol 66:698–704. [DOI] [PubMed] [Google Scholar]
- 36. Schirmer L, Antel JP, Bruck W, Stadelmann C (2011) Axonal loss and neurofilament phosphorylation changes accompany lesion development and clinical progression in multiple sclerosis. Brain Pathol 21:428–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sharp DJ, Beckmann CF, Greenwood R, Kinnunen KM, Bonnelle V, De BX et al (2011) Default mode network functional and structural connectivity after traumatic brain injury. Brain 134:2233–2247. [DOI] [PubMed] [Google Scholar]
- 38. Sherriff FE, Bridges LR, Gentleman SM, Sivaloganathan S, Wilson S (1994) Markers of axonal injury in post mortem human brain. Acta Neuropathol 88:433–439. [DOI] [PubMed] [Google Scholar]
- 39. Singleton RH, Povlishock JT (2004) Identification and characterization of heterogeneous neuronal injury and death in regions of diffuse brain injury: evidence for multiple independent injury phenotypes. J Neurosci 24:3543–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stadelmann C (2011) Multiple sclerosis as a neurodegenerative disease: pathology, mechanisms and therapeutic implications. Curr Opin Neurol 24:224–229. [DOI] [PubMed] [Google Scholar]
- 41. Stadelmann C, Kerschensteiner M, Misgeld T, Bruck W, Hohlfeld R, Lassmann H (2002) BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 125:75–85. [DOI] [PubMed] [Google Scholar]
- 42. Stagi M, Dittrich PS, Frank N, Iliev AI, Schwille P, Neumann H (2005) Breakdown of axonal synaptic vesicle precursor transport by microglial nitric oxide. J Neurosci 25:352–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sternberger LA, Sternberger NH (1983) Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ . Proc Natl Acad Sci USA 80:6126–6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tetzlaff W, Zwiers H, Lederis K, Cassar L, Bisby MA (1989) Axonal transport and localization of B‐50/GAP‐43‐like immunoreactivity in regenerating sciatic and facial nerves of the rat. J Neurosci 9:1303–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L (1998) Axonal transection in the lesions of multiple sclerosis. N Engl J Med 338:278–285. [DOI] [PubMed] [Google Scholar]
- 46. Vogt J, Paul F, Aktas O, Muller‐Wielsch K, Dorr J, Dorr S et al (2009) Lower motor neuron loss in multiple sclerosis and experimental autoimmune encephalomyelitis. Ann Neurol 66:310–322. [DOI] [PubMed] [Google Scholar]
- 47. Wegner C, Esiri MM, Chance SA, Palace J, Matthews PM (2006) Neocortical neuronal, synaptic, and glial loss in multiple sclerosis. Neurology 67:960–967. [DOI] [PubMed] [Google Scholar]
- 48. Wilkinson AE, Bridges LR, Sivaloganathan S (1999) Correlation of survival time with size of axonal swellings in diffuse axonal injury. Acta Neuropathol 98:197–202. [DOI] [PubMed] [Google Scholar]