

Abstract
In Creutzfeldt–Jakob disease (CJD), molecular typing based on the size of the protease resistant core of the disease‐associated prion protein (PrPSc) and the M/V polymorphism at codon 129 of the PRNP gene correlates with the clinico‐pathologic subtypes. Approximately 95% of the sporadic 129MM CJD patients are characterized by cerebral deposition of type 1 PrPSc and correspond to the classic clinical CJD phenotype. The rare 129MM CJD patients with type 2 PrPSc are further subdivided in a cortical and a thalamic form also indicated as sporadic fatal insomnia.
We observed two young patients with MM2‐thalamic CJD. Main neuropathological features were diffuse, synaptic PrP immunoreactivity in the cerebral cortex and severe neuronal loss and gliosis in the thalamus and olivary nucleus. Western blot analysis showed the presence of type 2A PrPSc. Challenge of transgenic mice expressing 129MM human PrP showed that MM2‐thalamic sporadic CJD (sCJD) was able to transmit the disease, at variance with MM2‐cortical sCJD. The affected mice showed deposition of type 2A PrPSc, a scenario that is unprecedented in this mouse line. These data indicate that MM2‐thalamic sCJD is caused by a prion strain distinct from the other sCJD subtypes including the MM2‐cortical form.
Keywords: 12B2 antibody, Creutzfeldt–Jakob disease, MM2‐thalamic, PrPSc typing, sporadic fatal insomnia, transmission studies
INTRODUCTION
Prion diseases are neurodegenerative disorders of humans and animals that are sporadic or inherited in origin and can be transmitted. In humans, the most common form is Creutzfeldt–Jakob disease (CJD). Despite remarkable differences in phenotypic characteristics, these disorders share a similar pathogenic mechanism, that is a posttranslational modification of the prion protein (PrP) from a normal cellular isoform (PrPC) to insoluble and protease‐resistant disease‐specific species (PrPSc) (23).
After limited proteolytic digestion and deglycosylation, PrPSc exists as ∼21 kDa (type 1) and ∼19 kDa (type 2) C‐terminal fragments (20). The PrPSc type and the status of methionine (M) / valine (V) polymorphism at codon 129 of the gene coding PrP (PRNP) define different molecular types of sporadic CJD (sCJD). There is wide agreement that a correlation links these molecular subtypes with specific clinical and neuropathological features of sCJD (18).
Focusing on sCJD patients homozygous for M at PRNP codon 129 (129MM), the presence of type 1 PrPSc defines the most typical and common CJD phenotype, clinically characterized by rapidly progressive dementia, cerebellar, pyramidal, extrapyramidal signs and cortical visual signs, myoclonus, and pseudoperiodic sharp wave complexes (PSWC) at electroencephalogram (EEG). Neuronal loss, astrogliosis and spongiform changes involve the cerebral cortex, striatum, thalamus and cerebellum; PrPSc accumulates mostly as diffuse, finely granular, synaptic‐like deposits 5, 18. Conversely, the MM2 type is very rare and is further subclassified into MM2‐cortical and MM2‐thalamic.
MM2‐cortical CJD has a clinical profile similar to that of MM1 but a slower progression, and is neuropathologically characterized by the presence of large clusters of coarse, confluent vacuoles in the cerebral cortex associated with a peculiar pattern of perivacuolar deposition of PrPSc. It is noteworthy that the distinction between MM1 and MM2‐cortical is not sharp, as in a consistent percentage of cases the two types of PrPSc co‐occur in the same cortical region. Following the original reports 18, 24, this phenomenon was confirmed and expanded by studies using PrP immunoreagents specific for type 1 PrPSc and extensive brain sampling 8, 19, 22.
MM2‐thalamic CJD is characterized by neurological and cognitive deterioration associated with early and prominent insomnia. Spongiform changes are absent or very subtle, and the most severe levels of neuronal loss and gliosis are in the thalamus and inferior olive (18). Immunohistochemistry for PrP ranges from very scanty and focal to undetectable. This disease form is also indicated as sporadic fatal insomnia (SFI) 13, 17 as most of the few reported patients share several features with fatal familial insomnia (11).
Transmission studies of the six sCJD subgroups (MM1, MM2, MV1, MV2, VV1, VV2) to a panel of transgenic (Tg) mice expressing different forms of human PRNP (HuMM, HuVV, HuMV) have been carried out (1). The MM2‐CJD specimen tested, which was from a patient with the MM2‐cortical form, did not elicit clinical disease or spongiform changes in any of the three lines of mice, which distinguish this sCJD subgroup from all of the others.
In the present study we describe two young patients with CJD (age at death 25 and 34 years) who neither have the characteristics of variant CJD (28) nor a history of risk factors for iatrogenic transmission (2). Both were MM at codon 129, without mutation of the PRNP gene. The clinical course was rather long (26 and 28 months), dominated by behavioral changes in the early stages and remarkable for the absence of hallmarks of CJD at cerebrospinal fluid (CSF) analysis, magnetic resonance imaging (MRI) and EEG. Even if sleep disturbances were not prominent, these patients should be classified as MM2‐thalamic by current criteria.
Main findings were the extremely low level of type 1 PrPSc, confirmed by a highly specific immunoreagent, and the ability to transmit the disease to Tg mice expressing the human 129MM PRNP, strongly suggesting that MM2‐thalamic CJD is related to a prion strain distinct form that of MM2‐cortical CJD.
MATERIALS AND METHODS
Diagnostic protocol and tissue collection
The patients were investigated using a diagnostic protocol that included CSF examination, EEG polygraphic recording and MRI of the brain. The complete sequence of the PRNP open reading frame, including the region of signal peptide, was carried out as described (10).
The brains were taken at autopsy with a post‐mortem delay of 24 h for both patients. Coronal slices of the left cerebral hemisphere were alternately fixed or frozen at −80°C for biochemical analysis.
Transmission studies
Groups of gene‐targeted Tg mice expressing human PrP on a mouse PrP knockout background and carrying the 129MM genotype (1) (HuMM) were inoculated by a combination of intracerebral (20 µL) and intraperitoneal (100 µL) routes with 10% homogenate of cerebral cortex from the brain of a MM2‐thalamic case (patient #2) and from a MM2‐cortical case of sCJD. Clinically affected mice were sacrificed at the terminal stage of disease while all other mice were monitored for the entire predicted life span (up to 650 days) and then culled and subjected to autopsy.
Neuropathological study
The neuropathological study of the human brains was carried out on Alcolin (an alcoholic nonlinking fixative, Diapath) and formalin‐fixed sections of the cerebral hemispheres, cerebellum and brainstem, stained with hematoxylin‐eosin (H&E), cresyl violet for Nissl substance, Heidenhain‐Woelcke for myelin and thioflavine S for amyloid. Immunohistochemistry was performed with several anti‐PrP antibodies including 3F4 (epitope at residues 109–112 of human PrP, DakoCytomation, 1:800, Piero Portaluppi, Milano), 6H4 (residues 144–152, Prionics, 1:500, Prestinari, Milano) and a monoclonal antibody specific for type 1 PrPSc, 12B2 (epitope at residues 89–93 of PrP, 2 µg/mL) (29). Before overnight incubation with the primary antibody, the sections were pretreated with proteinase K (10 µg/mL, 5 minutes) and guanidine isothiocyanate (3 M, 20 minutes) as previously described (7).
Additional sections were immunostained with a polyclonal anti‐glial fibrillary acidic protein (GFAP) antibody (DakoCytomation, 1:1000) and the monoclonal antibody CR3/43 that labels activated human microglia (DakoCytomation 1:200). EnVision (DakoCytomation) was used as detection system and 3‐3′‐diaminobenzidine (DAB) as chromogen.
Immunohistochemistry on mice brains was carried out as described previously. At autopsy, the left lateral two‐thirds of each mouse brain was fixed in Carnoy solution (ethanol : chloroform : acetic acid, 6:3:1) and serial 5‐µm‐thick sections from paraffin‐embedded blocks were stained with H&E and thioflavine S, or probed with 6H4 (Prionics, 1:1000) and a polyclonal GFAP antibody (DakoCytomation, 1:1000). Lesion profiles were established following the method described previously (4) using nine standard gray matter areas of the brains. Immunoreactions were revealed with the Envision System (DakoCytomation) for the polyclonal antibody or the Animal Research Kit Peroxidase (DakoCytomation) for the monoclonal antibody, using DAB as chromogen.
Biochemical analysis
Ten different areas from frozen human brains (frontal, temporal, parietal and occipital cortices, gyrus cinguli, hippocampus, caudate nucleus, putamen, thalamus and cerebellum) and samples from frozen mouse brains were homogenized in nine volumes of cold lysis buffer (100 mM sodium chloride, 10 mM EDTA, 0.5% Nonited P‐40, 0.5% sodium deoxycholate in 10 mM Tris‐hydrochloride, pH 7.4). The clear lysate was digested with proteinase K (50 µg/mL, 1 h) and the reaction was stopped by the addition of phenylmethanesulfonyl fluoride (PMSF, 5 mM). A total of 100 µg brain homogenate was loaded on 12.5% sodium dodecyl sulphate‐polyacrylamide gels. Where tissue samples were found to give scant or no signal, 0.5 mL of homogenate digested with PK was precipitated with sodium phosphotungstic acid (PTA) as previously described (27). Proteins were then transferred to polyvinylidene difluoride membranes and probed with the previously mentioned anti‐PrP antibodies 3F4 (1:25000), 6H4 (1:1000) and 12B2 (0.1 µg/mL). The immunoreaction was visualized by enhanced chemoluminescence system (Amersham, GE‐Healthcare Ltd., St. Giles, UK).
RESULTS
Clinical findings
Patient 1
At the age of 24 years, the patient started to complain unsteadiness, vertigo, weakness and occasional diplopia associated with depression that did not improve with pharmacological treatment. Eight months later, she was admitted at hospital with the clinical suspect of multiple sclerosis. Neurological examination evidenced diplopia, hyperreflexia at the four limbs and mild ataxia. Cognitive status was normal. MRI, CSF examination and EEG were unrevealing. The clinical picture gradually worsened, and at the age of 25 the patient showed marked symmetrical pyramidal signs at inferior limbs, bradykinesia, disequilibrium and diffuse muscular atrophy. Neuropsychological examination revealed deficits of attention and memory of moderate severity. MRI showed slight cerebellar atrophy without signal abnormalities. CSF analysis was unremarkable with absence of 14.3.3 protein and normal tau level (170 pg/mL).
Hematological and CSF examinations as well as genetic analyses ruled out a series of pathologic conditions that were considered in the diagnostic process, including Friedreich ataxia, SCA 1,2,3,6, 7,17, DRPLA, Huntington disease, viral and Treponema infections, Whipple's disease and Lyme disease.
The clinical picture started to worsen more rapidly and after a few other months the patient could not walk or stand without help and cognitive decline became severe. EEG showed slowing of the activity; F‐18‐ fluorodeoxyglucose (FDG) uptake in positron emission tomography (PET) documented bilateral frontal and thalamic hypometabolism; and DAT‐scan showed a significant symmetrical reduction of tracer in the basal ganglia.
A few months before death, the MRI showed olivopontocerebellar atrophy with normal DWI sequence, the EEG diffuse slowing without PSWC, and CSF the absence of 14.3.3 and normal tau (150 pg/mL).
The patient died at age 26 of pneumonia, 28 months after the onset of symptoms.
Patient 2
At the age of 32 years the patient presented with apathy, irritability and loss of appetite. A diagnosis of depressive syndrome was made that did not improve with pharmacological treatment. Some months later, she presented an episode of confusion (she did not recognize close relatives). At this time, memory deficits became evident. At age 33, she developed dysarthria and difficulty in walking for loss of balance, weight loss (12 kg in 1 year) and sphincter disturbances. MRI showed unspecific leukoencephalopathy and EEG slowing of the activity. The clinical picture continuously and gradually worsened to a severe cognitive and motor deterioration, and finally to a state of akinetic mutism. The patient died 26 months after the onset.
Neuropathological findings
Neuropathological, immunohistochemical and biochemical results showed remarkable common findings in the two patients. In particular, we detected type 2A PrPSc by immunoblot analysis, with the immunohistochemical counterpart of diffuse, finely granular, synaptic pattern of staining, without perivacuolar PrP deposition, with sparing of basal ganglia and cerebellum (Figure 1).
Figure 1.
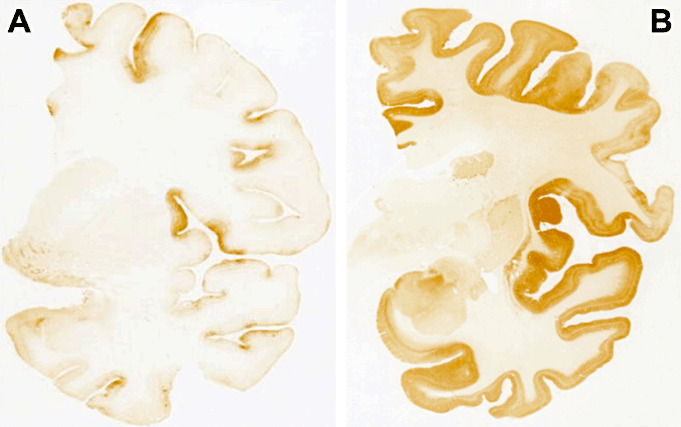
Prion protein (PrP) immunohistochemistry (3F4 antibody) of patient 1 (A) and patient 2 (B). Patient 1 showed faint PrP deposition mainly involving the cerebral cortex with focal areas of increased intensity such as the middle frontal gyrus and the superior part of the insula. Patient 2 displayed a diffuse, synaptic‐like pattern of PrP deposition that was intense in the cerebral cortex and weaker in striatum, closely resembling a typical MM1 case.
Patient 1 showed mild spongiform changes, neuronal loss and gliosis in the cerebral cortex (Figure 2A,D). The bulk of the pathology was centered in the mediodorsal thalamic and olivary nuclei that showed severe neuronal loss (Figure 2B,C), astrogliosis (Figure 2E) and microglia activation (Figure 2F) without spongiform degeneration. Neuronal depletion was also evident in the cerebellum, with loss of Purkinje cells accompanied by Bergmann gliosis and moderate spongiform changes.
Figure 2.
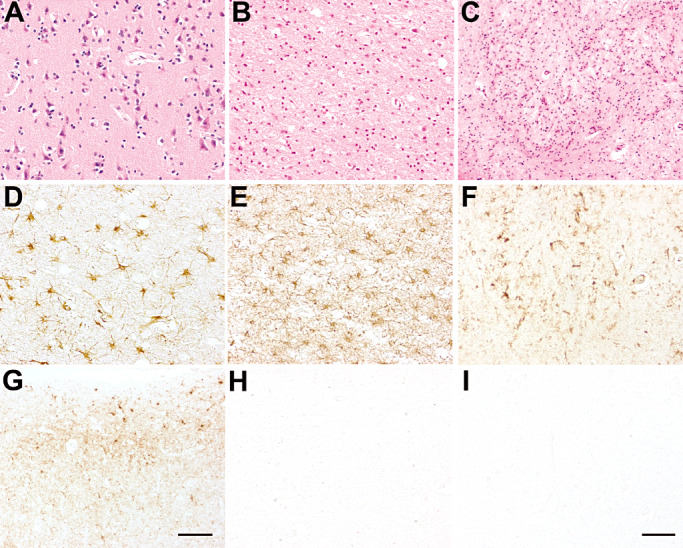
Patient 1: neuropathological findings in the cerebral cortex (A,D,G), dorsal nuclei of the thalamus (B,E,H) and olivary nucleus (C,F,I). Hematoxylin‐eosin (H&E) staining showed mild spongiform changes in the cerebral cortex (A), and severe neuronal loss in the thalamus (B) and olivary nucleus (C). Astrogliosis [glial fibrillary acidic protein (GFAP) immunostaining] was moderate in the cerebral cortex (D) and severe in the thalamus (E) and olivary nucleus (not shown), and was accompanied by marked microglial activation (F, olivary nucleus, CR3/43 immunostaining). Prion protein (PrP) immunoreactivity (3F4 immunostaining) exhibited a synaptic pattern with sub‐pial focal deposits in the cerebral cortex (G) but spared the thalamus (H), cerebellum (not shown) and brainstem (I, olivary nucleus). Scale bar in I = 5 µm. A, B, C, D, E, F, H, I are at the same magnification. Scale bar in G = 15 µm.
PrP immunoreactivity was present but globally scanty and mainly involved the cerebral cortex (Figure 1A), with clusters of deposits in the sub‐pial region (Figure 2G). Focal areas of the cerebral cortex (i.e. the middle frontal gyrus and the superior part of the insula) showed consistent PrP immunostaining (Figure 1A), while PrP immunoreactivity was absent in the striatum, thalamus (Figure 2H), olivary nuclei (Figure 2I) and cerebellum.
Patient 2 showed significant spongiform changes in the cerebral cortex but not in the thalamus (Figure 3A,B). Cerebellum was less involved (Figure 3C). PrP deposition was characterized by a diffuse, synaptic‐like pattern. It was intense in the cerebral cortex, much weaker in the striatum (Figure 1B), and absent in the thalamus (1, 3), cerebellum (Figure 3I) and brainstem. As for patient 1, small focal PrP deposits were observed in the sub‐pial region of the cerebral cortex (Figure 3G), but no perivacuolar pattern of PrP‐immunoreactivity was present. Severe astrogliosis and microglial activation were detected in the cerebral cortex (Figure 3D), striatum, thalamus (Figure 3E) and cerebellum (Figure 3F).
Figure 3.
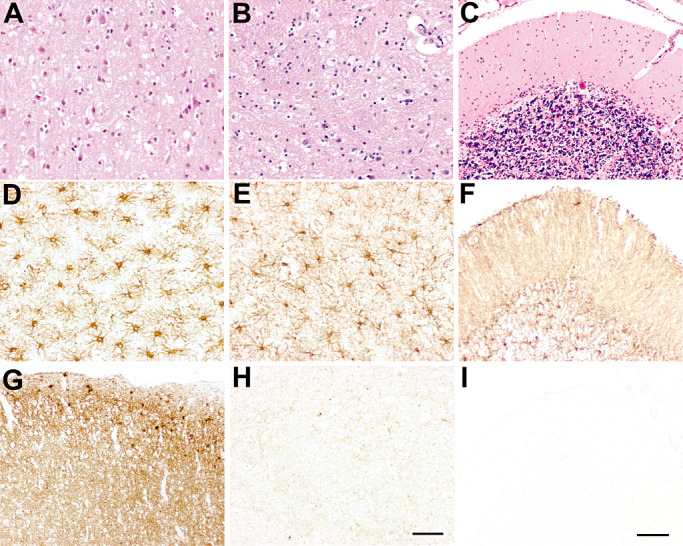
Patient 2: neuropathological findings in the cerebral cortex (A,D,G), dorsal nuclei of the thalamus (B,E,H) and cerebellum (C,F,I). Hematoxylin‐eosin (H&E) staining showed spongiform changes in the cerebral cortex (A). Neuronal loss was moderate in the cerebral cortex (A), prominent in the thalamus (B) and involved mostly the Purkinje cells in the cerebellum (C). Reactive gliosis [glial fibrillary acidic protein (GFAP) immunostaining] was severe in the cerebral cortex (D), thalamus (E) and cerebellum (F). Synaptic pattern of prion protein (PrP) deposition (3F4 immunostaining) was intense in the cerebral cortex (G), while absent in the thalamus (H) and the cerebellum (I). Scale bar in A, B, D, E and H = 5 µm; scale bar in C, F, G and I = 10 µm.
Biochemical and PRNP genetic analysis
Both patients were homozygous for methionine at codon 129, without PRNP mutation.
Western blot analysis with 3F4 antibody showed the presence of PrPSc in all brain areas examined except the cerebellum (Figure 4A) in both patients. The glycoform pattern of PrPSc was characterized by overrepresentation of the monoglycosilated band (Figure 4A,B) and the nonglycosylated band had an electrophoretic mobility of 19 kD, corresponding to type 2A PrPSc by Parchi et al (16).
Figure 4.
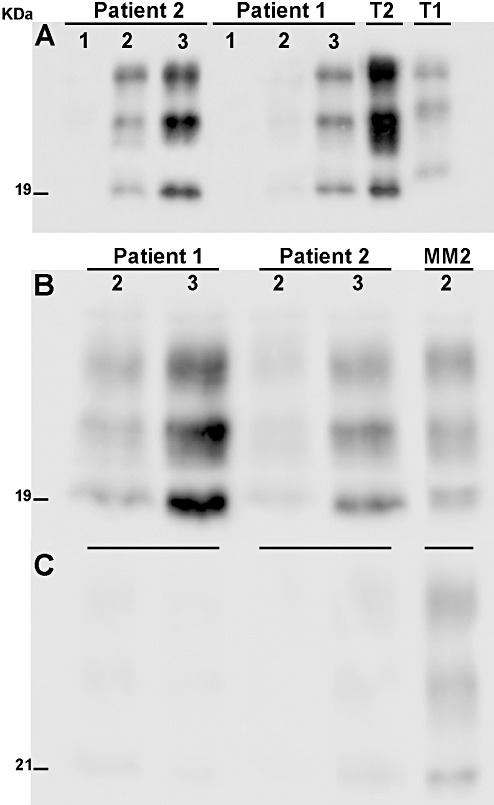
Biochemical findings. A. Western blot analysis with the 3F4 antibody showed type 2A protease resistant core of the disease‐associated prion protein (PrPSc) in both patients. Samples of the following areas are shown: 1 = cerebellum, 2 = thalamus, 3 = gyrus cinguli. No PrPSc immunoreactivity was detected in the cerebellum. The glycoform pattern of PrPSc of both our patients was indistinguishable from that of an MM2 sporadic Creutzfeldt–Jakob disease (CJD) case, with overrepresentation of the monoglycosylated band. T1 and T2 correspond to type 1 and type 2 PrPSc from control CJD cases. B,C. Comparison of Western blots probed with 3F4 antibody (B) and with the 12B2 antibody (C) which is specific for type 1 PrPSc and recognizes an epitope at residues 89–93 of prion protein (PrP). In both MM2‐thalamic patients, a faint signal was detected after incubation with 12B2, while control samples from MM2‐cortical CJD patients (lane MM2) showed strong immunoreactivity with overrepresentation of the monoglycosylated band. Samples of the following areas are shown: 2 = thalamus, 3 = gyrus cinguli; MM2 = MM2‐cortical case.
All samples examined of the two patients were negative under standard conditions (100 µg total protein loaded in the gel) when probed with 12B2 antibody that specifically recognizes type 1 PrPSc in proteinase K‐treated samples. When higher quantities of protein were loaded in the gel, a faint signal was detectable with 12B2 (Figure 4C).
Transmission studies
To evaluate the transmission properties of the MM2‐thalamic CJD, HuMM Tg mice were infected with brain homogenates from patient 2 and the results were compared with those obtained with a MM2‐cortical case.
The MM2‐thalamic case transmitted the disease to 13 out of 14 inoculated mice, with an incubation time of 535 ± 32 days (mean ± SEM) and a survival period of 557 ± 23 (mean ± SEM) days. On the contrary, no clinical signs were observed in mice (n = 16) injected with the MM2‐cortical case, and that were culled and subjected to autopsy after spontaneous death (up to 650 days postinoculation). All the clinically affected mice injected with the MM2‐thalamic case showed mild spongiform changes and PrP immunoreactivity in the brain, with a punctuate and coarse deposition pattern in the stratum lacunosum of the hippocampus and in the cerebral cortex (Figure 5A,C). Western blot analysis revealed the presence of type 2 PrPSc (Figure 5D). Conversely, the mice challenged with the MM2‐cortical inoculum showed no evidence of vacuolation or PrPSc deposition in the brain, either by immunohistochemistry or Western blot analysis without or with PTA precipitation (Figure 5B).
Figure 5.
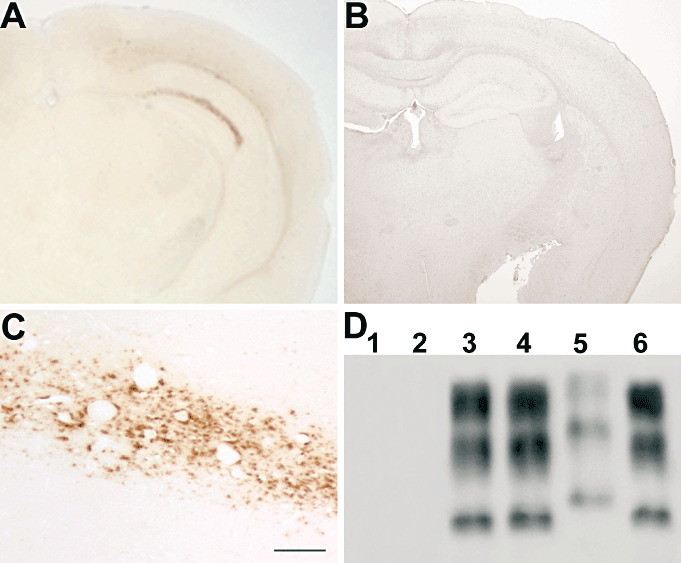
Transmission studies. Transgenic (Tg) HuMM mice challenged with brain homogenate of the MM2‐thalamic patient showed a punctuate and coarse pattern of protease resistant core of the disease‐associated prion protein (PrPSc) deposition, mainly confined to the stratum lacunosum of the hippocampus and the cerebral cortex (A,C). The same Tg HuMM mouse line did not show prion protein (PrP) immunoreactivity in the brain after inoculation with brain homogenate of the MM2‐cortical patient (B). Western blot analysis following PTA precipitation did not reveal the presence of PrPSc in brain samples from HuMM mice challenged with the MM2‐cortical case (D, lanes 1 and 2) but showed type 2 PrPSc in mice inoculated with the MM2‐thalamic case (D, lanes 3 and 4). Lane 5 and lane 6 correspond to sporadic CJD (sCJD) type 1 and type 2 PrPSc, respectively, used as controls. Scale bar in C = 100 µm.
DISCUSSION
The molecular typing of sCJD is based on PRNP polymorphism at codon 129 and the size and glycoform ratio of PrPSc core fragments generated by PK digestion. Among the different molecular subtypes of sCJD identified on this ground, patients homozygous for methionine at codon 129 of PRNP and type 2 PrPSc (MM2) are unique as they encompass two distinct categories: MM2‐cortical and MM2‐thalamic.
Patients with the MM2‐cortical subtype present a clinical picture dominated by dementia and are characterized by widespread degeneration of the cerebral cortex, with large confluent vacuoles framed by rims of tissue with intense PrP immunostaining (18). The tight correspondence between type 2 and this perivacuolar pattern of PrP‐immunoreactivity has been underlined and confirmed by extensive analysis, and it has been recently proposed that the detection of this histopathological profile allows to classify a CJD case as type 2 or mixed type even without the final proof of type 2 detection by Western blot (19). On the other hand, patients identified as MM2‐thalamic are characterized by major involvement of subcortical structures such as the medial thalamic nuclei and the inferior olives where neuronal loss and gliosis are severe and are not accompanied by spongiosis. They are peculiar also because PrPSc is detected in these and others brain structures by immunoblot, but not or only very faintly by immunohistochemistry 17, 25.
The co‐occurrence of PrPSc types 1 and 2 in the same brain 17, 24 is another intriguing aspect of CJD molecular pathology. The issue has been addressed using antibodies to an epitope between residues 82 and 97 of PrP which recognize the PK‐resistant fragment of type 1 PrPSc, but not type 2. Different groups of investigators found that all type 2 CJD samples also contain PrP fragments recognized by type 1‐specific immunoreagents. Some of them interpreted this results as a proof that type 1 is actually present in all CJD cases, also those classified as type 2 by conventional methods 9, 22. Other researchers advanced that material positive for “type 1 selective antibodies” may represent incompletely digested type 2 PrPSc (14).
Here we present two patients diagnosed as MM2‐thalamic on the basis of the neuropathological, immunohistochemical, biochemical and genetic findings, even if they show significant differences from the few patients previously reported under this phenotypic variant, especially for the fact that insomnia was not a prominent feature.
These two patients exhibited quite different PrP immunohistochemical pictures contributing to widen the spectrum of MM2‐thalamic CJD. The cerebral cortex showed only weak PrP immunoreactivity in patient 1, while it exhibited robust PrP labeling in patient 2, characterized by diffuse, finely granular, synaptic‐like PrP deposition indistinguishable from sCJD MM1 type without large vacuoles with PrP labeled rims (i.e. perivacuolar pattern). The protocol of fixation that we adopt, based on a nonlinking fixative (7), may have contributed to allow a more consistent PrP immunoreativity compared with previously reported cases of MM2 thalamic CJD/SFI fixed in formalin 12, 21, 25.
A main finding was that in both patients, the biochemical analyses demonstrated the presence of PrPSc type 2A with extremely low levels of immunoreaction with the 12B2 antibody. This makes these cases different from 30 sCJD patients of our series who had been classified as MM2‐cortical (n = 5), VV2 (n = 13) and MV2 (n = 12), and were all positive for 12B2 under standard conditions (26). On the other hand, other studies showed that there are sCJD cases in which only one PrPSc is detectable 3, 19.
Bishop et al (1) analyzed the transmission properties of the six subgroups of sCJD to a panel of gene‐targeted Tg mice expressing human PRNP with the three different codon 129 polymorphisms and identified four discrete transmission strains: (i) MM1 and MV1 (M1CJD); (ii) MV2 and VV2 (V2CJD); (iii) MM2 (M2CJD); and (iv) VV1 (V1CJD). It is noteworthy that the identification of the M2CJD strain was based on inoculation of an MM2‐cortical patient and derived partly from its inefficiency of transmission to HuMM mice. Brain homogenates from the patient with MM2‐thalamic CJD (patient 2 of this report) demonstrated the ability to transmit the disease to the gene‐targeted Tg mouse line HuMM with an attack rate of 90%, clearly distinguishing this subgroup from the MM2‐cortical CJD. Furthermore, infected mice showed deposition of type 2A PrPSc, a scenario that is unprecedented in transmission of different sCJD subtypes to HuMM mice (1).
Analyzing our results in the light of literature data, it can therefore be proposed that MM2‐thalamic is a distinct strain from the MM2‐cortical CJD subtype. The finding that the same 129 polymorphism and the same PrP type may be associated to two different prion strains should be taken into consideration when debating the question as to whether PrPSc subtypes alone encipher prion strain properties (6). It is noteworthy, in this regard, that diversity of the PrPSc species in MM2‐cortical and MM2‐thalamic sCJD has been reported previously (15) by two dimension gel electrophoresis that evidenced significant differences in glycosylation pattern.
In conclusion, our findings indicate that patients with MM2‐thalamic CJD show a unique combination of neuropathological, immunohistochemical, biochemical features and transmission properties, pointing to a distinct strain from other CJD molecular subtypes and in particular from the MM2‐cortical form.
ACKNOWLEDGMENTS
The project was funded by the Italian Ministry of Health, the European Community (Neuroprion FOOD‐CT‐2004‐506579) and by the Department of Health (England). The views expressed in the publication are those of the authors and not necessarily those of the Department of Health.
The authors thank Mrs. Francesca Cacciatore and Mrs. Sonia Spinello for their skillful technical assistance.
REFERENCES
- 1. Bishop MT, Will RG, Manson JC (2010) Defining sporadic Creutzfeldt‐Jakob disease strains and their transmission properties. Proc Natl Acad Sci U S A 107:12005–12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Brown P, Brandel JP, Preece M, Sato T (2006) Iatrogenic Creutzfeldt‐Jakob disease. The waning of an era. Neurology 67:389–393. [DOI] [PubMed] [Google Scholar]
- 3. Cali I, Castellani R, Alshekhlee A, Cohen Y, Blevins J, Yuan J et al (2009) Co‐existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt‐Jakob disease: its effect on the phenotype and prion‐type characteristics. Brain 132:2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fraser H, Dickinson AG (1968) The sequential development of the brain lesion of scrapie in three strains of mice. J Comp Pathol 78:301–311. [DOI] [PubMed] [Google Scholar]
- 5. Gambetti P, Kong Q, Zou W, Parchi P, Chen SG (2003) Sporadic and familial CJD: classification and characterisation. Br Med Bull 66:213–239. [DOI] [PubMed] [Google Scholar]
- 6. Gambetti P, Cali I, Notari S, Kong Q, Zou W, Surewicz WK (2011) Molecular biology and pathology of prion strains in sporadic human prion diseases. Acta Neuropathol (Berl) 121:79–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Giaccone G, Canciani B, Puoti G, Rossi G, Goffredo D, Iussich S et al (2000) Creutzfeldt‐Jakob disease: Carnoy's fixative improves the immunohistochemistry of the proteinase K‐resistant prion protein. Brain Pathol 10:31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Head MW, Bunn TJ, Bishop MT, McLoughlin V, Lowrie S, McKimmie CS et al (2004) Prion protein heterogeneity in sporadic but not variant Creutzfeldt‐Jakob disease: U.K. Cases 1991‐2002. Ann Neurol 55:851–859. [DOI] [PubMed] [Google Scholar]
- 9. Head MW, Ritchie D, Smith N, McLoughlin V, Nailon W, Samad S et al (2004) Peripheral tissue involvement in sporadic, iatrogenic, and variant Creutzfeldt‐Jakob disease: an immunohistochemical, quantitative, and biochemical study. Am J Pathol 164:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hsiao K, Baker HF, Crow TJ, Poulter M, Owen F, Terwilliger JD et al (1989) Linkage of a prion protein missense variant to Gerstmann‐Sträussler syndrome. Nature 338:342–345. [DOI] [PubMed] [Google Scholar]
- 11. Lugaresi E, Medori R, Montagna P, Baruzzi A, Cortelli P, Lugaresi A et al (1986) Fatal familial insomnia and dysautonomia with selective degeneration of thalamic nuclei. N Engl J Med 315:997–1003. [DOI] [PubMed] [Google Scholar]
- 12. Mastrianni JA, Nixon R, Layzer R, Telling GC, Han D, DeArmond SJ, Prusiner SB (1999) Prion protein conformation in a patient with sporadic fatal insomnia. N Engl J Med 340:1630–1638. [DOI] [PubMed] [Google Scholar]
- 13. Montagna P, Gambetti P, Cortelli P, Lugaresi E (2003) Familial and sporadic fatal insomnia. Lancet Neurol 2:167–176. [DOI] [PubMed] [Google Scholar]
- 14. Notari S, Capellari S, Langeveld J, Giese A, Strammiello R, Gambetti P et al (2007) A refined method for molecular typing reveals that co‐occurrence of PrPSc types in Creutzfeldt‐Jakob disease is not the rule. Lab Invest 87:1103–1112. [DOI] [PubMed] [Google Scholar]
- 15. Pan T, Colucci M, Wong BS, Li R, Liu T, Petersen RB et al (2001) Novel differences between two human prion strains revealed by two‐dimensional gel electrophoresis. J Biol Chem 276:37284–37288. [DOI] [PubMed] [Google Scholar]
- 16. Parchi P, Castellani R, Capellari S, Ghetti B, Young K, Chen SG et al (1996) Molecular basis of phenotypic variability in sporadic Creutzfeldt‐Jakob disease. Ann Neurol 39:767–778. [DOI] [PubMed] [Google Scholar]
- 17. Parchi P, Capellari S, Chin S, Schwarz HB, Schecter NP, Butts JD et al (1999) A subtype of sporadic prion disease mimicking fatal familial insomnia. Neurology 52:1257–1263. [DOI] [PubMed] [Google Scholar]
- 18. Parchi P, Giese A, Capellari S, Brown P, Schulz‐Schaeffer W, Windl O et al (1999) Classification of sporadic Creutzfeldt‐Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol 46:224–233. [PubMed] [Google Scholar]
- 19. Parchi P, Strammiello R, Notari S, Giese A, Langeveld JP, Ladogana A et al (2009) Incidence and spectrum of sporadic Creutzfeldt–Jakob disease variants with mixed phenotype and co‐occurrence of PrPSc types: an updated classification. Acta Neuropathol (Berl) 118:659–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Parchi P, Zou W, Wang W, Brown P, Capellari S, Ghetti B et al (2000) Genetic influence on the structural variations of the abnormal prion protein. Proc Natl Acad Sci U S A 97:10168–10172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Piao YS, Kakita A, Watanabe H, Kitamoto T, Takahashi H (2005) Sporadic fatal insomnia with spongiform degeneration in the thalamus and widespread PrPSc deposits in the brain. Neuropathology 25:144–149. [DOI] [PubMed] [Google Scholar]
- 22. Polymenidou M, Stoeck K, Glatzel M, Vey M, Bellon A, Aguzzi A (2005) Coexistence of multiple PrPSc types in individuals with Creutzfeldt‐Jakob disease. Lancet Neurol 4:805–814. [DOI] [PubMed] [Google Scholar]
- 23. Prusiner SB (1998) Prions. Proc Natl Acad Sci U S A 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F (1999) Sporadic Creutzfeldt‐Jakob disease: co‐occurrence of different types of PrPSc in the same brain. Neurology 53:2173–2176. [DOI] [PubMed] [Google Scholar]
- 25. Scaravilli F, Cordery RJ, Kretzschmar H, Gambetti P, Brink B, Fritz V et al (2000) Sporadic fatal insomnia: a case study. Ann Neurol 48:665–668. [PubMed] [Google Scholar]
- 26. Suardi S, Mangieri M, Limido L, Di Fede G, Capobianco R, Van Zijderveld F et al (2005) A subpopulation of Type 1 PrPres is present in all CJD patients with type 2 PrPres. NeuroPrion Abstract 267. [Google Scholar]
- 27. Wadsworth JD, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J (2001) Tissue distribution of protease resistant prion protein in variant Creutzfeldt‐Jakob disease using a highly sensitive immunoblotting assay. Lancet 358:171–180. [DOI] [PubMed] [Google Scholar]
- 28. Will RG, Ironside JW, Zeidler M, Cousens SN, Estibeiro K, Alperovitch A et al (1996) A new variant of Creutzfeldt‐Jakob disease in the UK. Lancet 347:921–925. [DOI] [PubMed] [Google Scholar]
- 29. Yull HM, Ritchie DL, Langeveld JPM, van Zijderveld FG, Bruce ME, Ironside JW, Head MW (2006) Detection of type 1 prion protein in variant Creutzfeldt‐Jakob disease. Am J Pathol 168:151–157. [DOI] [PMC free article] [PubMed] [Google Scholar]