

Abstract
Twenty‐five percent of ischemic strokes are lacunar in type, but the cause remains unclear. Pathological descriptions of lacunar lesions are available but have not been systematically assessed. We therefore systematically summarized studies describing lacunar lesions by extracting data on the number of patients and lesions, clinical details, pathological methods, brain regions and/or vessels examined, and both parenchymal and vascular findings.
Among 39 papers describing >4000 lesions (>50% from one study), 15 papers examined patients with a clinical lacunar syndrome. Terminology varied, many studies only reported macroscopic pathology and many lesions were cavitated (ie, old). Aside from symptomatic lesions occurring more often in the internal capsule or caudate nucleus, we found no other differences between symptomatic and asymptomatic patients. Perivascular edema and thickening, inflammation and disintegration of the arteriolar wall were common, whereas vessel occlusion was rare.
The causal mechanisms of lacunar stroke remain poorly defined because of methodological inconsistencies and challenges. Standardised pathological definitions based on well‐characterized post‐mortem derived material supported by detailed clinical and imaging data are needed.
Keywords: cerebral small vessel disease, lacunar infarct, lacune, lipohyalinosis, pathology
INTRODUCTION
About 20% of all strokes (about 25% of all ischemic stroke) are lacunar in type (57). These small lesions (<2 cm in diameter, <1.5 cm in some definitions) affect the white matter and deep gray matter of the cerebral hemispheres and brainstem and are associated with several discrete neurological syndromes 4, 25, 26. Lacunar lesions are associated with an abnormal deep perforating arteriole but the small size of the brain and vascular lesions make pathological examination of lacunar lesions technically difficult. In addition, lacunar stroke is rarely fatal and the autopsy rate is declining.
A previous review of lacunar stroke pathology found only 10 studies (11 including their own), most from before 1980 (3). Various descriptive reviews of lacunar stroke pathology have been published since then 2, 35, 48, but there has not been an in‐depth systematic analysis of the quality, breadth and reliability of the published knowledge. Other reviews focused more generally on ageing rather than lacunar stroke (29) or compared various features of small vessel disease seen on imaging with pathology (28). Hence, we performed a systematic examination to summarize all available published studies of lacunar stroke pathology.
METHODS
Search strategy
We searched for studies describing lacunar stroke pathology using, at least, a macroscopic assessment of brain slices. We developed a comprehensive electronic search strategy using terms such as lacun$, patholo$, stroke and autopsy in Embase and Medline from inception to February 2011, and hand searched reference lists in review papers, textbooks and two relevant journals (Stroke and Acta Neuropathologica). We excluded editorials, conference abstracts and review papers.
Inclusion/exclusion criteria
We included studies published in full, in any language, that described the pathology of human lacunar lesions, whether symptomatic (the patient had a documented lacunar syndrome and the lesion was located in the area of brain consistent with the symptoms) or asymptomatic (including in patients with no prior stroke symptoms and patients where the initial stroke was of uncertain or unknown subtype). We included lacunar lesions of any age (from acute to old) and appearance (cavitated or not), studies of brain‐banked tissue if they specifically described lacunar lesions, as well as lesions we considered to be lacunar from the descriptions given, even if the term “lacunar” was not specifically used. We defined lacunar lesions based on the classifications of Poirier and Lammie 14, 36 where a type 1a lacune is a cavitated lesion with a ring of gliosis and a type 1b lacune is a partially cavitated lesion, but also included noncavitated lacunar ischemic stroke lesions to avoid undue focus on cavitated lesions. Types II (old hemorrhage), III and IV lacunes (dilated and very dilated perivascular spaces, respectively) were not relevant.
We excluded studies of: leukoaraiosis, Binswanger's disease or vascular dementia (unless pathological descriptions of any lacunar lesions within these studies were given); patients with primary intracerebral hemorrhage (as our focus was on ischemic lesions); cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) and other monogenic disorders; and striatocapsular infarcts [which are larger than lacunar infarcts and due to middle cerebral artery (MCA) atherothromboembolic occlusion](68).
Data extraction
One reviewer extracted information on study population, number of patients, symptoms, vascular risk factors, previous stroke, age of patients at time of stroke and death or time lapse from stroke to death, cause of death, use of imaging, pathological methods, brain regions and vessels examined and brain/vascular lesion findings (see Supporting Information Table S2), including lesion location and size and specific evidence of ischemia, inflammation or blood–brain barrier disruption. We noted any other terms used by authors to describe what we considered to be lacunar lesions. A second reviewer checked the extracted information. Differences were resolved by consensus with a third reviewer.
Study quality
We assessed study quality using a checklist based on the STrengthening the Reporting of OBservational studies in Epidemiology (STROBE) statement (63) and the Meta‐analysis Of Observational Studies in Epidemiology (MOOSE) criteria (56) with factors such as: study population (eg, an aging cohort, stroke population, brain bank study, etc.), number of patients, reporting of risk factors (eg, hypertension, smoking, diabetes, etc.), the number and expertise of observers (eg, neuropathologist, neurologist, radiologist, etc.), whether the observers were blinded to clinical data and whether lesion size (in any dimension) and/or lesion location was reported. For a full checklist, see Supporting Information Table S1). Factors such as whether the pathology interpretation was appropriately blinded to clinical data are important as they highlight the potential problem that knowledge of risk factors may influence the examination of the specimen and bias the results.
Data analysis
We entered data into a spreadsheet and performed summary statistical analysis. We planned to perform a formal meta‐analysis of the association of lesion location and size with symptoms, and of the frequency of features indicating ischemia, inflammation or blood–brain barrier leakage.
RESULTS
Search results
Our initial search produced 1796 studies (Figure 1); excluding animal studies, non‐cerebral lacunes, duplicates and review papers left 1240 studies. Of these, 672 were full journal articles. We excluded 362/673 studies which only reported on vascular dementia and/or Alzheimer's pathology, 165 studies of imaging or retinopathy only and 106 studies of leukoaraiosis, none of which included data on lacunar lesions, of CADASIL, two studies that looked relevant but provided no data on lacunar pathology 61, 69 and the early reports of Dechambre (13) and Durand‐Fardel (16) whose patient populations are not specific and do not provide enough detailed pathological information. This left 39 relevant studies (see Supporting Information Table S2).
Figure 1.
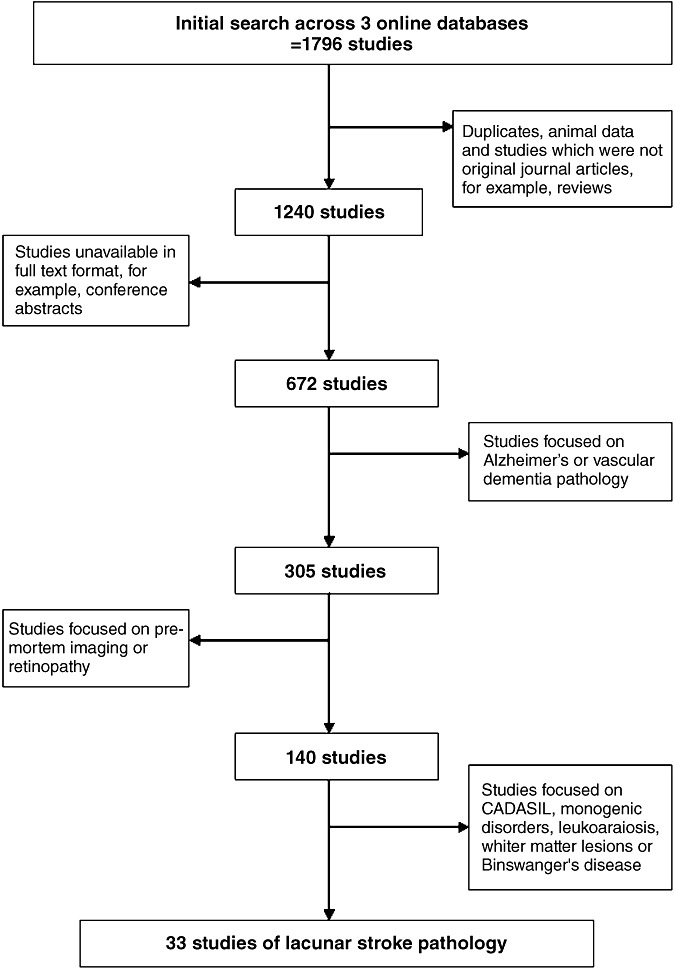
The search strategy used to identify studies of lacunar stroke pathology. CADASIL = cerebral autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy.
Study characteristics
There were 38/39 studies that included a total of 2340 subjects (one study did not report the number of subjects), with a median of 14 subjects per study (interquartile range 2–45, range 1 to >1000) (15). Twenty‐two studies reported the number of lesions studied (total lesions 4110), whereas 17 studies did not. Nine studies reported the median number of lesions studied per patient (two, range 1–5).
Fifteen studies (38%) described symptomatic brain lesions and six studies (15%) reported asymptomatic lacunar lesions on pre‐mortem imaging followed by autopsy in studies of aging. The remaining 18/39 studies (46%) described lacunar lesions in patients with a history of stroke (subtype unspecified and not clearly linked to a lesion on pathology, 10 studies) or who had stroke risk factors but had died of non‐neurological causes (eight studies), all of which we considered to be “asymptomatic” lesions because they were not clearly linked to a discrete symptomatic lesion. The time interval between symptoms and death ranged from 1 month to approximately 10 years. There were 29/39 studies (74%) that reported on hypertension (which affected >50% of subjects in those studies), 14/39 studies reported on diabetes (less frequent than hypertension) and a few studies reported other risk factors, for example, atrial fibrillation or serum cholesterol (Supporting Information Table S2).
The 15 studies of symptomatic lesions included 554 patients but the actual number of individual lesions assessed was unclear (some gave the total and some the mean per patient); six studies did not state any number. From those studies that gave numerical data, we calculated that at least 1200 lesions were assessed in patients with a lacunar syndrome, although it was unclear how many of these were truly symptomatic. In patients with multiple lacunar lesions, it was noted that at least half the patients had lesions which did not fit their symptoms (52).
Twenty‐four studies of subjects with asymptomatic lacunar lesions included six aging cohorts with asymptomatic lacunar lesions on pre‐mortem imaging (71 subjects) and 18 studies of autopsy brains from patients without neurological symptoms in life or who had had a previous undefined stroke. There were 9/24 studies that reported the number of lesions (total approximately 2700), 2567 of which came from one study with 1086 subjects (15). Nine studies did not report the total number of lesions. Sixteen studies used brain imaging [nine computed tomography (CT), four magnetic resonance imaging (MRI), and two used CT and MRI], nine before death, one after death, and four both before and after death, to match lesions in life to those at autopsy—none of these lesions were symptomatic (Supporting Information Table S2).
Pathological assessments
Ten studies provided only a macroscopic assessment of brain slices. Among the 29/39 studies reporting microscopic data, 25 different histological stains were used, the most common being hematoxylin and eosin (22 studies) then luxol fast blue (seven studies). Sixteen different antibodies for immunohistochemistry were used, of which glial fibrillary acidic protein was the most common (nine studies). Sixteen studies (41%) reported findings of typical type I cavitated lacunar lesions without reference to the lesion age, nine studies (23%) reported cavitated lacunar lesions with some reference to lesion age (ie, subacute or old), and the remaining 14 studies (36%) reported on lesions in varying stages of development describing softenings, partially cavitated lesions, gliosis and associated perivascular space dilatations.
Terminology and definitions
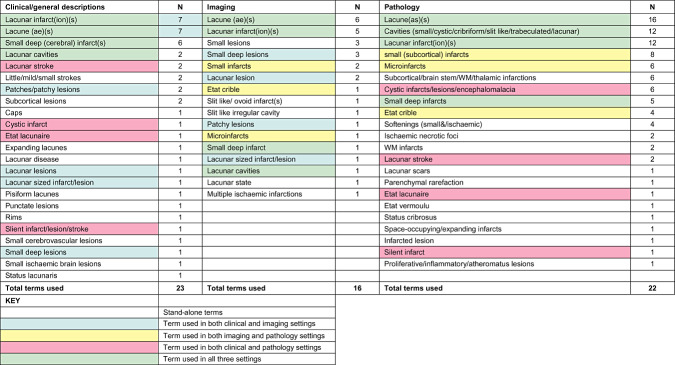
Descriptions of lacunar lesions and terminology varied widely (Table 1). Although 23/39 (59%) studies defined what they meant by a lacunar lesion, descriptions ranged from only the lesion size and location to more detailed pathological descriptions; six studies used terms such as “small deep infarcts” or “small subcortical infarcts,” and 10 studies did not define what they meant by lacunar lesions at all. Terms used for clinical or imaging features included lacunar infarct(ion), lacune, small deep infarcts and lacunar cavities. Pathological descriptions favored the term “lacune,” followed by various descriptions of cavities (small, lacunar, trabeculated, etc.). “Softening” was most often used to describe more acute lesions 20, 32. Terms used to describe vessel lesions included lipohyalinosis, hyalinization, hyaline arteriosclerosis and segmental arteriolar disorganization, all of which described apparently similar lesions.
Table 1.
The varying terminology used to describe lacunar lesions in both clinical and pathology settings. Abbreviations: WM = white matter; N = number of studies using the term
|
Study quality score
The average study quality score was 6/9 (range 4–9). The most common items omitted were blinding between clinical and pathological data, the number and expertise of observers, and lesion size. Where reported, neuropathologists were the most common observers (7/15 studies), but others were reported as simply “pathologists” and 24 studies (62%) did not report the specialty of the person performing the pathology. Only five studies blinded the observer to potentially confounding clinical data (13%).
Pathological findings
Twenty‐nine studies (74%) examined both parenchymal and associated vascular pathology, nine studies (23%) reported only parenchymal pathology, and one study only reported vascular pathology (47).
Parenchymal lesions
Location: Most studies sampled several areas of the brain, if not the entire brain, with little evidence of sampling bias, but data were not provided on the exact number of lesions found in each brain structure. Studies of symptomatic lesions more often reported precise locations (eg, caudate or lentiform nucleus) than those of asymptomatic lesions (eg, “basal ganglia”). Most lesions were in the thalamus (18/39), putamen (12/39), caudate and internal capsule (11/39). Studies of symptomatic subjects reported almost double the number of lesions in the internal capsule (seven vs. four studies) and almost three times as many studies reported lesions in the caudate nucleus (eight vs. three studies), compared with studies in asymptomatic subjects.
Size: Twenty‐five studies (64%) reported lacunar lesion size by: diameter (<5 to 30 mm, 17 studies); volume (0.1 to 525 m3, eight studies); area (0.1 to 28 mm2, six studies); and some used more than one unit of measurement. Many studies used 1 cm as the maximum diameter for a lacunar lesion. There was no difference in size between symptomatic and asymptomatic lesions with a range of 1–30 mm diameter encompassing both types of lesion. Determining the mean size of symptomatic vs. asymptomatic lesions was impossible because of the different types of measurements reported.
Macroscopic appearance: Lesions were generally bilateral, in the territory of the lenticulostriate arterioles (>75%) and a mixture of old and more acute 19, 40. Those in the deep gray matter were often more irregular shaped and collapsed than those in the white matter (20). Occasional hemorrhage and Charcot–Bouchard/saccular aneurysms (or microaneursyms) were also found close to lacunar lesions 6, 12, 22, 53, 59.
Microscopic appearance: Old lesions were described as small pale cavities with irregular borders, often trabeculated with connective tissue and accompanied by a ring of gliosis 7, 8, 33. Macrophages and fibrous astrocytes were observed at the lesion border 11, 39, 43. Acute lesions more often had plump macrophages, liquefaction necrosis and little gliosis (21), and were often described as “softenings”(12). The surrounding parenchyma showed varying degrees of edema and spongiosis as well as reactive astrocytes 36, 39, but little direct specific evidence of infarction. Small capillaries/arterioles often lay within or crossed the lesion 7, 21.
Some lacunar lesions had perivascular rarefaction without full cavitiation 36, 38 and were labeled as “edema associated lesions,” reflecting the impression that edema was a key factor in their formation 36, 38. Dilated perivascular spaces with a round regular border surrounded by rarefied and gliotic nervous tissue were often found close to lacunar lesions 7, 8, 37, 39, for example, in the putamen at the point of entry of the lenticulostriate arteries (37). This appearance was common in normal aging brains (9) and in those with vascular dementia 53, 62 rather than with clinical lacunar syndromes. Neuronal loss around the lesions was rare (32), but increased levels of inflammatory markers were common (58).
Vessel abnormalities
Fourteen studies (44%) attempted to trace the arteriole thought to be responsible for the symptomatic lesion. Nine studies that measured the affected arteriole reported diameters ranging from 14 to 400 µm. No arteriole >400 µm diameter was cited as having directly caused a lacunar lesion. Changes in arteries, arterioles and capillaries were reported; veins and/or venules were rarely mentioned (1). In general, the size of a lacunar lesion was proportional to the size of the affected vessel.
Large vessels—circle of Willis and major branches: We found similar descriptions of large vessel pathology in both symptomatic and asymptomatic patients, including atherosclerosis and dolichoectasia. Ferrand (19) described the basilar artery (BA) of patients with lacunes at autopsy as being “atheromatous,” with alterations of the internal membrane, proliferation of elastic tissue and infiltration of macrophages. Atherosclerosis of the circle of Willis, internal carotid artery, BA, posterior cerebral, MCA and anterior cerebral arteries in symptomatic lacunar stroke patients was reported in several studies as either “moderate” or “severe” in at least 50% of patients (“moderate” and “severe” were not defined) and more commonly in patients with hypertension 20, 59. Mancardi (39) described atherosclerotic changes consisting of collagen or fibrin deposits, cholesterol crystals, duplication and splitting of the elastic lamina and thickening of the adventitia in patients with lacunar syndromes, and other neurological symptoms (but did not provide information on hypertension). Patients with incomplete lacunar lesions (not clearly associated with symptoms) showed mild (60% of patients) to moderate (30% of patients) atheroma in the ipsilateral internal carotid arterys (ICAs) (36), but these patients had various previous stroke episodes, not just lacunar. Asymptomatic patients showed similar changes, with more atherosclerosis typically associated with more lacunes (8), particularly in patients with vascular dementia 53, 62. Widening of the internal and external diameters of the vertebral, ICA and MCAs (dolichoectasia) was also described, for example the median diameter of the BA was almost doubled in patients with asymptomatic lacunar lesions and those with a “history of stroke”(49).
Small penetrating arteries, arterioles and capillaries: We found no quantitative differences in the small vessel lesions in symptomatic vs. asymptomatic patients. Ferrand described small penetrating arterioles in proximity to symptomatic lacunar lesions as having thick walls and dilated perivascular spaces. This was also true in areas of the ipsilateral hemisphere devoid of lacunes (19). Hughes et al (32) described perforating arterioles as being sclerotic and without occlusion, but it was unclear whether these arterioles were directly related to lacunar lesions.
The first detailed description of small arteriolar changes directly related to lacunar lesions came from Fisher, who found abnormal penetrating arterioles or their branches in six patients, some of which were occluded, and which he stated was not due to atheroma (20). In a separate study, he identified “segmental arteriolar disorganisation” in 45/50 lacunar lesions from four patients (21). The “segmental arteriolar disorganization” was distributed along the length of arterioles, 40–200 µm in diameter and included a disintegrating arteriolar wall consisting of a loose mesh of collagenous strands, infiltration of fatty macrophages and/or foam cells, and deposition of fibrinoid material (25). “Lipohyalinosis” was not mentioned until 1978, when he described thalamic infarcts in two hypertensive patients (22) in whom the external diameters of the small arterioles were enlarged to twice the diameter of any adjacent normal artery, the wall architecture was lost and the lumenae were occluded by a fine trabecular meshwork of hyaline material (22).
Intimal thickening and hyalinization, fibrosis of the wall, and occasional microaneurysms were seen in the putamen 7, 42, thalamoperforating and lenticulostriate arterioles (39), and other small intraparenchymal vessels 6, 59. Intraluminal thrombi were rare and the arterioles were assumed to have recanalized (39). Ferrand described leukocytes infiltrating the walls of small arterioles and macrophages and active microglia in perivascular parenchymal lesions (19). One exception to the aforementioned is a single case report describing a patient with multiple cholesterol emboli in the perforating arteries with no evidence of vascular hyalinosis (34).
There were additional comments pertaining to arterioles in asymptomatic patients. Most lacunes occurred on the 1st or 2nd order branches of the penetrating arterioles of hypertensive patients (10). Arteriosclerosis with medial hyalinization 10, 52, 55, dense adventitial fibrosis and foamy macrophages in the vessel wall were common in patients with lacunar lesions dying of non‐stroke neurologic or nonneurologic causes (62). The degree of small arteriolar pathology increased with the number of lacunes visible pathologically in an ageing hypertensive population (15) or with the number of lacunar lesions and leukoaraiosis visible on imaging 30, 54. One study suggested that fibrinoid necrosis was commoner in patients with diastolic hypertension than in those with systolic and borderline hypertension (42).
Intimal hyalinization and thickening were more frequent in patients with lacunar ischemic stroke than in those with hemorrhage (52). In noncavitated (type 1b) lacunar lesions, hyaline arteriolar wall thickening was present but fibrinoid necrosis, luminal thrombi and foam cells were not 36, 62. There was no cerebral amyloid angiopathy on staining with Congo Red 41, 49.
DISCUSSION
Summary
Lacunar stroke is part of the spectrum of cerebral small vessel disease and represents an expanding global public health problem (31). Although methodological inconsistencies were present throughout the studies, we found 39 studies which described approximately 4000 lacunar lesions in 2300 subjects, with consistent descriptions of segmental nonatheromatous thickening of small artery/arteriole walls, along with breakdown of muscular and elastic fibres. However, information on venular pathology was rare. There were signs of inflammation in the arteriolar wall [noted as early as 1902 (19)] and perivascular tissue, but little evidence of infarction or arteriolar occlusion except as an occasional late‐stage phenomenon secondary to extreme vessel wall damage (46). This absence of true infarction could be because most studies were performed late in the disease course by which time the initiating features could have been obliterated. However, there were many studies that reported on the various evolutionary stages of lacunar lesions, including areas of initial softening and edema and/or partially cavitated lesions. The available data suggest that symptomatic lesions may occur in the internal capsule and caudate nucleus more often than asymptomatic ones, but otherwise we found little difference in the size or location of parenchymal or vascular lesions between documented symptomatic and asymptomatic patients. However, this apparent difference may reflect differences in sampling between studies rather than a true difference between symptomatic and asymptomatic lesions. The notion that symptomatic lesions were larger than asymptomatic ones may have originated from studies of striatocapsular infarcts (23). Striatocapsular infarcts occur due to MCA main stem occlusion in patients with good collateral pathways to protect the peripheral MCA territory and are due to large artery atherothromboembolic disease (45). We found that microvascular changes were spread throughout the deep gray and white matter (ie, not just around a symptomatic lesion), moderate large artery atheroma and dolichoectasia were common, and, where stated, many of the patients were hypertensive. However, the absence of comparisons between patients with other stroke subtypes, for example, cortical ischemic stroke, makes it difficult to say whether the large artery atheroma is relevant, or whether it reflects exposure to vascular risk factors such as hypertension.
Strengths and weaknesses of the review and primary studies
This is the first review of this topic to take a systematic approach. Hence, we hope that it provides additional insights. There was little difference, possibly apart from location, between symptomatic and asymptomatic lacunar lesions, and we found little objective evidence to suggest that the lack of difference was due to sampling bias, but there were many differences in examination techniques, patients and other factors between the studies so we cannot exclude the possibility of true differences in other features such as size. Furthermore, it was difficult to be certain we had found all the relevant papers and publication bias is almost certain to affect this topic, but in the absence of much quantitative data, we did not test formally for it. We excluded studies of leukoaraiosis, Binswanger's encephalopathy, Alzheimer's disease and vascular dementia (unless they specifically described lacunar lesions). However, the reliance on the author's description and interpretation of lacunar lesions, even with our inclusive approach, may have inadvertently excluded papers or introduced bias.
There were other limitations. Studies of symptomatic patients often did not specify the relevant lacunar lesion as efforts to do this, and/or to match a lacunar lesion to the responsible vessel lesion/s, were often futile, due to normal imaging appearances (28), difficulties singling out one lesion often among many, difficulties tracing vascular trees, and the delay between clinical diagnosis and autopsy (44). As much patient information as possible is needed here in order to distinguish clinically relevant lesions from asymptomatic findings, for example, asymptomatic lacunes, white matter lesions or dilated perivascular spaces. Reporting of risk factors varied precluding attempts to differentiate causative from co‐associated findings such as large artery atheroma and lacunar stroke.
Changes resulting from fixation (eg, shrinkage and changes in tissue proton relaxation characteristics) must also be considered when trying to correlate post‐mortem pathology or imaging with in vivo MRI findings (28). The exact brain regions examined were often poorly described. Most, after cutting 2 cm coronal sections, only took regions which were macroscopically interesting, potentially missing many smaller lacunar lesions and other primary pathology. Studies differed in their diameter definition of lacunar lesions, some classifying lesions <1 cm diameter as lacunar and other studies including lesions of up to 2 cm diameter.
Interpretation
This in‐depth, systematic review of the literature highlights the limitations of the current knowledge base surrounding lacunar stroke pathology, and concludes that many of these limitations are caused by methodological issues, in particular inconsistency of terminology and a lack of correlation between clinical, radiological and pathological data. However, the data available challenge the notion that most lacunar stroke is caused by occlusion of small perforating arterioles, whether due to intrinsic atherosclerotic occlusion, embolic atherothromboembolism or vessel occlusion due to other cause. Although some lacunar lesions were associated with narrowed or occluded arterioles, many were not and the arteriolar abnormality included luminal dilation as well as narrowing.
Investigators may have assumed that, when not occluded, arterioles must have recanalized as do thromboembolic occlusions of large arteries, rather than that the arterioles might never have been occluded in the first place.
There are many parallels between the findings described in lacunar stroke and findings in other forms of small vessel disease 5, 18, 29, 60, 62, 64. In leukoariaosis and indeed normal aging, atherosclerosis of large intracranial arteries has been reported accompanied by changes in the small perforating arterioles including vessel wall and/or perivascular macrophage infiltration and activated microglia (indicating inflammation) 29, 62. These features are also associated with hypertension. We offer the following as a hypothesis at this stage: the association between the pathological correlates of small vessel disease and large artery atheroma could be a consequence of both being associated with hypertensive damage rather than as a direct consequence of large artery atheroma causing cerebral small vessel disease. There is some support for this hypothesis from the radiological literature: the blood–brain barrier becomes increasingly permeable with advancing age, even more so in vascular dementia and leukoaraiosis (17), and patients with lacunar stroke have increased permeability of the blood–brain barrier compared with those with cortical stroke, and increased numbers of enlarged perivascular spaces are associated with further increases in blood–brain barrier permeability (65). The mechanism remains uncertain, and further investigation in order to clarify the molecular processes and to identify specific contributory biomarkers is required, with a particular need for translation to human post‐mortem tissue.
Future directions and recommendations
Based on the findings of this review, we propose the following recommendations to improve the quality of studies looking at the etiology of lacunar stroke;
Clinical studies need to subtype the stroke carefully so that patients with lacunar stroke can be differentiated from and compared with other stroke subtypes, for example, cortical ischemic stroke. Age‐matched nonneurological controls are generally not the most appropriate controls.
Reporting of risk factors needs to be prospective and thorough.
Studies where patients present with a clinically defined lacunar stroke syndrome should attempt to pinpoint the relevant lacunar lesion on imaging for better correlation with autopsy findings.
The difficulty of preserving and dissecting subcortical small vessels means that better use should be made of high‐field strength MR scanners that are now much more widely available (eg, 7T scanners). Post‐mortem MRI of the fresh or fixed brain, ideally at higher field strengths, particularly if this could be compared with MRI obtained in life at the time of the stroke, would help to localize the symptomatic lacunar lesion at autopsy and quantify associated features of small vessel disease 27, 67.
Studies of lacunar lesions in general lack histology targeting specific pathological processes, for example, using newer methods such as immunohistochemistry or mRNA expression analysis. Given the continually dwindling autopsy rate and difficulties in retaining material for research, perhaps some material from previous studies could even be reexamined with these methods.
Brain tissue banks are an essential resource but are of limited use without detailed clinical and other in vivo information. More detailed information on the patient in life especially stroke symptoms, subtype and vascular risk factors, should be recorded in primary research on lacunar disease or kept with brain tissue in brain banks (28).
In all pathological studies, the exact brain regions examined should be well described and reasons to justify the selection should be given. Lesion size, location and appearance (preferably with assigned lacunar classification) should also be given.
Finally, terminology and definitions used for lacunar disease in pathology have been highlighted as a continuing problem in this field since the 1960s 24, 48, but also affect descriptions of lacunar lesions clinically and on imaging 50, 66. These need to be addressed preferably with an agreed, defined list of terms beingproposed from those representing clinical practice, radiology and pathology.
DISCLOSURE STATEMENT
The authors declare that they have no conflict of interest.
Supporting information
Table S1. The 9‐point study quality checklist applied to all included studies. It was devised using influences from the STROBE statement and the MOOSE criteria.
Table S2. A full list of studies which met our inclusion criteria, arranged in chronological order. Number of patients refers to the number of patients with lacunar lesions (therefore not necessarily the total number in the study). Abbreviations: HT = hypertension; DM = diabetes mellitus; AF = atrial fibrillation; CVD = cerebrovascular disease; AD = Alzheimer‘s disease; IC = internal carotids; H&E = hematoxylin and eosin; EVG = elastic van gieson; PAS = periodic acid‐Schiff; PTAH = phosphotungstic acid hematoxylin; LFB = luxol fast blue; GFAP = glial fibrillary acidic protein; AP = alkaline phosphatase; IgG = immunoglobulin G; CV = cresyl violet; MSB = martius scarlet blue.
Supporting info item
ACKNOWLEDGMENTS AND FUNDING SOURCES
EB is funded by a Medical Research Council PhD studentship. JMW is funded by the Scottish Funding Council through the SINAPSE Collaboration (Scottish Imaging Network, A Platform for Scientific Excellence, http://www.sinapse.ac.uk). CLMS is funded by the Scottish Funding Council.
REFERENCES
- 1. Alex M, Baron EK, Goldenburg S, Blumenthal HT (1962) An autopsy study of cerebrovascular accident in diabetes mellitus. Circulation 25:663–673. [DOI] [PubMed] [Google Scholar]
- 2. Arboix A, Marti‐Vilalta J‐L (2004) New concepts in lacunar stroke etiology: the constellation of small vessel arterial disease. Cerebrovasc Dis 17:58–62. [DOI] [PubMed] [Google Scholar]
- 3. Arboix A, Ferrer I, Marti‐Vilalta JL (1996) Clinico‐anatomopathologic analysis of 25 patients with lacunar infarction. Rev Clin Esp 196:370–374. [PubMed] [Google Scholar]
- 4. Arboix A, Lopez‐Grau M, Casasnovas C, Garcia‐Eroles L, Massons J, Balcells M (2006) Clinical study of 39 patients with atypical lacunar syndrome. J Neurol Neurosurg Psychiatry 77:381–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baloh RW, Vinters HV (1995) White matter lesions and disequilibrium in older people: II. Clinicopathologic correlation. Arch Neurol 52:975–981. [DOI] [PubMed] [Google Scholar]
- 6. Benhaiem‐Sigaux N, Gherardi R, Salama J (1986) Thrombosis of a saccular microaneurysm causing cerebral (pontine) lacunae. Acta Neuropathol (Berl) 69:332–336. [DOI] [PubMed] [Google Scholar]
- 7. Benhaiem‐Sigaux N, Gray F, Gherardi R, Roucayrol AM, Poirier J (1987) Expanding cerebellar lacunes due to dilatation of the perivascular space associated with Binswanger's subcortical arteriosclerotic encephalopathy. Stroke 18:1087–1092. [DOI] [PubMed] [Google Scholar]
- 8. Bokura H, Kobayashi S, Yamaguchi S (1998) Distinguishing silent lacunar infarction from enlarged Virchow‐Robin spaces: a magnetic resonance imaging and pathological study. J Neurol 245:1432–1459. [DOI] [PubMed] [Google Scholar]
- 9. Braffman BH, Zimmerman RA, Trojanowski JQ, Gonatas NK, Hickey WF, Schlaepfer WW (1988) Brain MR: pathologic correlation with gross and histopathology. 1. Lacunar infarction and Virchow‐Robin spaces. AJR 151:551–558. [DOI] [PubMed] [Google Scholar]
- 10. Challa VR, Bell MA, Moody DM (1990) A combined hematoxylin‐eosin, alkaline phosphatase and high‐resolution microradiographic study of lacunes. Clin Neuropathol 9:196–204. [PubMed] [Google Scholar]
- 11. Chimowitz MI, Estes ML, Furlan AJ, Awad IA (1992) Further observations on the pathology of subcortical lesions identified on magnetic resonance imaging. Arch Neurol 49:747–752. [DOI] [PubMed] [Google Scholar]
- 12. Cole FM, Yates PO (1967) The occurrence and significance of intracerebral micro‐aneurysms. J Pathol Bacteriol 93:393–411. [DOI] [PubMed] [Google Scholar]
- 13. Dechambre A (1838) Mémoire sur la curabilité du ramollissement cérérebral. Gaz Méd Paris 6:305–314. [Google Scholar]
- 14. Derouesne C, Poirier J (1999) Cerebral lacunae: still under debate. Rev Neurol (Paris) 155:823–831. [PubMed] [Google Scholar]
- 15. Dozono K, Ishii N, Nishihara Y, Horie A (1991) An autopsy study of the incidence of lacunes in relation to age, hypertension and arteriosclerosis. Stroke 22:993–996. [DOI] [PubMed] [Google Scholar]
- 16. Durand‐Fardel M (1843) Traité du Remollissement du Cerveau . Bailliere: Paris. [Google Scholar]
- 17. Farrall AJ, Wardlaw JM (2009) Blood‐brain barrier: Ageing and microvascular disease—systematic review and meta‐analysis. Neurobiol Aging 30:337–352. [DOI] [PubMed] [Google Scholar]
- 18. Fazekas F, Kleinert R, Offenbacher H, Schmidt R, Kleinert G, Payer F et al (1993) Pathologic correlates of incidental MRI white matter signal hyperintensities. Neurology 43:1683–1689. [DOI] [PubMed] [Google Scholar]
- 19. Ferrand J (1902) Éssai sur l’hémiplegie des vieillards: Les Lacunes de Désintegration Cérébrale . Jules Rousset: Paris. [Google Scholar]
- 20. Fisher CM (1965) Lacunes: small deep cerebral infarcts. Neurology 15:774–784. [DOI] [PubMed] [Google Scholar]
- 21. Fisher CM (1968) The arterial lesions underlying lacunes. Acta Neuropathol (Berl) 12:1–15. [DOI] [PubMed] [Google Scholar]
- 22. Fisher CM (1978) Thalamic pure sensory stroke: a pathologic study. Neurology 28:1141–1144. [DOI] [PubMed] [Google Scholar]
- 23. Fisher CM (1979) Capsular infarcts: the underlying vascular lesions. Arch . Neurology 36:65–73. [DOI] [PubMed] [Google Scholar]
- 24. Fisher CM (1982) Lacunar strokes and infarcts: a review. Neurology 32:871–876. [DOI] [PubMed] [Google Scholar]
- 25. Fisher CM (1982) Pure sensory stroke and allied conditions. Stroke 13:434–447. [DOI] [PubMed] [Google Scholar]
- 26. Ghandehari K, Izadi ZM (2009) Clinical evaluation of 625 lacunar syndrome patients. Turk J Med Sci 39:607–612. [Google Scholar]
- 27. Gorelick PB, Scuteri A, Black SE, DeCarli C, Greenberg SM, Iadecola C et al (2011) Vascular contributions to cognitive impairment and dementia. Stroke 42:2672–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, Geurts J (2011) Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry 82:126–135. [DOI] [PubMed] [Google Scholar]
- 29. Grinberg L, Thal DR (2010) Vascular pathology in the aged human brain. Acta Neuropathol (Berl) 119:277–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gupta SR, Naheedy MH, Young JC, Ghobrial M, Rubino FA, Hindo W (1988) Periventricular white matter changes and dementia. Clinical, neuropsychological, radiological, and pathological correlation. Arch Neurol 45:637–641. [DOI] [PubMed] [Google Scholar]
- 31. Hachinski V (2002) Stroke: the next 30 years. Stroke 33:1–4. [PubMed] [Google Scholar]
- 32. Hughes W, Dodgson M, Maclennan D (1954) Chronic cerebral hypertensive disease. Lancet 264:770–774. [DOI] [PubMed] [Google Scholar]
- 33. Kawamoto Y, Akiguchi I, Tomimoto H, Shirakashi Y, Honjo Y, Budka H (2006) Upregulated expression of 14‐3‐3 proteins in astrocytes from human cerebrovascular ischemic lesions. Stroke 37:830–835. [DOI] [PubMed] [Google Scholar]
- 34. Laloux P, Brucher JM (1991) Lacunar infarctions due to cholesterol emboli. Stroke 22:1440–1444. [DOI] [PubMed] [Google Scholar]
- 35. Lammie GA (2000) Pathology of small vessel stroke. Br Med Bull 56:296–306. [DOI] [PubMed] [Google Scholar]
- 36. Lammie GA, Brannan F, Wardlaw JM (1998) Incomplete lacunar infarction (type 1b lacunes). Acta Neuropathol (Berl) 96:163–171. [DOI] [PubMed] [Google Scholar]
- 37. Loeb C, Gandolfo C, Caponnetto C, Del SM (1990) Pseudobulbar palsy: a clinical computed tomography study. Eur Neurol 30:42–46. [DOI] [PubMed] [Google Scholar]
- 38. Ma K‐C, Olsson Y (1997) The role of chronic brain edema in the formation of lacunes in Binswanger's encephalopathy. Cerebrovasc Dis 7:324–331. [Google Scholar]
- 39. Mancardi GL, Romagnoli P, Tassinari T, Gandolfo C, Primavera JM, Loeb C (1988) Lacunae and cribriform cavities of the brain. Eur Neurol 28:11–17. [DOI] [PubMed] [Google Scholar]
- 40. Marie P (1901) Des foyers lacunaires de désintégration et des differents autres états cavitaires du cerveau. Rev Méd 21:281–298. [Google Scholar]
- 41. Marshall VG, Bradley J, Marshall CE, Bhoopat T, Rhodes RH (1988) Deep white matter infarction: correlation of MR imaging and histopathologic findings. Radiology 167:517–522. [DOI] [PubMed] [Google Scholar]
- 42. Masuda J, Tanaka K, Omae T, Ueda K, Sadoshima S (1983) Cerebrovascular diseases and their underlying vascular lesions in Hisayama, Japan—a pathological study of autopsy cases. Stroke 14:934–940. [DOI] [PubMed] [Google Scholar]
- 43. Mayer SA, Tatemichi TK, Hair LS, Goldman JE, Camac A, Mohr JP (1993) Hemineglect and seizures in Binswanger's disease: clinical‐pathological report. J Neurol Neurosurg Psychiatry 56:816–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Nelson RF, Pullicino P, Kendall BE, Marshall J (1980) Computed tomography in patients presenting with lacunar syndromes. Stroke 11:256–261. [DOI] [PubMed] [Google Scholar]
- 45. Nishida N, Ogata J, Yutani C, Minematsu K, Yamaguchi T (2000) Cerebral artery thrombosis as a cause of striatocapsular infarction. A histopathological case study. Cerebrovasc Dis 10:151–154. [DOI] [PubMed] [Google Scholar]
- 46. Ogata J (1999) The arterial lesions underlying cerebral infarction. Neuropathology 19:112–118. [DOI] [PubMed] [Google Scholar]
- 47. Ogata J, Yutani C, Otsubo R, Yamanishi H, Naritomi H, Yamaguchi T, Minematsu K (2008) Heart and vessel pathology underlying brain infarction in 142 stroke patients. Ann Neurol 63:770–781. [DOI] [PubMed] [Google Scholar]
- 48. Pantoni L, Sarti C, Alafuzoff I, Jellinger K, Munoz DG, Ogata J, Palumbo V (2006) Postmortem examination of vascular lesions in cognitive impairment: a survey among neuropathological services. Stroke 37:1005–1009. [DOI] [PubMed] [Google Scholar]
- 49. Pico F, Labreuche J, Seilhean D, Duyckaerts C, Hauw J, Amarenco P (2007) Association of small‐vessel disease with dilatative arteriopathy of the brain: neuropathologic evidence. Stroke 38:1197–1202. [DOI] [PubMed] [Google Scholar]
- 50. Potter G, Doubal F, Jackson C, Sudlow C, Dennis MS, Wardlaw J (2010) Associations of clinical stroke misclassification (“clinical‐imaging dissociation”) in acute ischemic stroke. Cerebrovasc Dis 29:395–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Pullicino P, Nelson RF, Kendall BE, Marshall J (1980) Small deep infarcts diagnosed on computed tomography. Neurology 30:1090–1096. [DOI] [PubMed] [Google Scholar]
- 52. Reed D, Jacobs J, Hayashi T, Konishi M, Nelson J, Iso H, Strong J (1994) A comparison of lesions in small intracerebral arteries among Japanese men in Hawaii and Japan. Stroke 25:60–65. [DOI] [PubMed] [Google Scholar]
- 53. Revesz T, Hawkins CP, du Boulay EP, Barnard RO, McDonald WI (1989) Pathological findings correlated with magnetic resonance imaging in subcortical arteriosclerotic encephalopathy (Binswanger's disease). J Neurol Neurosurg Psychiatry 52:1337–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Rossi R, Joachim C, Geroldi C, Combrinck M, Esiri MM, Smith AD, Frisoni GB (2004) Association between subcortical vascular disease on CT and neuropathological findings. Int J Geriatr Psychiatry 19:690–695. [DOI] [PubMed] [Google Scholar]
- 55. Schneider JA, Bienias JL, Wilson RS, Berry‐Kravis E, Evans DA, Bennett DA (2005) The apolipoprotein E epsilon4 allele increases the odds of chronic cerebral infection detected at autopsy in older persons. Stroke 36:954–959. [DOI] [PubMed] [Google Scholar]
- 56. Stroup DF, Berlin JA, Morton SC, Olkin I, Williamson GD, Rennie D et al, for the Meta‐analysis Of Observational Studies in Epidemiology Group (2000) Meta‐analysis of observational studies in epidemiology: a proposal for reporting. JAMA 283:2008–2012. [DOI] [PubMed] [Google Scholar]
- 57. Sudlow CLM, Warlow CP (1997) Comparable studies of the incidence of stroke and its pathological types: results from an international collaboration. Stroke 28:491–499. [DOI] [PubMed] [Google Scholar]
- 58. Tomimoto H, Shibata M, Ihara M, Akiguchi I, Ohtani R, Budka H (2002) A comparative study on the expression of cyclooxygenase and 5‐lipoxygenase during cerebral ischemia in humans. Acta Neuropathol (Berl) 104:601–607. [DOI] [PubMed] [Google Scholar]
- 59. Tuszynski MH, Petito CK, Levy DE (1989) Risk factors and clinical manifestations of pathologically verified lacunar infarctions. Stroke 20:990–999. [DOI] [PubMed] [Google Scholar]
- 60. Udaka F, Sawada H, Kameyama M (2002) White matter lesions and dementia. Ann N Y Acad Sci 977:411–415. [DOI] [PubMed] [Google Scholar]
- 61. van Swieten JC, van den Hout J, van Ketel BA, Hijdra A, Wokke J, van Gijn J (1991) Periventricular lesions in the white matter on magnetic resonance imaging in the elderly. Brain 114:761–774. [DOI] [PubMed] [Google Scholar]
- 62. Vinters HV, Ellis W, Jagust W, Ellis C, Zaias B, Chui H, Mack W (2000) Neuropathologic substrates of ischemic vascular dementia. J Neuropathol Exp Neurol 59:931–945. [DOI] [PubMed] [Google Scholar]
- 63. von Elm E, Altman DG, Egger M, Pocock SJ, Gotzsche PC, Vandenbroucke JP (2007) The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) Statement: guidelines for reporting observational studies. Prev Med 45:247–251. [DOI] [PubMed] [Google Scholar]
- 64. Wang L, Larson E, Sonnen J, Shofer J, McCormick W, Bowen J et al (2009) Blood pressure and brain injury in older adults: findings from a community‐based autopsy study. J Am Geriatr Soc 57:1975–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Wardlaw J, Doubal F, Armitage P, Chappell F, Carpenter T, Munoz Maniega S et al (2009) Lacunar stroke is associated with diffuse blood brain barrier dysfunction. Ann Neurol 65:194–202. [DOI] [PubMed] [Google Scholar]
- 66. Wardlaw JM (2008) What is a lacune? Stroke 39:2921–2922. [DOI] [PubMed] [Google Scholar]
- 67. Wardlaw JM, Doubal FN, Eadie E, Chappell F, Shuler K, Cvoro V (2011) Little association between intracranial arterial stenosis and lacunar stroke. Cerebrovasc Dis 31:12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Warlow C, van Gijn J, Dennis MS, Wardlaw J, Bamford JM, Hankey GJ et al (2008) Is it a vascular event and where is the lesion? Identifying and interpreting the symptoms and signs of cerebrovascular disease. In: Stroke: Practical Management, Dennis M, Langhorne P (eds), pp. 28–105. Blackwell Science: Oxford. [Google Scholar]
- 69. Yamada T, Miyazaki K, Koshikawa N, Takahashi M, Akatsu H, Yamamoto T (1995) Selective localization of gelatinase A, an enzyme degrading β‐amyloid protein, in white matter microglia and in Schwann cells. Acta Neuropathol (Berl) 89:199–203. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. The 9‐point study quality checklist applied to all included studies. It was devised using influences from the STROBE statement and the MOOSE criteria.
Table S2. A full list of studies which met our inclusion criteria, arranged in chronological order. Number of patients refers to the number of patients with lacunar lesions (therefore not necessarily the total number in the study). Abbreviations: HT = hypertension; DM = diabetes mellitus; AF = atrial fibrillation; CVD = cerebrovascular disease; AD = Alzheimer‘s disease; IC = internal carotids; H&E = hematoxylin and eosin; EVG = elastic van gieson; PAS = periodic acid‐Schiff; PTAH = phosphotungstic acid hematoxylin; LFB = luxol fast blue; GFAP = glial fibrillary acidic protein; AP = alkaline phosphatase; IgG = immunoglobulin G; CV = cresyl violet; MSB = martius scarlet blue.
Supporting info item