

Abstract
Progressive multifocal leukoencephalopathy (PML) and JCV granular cell neuronopathy occur secondary to JCV polyomavirus (JCV) infection of oligodendrocytes and cerebellar granular cell neurons (CGNs) during immunosuppression. Pure populations of astrocytes, oligodendrocytes, CGNs and microglia from frontal cortex and cerebellum of 17 non‐PML patients (9 immunocompetent; 8 immunosuppressed) were isolated by laser capture microdissection (LCM). JCV large T (LT) antigen DNA was detected by triple nested polymerase chain reaction (PCR). Sequence analysis was performed to assess LT gene variation. JCV DNA was detected in oligodendrocytes, astrocytes and CGNs of non‐PML brains. The most common site for viral latency was cortical oligodendrocytes (65% of samples analyzed). Immunosuppressed patients were significantly more likely to harbor JCV DNA in CGN populations than immunocompetent patients (P = 0.01). Sequence analysis of the LT region revealed eight novel single nucleotide polymorphisms (SNPs) in four immunosuppressed patients. Of the eight novel SNPs detected, six were silent and two resulted in amino acid changes. JCV DNA is present within cells of the non‐PML brain, known to be infected during PML and granular cell neuronopathy. This supports the argument for a brain only reservoir of JCV and supports the hypothesis that reactivation of latent brain JCV may be central to disease pathogenesis.
Keywords: immunosuppression, JC virus, laser capture microdissection, progressive multifocal leukoencephalopathy, viral latency
INTRODUCTION
Progressive multifocal leukoencephalopathy (PML) is a disease of the central nervous system (CNS) caused by demyelination secondary to infection with JC polyomavirus (JCV). The disease most often occurs at times of severe immunosuppression and more recently has been seen in patients receiving monoclonal antibody therapy (5).
PML arises because of the lytic infection of oligodendrocytes by JCV. In some cases, there may also be productive infection of astrocytes 41, 43, but infection of these cells is generally considered to be abortive. There may also be microglial cells present at the periphery of PML lesions; however, these cells are not known to permit productive JCV infection (41). While PML results from infection of oligodendrocytes, a related disorder, JCV granular cell neuronopathy is caused by the infection and subsequent lysis of cerebellar granular neurons (CGNs). JCV granular cell neuronopathy can occur in isolation from or in conjunction with PML (44) and has been reported in HIV/AIDS patients 13, 21 and other individuals on high‐dose immunosuppressants (17). It has been suggested that the incidence of JCV granular cell neuronopathy may be higher than previously reported, with retrospective analysis of cases diagnosed as PML indicating that up to half of these patients also showed evidence of JCV within granular neurons (44), highlighting a potential role for CGNs in the pathogenesis of PML.
Infection with JCV is presumed to occur during childhood, most likely via respiratory transmission (25). By adulthood, approximately 85% of the population is seropositive for JCV (14), and there is evidence to suggest that following primary exposure a lifelong infection is established at multiple sites throughout the body 6, 28, 36, 37, 42. JCV reactivation from virus within the kidney is common and can be easily detected by intermittent viruria 8, 14.
Whether PML or JCV granular cell neuronopathy, occur subsequent to primary entry of JCV into the CNS via infected lymphocytes or as unbound virus, or whether they are a result of reactivation of a preexisting CNS infection following immunosuppression is unknown. Previous studies have identified JCV DNA in healthy brain tissue 12, 37, 42; however, the possibility of presence of JCV‐infected lymphocytes has not been excluded. In a recent report, Perez‐Liz et al used laser capture microdissection (LCM) and Southern blot hybridization to isolate JCV DNA fragments from oligodendrocytes and, to a lesser extent, astrocytes from the frontal cortex of a single non‐PML patient (27).
The supercoiled, double‐stranded JCV genome encodes early, late and regulatory genes (23). The regulatory region (RR) and early phase large T (LT) antigen are commonly targeted in polymerase chain reaction (PCR) studies 27, 42. The RR contains a bi‐directional promoter/enhancer, which controls transcription and may be of an “archetypal” or “rearranged” formation (45). Presence of a rearranged RR has been documented to markedly increase viral transcription and replication levels (11). Whilst establishment of an efficient lytic infection seems to be dependent on host cell nuclear factor binding to rearranged RR, this region does not control cellular tropism (33). Repeated detection of rearranged RR DNA in systemic organs in individuals with no evidence of PML 25, 36, 39 has stimulated arguments that other factors are involved in determining JCV latency, reactivation and progression to PML.
The LT gene can direct DNA transcription and replication (23). Previous studies have demonstrated helicase activity of LT protein and its ability to drive viral replication through recruitment of host cell DNA polymerases and LT‐mediated binding to the viral origin of DNA replication 22, 29. The LT gene shows greater than 80% homology across the species of the polyomavirus family (19), highlighting its importance in the viral life cycle. Viral protein 1 (VP1) is a late gene, encoding the major capsid protein, defining viral strain and is also targeted in some PCR studies 6, 28. Single nucleotide polymorphisms (SNPs) encoding variations to the consensus prototype JCV sequence (NC_001699.1) have been reported in the RR, LT and VP1 genes 37, 46, 47. Previous data suggest that detection rates of JCV infection may be underestimated when RR and VP1 DNA are targeted as opposed to LT DNA 1, 4.
In this study, we have investigated the cellular localization of JCV in 17 non‐PML cases through the use of LCM and triple nested PCR for JCV LT DNA. This technique allows the isolation of highly pure cell populations from a heterogenous tissue section via direct cell visualization, minimizing the possibility of contamination by surrounding matrix or other cells.
MATERIALS AND METHODS
Patient selection
Formalin‐fixed, paraffin‐embedded (FFPE) brain tissue from 17 non‐PML patients (10 male and 7 female) was obtained from the Alfred Hospital Autopsy Tissue Archive. All patients were selected for inclusion in this study on the basis of having returned JCV LT and JCV VP1 DNA‐positive sections of frontal cortex or cerebellum as previously reported, from a larger group of immunosuppressed and immunocompetent cases selected at random from those brought to autopsy at the Alfred Hospital (4). Non‐PML cases were diagnosed on the basis of full neuropathologic examination showing no evidence of demyelination, oligodendrocyte or granular neuron inclusions or any evidence of JC viral protein expression on immunoperoxidase reaction (4). The non‐PML group included five HIV positive patients (mean CD4 count 120 cells/mm3; mean viral load >65 000 copies/mL), one lung transplant recipient and 11 patients who died of other, unrelated, nonneurological causes. Patients were classified as immunocompetent (n = 9) or immunosuppressed (n = 8) (1a, 1b). The latter were considered immunosuppressed if they were on immunosuppressive medication, were HIV positive with CD4 count below 500 cells/mm3, or if they had widespread metastatic tumor or multisystems failure in conjunction with a significantly decreased lymphocyte count (<0.5 × 109 cells/L) (normal lymphocyte count is in the range of 1.0–4.0 × 109 cells/L). Frontal white matter from a patient with pathological evidence of PML as diagnosed at autopsy was included as a positive control and frontal white matter from two immunocompetent patients with no pathological evidence of PML who had previously returned JCV LT and VP1 DNA negative sections of frontal cortex and cerebellum (4) were included as negative controls (results not shown). Ethics approval for this study was given by the Alfred Hospital Human Research Ethics Committee (237/09).
Table 1a.
Patient demographics (immunocompetent).
| ID | Gender | Age | Cause of death | LC* | JCV DNA | |
|---|---|---|---|---|---|---|
| Kidney | Brain | |||||
| 2 | F | 83 | Acute myocardial infarction | 1.34 | × | |
| 4 | F | 69 | Acute myocardial infarction | 1.2 | × | |
| 7 | M | 87 | Bronchopneumonia | 0.63 | × | × |
| 9 | F | 87 | Metastatic squamous cell carcinoma | 1.65 | × | |
| 10 | M | 90 | Bronchopneumonia | 0.8 | × | |
| 11 | M | 62 | Bronchopneumonia | 1.35 | × | |
| 12 | M | 60 | Sub arachnoid haemorrhage | 3.28 | × | |
| 16 | F | 74 | Acute myocardial infarction | 3.12 | × | |
| 17 | F | 80 | Liver failure | 1.64 | × | |
Table 1b.
Patient demographics (immunosuppressed).
| ID | Gender | Age | Cause of death | LC* | JCV DNA | |
|---|---|---|---|---|---|---|
| Kidney | Brain | |||||
| 1 | M | 63 | HIV/AIDS | 0.34 | × | |
| 3 | M | 44 | HIV/AIDS | 0.36 | × | × |
| 5 | M | 62 | Candidal sepsis | 0.08 | × | |
| 6 | M | 34 | HIV/AIDS | 0.2 | × | |
| 8 | F | 67 | Vascular rejection in lung transplant | 1.2 | × | |
| 13 | M | 70 | Chronic lymphocytic leukaemia | 0.35 | × | |
| 14 | M | 47 | HIV/AIDS | 0.07 | × | |
| 15 | F | 59 | HIV/AIDS | 1.0 | × | |
JCV DNA = presence of JCV DNA on whole tissue homogenates as described in Bayliss et al (4).
Lymphocyte count (LC) measured in 109 cells/L.
Cell identification
Brain tissue sections (4 µM) were obtained from paraffin‐embedded tissue blocks from the frontal cortex and incorporating frontal white matter and the cerebellar white matter with an intact granular cell neuronal layer. Sections were mounted on “Superfrost plus” charged glass slides (Fisher Scientific, ON, Canada) as previously described (38). Morphologic examination of hematoxylin‐stained and immunohistochemically reacted sections was performed for the detection of astrocytes, oligodendrocytes, cerebellar granular neurons and activated microglia.
Specifically, a rabbit anti‐human glial fibrillary acidic protein (GFAP) polyclonal antibody was used for the detection of astrocytes within the frontal white matter and cerebellar white matter (1:50; Zymed, San Francisco, CA, USA) (Figure 1A). Immunoreactivity with a mouse anti‐human CD68 monoclonal antibody was used to detect parenchymal microglial cells (clone KP1, 1:200; Dako, Glostrup, Denmark) within the frontal white matter and cerebellar white matter, distinguished by a rod‐shaped nucleus with ramified cytoplasmic processes 2, 18 (Figure 1B). Oligodendrocytes from frontal and cerebellar white matter and CGNs from the cerebellar granular layer were identified using hematoxylin and eosin staining, based on cell morphology and microanatomical location. Specifically, oligodendrocytes were identified as cells with a round nucleus with dense chromatin, an artificial perinuclear halo and scanty, opaque cytoplasm, existing in short, parallel chains within the frontal white cortex and cerebellar white matter 16, 32. CGNs were distinguished as those neurons with a 5–8 µM rounded, dense nucleus, residing in the granular layer of the cerebellum 16, 32 (Figure 1C,D). Stained sections were dehydrated for 20–30 minutes in a fume hood and stored overnight in a desiccator and LCM was performed the following day.
Figure 1.
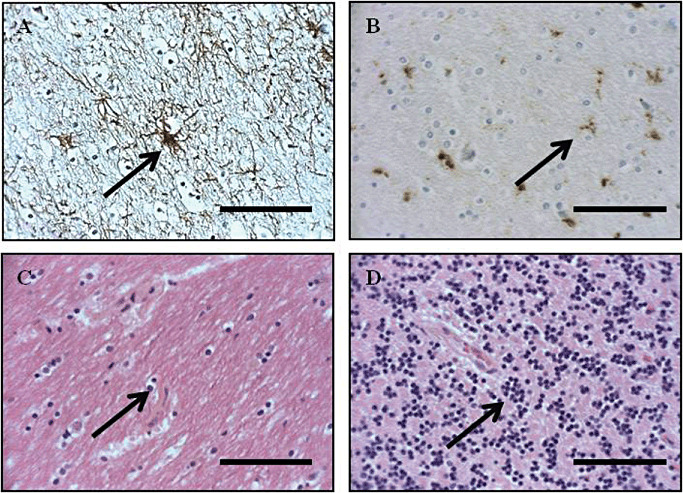
Identification of astrocytes, microglia, oligodendrocytes and granular cell neurons for laser capture microdissection via immunohistochemistry. A. A rabbit anti‐human glial fibrillary acidic protein (GFAP) polyclonal antibody was used for the detection of astrocytes (arrowed). B. A mouse anti‐human CD68 monoclonal antibody was for the detection of parenchymal ramified microglial cells. Ramified parenchymal microglia were distinguished as cells with slender, elongated nuclei, ramified cytoplasmic processes and a rod‐shaped nucleus within the neuropil. Activated parenchymal microglial cells displayed up‐regulated expression of the CD68 antigen, with more prominent ramification of processes and a more oval nucleus (arrowed). C,D. Oligodendrocytes (C) were identified with hematoxylin and eosin stain as cells with a round nucleus with dense chromatin, an artificial perinuclear halo and scanty, opaque cytoplasm, existing in short, parallel chains within the white matter of the frontal cortex and cerebellum (arrowed) 16, 32. Cerebellar granular neurons (D) were distinguished as those neurons with a soma of 5–8 µM residing in the granular layer of the cerebellum 16, 32 using a hematoxylin and eosin stain (arrowed). Sections captured at ×400 magnification. Scale bars represent 40 µM.
LCM
Astrocytes, oligodendrocytes and microglia from the frontal white matter, astrocytes, oligodendrocytes and microglia from the cerebellar white matter, and CGNs from the cerebellar granular layer were laser dissected from the uncovered stained slide sections as pure cell populations for downstream PCR analysis. PALM Microlaser system (PALM Mikrolaser Technologie, Göttingen, Germany) was utilized for laser microdissection. By combining the intense focal cutting tool with substage robotics, the laser beam was positioned to cut around the selected area on the tissue section and microdissect single cells, creating a clear‐cut gap between the selected and non‐selected areas (Figure 2).
Figure 2.
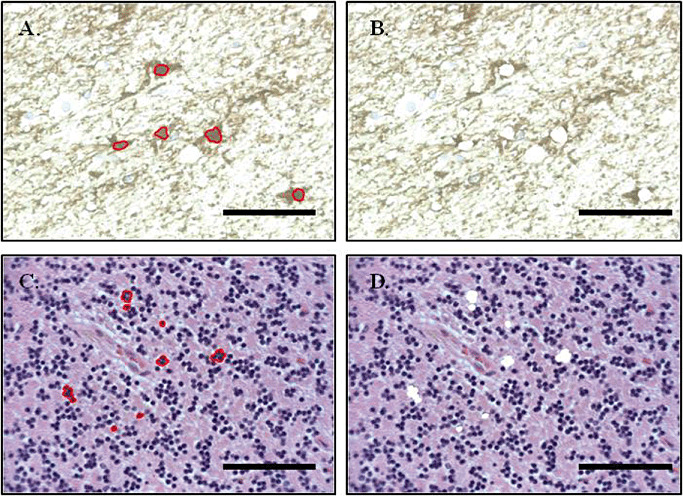
Before and after images of astrocytes and cerebellar granular cell excision from 4‐µM sections of formalin‐fixed paraffin‐embedded tissue via laser capture microdissection. Nuclei from specific cells of interest are recognized through characteristic appearance under light microscopy and co‐localization of antibody staining with hematoxylin counterstain. Individual nuclei are outlined using the PALM software and an intense focal cutting tool with substage robotics to allow laser positioning is used to cut around the selected area and microdissect single cells, creating a clear‐cut gap between the selected and non‐selected areas. Sections captured at ×400 magnification. Scale bars represent 40 µM.
The LCM protocol was specifically optimized for use in 4 µM FFPE brain tissue sections. To prevent excision of horizontally adjacent cell processes there was specific targeting of the nuclei of only those cells with the appropriate morphological appearance and (where appropriate) specific co‐localization of immunohistochemistry and hematoxylin staining (Figure 2). Presence of vertically (as viewed down the microscope) adjacent cells processes was minimized through specific targeting of only those cells with a fully rounded nucleus with a specific diameter of 5–10 µM (depending on cell type).
Once the cells of interest were isolated, the high energy generated by the focused laser light was used to catapult the dissected cells into a cap of an Eppendorf‐type tube. Using this technology, PALM was able to microdissect and capture cells without the use of manual manipulation, preventing contamination (31). For each sample section, pure cell populations of approximately 200 cells of each subtype were isolated in triplicate.
PCR detection of JCV LT DNA
DNA was extracted using the QuickExtract™ FFPE DNA Extraction Kit (Epicentre Biotechnologies, Madison, WI, USA) according to the manufacturer's instructions. DNA yield and purity were measured spectrophotometrically and adjusted through dilution. DNA integrity was tested through amplification of a 240 bp fragment of the glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) gene as previously described (4).
JCV LT PCR was performed via triple‐nested touchdown PCR using the HotStarTaq Plus Kit (QIAGEN, Hilden, Germany). The LT gene was targeted due to its relative conservation between JCV isolates to ensure the highest possible assay sensitivity (23). Our laboratory has previously demonstrated a highly sensitive nested PCR for JCV LT DNA in FFPE tissue (4). A triple‐nested approach was chosen to further increase assay sensitivity and to overcome the combined issues of PCR inhibition and fragmentation of template DNA caused by extended formalin‐fixation and low abundance of viral DNA. Triple‐nested PCR has been successfully used by our laboratory (38) and others 9, 10 in conjunction with LCM for the amplification and characterization of low‐abundance DNA viruses from FFPE brain tissues.
Each round of PCR was prepared in a 25‐µL touchdown nested PCR reaction containing 15 ng of template DNA [3 µL of Round 1 or Round 2 PCR product served as the template for the subsequent (rounds 2 and 3) reaction], 1 U HotStar Taq Plus enzyme and 0.25 µM of primer (Round 1; JC‐F2 and JC‐R2: Round 2; F3‐tjk and R3‐tjk: Round 3; EHJCF and EHJCR) (Table 2). The cycling conditions for all three rounds of this PCR were denaturation at 94°C for 1 minute, annealing at 60°C for 1 minute (−1°C/cycle for 10 cycles), then 30 cycles at 50°C for 1 minute and extension at 72°C for 1 minute.
Table 2.
Primer sequences used in PCR reactions in this study.
| Name | Sequence | Nucleotide position* |
|---|---|---|
| GAPDH‐F | GGTGAAGGTCGGAGTCAACGGA | |
| GAPDH‐R | GAGGGATCTCGCTCCTGGAAGA | |
| JC‐F2 | TGCTACAGTATCAACAGCCT | 3632–3651 |
| JC‐R2 | AGGAGCATGACTTTAACCCA | 3890–3909 |
| F3‐tjk | CAGCCTGCTGGCAAATCGT | 3646–3664 |
| R3‐tjk | TGGAMACCAAGTGTGAGG | 3817–3834 |
| EHJCF | AGCCTGCTGGCAAATGCT | 3647–3664 |
| EHJCR | CTTTCAGGAAAACCCACAGC | 3766–3785 |
Nucleotide position relative to consensus prototype JCV sequence (NC_001699.1).
Note that the R3‐tjk primer contains a degenerate base M to allow amplification of multiple genotypes containing different alleles at these sites.
No template controls (NTCs) consisting of PCR mastermix without template DNA, were used as negative controls and carried across between each round of PCR such that each experiment included a first, second and third nested round NTC. DNA extracted from areas of active demyelination within the PML control was diluted 1:100 to serve as a positive control. The lower limit of detection for the triple‐nested PCR assay was determined through serial dilution and PCR amplification of a JCV positive control with viral load previously quantified via real‐time PCR (3). The inhibitory effect of formalin‐fixation was determined by spiking a JCV negative FFPE sample with the serially diluted positive control and determined to decrease the sensitivity by 50%, resulting in a limit of detection of approximately two copies of viral DNA. The specificity of the triple‐nested PCR was proven through amplification and subsequent sequencing (see later) of a 139 bp product after three rounds of PCR (Figure 3). Samples were considered to contain JCV LT DNA if at least one of the three triplicates returned a positive result.
Figure 3.
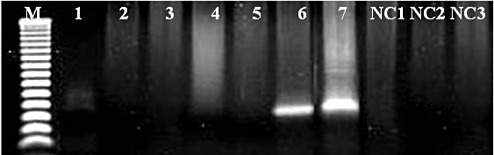
JCV LT triple nested PCR specificity. The specificity of the triple‐nested PCR was proven through amplification and subsequent sequencing of a 139 bp product after three rounds of PCR. Lanes 1–3; FFPE JCV negative tissue, Lanes 4–6; FFPE JCV positive tissues Lane 7; JCV control. M; 50 bp molecular weight ladder (Promega, Melbourne, Australia). NC = negative controls from the first (NC1) second (NC2) and third (NC3) rounds of PCR; PCR = polymerase chain reaction; FFPE = formalin‐fixed, paraffin‐embedded; LT = large T antigen; JCV = JC polyomavirus.
Sequencing of JCV LT DNA
After purification with the Exo‐SAP IT kit (GE Biosciences, Little Chalfont, UK), 20 µL PCR product was sequenced using the ABI 3100 Genetic Analyser. To confirm presence of JCV DNA, Basic Local Alignment Search Tool (BLAST) analysis was used to align the sequenced amplicons to the consensus prototype of JCV (NCBI Reference Sequence NC_001699.1). Nucleotide alignments were generated using CLUSTALW for multiple sequence alignments.
Bioinformatics of JCV LT DNA
Functional analyses of mutations within the JCV LT DNA sequence were performed using Swiss PDB Viewer Version 4.0.1 for Windows (Swiss Institute of Bioinformatics, Basel, Switzerland). The model for the highly related SV40 LT protein (PDB entry 1n25) was accessed from the Worldwide Protein Data Bank (http://www.wwpdb.org/ccd.html) and the JCV LT sequence was modeled from this overlay.
Statistical analyses
Statistical analyses were performed using GraphPad Version 5.03 for Windows (GraphPad Software Inc, La Jolla, CA, USA). Univariate analyses of outcome variables were conducted using t‐tests and Fisher's exact tests for equal proportion where appropriate. Normally distributed continuous data was reported as mean ± standard error. Categorical data was reported as number (percentage). Tissues from patients with PML served as positive controls and were excluded from all statistical analysis. In all cases a P‐value of 0.05 was considered statistically significant.
RESULTS
Cellular localization of JCV DNA
JCV DNA was detected in cortical and cerebellar astrocytes, oligodendrocytes and CGNs from immunosuppressed and immunocompetent patients (Table 3). The most common site for viral latency was within cortical oligodendrocytes (65%). There were no statistically significant differences in JCV DNA in astrocytes or oligodendrocytes from either the frontal cortex or cerebellum from immunosuppressed as compared with immunocompetent patients (Table 3). JCV LT DNA could not be detected in any of the microglia.
Table 3.
Summary of JCV LT triple nested PCR data showing frequency of viral infection in LCM‐acquired cell populations.
| Patient group | n | Age | Male n (%) | LC* | Frontal cortex n (%) | Cerebellum n (%) | |||
|---|---|---|---|---|---|---|---|---|---|
| Astrocytes | Oligodendrocytes | Astrocytes | Oligodendrocytes | Neurons† | |||||
| Immunocompetent | 9 | 76.89 ± 3.73 | 4 (44) | 1.67 ± 0.31 | 5 (56) | 7 (78) | 2 (22) | 1 (11) | 1 (11) |
| Immunosuppressed | 8 | 55.75 ± 4.75 | 6 (75) | 0.52 ± 0.16 | 3 (38) | 4 (50) | 5 (63) | 4 (50) | 6 (75) |
| Total | 17 | 66.94 ± 3.84 | 10 (59) | 1.13 ± 0.22 | 8 (47) | 11 (65) | 7 (41) | 5 (29) | 7 (41) |
Lymphocyte count (LC) measured in 109 cells/L.
Indicates cerebellar granular neurons.
Immunosuppressed patients were significantly more likely to harbour JCV DNA in CGN populations than immunocompetent patients [75% (6/8) vs. 11% (1/9) respectively; P = 0.01]. The immunosuppressed patients which harboured JCV DNA in the CGNs included all of the HIV patients making this patient group more likely to harbour virus at this site than any other patient group [100% (5/5) HIV vs. 33% (1/3) other immunosuppressed patients vs. 11% (1/9) immunocompetent patients; P = 0.1 and P = 0.003, respectively].
Sequence analysis of JCV DNA
In 13 of 17 patients, sequence analysis of the third‐round JCV amplicons from each of the positive cell populations did not differ from the consensus sequence (NC_001699.1). Of the remaining four patients, Px1 (HIV+) demonstrated one SNP in each of the astrocytes (A3724T) and oligodendrocytes (T3679C) from the frontal cortex and Px8 (lung transplant) also demonstrated one SNP in the astrocytes (C3722T) from the frontal cortex (Table 3). Px14 (HIV+) demonstrated two SNPs (T3701C and A3746G) in the CGNs and Px15 (multisystems failure) demonstrated three SNPs (C3695T, A3744G and T3763G) two of which were in similar regions to those of Px14 (Figure 4). Of the eight novel SNPs which were detected, only two resulted in amino acid changes (Table 4). Functional analysis of the locations of the two missense mutations revealed localization within the helicase domain of the LT protein.
Figure 4.

Results of CLUSTALW alignment of selected third round PCR products. Polymorphisms varying from the consensus sequence (NC_001699.1) were noted in samples from four immunosuppressed patients (Table 2). 1AstroFC, Patient 1 astrocytes from frontal cortex; 1OligoFC, Patient 1 oligodendrocytes from frontal cortex; 8AstroFC, Patient 8 astrocytes from frontal cortex; 14CGNCBM, Patient 14 cerebellar granular neurons from cerebellum; 15CGNCBM, Patient 15 cerebellar granular neurons from cerebellum. Underlined regions refer to primers as listed in 1a, 1b. The figure above corresponds to nucleotides 3647–3785 of the anti sense strand of the viral genome (LT is encoded on the anti sense strand; NCBI Reference Sequence NC_001699.1). PCR = polymerase chain reaction; LT = large T antigen.
Table 4.
Locations of amino acid substitutions in the LT gene.
| SNP* | Patient | Con. codon | Ob. codon | AA substitution |
|---|---|---|---|---|
| T3679C | 1OligoFC | GAT | GAC | Asp (no change) |
| C3695T | 15CGNCBM | GCC | GCA | Ala (no change) |
| T3701C | 14CGNCBM | TAT | TAC | Tyr (no change) |
| C3722T | 8AstroFC | AAC | AAT | Asn (no change) |
| A3724T | 1AstroFC | AAC | TAC | Asn → Tyr |
| G3744A | 15CGNCBM | AAG | AAA | Lys (no change) |
| A3746G | 14CGNCBM | AAA | AAG | Lys (no change) |
| T3763G | 15CGNCBM | TGC | GGC | Cys → Gly |
SNPs are listed according to position and substitution on the anti sense strand (LT is encoded on the anti sense strand; NCBI Reference Sequence NC_001699.1).
Abbreviations: AA = amino acid; Astro = astrocytes; CBM = cerebellum; Con. = consensus sequence NC_001699.1; FC = frontal cortex; GCN = granular cell neuron; Ob. = observed; Oligo = oligodendrocyte.
All of the patients who demonstrated SNPs (silent or missense) were immunosuppressed, making immunosuppressed patients significantly more likely to have variations within the LT gene as compared with immunocompetent patients [50% (4/8) vs. 0, respectively; P = 0.02].
DISCUSSION
This study represents the first detailed investigation of the specific cellular localization of brain JCV DNA in immunocompetent and immunosuppressed patients. The specificity of LCM was combined with the sensitivity of triple‐nested PCR to identify JCV DNA within the brain.
In these 17 non‐PML individuals JCV DNA was present in multiple cell populations of the frontal cortex and cerebellum; astrocytes, oligodendrocytes and CGNs. JCV DNA was most frequently detected in cortical oligodendrocytes (65% of samples analyzed) and commonly in astrocytes (approximately 40% of samples analyzed). A previous study of a single non‐PML case also identified JCV LT DNA in oligodendrocytes and astrocytes (27). Pathogenesis of PML is well documented and implicates JCV infection of oligodendrocytes, and subsequent viral replication and lysis of these cells (35). We have previously documented presence of JCV LT and VP1 DNA in the cases used in this study (4), suggesting that fully formed virus, capable of reactivation and lytic infection, is present within the cellular reservoirs identified in the current study. The finding of JCV DNA within oligodendrocytes in both immunosuppressed and immunocompetent patients in this study lends support to the hypothesis that PML could be due to reactivation of latent brain infection.
This study is the first to demonstrate JCV infection of CGNs in the absence of PML. JCV DNA was detected in CGNs from 75% of immunosuppressed patients. Wüthrich et al previously, suggested that CGNs may be an important site of JCV infection following immunosuppression (44). The majority of patients with infection of CGNs in our study (5/7) were HIV positive. An increased propensity for JCV latency in CGNs of HIV patients may be relevant to the relatively high incidence of JCV granular cell neuronopathy in HIV.
Previous histological studies have identified JCV protein in macrophage and microglia during PML 24, 40. Our study shows no evidence of JCV DNA within microglia in non‐PML cases, suggesting that microglia are unlikely to be sites for JCV infection prior to disease development; their involvement during disease being explained by phagocytosis of excess virus and cellular debris during active infection.
This study identified several novel genetic polymorphisms in the helicase region of the LT gene suggesting increased genetic diversity among circulating viral strains particular in those taken from immunosuppressed patients. Previous studies have focused on viral classification through polymorphisms within the VP1 (capsid) region of JCV 20, 34, 45, few have examined variation within the LT gene.
Six of the eight novel polymorphisms detected within the LT gene in this study resulted in silent mutations. Historically considered biologically irrelevant, the impact of silent mutations on disease has only recently been realized. Silent mutations have the potential to impact or alter any stage of protein production, from DNA transcription rates, to misfolding of mRNA products and translation into protein (7). A study of the LT gene sequence in 44 PML patients reported complete homology (15), suggesting that sequence conservation in this region is a prerequisite for completion of the viral lifecycle. Further work is required to determine the biological impact of the novel LT gene mutations identified in this study with respect to viral latency and progression to PML.
There are limitations associated with DNA extraction from FFPE tissue following LCM. Previous studies, with the exception of Perez‐Liz et al (27) have utilized a triple‐nested approach to overcome the issues associated with detection of extremely low‐abundance viral nucleic acids 9, 10, 30. Formalin‐fixation of tissue also induces cross‐linking and fragmentation of DNA and inhibit molecular studies (26), which may result in an underestimation of the frequency of JCV LT DNA. We acknowledge the possibility that vertically adjacent processes from neighboring cells may have been inadvertently collected at the time of LCM, particularly during collection of smaller microglial nuclei. However, we found no evidence of JCV in any of the captured microglia from non‐PML brain, making vertical process contamination highly unlikely.
Fifteen of the 17 non‐PML patients included in this study did not have any evidence of JCV LT or VP1 DNA on PCR analysis of whole tissue extracts of kidney (4). In these 15, JCV DNA was demonstrated in resident brain cells of immunocompetent and immunosuppressed individuals, strengthening the argument for a CNS‐only cellular reservoir of JCV. We have previously reported increased JCV DNA detection within the brain tissue homogenates with immunosuppression and suggested a change in the latency profile may occur with immunosuppression (4). This current cell‐specific study further supports a role for immunosuppression and viral site specificity with significantly more individuals with HIV harboring JCV DNA in cerebellar granular neurons. Future studies using fresh tissue should aim to delineate whether viral DNA within specific cells is capable of establishing productive infection upon immunosuppression.
ACKNOWLEDGMENTS
The authors acknowledge the contributions of Dr Katherine Thompson in the preparation of this manuscript.
REFERENCES
- 1. Agostini HT, Ryschkewitsch CF, Mory R, Singer EJ, Stoner GL (1997) JC virus (JCV) genotypes in brain tissue from patients with progressive multifocal leukoencephalopathy (PML) and in urine from controls without PML: increased frequency of JCV type 2 in PML. J Infect Dis 176:1–8. [DOI] [PubMed] [Google Scholar]
- 2. Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005) Does drug abuse alter microglial phenotype and cell turnover in the context of advancing HIV infection? Neuropathol Appl Neurobiol 31:325–338. [DOI] [PubMed] [Google Scholar]
- 3. Bayliss J, Moser R, Bowden S, McLean CA (2010) Characterisation of single nucleotide polymorphisms in the genome of JC polyomavirus using MALDI TOF mass spectrometry. J Virol Methods 164:63–67. [DOI] [PubMed] [Google Scholar]
- 4. Bayliss J, Karasoulos T, Bowden S, Glogowski I, McLean CA (2011) Immunosuppression increases latent infection of brain by JC polyomavirus. Pathology 43:362–367. [DOI] [PubMed] [Google Scholar]
- 5. Berger JR, Houff SA, Major EO (2009) Monoclonal antibodies and progressive multifocal leukoencephalopathy. mAbs 1:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caldarelli‐Stefano R, Vago L, Omodeo‐Zorini E, Mediati M, Losciale L, Nebuloni M et al (1999) Detection and typing of JC virus in autopsy brains and extraneural organs of AIDS patients and non‐immunocompromised individuals. J Neurovirol 5:125–133. [DOI] [PubMed] [Google Scholar]
- 7. Chamary JV, Hurst LD (2009) The price of silent mutations. Sci Am 300:46–53. [DOI] [PubMed] [Google Scholar]
- 8. Chen Y, Bord E, Tompkins T, Miller J, Tan CS, Kinkel RP et al (2009) Asymptomatic reactivation of JC virus in patients treated with natalizumab. N Eng J Med 361:1067–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Churchill M, Gorry P, Cowley D, Lal L, Sonza S, Purcell DFJ et al (2006) Use of laser capture microdissection to detect integrated HIV‐1 DNA in macrophages and astrocytes from autopsy brain tissues. J Neurovirol 12:146–152. [DOI] [PubMed] [Google Scholar]
- 10. Churchill MJ, Wesselingh SL, Cowley D, Pardo CA, McArthur JC, Brew BJ, Gorry PR (2009) Extensive astrocyte infection is prominent in human immunodeficiency virus–associated dementia. Ann Neurol 66:253–258. [DOI] [PubMed] [Google Scholar]
- 11. Daniel AM, Swenson JJ, Mayreddy RP, Khalili K, Frisque RJ (1996) Sequences within the early and late promoters of archetype JC virus restrict viral DNA replication and infectivity. Virology 216:90–101. [DOI] [PubMed] [Google Scholar]
- 12. Delbue S, Branchetti E, Boldorini R, Vago L, Zerbi P, Veggiani C et al (2008) Presence and expression of JCV early gene large T antigen in the brains of immunocompromised and immunocompetent individuals. J Med Virol 80:2147–2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Du Pasquier RA, Corey S, Margolin DH, Williams K, Pfister L‐A, De Girolami U et al (2003) Productive infection of cerebellar granule cell neurons by JC virus in an HIV+ individual. Neurology 61:775–782. [DOI] [PubMed] [Google Scholar]
- 14. Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C et al (2009) Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J Infect Dis 199:837–846. [DOI] [PubMed] [Google Scholar]
- 15. Glass AJ, Venter M (2009) Improved detection of JC virus in AIDS patients with progressive multifocal leukoencephalopathy by T‐antigen specific fluorescence resonance energy transfer hybridization probe real‐time PCR: evidence of diverse JC virus genotypes associated with progressive multifocal leukoencephalopathy in Southern Africa. J Med Virol 81:1929–1937. [DOI] [PubMed] [Google Scholar]
- 16. Graham DI, Lantos PL (2002) Greenfield's Neuropathology, 7th edn. Arnold Publishers: New York. [Google Scholar]
- 17. Granot R, Lawrence R, Barnett M, Masters L, Rodriguez M, Theocharous C et al (2009) What lies beneath the tent? JC‐virus cerebellar granule cell neuronopathy complicating sarcoidosis. J Clin Neurosci 16:1091–1092. [DOI] [PubMed] [Google Scholar]
- 18. Hulette CM, Downey BT, Burger PC (1992) Macrophage markers in diagnostic neuropathology. Am J Surg Pathol 16:493–499. [DOI] [PubMed] [Google Scholar]
- 19. Imperiale MJ (2001) Oncogenic transformation by the human polyomaviruses. Oncogene 20:7917–7923. [DOI] [PubMed] [Google Scholar]
- 20. Jobe DV, Friedlaender JS, Mgone CS, Agostini HT, Koki G, Yanagihara R et al (2001) New JC virus (JCV) genotypes from papua new guinea and micronesia (type 8 and type 2E) and evolutionary analysis of 32 complete JCV genomes. Arch Virol 146:2097–2113. [DOI] [PubMed] [Google Scholar]
- 21. Koralnik IJ, Wüthrich C, Dang X, Rottnek M, Gurtman A, Simpson D, Morgello S (2005) JC virus granule cell neuronopathy: a novel clinical syndrome distinct from progressive multifocal leukoencephalopathy. Ann Neurol 57:576–580. [DOI] [PubMed] [Google Scholar]
- 22. Lepik D, Ustav M (2000) Cell‐specific modulation of papovavirus replication by tumor suppressor protein p53. J Virol 74:4688–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Maginnis MS, Atwood WJ (2009) JC Virus: an oncogenic virus in animals and humans? Semin Cancer Biol 19:261–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Major EO, Amemiya K, Tornatore CS, Houff SA, Berger JR (1992) Pathogenesis and molecular biology of progressive multifocal leukoencephalopathy, the JC virus‐induced demyelinating disease of the human brain. Clin Microbiol Rev 5:49–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Monaco MC, Jensen PN, Hou J, Durham LC, Major EO (1998) Detection of JC virus DNA in human tonsil tissue: evidence for site of initial viral infection. J Virol 72:9918–9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Namimatsu S, Ghazizadeh M, Sugisaki Y (2005) Reversing the effects of formalin fixation with citraconic anhydride and heat: a universal antigen retrieval method. J Histochem Cytochem 53:3–11. [DOI] [PubMed] [Google Scholar]
- 27. Perez‐Liz G, Del Valle L, Gentilella A, Croul S, Khalili K (2008) Detection of JC virus DNA fragments but not proteins in normal brain tissue. Ann Neurol 64:379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pietropaolo V, Fioriti D, Simeone P, Videtta M, Di Taranto C, Arancio A et al (2003) Detection and sequence analysis of human polyomaviruses DNA from autoptic samples of HIV‐1 positive and negative subjects. Int J Immunopathol Pharmacol 16:269–276. [DOI] [PubMed] [Google Scholar]
- 29. Prins C, Frisque RJ (2001) JC virus T′ proteins encoded by alternatively spliced early mRNAs enhance T antigen‐mediated viral DNA replication in human cells. J Neurovirol 7:250–264. [DOI] [PubMed] [Google Scholar]
- 30. Rhodes DI, Ashton L, Solomon A, Carr A, Cooper D, Kaldor J, Deacon N (2000) Characterization of Three nef‐Defective Human Immunodeficiency Virus Type 1 Strains Associated with Long‐Term Nonprogression. J Virol 74:10581–11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schutze K, Becker I, Becker KF, Thalhammer S, Stark R, Heckl WM et al (1997) Cut out or poke in–the key to the world of single genes: laser micromanipulation as a valuable tool on the look‐out for the origin of disease. Genet Anal 14:1–8. [DOI] [PubMed] [Google Scholar]
- 32. Shepherd GM (2004) The Synaptic Organization of the Brain. Oxford University Press: New York. [Google Scholar]
- 33. Shishido‐Hara Y (2010) Progressive multifocal leukoencephalopathy and promyelocytic leukemia nuclear bodies: a review of clinical, neuropathological, and virological aspects of JC virus‐induced demyelinating disease. Acta Neuropathol 120:403–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Stoner GL, Jobes DV, Fernandez Cobo M, Agostini HT, Chima SC, Ryschkewitsch CF (2000) JC virus as a marker of human migration to the Americas. Microbes Infect 2:1905–1911. [DOI] [PubMed] [Google Scholar]
- 35. Tan CS, Koralnik IJ (2010) Progressive multifocal leukoencephalopathy and other disorders caused by JC virus: clinical features and pathogenesis. Lancet Neurol 9:425–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Tan CS, Dezube Bruce J, Bhargava P, Autissier P, Wuthrich C, Miller J, Koralnik Igor J (2009) Detection of JC virus DNA and proteins in the bone marrow of HIV positive and HIV negative patients: implications for viral latency and neurotropic transformation. J Infect Dis 199:881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tan CS, Ellis LC, Wuthrich C, Ngo L, Broge TA Jr, Saint‐Aubyn J et al (2010) JC virus latency in the brain and extraneural organs of patients with and without progressive multifocal leukoencephalopathy. J Virol 84:9200–9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Thompson KA, Varrone JJ, Jankovic‐Karasoulos T, Wesselingh SL, McLean CA (2009) Cell‐specific temporal infection of the brain in a simian immunodeficiency virus model of human immunodeficiency virus encephalitis. J Neurovirol 15:300–311. [DOI] [PubMed] [Google Scholar]
- 39. Tornatore C, Berger JR, Houff SA, Curfman B, Meyers K, Winfield D, Major EO (1992) Detection of JC virus DNA in peripheral lymphocytes from patients with and without progressive multifocal leukoencephalopathy. Ann Neurol 31:454–462. [DOI] [PubMed] [Google Scholar]
- 40. Vendrely A, Bienvenu B, Gasnault J, Thiebault JB, Salmon D, Gray F (2005) Fulminant inflammatory leukoencephalopathy associated with HAART‐induced immune restoration in AIDS‐related progressive multifocal leukoencephalopathy. Acta Neuropathol 109:449–455. [DOI] [PubMed] [Google Scholar]
- 41. von Einsiedel RW, Samorei IW, Pawlita M, Zwissler B, Deubel M, Vinters HV (2004) New JC virus infection patterns by in situ polymerase chain reaction in brains of acquired immunodeficiency syndrome patients with progressive multifocal leukoencephalopathy. J Neurovirol 10:1–11. [DOI] [PubMed] [Google Scholar]
- 42. White FA, Ishaq M, Stoner GL, Frisque RJ (1992) JC virus DNA is present in many human brain samples from patients without progressive multifocal leukoencephalopathy. J Virol 66:5726–5734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wüthrich C, Dang X, Westmoreland S, McKay J, Maheshwari A, Anderson MP et al (2009) Fulminant JC virus encephalopathy with productive infection of cortical pyramidal neurons. Ann Neurol 65:742–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wüthrich C, Cheng YM, Joseph JT, Kesari S, Beckwith C, Stopa E et al (2009) Frequent infection of cerebellar granule cell neurons by polyomavirus JC in progressive multifocal leukoencephalopathy. J Neuropathol Exp Neurol 68:15–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yogo Y, Sugimoto C, Zheng H‐Y, Kitamura T (2007) Genetic changes in JC virus possibly associated with progressive multifocal leukoencephalopathy. No To Shinkei 59:109–118. [PubMed] [Google Scholar]
- 46. Zheng HY, Kitamura T, Takasaka T, Chen Q, Yogo Y (2004) Unambiguous identification of JC polyomavirus strains transmitted from parents to children. Arch Virol 149:261–273. [DOI] [PubMed] [Google Scholar]
- 47. Zheng HY, Ikegaya H, Takasaka T, Matsushima‐Ohno T, Sakurai M, Kanazawa I et al (2005) Characterization of the VP1 loop mutations widespread among JC polyomavirus isolates associated with progressive multifocal leukoencephalopathy. Biochem Biophys Res Commun 333:996–1002. [DOI] [PubMed] [Google Scholar]