

Abstract
The locus ceruleus is among the earliest affected brain regions in Parkinson's disease (PD) showing Lewy body pathology and neuronal loss. To improve our understanding of the pathogenesis of PD, we performed the first proteomic analysis ever of post‐mortem locus ceruleus tissue of six pathologically confirmed PD patients, and six age‐ and gender‐matched non‐neurological controls. In total 2495 proteins were identified, of which 87 proteins were differentially expressed in the locus ceruleus of PD patients compared with controls. The majority of these differentially expressed proteins are known to be involved in processes that have been implicated in the pathogenesis of PD previously, including mitochondrial dysfunction, oxidative stress, protein misfolding, cytoskeleton dysregulation and inflammation. Several individual proteins were identified that have hitherto not been associated with PD, such as regucalcin, which plays a role in maintaining intracellular calcium homeostasis, and isoform 1 of kinectin, which is involved in transport of cellular components along microtubules. In addition, pathway analysis suggests a pathogenetic role for aminoacyl‐tRNA‐biosynthesis. These findings indicate that the proteome of the locus ceruleus of PD patients and non‐neurological controls provides data that are relevant to the pathogenesis of PD, reflecting both known and potentially novel pathogenetic pathways.
Keywords: Neurodegenerative diseases, aminoacyl‐tRNA‐synthetases, brain proteins, human post‐mortem tissue, noradrenalin
INTRODUCTION
Parkinson's disease (PD) is neuropathologically characterized by intraneuronal protein aggregates called Lewy bodies. Lewy body pathology within the central nervous system coincides with neuronal loss and affects specific brain regions in a predictive caudal to rostral order, ascending from the lower brainstem to neocortical areas (14). The underlying pathogenetic mechanisms have not yet been fully clarified, although several processes have been implicated. Both genetic studies and post‐mortem analyses of substantia nigra tissue are in support of an important role of mitochondrial dysfunction, oxidative stress and inflammation in PD pathogenesis 24, 46, whereas impaired protein degradation and apoptosis‐mediated cell death appear to be other major pathogenetic factors (55).
The necessity to further improve our understanding of the pathogenetic mechanisms underlying PD results from the current lack of an effective causal treatment. Until now, the treatment of PD is largely limited to symptomatic dopaminergic substitution therapy. Increased insight into pathogenetic pathways and cellular processes involved in PD pathology is a prerequisite for the development of disease‐modifying therapeutic strategies. Important sources of diagnostic biomarkers or therapeutic targets are proteins, which are abundantly present in brain tissue and likely reflect disturbed brain mechanisms. The recent application of proteomics—a protein profiling technique that enables to study disease mechanisms in an unbiased way—has already revealed additional proteins and molecular mechanisms involved in PD pathogenesis, such as cytoskeletal dysregulation and neuronal plasticity 2, 35. So far, the rare comparative proteomic studies of post‐mortem brain tissue in PD patients have been confined to substantia nigra and frontal cortical regions 7, 8, 25, 30, 49, 50, 56. Tissue proteomic analyses of potentially interesting early‐affected brain regions have, as yet, not been described.
One of the early‐affected brain regions in PD is the locus ceruleus (LC), which may already be involved in preclinical stages of the disease, that is, before the appearance of the characteristic motor symptoms (14). The average loss of noradrenergic neurons in this region is around 70% in post‐mortem brain tissue of PD patients compared with controls (10). The LC is the principal site for the synthesis of noradrenaline and its neurons extensively project to the entire brain (21). It is functionally related to arousal, autonomic function and cognition 45, 58. The involvement of the LC in PD may therefore contribute to some of the prodromal non‐motor symptoms, in particular sleep disorders and autonomic dysfunction. Neuronal loss in the LC is not specific for PD. It can also be observed in other neurological disorders, including Alzheimer's disease (AD) and multiple system atrophy (MSA) 9, 20.
In the present study, we performed the first proteomic analysis on human post‐mortem LC tissue to date. By profiling the LC proteomes of PD patients and comparing them with non‐neurological controls, we aimed to identify proteins and processes that are involved in the pathogenesis of PD. In particular, we intended to identify novel pathogenetic mechanisms.
MATERIALS AND METHODS
Post‐mortem human LC tissue
Snap‐frozen post‐mortem LC tissue of six PD patients and six non‐neurological controls was obtained from the Netherlands Brain Bank (NBB; Amsterdam, the Netherlands). All donors gave written informed consent for brain autopsy and the subsequent use of their brains for research purposes. To ensure high quality post‐mortem tissue, only cases with a post‐mortem interval less than 12 h and a pH of ventricular cerebrospinal fluid between 6.3 and 7.1 were included. Only females were included, to have more homogeneous groups. PD patients were clinically and pathologically diagnosed with PD or PD dementia and had no history of cancer or concomitant disease of the central nervous system. The controls had no history of cancer, psychiatric or central nervous system diseases, and their neuropathological examinations revealed age‐related changes only. PD patients and controls were matched for age [mean ± standard deviation (SD) for PD 77.8 ± 4.4 years, for controls 77.8 ± 5.7 years; P = 1.0], post‐mortem interval (mean ± SD for PD 5:46 ± 2:20 h, for controls 7:09 ± 2:31 h; P = 0.35) and pH of ventricular cerebrospinal fluid (mean ± SD for PD 6.69 ± 0.19, for controls 6.75 ± 0.27; P = 0.67). The brainstem was dissected at the level of the inferior cerebral peduncles and directly caudally to the pons and cut into a right and left half. The left side of the brain stem including the LC was frozen in liquid nitrogen and stored at −80°C until further processing. In both PD patients and controls the presence and distribution of alpha‐synuclein and tangle pathology was evaluated and staged according to Braak 13, 14 and the BrainNet Europe protocol (1). Amyloid pathology was staged according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria (36). For immunohistochemical validation, we included two additional PD patients, one additional control and four AD patients. AD patients were clinically and pathologically diagnosed with AD and were included to evaluate the specificity of our findings. See Table 1 for the clinical and neuropathological characteristics of the included cases.
Table 1.
Clinical and neuropathological characteristics of included PD patients, AD patients and controls. Abbreviations: PD = Parkinson's disease; PDD = Parkinson's disease with dementia; CTRL = control; AD = Alzheimer's disease; PMI = post‐mortem interval in hours; CSF = cerebrospinal fluid; NA = not applicable; ND = not determined.
| Subject | Diagnosis | PD Braak stage | AD Braak stage | Amyloid load* | Disease duration (years)† | Sex | Age | PMI (h) | pH CSF | Cause of death |
|---|---|---|---|---|---|---|---|---|---|---|
| NBB06‐002 | PD | 5 | 2 | B | 12 | F | 84 | 7:25 | 6.85 | Old age and shortness of breath |
| NBB94‐092 | PD | 4 | 0 | O | 4 | F | 77 | 9:40 | 6.86 | Euthanasia |
| NBB98‐043 | PD | 5 | 1 | O | 17 | F | 81 | 4:10 | 6.86 | Pneumonia |
| NBB98‐064 | PDD | 6 | 1 | B | 16 | F | 74 | 4:35 | 6.5 | Pneumonia |
| NBB99‐014 | PDD | 6 | 2 | B | 7 | F | 72 | 3:25 | 6.46 | Pneumonia and heart failure |
| NBB99‐069 | PD | 5 | 1 | O | 15 | F | 79 | 5:20 | 6.62 | Kidney failure complicated by heart failure |
| NBB96‐078 | CTRL | 0 | 2 | ND | NA | F | 87 | 8:00 | 6.91 | Heart failure |
| NBB03‐040 | CTRL | 0 | 0 | B | NA | F | 73 | 4:00 | 6.47 | Pulmonary fibrosis, rhabdomyolysis and progressive renal insufficiency |
| NBB03‐013 | CTRL | 0 | 1 | O | NA | F | 82 | 11:30 | 6.77 | Congestive heart failure |
| NBB95‐101 | CTRL | 0 | 1 | O | NA | F | 73 | 5:30 | 6.38 | Cardiac arrest |
| NBB98‐104 | CTRL | 0 | 2 | A | NA | F | 74 | 7:25 | 6.95 | Intestinal necrosis secondary to thrombosis |
| NBB00‐032 | CTRL | 0 | 1 | A | NA | F | 78 | 6:30 | 7.02 | Heart failure |
| Additional subjects included for immunohistochemistry | ||||||||||
| NBB07‐090 | PD | 6 | 2 | B | 22 | F | 70 | 7:05 | 6.64 | Pneumonia |
| NBB07‐008 | PD | 6 | 1 | B | 6 | M | 80 | 7:05 | 6.34 | Pneumonia |
| NBB97‐043 | CTRL | 0 | ND | ND | NA | M | 68 | 10:10 | 7.08 | Cardiac arrest |
| NBB02‐050 | AD | 0 | 4 | B | 7 | F | 95 | 4:10 | 6.45 | Myocardial infarction |
| NBB02‐021 | AD | 0 | 6 | C | 6 | F | 64 | 3:40 | 6.65 | Pneumonia |
| NBB07‐027 | AD | 0 | 6 | C | 2 | M | 60 | 6:15 | 6.58 | Dehydration |
| NBB07‐080 | AD | 0 | 5 | C | 3 | F | 90 | 5:00 | 6.36 | Unknown |
The load of amyloid was scored according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) criteria (41).
Disease duration in years, starting from timepoint of clinical diagnosis.
Tissue dissection
Accurate dissection of the LC is essential to acquire useful proteomic data of this nucleus. Including non‐affected neighbouring tissue would reduce the fold change of differential proteins in the affected LC itself. In the current study we were able to accurately dissect the LC by a punch technique in a −20°C cryostat. With a 3‐mm punch, the LC was marked and a 20 µm thick section was made to microscopically verify whether the neuromelanin‐containing neurons of the LC were located within the punch. When all neuromelanin‐containing neurons of the LC were inside the punch, thicker sections were made and punches were collected in Eppendorf tubes. The punches were collected throughout the entire available nucleus, ranging from 4 to 16 mm. Sections for microscopic evaluation were made every 1.2 mm to verify the location of the punch.
Sample preparation
The punched LC of each case was homogenized in 300 µL sample buffer [50 mM Tris‐HCl pH 6.8, 8% glycerol, 1.6% sodium dodecyl sulfate (SDS), 0.002% bromophenol blue, 0.1 M dithiothreitol (DTT)] per approximately 10 mg of tissue. Samples were subsequently denatured by boiling for 5 minutes, centrifuged to remove insoluble fractions and 1.5 times diluted in sample buffer. Two samples were diluted two times in sample buffer (NBB94‐092 and NBB03‐040) and one sample was diluted three times (NBB98‐064), because of protein overloading on the gel using higher concentrations. The different dilutions did not affect the subsequent quantification, because the spectral counts were normalized on the sum of the spectral counts per case.
One dimensional gel electrophoresis and in‐gel tryptic digestion
In total 28 µL of homogenized and diluted sample per case was separated on 2 NuPAGE® Novex 4–12% Bis‐Tris gels (1.5 mm, 10 well; Invitrogen, Carlsbad CA, USA). The gels were stained with Coomassie Brilliant Blue solution (Coomassie Brilliant Blue R B‐0630; Sigma, St. Louis MO, USA; 17% ammonium sulfate, 3% phosphoric acid, 34% methanol), fixed with 50% ethanol/3% phosphoric acid, washed with MilliQ and scanned. Tryptic digestion was performed according to Shevchenko et al (48). Gels were washed and dehydrated once in 50 mM ammonium bicarbonate (ABC) and twice in 50 mM ABC/50% acetonitrile (ACN). Cysteine bonds were reduced by incubation with 10 mM DTT/50 mM ABC at 56°C for 1 h and alkylated with 50 mM iodoacetamide/50 mM ABC at room temperature (RT) in the dark for 45 minutes. After washing sequentially with ABC and ABC/50% ACN, we sliced the whole gel lane of each case into 10 parts. Gel parts were sliced up into approximately 1‐mm cubes and collected in tubes, washed in ABC/ACN and dried in a vacuum centrifuge. Gel cubes were incubated overnight at 23°C with 6.25 ng/mL trypsin and covered with ABC to allow digestion. Peptides were extracted once in 1% formic acid and twice in 5% formic acid/50% ACN. The volume was reduced to 50 µL in a vacuum centrifuge prior to LC‐MS analysis.
Nano‐LC separation and tandem mass spectrometry (MS/MS)
Peptides were separated by an Ultimate 3000 nano‐LC system (Dionex LC‐Packings, Amsterdam, the Netherlands) as described in Piersma et al (43). Intact peptide MS spectra and MS/MS spectra were acquired on an LTQ‐FT hybrid mass spectrometer (Thermo Fisher, Bremen, Germany) as described previously 3, 43. Intact masses were measured at 50.000 resolution and the top five most intense signals were subjected to collision‐induced dissociation (CID) in the linear ion trap.
Protein identification, quantification, statistics, pathway analysis and protein classification
MS/MS spectra were searched against the human International Protein Index (IPI) database 3.59 (80, 128 entries) using Sequest (version 27, rev 12, Thermo Fisher Scientific, San Jose, CA, USA) with a maximum allowed deviation of 10 ppm for the precursor mass and 1 amu for fragment masses. Scaffold 2.02.03 (Proteomesoftware, Portland, OR, USA) was used to organize the gel‐slice data and to validate peptide identifications using the PeptideProphet 27, 37 algorithm; only identifications with a probability >95% were retained. Subsequently, the ProteinProphet 27, 37 algorithm was applied to group peptide identifications into protein identifications. Protein identifications with a probability of >99% with two peptides or more in at least one of the samples were retained (P < 0.01). For each protein identified, the number of spectral counts (the number of MS/MS spectra associated with an identified protein) was exported to Excel. For quantitative analysis across samples, spectral counts were normalized to the sum of the spectral counts per biological sample. Each sample was separated in 10 fractions that were subjected to nano‐LC‐MS/MS. Spectral counts for identified proteins in a sample were summed across all fractions for each sample and were normalized to the total sum of spectral counts for that sample (42). This gives the relative spectral count contribution of a protein to all spectral counts in the sample. When comparing different datasets, these normalized spectral counts were used to calculate ratios. In this way, we were able to correct for loading differences between samples. Differential analysis of samples was performed using the beta‐binominal test, which takes into account within‐ and between‐sample variations, giving fold‐change values and associated P‐values for all identified proteins (42). Further protein identification and quantification details can be found in 42, 43. Protein cluster analysis of the differentially expressed proteins was performed using hierarchical clustering in R. The protein abundances were normalized to zero mean and unit variance for each individual protein. Subsequently, the Euclidean distance measure was used for protein clustering. The differentially expressed proteins were imported in the online software package Ingenuity Pathway Analysis (Ingenuity IPA, version 8.8, Ingenuity Systems, Redwood City, CA, USA). Pathway analysis was performed with only direct relationships and P‐values were obtained and adjusted under Benjamini‐Hochberg (B‐H) multiple testing corrections. Proteins were functionally classified according to the PANTHER classification (http://www.pantherdb.org). They were assigned to hypothesized pathogenetic pathways for PD based on their GO annotation (6) and Entrez Gene information (http://www.ncbi.nlm.nih.gov/gene) and compared with the proteomic data of previous human brain tissue studies in PD 7, 8, 25, 30, 49, 50, 56.
Western blotting
The expression levels of selected proteins were validated using Western blotting in LC homogenates of the six PD patients and six controls that were also included in our proteomic analysis (for the case characteristics see Table 1). The selected proteins were tyrosine hydroxylase (TH), gammabutyrobetaine dioxygenase (BBOX1), phenylalanyl‐tRNA synthetase alpha chain (FARSA), isoform 1 of kinectin (KTN1) and complement component C4B (C4B). Total protein amounts were measured with a bicinchoninic acid assay (BCA; Pierce BCA Protein Assay Kit #23225, Thermo Scientific, Rockford IL, USA). Ten micrograms of protein per case were loaded and separated on SDS‐12% or SDS‐8% polyacrylamide gels under reduced conditions and blotted onto a PVDF membrane (Immobilon, Millipore, Billerica MA, USA). After blockage with 5% (w/v) non‐fat dry milk for 30 minutes, the blots were probed with: (i) mouse monoclonal antibody against TH (1:2000, MAB318, Chemicon, Billerica MA, USA); (ii) rabbit polyclonal antibody against BBOX1 (1:250, HPA007600, Atlas Antibodies, Stockholm, Sweden); (iii) rabbit polyclonal antibody against FARSA (1:1000, HPA001911, Atlas Antibodies); (iv) rabbit polyclonal antibody against KTN1 (1:250, HPA003178, Atlas Antibodies); (v) rabbit polyclonal antibody against C4B (1:1000, GTX110502, Genetex, Irvine, CA, USA) or mouse monoclonal antibody against actin (1:200.000, Ab6276, Abcam, Cambridge, MA, USA) overnight at 4°C and incubated for 1 h at RT with horseradish peroxidase‐conjugated secondary antibody. Actin was included as a reference protein that should not change in expression level between samples or groups. Actin was loaded on a separate gel because of the overlap of its molecular weight with the other measured proteins. Labeling was detected using a chemiluminescent substrate (Supersignal West Dura #34076, Thermo Scientific) and quantified with Chemidoc XRS Imaging System (Bio‐Rad, Hercules CA, USA) and Quantity One Analysis Software (Bio‐Rad) after background correction. If more than one band was present, we selected the band that corresponded with the expected molecular weight for quantification. Statistical analysis was performed using unpaired, one‐tailed Student's t‐tests.
Immunohistochemical validation
Immunohistochemical stainings of alpha‐synuclein and two selected proteins were performed on paraffin embedded post‐mortem LC tissue of four PD patients, four controls and four AD patients obtained from the NBB. For this validation we included post‐mortem LC tissue of two PD patients (NBB06‐002, NBB99‐014) and three controls (NBB00‐032, NBB03‐040, NBB03‐013) that were also included in the proteome discovery, with two additional PD patients (NBB07‐090, NBB07‐008) and one additional non‐neurological control (NBB97‐043; see Table 1). AD patients were included to evaluate the expression of the two selected proteins in another neurodegenerative disease in which LC neuronal loss occurs, which could inform us about the specificity of our findings.
We focused on one protein of which the level was significantly increased in the LC of PD patients compared with controls, that is, BBOX1, and one protein of which the level was significantly decreased, that is, FARSA. By examining their localizations—guided by the Nissl staining—we aimed to find explanations for the altered levels of these proteins and their relationship to the alpha‐synuclein pathology and cell death in the LC.
Ten‐micrometer‐thick transversal sections were collected throughout the LC using a paraffin microtome (Microm HM340E, Mikron Instruments Inc., San Marcos, CA, USA).
Sections for alpha‐synuclein immunostaining were deparaffinized, rehydrated and washed in 95% formic acid, aquadest and Tris‐buffered saline (TBS), followed by incubation in 0.3% hydrogen peroxide diluted in ethanol for 15 minutes in order to eliminate endogenous peroxidase activity. Subsequently, the sections were incubated with mouse monoclonal antibody against alpha‐synuclein (1:2000, #610786, BD Transduction Laboratories, Franklin Lakes, NJ, USA) diluted in TBS‐Triton with 2% BSA for two nights at 4°C. After extensive washes with TBS, we incubated the sections for 1 h at RT with horseradish peroxidase‐conjugated anti‐mouse secondary antibody (1:200) and for 1 h with avidin‐biotin‐peroxidase complex (1:200; ABC Kit, Vector Laboratories, Burlingame, CA, USA). Staining was visualized using 3,3′‐diaminobenzidine (DAB) as chromogen. Sections were counterstained with thionine and coverslipped using Entellan (Merck Chemicals, Darmstadt, Germany).
Sections for BBOX1 and FARSA were deparaffinized, rehydrated and washed in PBS followed by heat‐induced antigen retrieval in citrate‐buffer pH 6. Subsequently, the sections were depleted of neuromelanin using potassium permanganate, potassium metabisulfite and oxalic acid in order to evaluate the cellular localization. After the above‐described antigen retrieval, sections were washed in TBS and incubated in rabbit polyclonal antibody against BBOX1 (1:100, HPA007600, Atlas Antibodies) or rabbit polyclonal antibody against FARSA (1:4000, HPA001911, Atlas Antibodies) diluted in TBS‐Triton with 1% BSA and 5% normal donkey serum for 1 h at RT and two nights at 4°C. After extensive washes with TBS, sections were incubated in 0.3% hydrogen peroxide to eliminate endogenous peroxidase activity, again washed with TBS and incubated with horseradish peroxidase‐conjugated secondary antibody for 1 h and an additional hour with ABC (Vector Laboratories). Staining was visualized using DAB and sections were dehydrated and coverslipped using Entellan. Negative control sections were treated identically, except for the primary antibody that was omitted. There was no immunostaining observed in these negative control sections. Photographs (200× and 630× magnification) were captured using a Leica microscope (LEICA DMR, Leica Microsystems BV, Rijswijk, The Netherlands) equipped with a CCD Microfire (Optronics, Goleta, CA, USA) color video camera using Neurolucida software version 9 (Microbrightfield Inc., Colchester, VT, USA).
RESULTS
Differential protein levels and pathogenetic pathways in the LC in PD
One‐dimensional gel‐electrophoresis of LC homogenates followed by in‐gel tryptic digestion, nano‐LC‐FTMS/MS and database searching resulted in the identification of 2495 proteins. The overall protein profiles of the LC in PD patients and controls were very similar. Functional protein classification of all LC proteins using PANTHER classification indicated that the most represented functional classes were transferases (8.7% of 2495 proteins), nucleic acid binding proteins (8.6%), oxidoreductases (7.5%), enzyme modulators (7.1%) and transporters (7.1%). We refer to the supplementary material for the Coomassie Blue‐stained protein gel images of LC lysates (Supporting Information Figure S1), the full list of identified proteins including the raw and normalized spectral counts (Supporting Information Table S1) and a pie chart of all represented functional protein classes (Supporting Information Figure S2).
Quantitative analysis of normalized protein spectral counts and statistical analysis yielded 87 proteins that had significantly different expression levels when comparing PD patients with controls, with 33 upregulated and 54 downregulated proteins (Table 2). We refer to Supporting Information Figure S3 for a heat map view of the cluster analysis using the differentially regulated proteins. Based on the GO annotation of cellular component (6), the differentially expressed proteins were predominantly localized in the cytoplasm (50.6%), cytosol (20.7%), mitochondrion (19.5%), membrane (general 19.5%; plasma membrane 18.4%; endoplasmic reticulum membrane 10.3%), nucleus (18.4%), endoplasmic reticulum (12.6%) or were attached to the membrane (17.2%). Note that the sum of percentages exceeds 100% because of multiple cellular components for certain proteins.
Table 2.
List of differentially expressed proteins in the locus ceruleus of PD patients compared with controls ranked on fold change. Abbreviations: IPI = International Protein Index; PD = Parkinson's disease.
| Protein name | IPI number | Gene symbol | Fold change | P‐value | Functional classification* |
|---|---|---|---|---|---|
| Dopamine beta‐hydroxylase | IPI00171678 | DBH | −∞ | 0.00014 | Hydroxylase |
| Isoform 2 of ELAV‐like protein 4 | IPI00395507 | ELAVL4 | −∞ | 0.0040 | mRNA processing factor |
| Isoform 1 of uncharacterized protein KIAA0513 | IPI00028516 | KIAA0513 | −∞ | 0.0040 | None |
| 28S ribosomal protein S6, mitochondrial | IPI00305668 | MRPS6 | −∞ | 0.0073 | Ribosomal protein |
| Serine/threonine‐protein kinase Nek7 | IPI00152658 | NEK7 | −∞ | 0.0086 | Protein kinase |
| Putative uncharacterized protein MRPL23 | IPI00658024 | MRPL23 | −∞ | 0.015 | Ribosomal protein |
| 7‐dehydrocholesterol reductase | IPI00294501 | DHCR7 | −∞ | 0.016 | Receptor |
| 28S ribosomal protein S34, mitochondrial | IPI00169413 | MRPS34 | −∞ | 0.021 | None |
| Galectin‐3 | IPI00465431 | LGALS3 | −∞ | 0.023 | Signaling molecule |
| Isoform 2 of tyrosine 3‐monooxygenase | IPI00218276 | TH | −23.965 | 0.00013 | Oxygenase |
| Neuroglobin | IPI00009758 | NGB | −13.079 | 0.0030 | Transporter |
| HESB like domain containing 2, isoform CRA_b | IPI00329615 | ISCA1 | −8.286 | 0.013 | None |
| Probable E3 ubiquitin‐protein ligase HERC1 | IPI00022479 | HERC1 | −6.165 | 0.048 | Chromatin/chromatin‐binding protein |
| Resistance to inhibitors of cholinesterase 8 homolog A | IPI00100106 | RIC8A | −5.577 | 0.048 | None |
| Isoform 2 of serine/threonine‐protein kinase PAK 3 | IPI00027382 | PAK3 | −4.659 | 0.0046 | Protein kinase |
| Isoform 2 of small G protein signaling modulator 1 | IPI00514149 | SGSM1 | −4.623 | 0.045 | Hydrolase |
| Isoform 1 of regulator of nonsense transcripts 1 | IPI00034049 | UPF1 | −3.444 | 0.0018 | DNA helicase |
| Thioredoxin domain‐containing protein 4 | IPI00401264 | ERP44 | −3.426 | 0.033 | Isomerase |
| Gamma‐aminobutyric acid receptor‐associated protein | IPI00027253 | GABARAP | −3.418 | 0.043 | Non‐motor microtubule binding protein |
| Carbonyl reductase [NADPH] 3 | IPI00290462 | CBR3 | −3.352 | 0.048 | Dehydrogenase |
| Brefeldin A‐inhibited guanine nucleotide‐exchange protein 3 | IPI00514897 | KIAA1244 | −3.347 | 0.0011 | Guanyl‐nucleotide exchange factor |
| Rap guanine nucleotide exchange factor 2 | IPI00853219 | RAPGEF2 | −2.984 | 0.042 | Guanyl‐nucleotide exchange factor |
| Isoleucyl‐tRNA synthetase, cytoplasmic | IPI00644127 | IARS | −2.954 | 0.015 | Amino‐acyl tRNA synthetase |
| Isoform 1 of nitric oxide synthase, brain | IPI00217225 | NOS1 | −2.842 | 0.037 | Nuclease |
| Arginine‐rich, mutated in early stage tumors | IPI00328748 | MANF | −2.759 | 0.037 | None |
| Coatomer subunit beta | IPI00295851 | COPB1 | −2.718 | 0.013 | Vesicle coat protein |
| Isoform 1 of Rab3 GTPase‐activating protein non‐catalytic subunit | IPI00554590 | RAB3GAP2 | −2.539 | 0.0034 | G‐protein modulator |
| GrpE protein homolog 1, mitochondrial | IPI00029557 | GRPEL1 | −2.454 | 0.019 | Cytoskeletal protein |
| Phenylalanyl‐tRNA synthetase alpha chain | IPI00031820 | FARSA | −2.432 | 0.0091 | Amino‐acyl tRNA synthetase |
| Isoform Long of cold shock domain‐containing protein E1 | IPI00470891 | CSDE1 | −2.239 | 0.041 | None |
| Isoform 2 of protein tweety homolog 1 | IPI00010183 | TTYH1 | −2.113 | 0.048 | Anion channel |
| Protein FAM127A | IPI00816474 | FAM127A | −2.104 | 0.011 | None |
| Isoform 2 of ankyrin‐3 | IPI00472779 | ANK3 | −2.037 | 0.0061 | Cytoskeletal protein |
| Isoform 1 of acyl‐coenzyme A thioesterase 9, mitochondrial | IPI00220710 | ACOT9 | −2.006 | 0.0084 | Esterase |
| Phospholipase D3 | IPI00328243 | PLD3 | −1.983 | 0.038 | Phospholipase |
| Isoform 1 of kinectin | IPI00328753 | KTN1 | −1.972 | 0.0012 | Receptor |
| Dolichyl‐diphosphooligosaccharide‐protein glycosyltransferase subunit 1 precursor (Ribophorin I) | IPI00025874 | RPN1 | −1.910 | 0.020 | Glycosyltransferase |
| LYR motif‐containing protein 4 | IPI00006979 | LYRM4 | −1.836 | 0.030 | None |
| Tumor protein D52 isoform 2 | IPI00619951 | TPD52 | −1.808 | 0.037 | None |
| Isoform 1 of UDP‐glucose: glycoprotein glucosyltransferase 1 | IPI00024466 | UGGT1 | −1.790 | 0.015 | Glycosyltransferase |
| Protein kinase, cAMP‐dependent. regulatory, type II, alpha, isoform CRA_b | IPI00063234 | PRKAR2A | −1.783 | 0.040 | Kinase modulator |
| Flotillin‐1 | IPI00027438 | FLOT1 | −1.714 | 0.014 | Membrane traffic protein |
| Seipin | IPI00479357 | BSCL2 | −1.683 | 0.014 | None |
| FK506‐binding protein 4 | IPI00219005 | FKBP4 | −1.667 | 0.019 | Isomerase |
| Phenylalanyl‐tRNA synthetase beta chain | IPI00300074 | FARSB | −1.655 | 0.018 | Nucleic acid binding |
| Upregulated during skeletal muscle growth protein 5 | IPI00063903 | USMG5 | −1.554 | 0.017 | None |
| Prolow‐density lipoprotein receptor‐related protein 1 | IPI00020557 | LRP1 | −1.537 | 0.043 | Receptor |
| Isoform 1 of Mammalian ependymin‐related protein 1 | IPI00259102 | EPDR1 | −1.516 | 0.032 | None |
| cDNA FLJ56389, highly similar to Elongation factor 1‐gamma | IPI00000875 | EEF1G | −1.352 | 0.030 | Anion channel |
| A kinase (PRKA) anchor protein 12 isoform 2 | IPI00217683 | AKAP12 | −1.265 | 0.011 | None |
| Gamma‐synuclein | IPI00297714 | SNCG | −1.255 | 0.020 | Signaling molecule |
| cDNA FLJ56034, highly similar to 4‐aminobutyrate aminotransferase, mitochondrial | IPI00009532 | ABAT | −1.249 | 0.0095 | Transaminase |
| Isoform IA of synapsin‐1 | IPI00300568 | SYN1 | −1.208 | 0.034 | Membrane trafficking regulatory protein |
| Leucine‐rich PPR motif‐containing protein, mitochondrial | IPI00783271 | LRPPRC | −1.193 | 0.042 | Transporter |
| Isoform alpha‐enolase of alpha‐enolase | IPI00465248 | ENO1 | 1.152 | 0.049 | Lyase |
| Isoform M1 of pyruvate kinase isozymes M1/M2 | IPI00220644 | PKM2 | 1.155 | 0.00060 | Carbohydrate kinase |
| Creatine kinase B‐type | IPI00022977 | CKB | 1.178 | 0.015 | Amino acid kinase |
| gelsolin isoform c | IPI00647556 | GSN | 1.181 | 0.046 | Non‐motor actin binding protein |
| Carbonic anhydrase 2 | IPI00218414 | CA2 | 1.209 | 0.050 | Dehydratase |
| Isoform V0 of Versican core protein | IPI00009802 | VCAN | 1.258 | 0.046 | Extracellular matrix glycoprotein |
| Adenylate kinase isoenzyme 1 | IPI00018342 | AK1 | 1.277 | 0.033 | Nucleotide kinase |
| Ferritin heavy chain | IPI00554521 | FTH1 | 1.292 | 0.019 | Storage protein |
| Protein‐arginine deiminase type‐2 | IPI00294187 | PADI2 | 1.337 | 0.0010 | Hydrolase |
| Isoform 1 of cytosol aminopeptidase | IPI00419237 | LAP3 | 1.355 | 0.018 | Metalloprotease |
| Vimentin | IPI00418471 | VIM | 1.369 | 0.020 | Structural protein |
| Inositol monophosphatase | IPI00020906 | IMPA1 | 1.374 | 0.038 | Phosphatase |
| V‐crk sarcoma virus CT10 oncogene homolog isoform b | IPI00305469 | CRK | 1.926 | 0.016 | None |
| Isoform 1 of regucalcin | IPI00017551 | RGN | 1.930 | 0.047 | Esterase |
| Transgelin | IPI00216138 | TAGLN | 1.971 | 0.037 | Non‐motor actin binding protein |
| Gamma‐butyrobetaine dioxygenase | IPI00027416 | BBOX1 | 2.155 | 0.020 | Hydroxylase |
| Ribosyldihydronicotinamide dehydrogenase [quinone] | IPI00219129 | NQO2 | 2.191 | 0.046 | None |
| HLA class I histocompatibility antigen, B‐59 alpha chain | IPI00472073 | HLA‐B | 2.987 | 0.0071 | Immunoglobulin receptor family |
| Complement component 4B | IPI00887154 | C4B | 3.063 | 0.0078 | None |
| Wolframin | IPI00008711 | WFS1 | 3.373 | 0.015 | None |
| Putative uncharacterized protein DKFZp686I04196 (Fragment) | IPI00399007 | IGHG2 | 3.621 | 0.019 | Immunoglobulin |
| c‐Myc‐responsive protein Rcl | IPI00007926 | C6ORF108 | 4.234 | 0.048 | None |
| cDNA FLJ55177, highly similar to ras‐related protein Ral‐B | IPI00004397 | RALB | 4.374 | 0.027 | Small GTPase |
| Mitochondrial glutamate carrier 2 | IPI00027826 | SLC25A18 | 7.117 | 0.032 | Amino acid transporter |
| Isoform 1 of NHL repeat‐containing protein 2 | IPI00301051 | NHLRC2 | 7.357 | 0.024 | None |
| Aldehyde dehydrogenase X, mitochondrial | IPI00103467 | ALDH1B1 | 7.522 | 0.033 | Dehydrogenase |
| Methylmalonyl‐CoA epimerase, mitochondrial | IPI00107722 | MCEE | 7.905 | 0.012 | DNA glycosylase |
| Isoform 2 of endoplasmic reticulum aminopeptidase 1 | IPI00165949 | ERAP1 | 9.501 | 0.020 | Metalloprotease |
| SAT2 protein | IPI00744810 | SAT2 | ∞ | 0.022 | Acetyltransferase |
| Isoform 1 of catenin alpha‐1 | IPI00215948 | CTNNA1 | ∞ | 0.021 | Non‐motor actin binding protein |
| Hemopexin | IPI00022488 | HPX | ∞ | 0.020 | Transfer/carrier protein |
| Podocalyxin‐like protein 1 precursor | IPI00299116 | PODXL | ∞ | 0.0083 | None |
| HLA class II histocompatibility antigen, DRB1‐1 beta chain | IPI00738107 | HLA‐DRB1 | ∞ | 0.0021 | Major histocompatibility complex antigen |
According to functional PANTHER classification (http://www.pantherdb.org).
Sixty‐one out of 87 differentially expressed proteins were associated with processes that have previously been implicated in the pathogenesis of PD (Figure 1). These included oxidative stress (11.5% of the 87 differentially expressed proteins), mitochondrial dysfunction (10.3%), cytoskeleton structure and regulation (10.3%), inflammation and glial activation (8%) and synaptic neurotransmission (6.9%).
Figure 1.
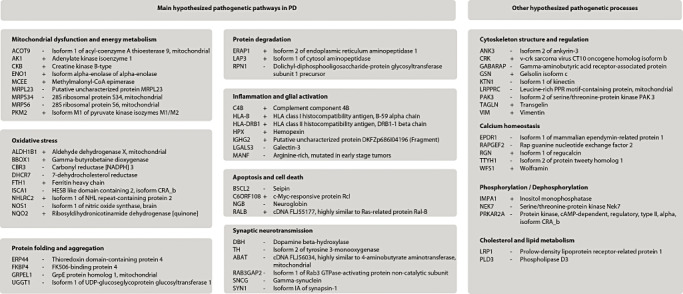
Listing of differentially expressed proteins associated with known pathogenetic pathways in Parkinson's disease (PD). Plus and minus signs indicate increased (+) or decreased (−) protein levels in PD patients compared with controls.
Ingenuity pathway analysis of the differentially expressed proteins led to three significantly altered pathways: aminoacyl‐tRNA‐biosynthesis (P = 0.005), arginine and proline metabolism (P = 0.005) and phenylalanyl, tyrosine and tryptophan biosynthesis (P = 0.005). The aminoacyl‐tRNA‐biosynthesis pathway included isoform 1 of Rab3 GTPase‐activating protein non‐catalytic subunit (RAB3GAP2), cytoplasmic isoleucyl‐tRNA synthetase (IARS), FARSA and phenylalanyl‐tRNA synthetase beta chain (FARSB) whereas creatine kinase B‐type (CKB), isoform 1 of nitric oxide synthase (NOS1), mitochondrial aldehyde dehydrogenase X (ALDH1B1), SAT2 protein (SAT2) and isoform 1 of cytosol aminopeptidase (LAP3) were involved in arginine and proline metabolism. The proteins implicated in phenylalanyl, tyrosine and tryptophan biosynthesis partly overlapped with the aminoacyl‐tRNA‐biosynthesis proteins: isoform alpha‐enolase of alpha‐enolase (ENO1), FARSA and FARSB.
Validation using Western blotting and immunohistochemistry
The expression of a selection of proteins was analyzed using both Western blotting and immunohistochemistry in order to confirm the direction of change of these proteins in PD patients compared with controls using a different, antibody‐based approach (2, 3; Supporting Information Figure S4). Proteins were selected for Western blotting and/or immunohistochemistry using the following criteria: (i) P‐value lower than 0.05; (ii) positive or negative fold change equal to or higher than 2.0; and (iii) commercial availability of antibodies. From the 48 proteins that fulfilled these criteria, we selected both up‐ and downregulated proteins that were involved in different pathways. We furthermore took into account the quality of antibodies and selected antibodies that were specific for the detected isoform. The selected proteins were TH, FARSA, BBOX1, KTN1 and C4B.
Figure 2.
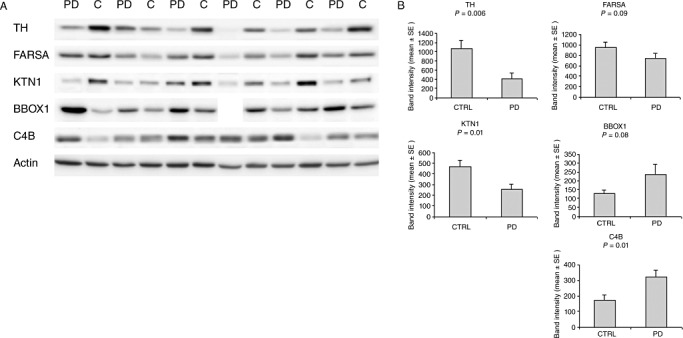
Confirmation of proteomic candidates by Western blotting. A. Western blots for the five proteins that were selected for technical validation. Actin was included as a control protein that should not change between samples. B. Bar graphs of average band intensities for the blotted proteins. Data for BBOX1 of the fourth PD patient from the left were excluded from quantitative analysis because of an artifact. Abbreviations: TH = tyrosine hydroxylase; FARSA = phenylalanyl‐tRNA synthetase alpha chain; KTN1 = isoform 1 of kinectin; BBOX1 = gammabutyrobetaine dioxygenase; C4B = complement component 4B.
Figure 3.
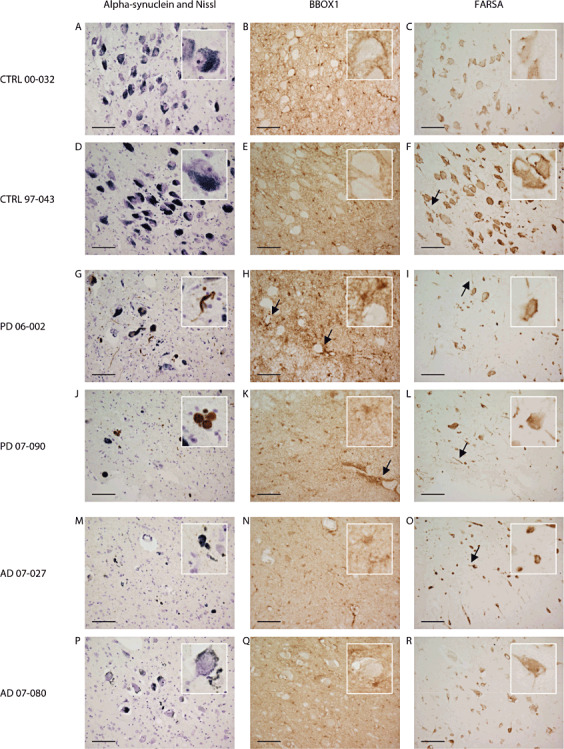
Localization of phenylalanyl‐tRNA synthetase alpha chain (FARSA) and gammabutyrobetaine dioxygenase (BBOX1) by immunohistochemistry in paraffin sections (10 µm) of the locus ceruleus of PD patients, controls and AD patients. Alpha‐synuclein and Nissl staining of the locus ceruleus (LC) of controls (A,D), PD patients (G,J) and AD patients (M,P) revealed neuronal loss in PD and AD patients and alpha‐synuclein immunostained Lewy bodies and Lewy neurites in PD patients. BBOX1 was detected in both soma and cellular processes of glial cells of the LC of controls (B,E), PD patients (H,K) and AD patients (N,Q); arrows in picture H indicate BBOX1 staining surrounding LC neurons in a PD patient with relatively little cell death; enriched staining pattern in blood vessel endothelium is pointed out by arrow in picture K. In controls (C,F), PD (I,L) and AD patients (O,R) FARSA staining in the LC was localized in the cytoplasm and cellular processes of both neuronal and glial cells. In the cytoplasm of neurons, FARSA staining was diffuse and granular, predominantly near the cell borders and the nuclear membrane; arrows denote FARSA staining in a number of glial and neuronal processes. Scale bar = 1 µm.
For all proteins analyzed, the Western blot data supported the results of the proteomic analysis: the levels of TH, FARSA, and KTN1 were decreased (TH −62% compared with controls, FARSA −23%, KTN1 −44%) whereas the levels of BBOX1 and C4B were increased in PD compared with controls (BBOX1 +85% compared with controls; C4B +88%; Figure 2). For TH (P = 0.006), KTN1 (P = 0.01) and C4B (P = 0.01) Western blot data were significantly different, P‐values for FARSA (P = 0.09) and BBOX1 (P = 0.08) indicated a trend. The expression level of the reference protein actin was not significantly different between the PD and control group (P = 0.44). These data confirmed the proteomic data for cytoplasmic actin (fold‐change PD compared with controls 1.07; P = 0.27).
Alpha‐synuclein and Nissl stainings on the sections of all PD patients and none of the controls revealed alpha‐synuclein‐immunoreactive Lewy bodies and Lewy neurites and neuronal loss in the LC. Neuronal loss in the LC was also observed in all AD patients, although the degree of cell loss was highly variable. Lewy bodies and Lewy neurites were not seen in the LC of AD patients.
Microscopical examination of BBOX1 immunostaining revealed that this protein was localized in both soma and cellular processes of glial cells, but not in neurons, in the LC of PD patients, AD patients and controls (Figure 3B,E,H,K,N,Q).
BBOX1 staining in glial cells surrounding LC neurons was observed in PD cases with relatively little neuronal loss in the LC (as in Figure 3H). This staining pattern was less pronounced in PD cases with more extensive neuronal death and in all four AD cases (as in Figure 3K,N,Q). In contrast to the PD cases, we did not observe a relation between BBOX1 expression and the extent of neuronal loss in the AD cases. BBOX1 was furthermore enriched in blood vessel endothelium and ependymal cells in all cases (pointed out by arrow in Figure 3K).
FARSA was localized in the cytoplasm and cellular processes of both neuronal and glial cells in PD patients as well as AD cases and controls (Figure 3C,F,I,L,O,R). The number of FARSA‐immunoreactive neurons in the LC of PD patients and AD patients was markedly decreased compared with the controls. Although localization in cellular processes was seen in all cases, including the controls, it appeared that this localization was more profound in PD and AD patients, which indicates that our findings for FARSA may not be specific for PD. The distribution of FARSA was diffuse and granular in the cytoplasm of neurons and appeared enriched near the cell membrane and the nuclear membrane.
DISCUSSION
By profiling protein expression of accurately dissected post‐mortem LC tissue in PD patients and comparing with LC tissue of non‐neurological controls, we aimed to identify proteins and processes that are involved in the pathogenesis of PD and to obtain new pathogenetic insights. To our knowledge this is the most comprehensive proteomic analysis of post‐mortem PD brain tissue to date, and the first analysis of the human LC proteome. Until now, comparative proteomic analysis of human post‐mortem tissue of PD patients had been restricted to substantia nigra and frontal cortical regions 7, 8, 25, 30, 49, 50, 56 and had largely been restricted to analysis of pooled patient samples 25, 30, 49, 50. Importantly, we analyzed a series of individual patient samples and matched controls. The most important finding of our study is that the LC proteome of PD patients not only reflects alterations in pathways and processes that were previously implicated in PD pathogenesis, most importantly mitochondrial dysfunction, oxidative stress and cytoskeletal dysregulation, but also reveals less known and hitherto unknown PD‐related proteins and pathogenetic pathways, in particular aminoacyl‐tRNA‐biosynthesis.
The most significantly downregulated proteins in our dataset, TH and dopamine beta‐hydroxylase (DBH) reflect the expected catecholaminergic neuronal loss in the LC of PD patients. Both proteins are enzymes involved in the synthesis of noradrenaline: TH is responsible for the conversion of tyrosine into L‐dopa and DBH converts dopamine into noradrenaline.
Three mitochondrial ribosomal proteins—proteins that play a central role in translation within the mitochondrion (28)—were present in the top‐10 list of downregulated proteins (see Table 1), which supports a pathogenetic role of mitochondrial dysfunction in PD (46): mitochondrial 28S ribosomal protein S6 (MRPS6), mitochondrial 28S ribosomal protein S34 (MRPS34) and mitochondrial ribosomal protein L23 (MRPL23). Interestingly, the gene encoding MRPS6, was previously identified in a gene expression study in post‐mortem PD and control brains as a dysregulated gene in PD (39).
The mitochondrial protein ALDH1B1, as well as several other differentially regulated proteins in our dataset, including ferritin heavy chain (FTH1), catalyze oxidation‐reduction reactions (6) that may link them with increased oxidative stress as previously observed in affected post‐mortem brain tissue in PD (46). The increased levels of FTH1 were in agreement with previous proteomic and gene expression data of the substantia nigra 29, 56.
The most upregulated protein in our dataset was the DRB1‐1 beta chain of HLA class II histocompatibility antigen (HLA‐DRB1), which plays a central role in the immune response by antigen processing and presentation (6). Several other inflammatory molecular markers were increased in the LC of PD patients in our dataset including HLA class I HLA B‐59 alpha chain (HLA‐B) and C4B. Inflammation may be a non‐specific secondary response to neuronal damage in the LC. As noradrenalin can suppress inflammation (19), the loss of noradrenergic neurons may contribute to increased levels of inflammatory markers. Inflammatory changes in the LC have been reported before, in particular mild astro‐ and microglia activation (10).
In the present study, we observed altered levels of three proteins that are responsible for protein degradation, a process that is impaired in PD (55). The levels of two aminopeptidases were increased in PD patients compared with controls: isoform 2 of endoplasmic reticulum aminopeptidase 1 (ERAP1) and LAP3. Aminopeptidases are high‐abundant proteolytic enzymes in the brain (17) and altered plasma levels of cystine‐ and aspartate‐aminopeptidases have previously been related to PD (18).
Several cytoskeletal proteins were dysregulated in our dataset, including ankyrin and vimentin. The elevated vimentin levels match with the higher vimentin gene expression previously observed in the PD substantia nigra 12, 29. We were furthermore intrigued by a more than fourfold decrease in the levels of the classic axon‐guidance molecule serine/threonine‐protein kinase PAK 3 (PAK3) in the LC of PD patients. Genome‐wide association datasets indicate that this protein is associated with a genetic risk for PD (34).
A cytoskeleton‐related protein that has not yet been described for PD is KTN1, which expression was decreased in our PD patients. KTN1 is an integral membrane protein primarily located in the endoplasmic reticulum (53) that binds to kinesin, a motor protein responsible for moving cellular components along microtubules in both axons and dendrites (54). The family of kinesins have previously been related to Alzheimer's and Huntington's disease (23). The possible involvement of the cytoskeleton in the pathogenesis of PD has also been put forward in recent proteomic studies on lymphocytes (35) and a PD cellular model (2).
Adding to alterations in cytoskeletal proteins, we also observed changes in proteins related to protein folding. Mitochondrial GrpE protein homolog 1 (GRPEL1), FK506‐binding protein 4 (FKBP4), thioredoxin domain‐containing protein 4 (ERP44) and isoform 1 of UDP‐glucose:glycoprotein glucosyltransferase 1 (UGGT1) were all downregulated in the PD patients. These proteins play a role in protein folding either as a component of the Hsp70 or Hsp90 chaperone machinery (GRPEL1, FKBP4) 11, 32, as a endoplasmic reticulum folding assistant (ERP44) (4) or by reglucosylating unfolded glycoproteins (UGGT1) (52).
Interestingly, we also found differences in expression levels of proteins involved in cellular calcium homeostasis, including regucalcin and the transmembrane endoplasmic reticulum protein wolframin. Wolframin was previously linked with PD: a single‐nucleotide polymorphism mutation in the gene that encodes wolframin was associated with a higher risk for the disease (47). According to our knowledge, regucalcin has not previously been linked with PD. Regucalcin plays a role in maintaining intracellular calcium homeostasis by activating Ca2+ pump enzymes (57). The general association between calcium and PD pathogenesis was suggested by the possible neuroprotective role of centrally acting L‐type calcium channel blockers in PD (44).
In contrary to most other differentially expressed proteins in our dataset, isoform 2 of ELAV‐like protein 4 (ELAVL4), which was detected in five out of six controls, but in none of the PD patients, could not be related to a known pathogenetic pathway. ELAVL4 is an mRNA binding protein and implicated in neuronal differentiation and maintenance (16). Genetic variants of ELAVL4 have been associated with the risk for PD 16, 22 and its age of onset 33, 38.
Finally, it is noteworthy to address the expression of the protein alpha‐synuclein, which is a major component of Lewy bodies. Alpha‐synuclein expression levels in the LC of PD patients were not different from the controls in our dataset. The literature has been contradictory on the total amounts of alpha‐synuclein in affected regions in PD patients compared with controls [reviewed in (51)]. A recent large post‐mortem study on alpha‐synuclein expression in PD, MSA and progressive supranuclear palsy indicated increased levels of membrane‐associated alpha‐synuclein in PD patients compared with controls, whereas the levels in the cytosolic fraction were normal (51).
In total, 70% of the differentially expressed proteins could be related to processes that have previously been implicated in the pathogenesis of PD. However, our pathway analysis did not detect significant alterations in these pathways. This may be explained by the small number of cases in combination with using conservative statistics, that is, P‐values of the pathways were adjusted under B‐H multiple testing corrections. Not correcting for multiple measurements did yield several previously implicated processes, including processes involved in energy metabolism and inflammation, such as pyruvate metabolism (P = 0.007), glycolysis/gluconeogenesis (P = 0.01) and the antigen presentation pathway (P = 0.02).
After multiple testing corrections, we detected significant alterations in three molecular pathways: (i) aminoacyl‐tRNA‐biosynthesis; (ii) arginine and proline metabolism; and (iii) phenylalanyl, tyrosine and tryptophan biosynthesis. All three pathways are implicated in the amino‐acid cycle. Two of these pathways were reported in a PD gene expression study (12): aminoacyl‐tRNA‐biosynthesis was one of the several significantly altered gene groups in the substantia nigra, and the expression of genes related to arginine and proline metabolism was altered in the putamen of PD patients compared with controls.
In our study, the expression levels of three aminoacyl‐tRNA‐synthetases (ARS) of the aminoacyl‐tRNA‐biosynthesis pathway were significantly different: FARSA, FARSB and IARS. ARS are enzymes responsible for the first step of protein synthesis; they join amino acids to their cognate tRNAs 5, 40. Several ARSs, however, do have secondary functions unrelated to tRNA charging including transcription, translation, inflammation and apoptosis [reviewed in 5, 40]. The ARSs are responsible for incorporation of the correct amino acids in proteins. Mouse studies have demonstrated that incorporation of the wrong amino acid results in misfolded or unfolded proteins, protein accumulation and cell death (31), which provides a possible pathogenetic link between ARSs and protein aggregation in PD. Consequently, our data suggest involvement of ARS in the pathogenesis PD. The biological basis of the alterations of ARS expression levels in the LC in PD remains to be evaluated in future studies.
Proteomic data of earlier comparative proteomic analyses of human PD post‐mortem brain tissue show little overlap with the 87 individual altered proteins in our dataset: 14 of these proteins were also differently expressed in previous proteomic studies involving PD substantia nigra and PD frontal cortex brain tissue 7, 8, 25, 30, 49, 50, 56 such as FTH1 (56), pyruvate kinase 25, 49 (PKM2) and gamma‐synuclein (49) (SNCG). A similar low concordance was also seen between other studies [reviewed in (15)]. However, the molecular pathways wherein these proteins are involved do overlap between studies and include mitochondrial dysfunction and oxidative stress. The lack of similarity between the individual proteins in the various tissue proteomics studies can be explained by the analysis of different brain regions and by differences in the fractions of the regions that were analyzed, for example, mitochondrial, nuclear or cytosolic fractions vs. whole brain tissue lysates. An alternative explanation is the use of a diversity of techniques, each with its own detection and resolution properties (15).
It may be argued that the alterations of protein expression observed in this study may be secondary to the loss of catecholaminergic neurons in the LC of PD patients, as the average loss of catecholaminergic neurons in this region is around 70% in post‐mortem brain tissue (10). This particularly applies to neuronal proteins of which the levels are decreased in PD, including DBH and TH that are both normally highly abundant in noradrenergic LC neurons and the proteins that cluster with DBH and TH, such as galectin‐3 (LGALS3) and UGGT1. On the other hand, it is quite possible that neuronal proteins that were not significantly different in our dataset actually have increased expression levels inside neurons that are masked by the lower number of neurons in the total sample.
It has been reported that a selection of proteins repeatedly appears on lists of differentially expressed proteins in proteomic analyses (41). Three out of a recently published top 15 list of most reported differentially expressed proteins in human proteomic studies of total cellular homogenates overlap with our dataset (41). The proteins include alpha‐enolase, pyruvate kinase M1/M2 and vimentin. Most probably, these proteins represent a general cellular stress response rather than a specific PD‐related pathogenetic process.
The immunohistochemical validation of FARSA and BBOX1 provides some explanation for the changed levels of these proteins. The immunohistochemical validation of FARSA suggests that lower levels of FARSA may be related to the reduced number of neurons in the LC, although increased amounts of glial cells in PD may partly compensate for the lower levels of neuronal FARSA. On the other hand, the localization of FARSA appeared to be more often present in the cellular processes in PD patients compared with controls, which may reflect a role in PD pathogenesis. A different localization of ARSs was previously observed for tyrosyl‐tRNA‐synthetase (YARS): in cell cultures expressing mutant YARS proteins, granular YARS immunoreactive structures were homogeneously distributed in most cells, in contrast to the preferential sorting to neuronal tips in non‐mutant cells (26). AD patients revealed an expression pattern of FARSA that was similar to the PD patients indicating that the findings for PD were not specific.
Immunohistochemical validation of BBOX1 showed that increased glia activation may explain the increased levels of this protein in the LC of PD patients. This glia activation was less pronounced in the AD patients, although the small number of patients makes it difficult to draw firm conclusions.
It remains unclear whether changes in FARSA and BBOX1 and other differentially expressed proteins are the consequence or actually the cause of neuronal loss or glial activation. One way to take away the influence of protein alterations caused by cell death is to analyze microdissected neurons. However, the full biological significance of the changes in the LC proteome in PD as well as the specificity of our findings relative to other neurodegenerative disorders can only be established in future functional studies.
Conclusion
The aim of this study was to identify proteins and processes in the human post‐mortem LC that are relevant to PD pathogenesis in order to obtain novel pathogenetic insights. We have demonstrated that the proteome of the affected LC: (i) reflects the main known molecular pathogenetic pathways in PD, most importantly mitochondrial dysfunction, oxidative stress and cytoskeleton dysregulation; (ii) emphasizes a potential pathogenetic role for previously identified candidate proteins such as the classic axon‐guidance molecule serine/threonine protein kinase PAK3 and the mitochondrial ribosomal protein MRPS6; (iii) indicates a potential pathogenetic role for individual proteins that have not yet been associated with PD, such as the calcium‐regulating protein regucalcin and the microtubule‐associated protein KTN1; and (iv) suggests a pathogenetic role for the amino acid cycle and for aminoacyl‐tRNA‐biosynthesis in particular. Further evaluation of the biological mechanisms underlying the observed alterations in protein expression levels and how they relate to protein aggregation and neuronal death may ultimately lead to new therapeutic targets and/or novel diagnostic biomarkers for PD.
FUNDING
This work was supported by Neuroscience Campus Amsterdam, VU University Medical Centre, the Netherlands.
Supporting information
Figure S1. Coomassie Blue‐stained protein gel images of locus ceruleus lysates. Precision Plus Protein standard (Bio‐Rad Laboratories, Hercules CA, USA) was used as a reference marker. Abbreviations below the lanes correspond to subject numbers in Table 1: C1 = NBB00‐032; C2 = NBB98‐104; C3 = NBB95‐101; C4 = NBB96‐078; C5 = NBB03‐013; C6 = NBB03‐040; PD1 = NBB99‐069; PD2 = NBB99‐014; PD3 = NBB98‐064; PD4 = NBB98‐043; PD5 = NBB94‐092; PD6 = NBB06‐002.
Figure S2. Pie chart of functional protein classes of all identified proteins in the locus ceruleus using PANTHER classification. Numbers used in pie chart correspond to numbers in attached table.
Figure S3. Heat map view of supervised cluster analysis using the differentially expressed proteins. Supervised clustering of 87 proteins (horizontal axis) that had significantly different expression levels between PD patients (PD) and controls (CTRL; vertical axis), with 33 upregulated and 54 downregulated proteins. The colors indicate the protein abundances, which were normalized to zero mean and unit variance for each protein. NBB numbers correspond to subject numbers in Table 1.
Figure S4. Proteomic and Western blot data of individual PD patients and controls for proteins included in validation. Numbers on X‐axis for PD patients and controls correspond to NBB subject numbers in Table 1. For proteomic data the normalized spectral counts are shown, for Western blot data the band intensity. Band intensity data for BBOX1 of PD patient 98‐064 were excluded from quantitative analysis because of an artifact.
Table S1. List of all identified proteins and their raw and normalized spectral counts in the locus ceruleus of PD patients compared with controls sorted according to P‐value. Spectral counts were normalized on the sum of the spectral counts per biological sample. NBB numbers correspond to subject numbers in Table 1; *IPI = International Protein Index.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGMENTS
We thank the Netherlands Brain Bank (Amsterdam, the Netherlands), especially Michiel Kooreman and Inge Huitinga, for providing excellent post‐mortem human brain tissue, and are grateful to all patients and controls that donated their brains. We also thank Annemieke J.M. Rozemuller and Wouter Kamphorst for pathologically diagnosing all included cases and the Department of Pathology of the VU University Medical Center Amsterdam for providing microscopical sections for antibody validation. We would also like to acknowledge the valuable work and input of Anke Dijkstra, Pieter Voorn, Angela Ingrassia, Yvonne Galis and John Bol (Department of Anatomy and Neurosciences, VU University Medical Center, Amsterdam).
REFERENCES
- 1. Alafuzoff I, Ince PG, Arzberger T, Al‐Sarraj S, Bell J, Bodi I et al (2009) Staging/typing of Lewy body related alpha‐synuclein pathology: a study of the BrainNet Europe Consortium. Acta Neuropathol 117:635–652. [DOI] [PubMed] [Google Scholar]
- 2. Alberio T, Bossi AM, Milli A, Parma E, Gariboldi MB, Tosi G et al (2010) Proteomic analysis of dopamine and alpha‐synuclein interplay in a cellular model of Parkinson's disease pathogenesis. FEBS J 277:4909–4919. [DOI] [PubMed] [Google Scholar]
- 3. Albrethsen J, Knol JC, Piersma SR, Pham TV, de Wit M, Mongera S et al (2010) Subnuclear proteomics in colorectal cancer: identification of proteins enriched in the nuclear matrix fraction and regulation in adenoma to carcinoma progression. Mol Cell Proteomics 9:988–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Anelli T, Alessio M, Mezghrani A, Simmen T, Talamo F, Bachi A et al (2002) ERp44, a novel endoplasmic reticulum folding assistant of the thioredoxin family. EMBO J 21:835–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Antonellis A, Green ED (2008) The role of aminoacyl‐tRNA synthetases in genetic diseases. Annu Rev Genomics Hum Genet 9:87–107. [DOI] [PubMed] [Google Scholar]
- 6. Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM et al (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Basso M, Giraudo S, Lopiano L, Bergamasco B, Bosticco E, Cinquepalmi A et al (2003) Proteome analysis of mesencephalic tissues: evidence for Parkinson's disease. Neurol Sci 24:155–156. [DOI] [PubMed] [Google Scholar]
- 8. Basso M, Giraudo S, Corpillo D, Bergamasco B, Lopiano L, Fasano M (2004) Proteome analysis of human substantia nigra in Parkinson's disease. Proteomics 4:3943–3952. [DOI] [PubMed] [Google Scholar]
- 9. Benarroch EE, Schmeichel AM, Low PA, Sandroni P, Parisi JE (2008) Loss of A5 noradrenergic neurons in multiple system atrophy. Acta Neuropathol 115:629–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertrand E, Lechowicz W, Szpak GM, Dymecki J (1997) Qualitative and quantitative analysis of locus coeruleus neurons in Parkinson's disease. Folia Neuropathol 35:80–86. [PubMed] [Google Scholar]
- 11. Borges JC, Fischer H, Craievich AF, Hansen LD, Ramos CH (2003) Free human mitochondrial GrpE is a symmetric dimer in solution. J Biol Chem 278:35337–35344. [DOI] [PubMed] [Google Scholar]
- 12. Bossers K, Meerhoff G, Balesar R, van Dongen JW, Kruse CG, Swaab DF et al (2009) Analysis of gene expression in Parkinson's disease: possible involvement of neurotrophic support and axon guidance in dopaminergic cell death. Brain Pathol 19:91–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Braak H, Braak E (1991) Neuropathological staging of Alzheimer‐related changes. Acta Neuropathol 82:239–259. [DOI] [PubMed] [Google Scholar]
- 14. Braak H, Del TK, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 15. Caudle WM, Bammler TK, Lin Y, Pan S, Zhang J (2010) Using “omics” to define pathogenesis and biomarkers of Parkinson's disease. Expert Rev Neurother 10:925–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. DeStefano AL, Latourelle J, Lew MF, Suchowersky O, Klein C, Golbe LI et al (2008) Replication of association between ELAVL4 and Parkinson disease: the GenePD study. Hum Genet 124:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Duran R, Barrero FJ, Morales B, Luna JD, Ramirez M, Vives F (2010) Oxidative stress and plasma aminopeptidase activity in Huntington's disease. J Neural Transm 117:325–332. [DOI] [PubMed] [Google Scholar]
- 18. Duran R, Barrero FJ, Morales B, Luna JD, Ramirez M, Vives F (2011) Oxidative stress and aminopeptidases in Parkinson's disease patients with and without treatment. Neurodegener Dis 8:109–116. [DOI] [PubMed] [Google Scholar]
- 19. Galea E, Heneka MT, Dello RC, Feinstein DL (2003) Intrinsic regulation of brain inflammatory responses. Cell Mol Neurobiol 23:625–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. German DC, Manaye KF, White CL III, Woodward DJ, McIntire DD, Smith WK et al (1992) Disease‐specific patterns of locus coeruleus cell loss. Ann Neurol 32:667–676. [DOI] [PubMed] [Google Scholar]
- 21. Gesi M, Soldani P, Giorgi FS, Santinami A, Bonaccorsi I, Fornai F (2000) The role of the locus coeruleus in the development of Parkinson's disease. Neurosci Biobehav Rev 24:655–668. [DOI] [PubMed] [Google Scholar]
- 22. Hicks AA, Petursson H, Jonsson T, Stefansson H, Johannsdottir HS, Sainz J et al (2002) A susceptibility gene for late‐onset idiopathic Parkinson's disease. Ann Neurol 52:549–555. [DOI] [PubMed] [Google Scholar]
- 23. Hirokawa N, Niwa S, Tanaka Y (2010) Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68:610–638. [DOI] [PubMed] [Google Scholar]
- 24. Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson's disease: a target for neuroprotection? Lancet Neurol 8:382–397. [DOI] [PubMed] [Google Scholar]
- 25. Jin J, Hulette C, Wang Y, Zhang T, Pan C, Wadhwa R et al (2006) Proteomic identification of a stress protein, mortalin/mthsp70/GRP75: relevance to Parkinson disease. Mol Cell Proteomics 5:1193–1204. [DOI] [PubMed] [Google Scholar]
- 26. Jordanova A, Irobi J, Thomas FP, Van DP, Meerschaert K, Dewil M et al (2006) Disrupted function and axonal distribution of mutant tyrosyl‐tRNA synthetase in dominant intermediate Charcot‐Marie‐Tooth neuropathy. Nat Genet 38:197–202. [DOI] [PubMed] [Google Scholar]
- 27. Keller A, Nesvizhskii AI, Kolker E, Aebersold R (2002) Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem 74:5383–5392. [DOI] [PubMed] [Google Scholar]
- 28. Kenmochi N, Suzuki T, Uechi T, Magoori M, Kuniba M, Higa S et al (2001) The human mitochondrial ribosomal protein genes: mapping of 54 genes to the chromosomes and implications for human disorders. Genomics 77:65–70. [DOI] [PubMed] [Google Scholar]
- 29. Kim JM, Lee KH, Jeon YJ, Oh JH, Jeong SY, Song IS et al (2006) Identification of genes related to Parkinson's disease using expressed sequence tags. DNA Res 13:275–286. [DOI] [PubMed] [Google Scholar]
- 30. Kitsou E, Pan S, Zhang J, Shi M, Zabeti A, Dickson DW et al (2008) Identification of proteins in human substantia nigra. Proteomics Clin Appl 2:776–782. [DOI] [PubMed] [Google Scholar]
- 31. Lee JW, Beebe K, Nangle LA, Jang J, Longo‐Guess CM, Cook SA et al (2006) Editing‐defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443:50–55. [DOI] [PubMed] [Google Scholar]
- 32. Li J, Richter K, Buchner J (2011) Mixed Hsp90‐cochaperone complexes are important for the progression of the reaction cycle. Nat Struct Mol Biol 18:61–66. [DOI] [PubMed] [Google Scholar]
- 33. Li YJ, Scott WK, Hedges DJ, Zhang F, Gaskell PC, Nance MA et al (2002) Age at onset in two common neurodegenerative diseases is genetically controlled. Am J Hum Genet 70:985–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lin L, Lesnick TG, Maraganore DM, Isacson O (2009) Axon guidance and synaptic maintenance: preclinical markers for neurodegenerative disease and therapeutics. Trends Neurosci 32:142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mila S, Albo AG, Corpillo D, Giraudo S, Zibetti M, Bucci EM et al (2009) Lymphocyte proteomics of Parkinson's disease patients reveals cytoskeletal protein dysregulation and oxidative stress. Biomark Med 3:117–128. [DOI] [PubMed] [Google Scholar]
- 36. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 37. Nesvizhskii AI, Keller A, Kolker E, Aebersold R (2003) A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem 75:4646–4658. [DOI] [PubMed] [Google Scholar]
- 38. Noureddine MA, Qin XJ, Oliveira SA, Skelly TJ, van der Walt J, Hauser MA et al (2005) Association between the neuron‐specific RNA‐binding protein ELAVL4 and Parkinson disease. Hum Genet 117:27–33. [DOI] [PubMed] [Google Scholar]
- 39. Papapetropoulos S, Ffrench‐Mullen J, McCorquodale D, Qin Y, Pablo J, Mash DC (2006) Multiregional gene expression profiling identifies MRPS6 as a possible candidate gene for Parkinson's disease. Gene Expr 13:205–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Park SG, Ewalt KL, Kim S (2005) Functional expansion of aminoacyl‐tRNA synthetases and their interacting factors: new perspectives on housekeepers. Trends Biochem Sci 30:569–574. [DOI] [PubMed] [Google Scholar]
- 41. Petrak J, Ivanek R, Toman O, Cmejla R, Cmejlova J, Vyoral D et al (2008) Deja vu in proteomics. A hit parade of repeatedly identified differentially expressed proteins. Proteomics 8:1744–1749. [DOI] [PubMed] [Google Scholar]
- 42. Pham TV, Piersma SR, Warmoes M, Jimenez CR (2010) On the beta‐binomial model for analysis of spectral count data in label‐free tandem mass spectrometry‐based proteomics. Bioinformatics 26:363–369. [DOI] [PubMed] [Google Scholar]
- 43. Piersma SR, Fiedler U, Span S, Lingnau A, Pham TV, Hoffmann S et al (2010) Workflow comparison for label‐free, quantitative secretome proteomics for cancer biomarker discovery: method evaluation, differential analysis, and verification in serum. J Proteome Res 9:1913–1922. [DOI] [PubMed] [Google Scholar]
- 44. Ritz B, Rhodes SL, Qian L, Schernhammer E, Olsen JH, Friis S (2010) L‐type calcium channel blockers and Parkinson disease in Denmark. Ann Neurol 67:600–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Samuels ER, Szabadi E (2008) Functional neuroanatomy of the noradrenergic locus coeruleus: its roles in the regulation of arousal and autonomic function part I: principles of functional organisation. Curr Neuropharmacol 6:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schapira AH (2008) Mitochondria in the aetiology and pathogenesis of Parkinson's disease. Lancet Neurol 7:97–109. [DOI] [PubMed] [Google Scholar]
- 47. Shadrina M, Nikopensius T, Slominsky P, Illarioshkin S, Bagyeva G, Markova E et al (2006) Association study of sporadic Parkinson's disease genetic risk factors in patients from Russia by APEX technology. Neurosci Lett 405:212–216. [DOI] [PubMed] [Google Scholar]
- 48. Shevchenko A, Wilm M, Vorm O, Mann M (1996) Mass spectrometric sequencing of proteins silver‐stained polyacrylamide gels. Anal Chem 68:850–858. [DOI] [PubMed] [Google Scholar]
- 49. Shi M, Jin J, Wang Y, Beyer RP, Kitsou E, Albin RL et al (2008) Mortalin: a protein associated with progression of Parkinson disease? J Neuropathol Exp Neurol 67:117–124. [DOI] [PubMed] [Google Scholar]
- 50. Shi M, Bradner J, Bammler TK, Eaton DL, Zhang J, Ye Z et al (2009) Identification of glutathione S‐transferase pi as a protein involved in Parkinson disease progression. Am J Pathol 175:54–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tong J, Wong H, Guttman M, Ang LC, Forno LS, Shimadzu M et al (2010) Brain alpha‐synuclein accumulation in multiple system atrophy, Parkinson's disease and progressive supranuclear palsy: a comparative investigation. Brain 133:172–188. [DOI] [PubMed] [Google Scholar]
- 52. Totani K, Ihara Y, Tsujimoto T, Matsuo I, Ito Y (2009) The recognition motif of the glycoprotein‐folding sensor enzyme UDP‐Glc:glycoprotein glucosyltransferase. Biochemistry 48:2933–2940. [DOI] [PubMed] [Google Scholar]
- 53. Toyoshima I, Yu H, Steuer ER, Sheetz MP (1992) Kinectin, a major kinesin‐binding protein on ER. J Cell Biol 118:1121–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vale RD (2003) The molecular motor toolbox for intracellular transport. Cell 112:467–480. [DOI] [PubMed] [Google Scholar]
- 55. van Dijk KD, Teunissen CE, Drukarch B, Jimenez CR, Groenewegen HJ, Berendse HW et al (2010) Diagnostic cerebrospinal fluid biomarkers for Parkinson's disease: a pathogenetically based approach. Neurobiol Dis 39:229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Werner CJ, Heyny‐von HR, Mall G, Wolf S (2008) Proteome analysis of human substantia nigra in Parkinson's disease. Proteome Sci 6:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yamaguchi M (2005) Role of regucalcin in maintaining cell homeostasis and function (review). Int J Mol Med 15:371–389. [PubMed] [Google Scholar]
- 58. Zweig RM, Cardillo JE, Cohen M, Giere S, Hedreen JC (1993) The locus ceruleus and dementia in Parkinson's disease. Neurology 43:986–991. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Coomassie Blue‐stained protein gel images of locus ceruleus lysates. Precision Plus Protein standard (Bio‐Rad Laboratories, Hercules CA, USA) was used as a reference marker. Abbreviations below the lanes correspond to subject numbers in Table 1: C1 = NBB00‐032; C2 = NBB98‐104; C3 = NBB95‐101; C4 = NBB96‐078; C5 = NBB03‐013; C6 = NBB03‐040; PD1 = NBB99‐069; PD2 = NBB99‐014; PD3 = NBB98‐064; PD4 = NBB98‐043; PD5 = NBB94‐092; PD6 = NBB06‐002.
Figure S2. Pie chart of functional protein classes of all identified proteins in the locus ceruleus using PANTHER classification. Numbers used in pie chart correspond to numbers in attached table.
Figure S3. Heat map view of supervised cluster analysis using the differentially expressed proteins. Supervised clustering of 87 proteins (horizontal axis) that had significantly different expression levels between PD patients (PD) and controls (CTRL; vertical axis), with 33 upregulated and 54 downregulated proteins. The colors indicate the protein abundances, which were normalized to zero mean and unit variance for each protein. NBB numbers correspond to subject numbers in Table 1.
Figure S4. Proteomic and Western blot data of individual PD patients and controls for proteins included in validation. Numbers on X‐axis for PD patients and controls correspond to NBB subject numbers in Table 1. For proteomic data the normalized spectral counts are shown, for Western blot data the band intensity. Band intensity data for BBOX1 of PD patient 98‐064 were excluded from quantitative analysis because of an artifact.
Table S1. List of all identified proteins and their raw and normalized spectral counts in the locus ceruleus of PD patients compared with controls sorted according to P‐value. Spectral counts were normalized on the sum of the spectral counts per biological sample. NBB numbers correspond to subject numbers in Table 1; *IPI = International Protein Index.
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item
Supporting info item