

Abstract
T cell immunoglobulin and mucin domain-containing-3 (TIM-3) is an immune checkpoint expressed mainly on CD4+ and CD8+ T cells. In addition to negatively regulating inflammatory T cell function, TIM-3 is a promising immunotherapy target. Herein, the aim is to develop an immuno-positron emission tomography (immunoPET) probe for noninvasively characterizing TIM-3 expression. Flow cytometry is used to detect the expression levels of TIM-3 in B16F10 cells. RMT3–23, a rat antimouse TIM-3-specific monoclonal antibody, is radiolabeled with 64Cu and the performance of 64Cu-NOTA-RMT3–23 is interrogated by immunoPET in murine melanoma models before and after radiation therapies. Biodistribution and immunofluorescent staining studies are carried out after the immunoPET imaging studies. TIM-3 is negatively expressed in B16F10 cells, and its expression is not induced by radiation therapies. ImmunoPET imaging with 64Cu-NOTA-RMT3–23 precisely tracks the unique distribution of TIM-3-positive lymphocytes in immunocompetent melanoma models, and tumor uptake of the radiotracer is not affected by either single-dose or fractionated radiation therapies. The 64Cu-NOTA-RMT3–23 immunoPET imaging results are further mirrored by the immunofluorescent staining studies. These results demonstrate the feasibility of 64Cu-NOTA-RMT3–23 immunoPET in tracking TIM-3 and highlight a new opportunity to optimize TIM-3-targeted immunotherapies with this novel imaging strategy.
Keywords: immune checkpoints, immuno-positron emission tomography, immunotherapy, melanomas, TIM-3
1. Introduction
With the clinical use of chimeric antigen receptor (CAR)- modified T cells and immune checkpoint inhibitors, cancer immunotherapy has resulted in durable or complete responses in patients with various types of cancers.[1] Immune checkpoint blockade (ICB) usually involves in inhibiting cytotoxic T lymphocyte-associated protein 4 (CTLA-4) and programmed cell death receptor 1 (PD-1)/programmed cell death ligand 1 (PD-L1) signaling axis. CTLA-4 is highly expressed in the regulatory T cells (Tregs).[2] Therefore, the antitumor activity of an anti-CTLA-4 monoclonal antibody (mAb) is mediated, in part, by ablation of the Tregs at tumor sites.[3] PD-1 is relatively overexpressed on effector tumor-infiltrating lymphocytes (TILs) and acts as a negative regulator of T cell antitumor response. After engaging with its major ligand PD-L1, PD-1 mediates its immune inhibitory function by tyrosine phosphatase SHP-2, which dephosphorylates the downstream signaling cascades of T cell receptor.[4] In patients with metastatic melanoma, immune checkpoint inhibitors targeting the CTLA-4 and/or the PD-1/PD-L1 signaling pathway have demonstrated significant improvements in progression-free survival and overall survival.[5] However, primary and adaptive resistance associated with immune checkpoint inhibitors hinders melanoma patients from sustained ICB treatment.[6] In this setting, identification of novel immune checkpoints negatively regulating the antimelanoma immune response is of clinical significance, since blockage of these novel immune checkpoints may enhance the antitumor activity or even overcome resistance of the clinically approved ICB regimens.
As a type I transmembrane protein, T cell immunoglobulin and mucin domain-containing-3 (TIM-3) was originally identified as a molecule that can help identify IFNγ-secreting Th1 cells.[7] While TIM-3 negatively controls the progression of several autoimmune diseases, it also acts as a pivotal immune checkpoint by regulating T cell exhaustion during tumor development.[8] Immune inhibitory receptors including TIM-3, PD-1, LAG-3, and TIGIT are tightly coexpressed on both CD8+ and CD4+ TILs,[9] of which TIM-3 and PD-1 double-positive TILs represent the most exhausted phenotypes.[10] TIM-3 is also overexpressed in several types of human malignancies including melanoma and cervical carcinoma.[8,11] Consequently, combinatorial immunotherapy consisting of anti-TIM-3 and anti-PD-1 or anti-CTLA-4 antibodies could significantly reduce tumor burden in preclinical settings.[12] Moreover, the expression of TIM-3 on CD8+ T cells and Tregs increased following radiation and anti-PD-L1 therapies, and the addition of an anti-TIM-3 antibody significantly delayed tumor growth in preclinical lung cancer models.[13] Currently, antibody therapeutics targeting TIM-3 or dually targeting TIM-3 and another immune checkpoint (e.g., PD-1 or PD-L1) are undergoing clinical investigation.[14] The therapeutic benefits of these novel immunotherapy regimens are urgently awaited.
Substantial clinical studies have revealed that the infiltration of CD8+ T cells and (or) pre-existing CD8+ T cells in the tumor microenvironment indicated better responses of ICB treatments.[15] Instead of using tumor and liquid biopsies, we previously reported that immuno-positron emission tomography (immunoPET) imaging is a powerful technique to noninvasively map T cells and oncoproteins.[16–21] However, methods that can capture the dynamics of TIM-3-positive lymphocytes are still lacking. Herein, we aim to develop a TIM-3-specific immunoPET probe and use this probe to delineate TIM-3 expression in murine melanoma models. By harnessing this TIM-3-targeted immunoPET imaging approach, we also characterize the alteration of TIM-3 expression following single-dose or fractionated radiation therapies in murine melanoma models.
2. Results
2.1. 64Cu-NOTA-RMT3–23 ImmunoPET Imaging of Nonirradiated Melanomas
In this study, a rat antimouse mAb RMT3–23 was used to screen TIM-3 expression and to conjugate the immunoPET imaging probe. RMT3–23 reacts with TIM-3 from both BALB/c mice and C57BL/6 mice.[22] We first assessed the expression of TIM-3 on the surface of B16F10 cells by performing flow cytometry and found that TIM-3 was negatively expressed on B16F10 cells (Figure 1A), indicating that TIM-3 is not a marker for B16F10 cell line. However, TIM-3 was found positively expressed in certain human melanoma tissues and melanoma cell lines.[23] Despite this dichotomous observation, the negative expression of TIM-3 on the surface of B16F10 cells precludes the TIM-3 signal contributed by B16F10 tumors in the following studies.
Figure 1.
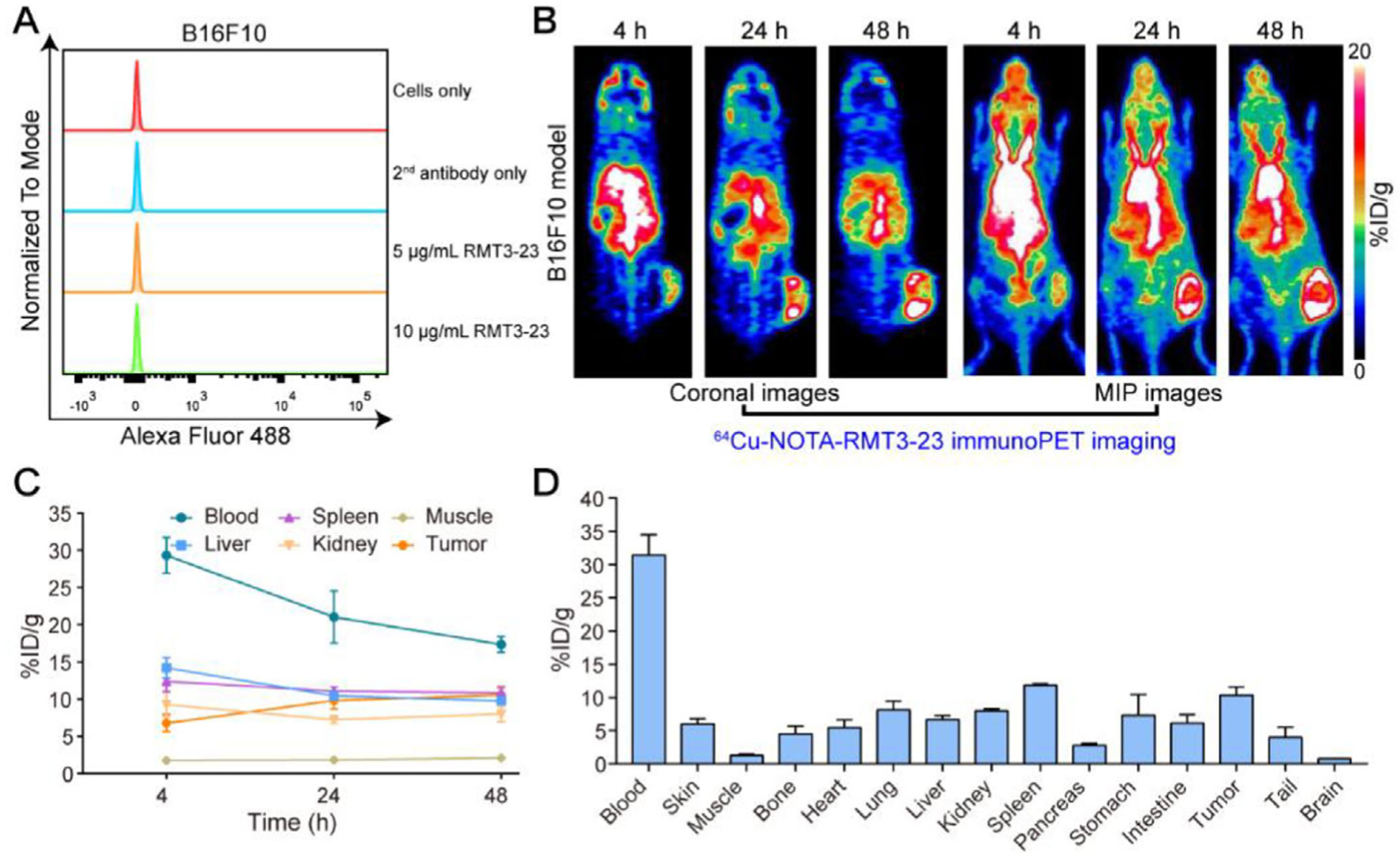
Flow cytometry assessment and 64Cu-NOTA-RMT3–23 immunoPET imaging of C57BL/6N mice bearing B16F10 melanomas. A) Flow cytometry assessing cell surface expression of TIM-3. B) Representative coronal and maximum intensity projection (MIP) images of 64Cu-NOTA-RMT3–23 immunoPET at different time-points after administration of the radiotracer. C) Quantitative analyses of 64Cu-NOTA-RMT3–23 immunoPET imaging data (n = 3). D) Biodistribution data obtained after the terminal immunoPET imaging (n = 3).
RMT3–23 was first conjugated to NOTA and then radiolabeled with 64Cu. In our hands, 64Cu-labeling of NOTA-RMT3–23 resulted in a radiolabeling yield of >80% and a radiochemical purity of >99%. The feasibility of 64Cu-NOTA-RMT3–23 immunoPET imaging was first interrogated in C57BL/6N mice bearing B16F10 melanomas (with an average tumor volume of ≈400 mm3). As shown by immunoPET images in Figure 1B, 64Cu-NOTA-RMT3–23 circulated well throughout the imaging period, which differed from other immune checkpoint-targeted immuno- PET probes that rapidly deposited in lymphatic tissues (e.g., spleen and lymph nodes).[18,19] Indeed, the expression of TIM-3 is largely restricted to IFNγ-producing CD4+ T cells, CD8+ T cells, and exhausted T cells.[7,24] Meanwhile, 64Cu-NOTA-RMT3–23 concentrated more obvious in the tumor edge than in the tumor center, which may indicate TIM-3-positive lymphocytes failed to penetrate deeply inside the tumor. Region of interest (ROI) analysis revealed the overall decline of 64Cu-NOTA-RMT3–23 uptake in the blood pool, liver, and spleen over the imaging period, with the uptake value at the terminal imaging time-point was 17.37 ± 1.08, 9.80 ± 0.36, and 10.87 ± 0.75%ID g−1 (n = 3), respectively. Tumor uptake of 64Cu-NOTA-RMT3–23 gradually increased with a peak uptake of 10.57 ± 1.17%ID g−1 (n = 3) reached at 48 h after administration of the radiotracer (Figure 1C). The ex vivo biodistribution study performed immediately after termination of the immunoPET imaging studies demonstrated higher retention of the radiotracer in the circulation (31.46 ± 3.03%ID g−1, n = 3) but a comparable tumor uptake (10.35 ± 1.25%ID g−1, n = 3) (Figure 1D). These results demonstrated that TIM-3 expression in the tumor microenvironment was readily accessed and visualized by TIM-3- targeted immunoPET imaging. However, this imaging approach is otherwise unable to detect other secondary lymphatic tissues except the spleen, due to either low expression of TIM-3 in these tissues or limited detection ability of the imaging technique.
2.2. 64Cu-NOTA-RMT3–23 ImmunoPET Imaging Following Single-Dose Radiation Therapy
Before exploring the potential effects of radiation on the expression level of TIM-3 in the tumor microenvironment, we irradiated B16F10 cells in vitro with increasing doses of irradiation (i.e., 2, 6, and 12 Gy in one fraction) and monitored these samples with flow cytometry. The results showed that cell surface TIM-3 of the irradiated cells did not increase when compared to the control groups (irradiated cells only, second antibody only, and nonirradiated cells; Figure 2A).
Figure 2.
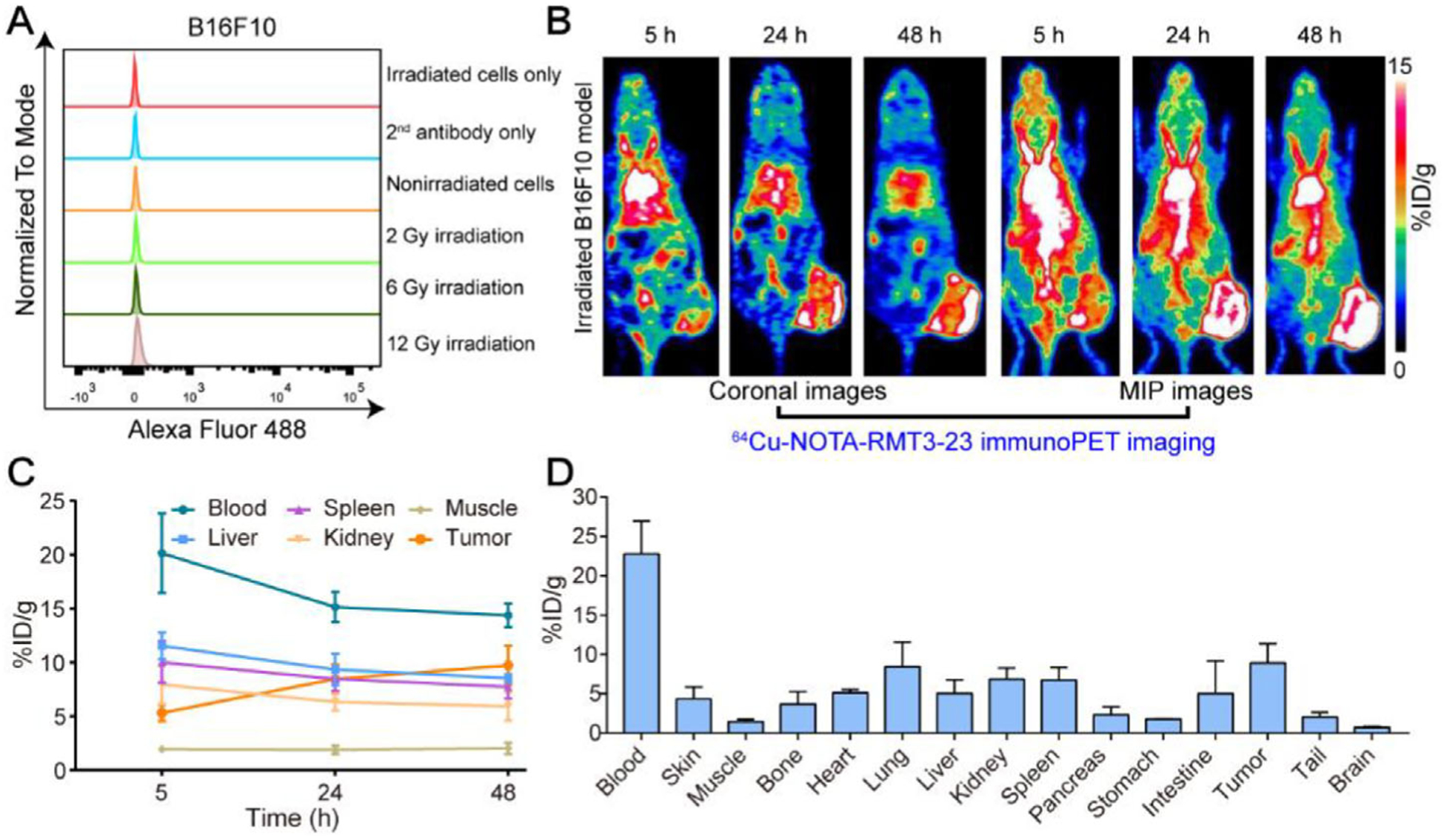
Flow cytometry assessment and 64Cu-NOTA-RMT3–23 immunoPET imaging of irradiated tumor-bearing C57BL/6N mice (12 Gy in one fraction). A) Flow cytometry assessing the influence of irradiation on the expression level of TIM-3. Irradiated cells only, second antibody only (irradiated cells incubated only with secondary antibody), and nonirradiated cells are control groups. B) Representative coronal and maximum intensity projection (MIP) images of 64Cu-NOTA-RMT3–23 immunoPET at different time-points after administration of the radiotracer. C) Quantitative analyses of 64Cu-NOTA-RMT3–23 immunoPET imaging data (n = 4). D) Biodistribution data obtained after the terminal immunoPET imaging (n = 4).
The average tumor volume reached 299.81 ± 135.04 mm3 (n = 4) seven days after the inoculation of 1 × 106 B16F10 cells to each C57BL/6N mouse. However, the high-dose radiation therapy, that is, 12 Gy in one fraction, did not suppress the growth of the tumors because the tumor volume expanded to 956.25 ± 131.44 mm3 (n = 4) ten days after the therapy. To understand whether TIM-3 infiltration is irradiation-responsive, we carried out TIM-3 immunoPET imaging using the irradiated mice. The results demonstrated a substantial uptake of 64Cu-NOTA-RMT3–23 around the growing tumors (Figure 2B). ROI analysis showed the tumor accumulation of 64Cu-NOTA-RMT3–23 was 9.75 ± 1.81%ID g−1 (n = 4; Figure 2C). This was further corroborated by the ex vivo biodistribution data (Figure 2D), which revealed a tumor uptake of 8.94 ± 2.41%ID g−1 (n = 4). However, uptake of 64Cu-NOTA-RMT3–23 in the irradiated tumors was not statistically different from that in the above nonirradiated tumors (p = 0.53 for tumor uptake at 48 h when comparing the ROI data and p = 0.42 when comparing the biodistribution data).
2.3. 64Cu-NOTA-RMT3–23 ImmunoPET Imaging Following Fractionated Radiation Therapy
A previous study elucidated that fractionated, rather than single-dose irradiation, was effective in inducing infiltration of CD4+ and CD8+ T cells and abscopal effect.[25] To test the influence of fractionated irradiation therapy on the immigration of TIM-3-positive lymphocytes, we prepared melanoma models by subcutaneously injecting 5 × 105 cells to each mouse. Following the inoculation, we initiated fractionated irradiation (12 Gy in six fractions, one fraction day−1) on day 2 when the tumors were still unmeasurable. However, the fractionated radiation therapy failed to delay the tumor growth as the average tumor volume reached 130.68 ± 25.73 (n = 6) on day 7 after the implantation. To explore whether tumor accumulation of the radiotracers increased after the fractionated irradiation therapy, we subjected the irradiated mice to immunoPET imaging on day 8. For the mice receiving the 64Cu-NOTA-RMT3–23 injection, the overall distribution patterns were similar to those seen in mice receiving single-dose radiation therapy (Figure 3A–D). Note that the distribution of the radiotracer was heterogeneous across the tumor (Figure 3A,B). Quantitative analyses of the immunoPET data showed an average tumor uptake of 10.03 ± 0.78%ID g−1 (n = 3) at 48 h post-injection of the radiotracer (Figure 3C), which was comparable to that of the nonirradiated group (10.03 ± 0.78%ID g−1, n = 3 versus 10.57 ± 1.17%ID g−1, n = 3; p = 0.85) or to that of the single-dose irradiation group (10.03 ± 0.78%ID g−1, n = 3 versus 9.75 ± 1.81%ID g−1, n = 4; p = 0.95). Statistical analyses also showed no difference in tumor uptake of the 64Cu-NOTA-RMT3–23 at the other two time-points (i.e., 4 and 24 h). Biodistribution data revealed that the majority of the radiotracer was still in the circulation (23.01 ± 2.05%ID g−1, n = 3) at the terminal imaging time-point, accompanied by the second-highest uptake in the tumor (7.88 ± 2.36%ID g−1, n = 3; Figure 3D).
Figure 3.
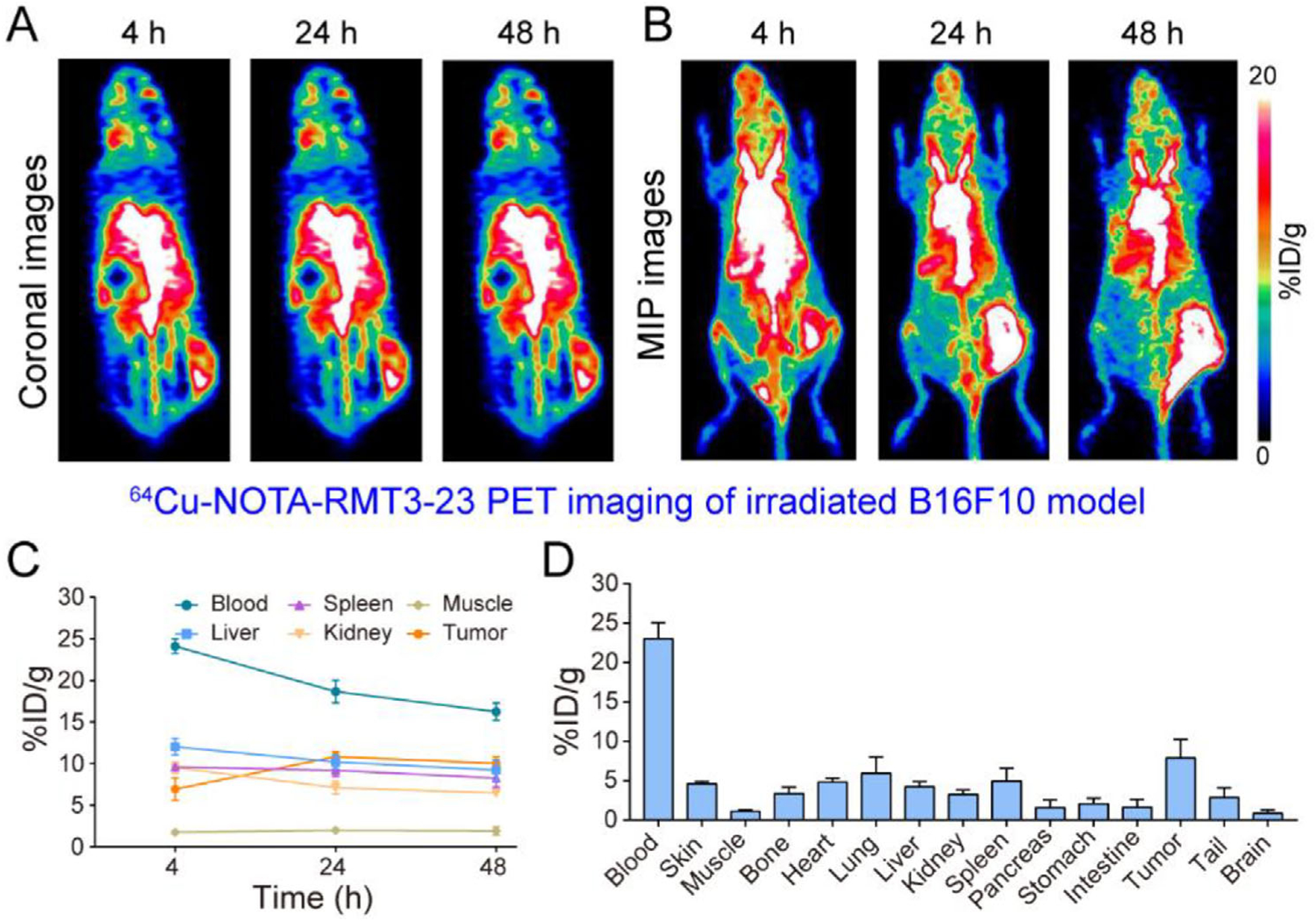
64Cu-NOTA-RMT3–23 immunoPET imaging of irradiated tumor-bearing C57BL/6N mice (12 Gy in six fractions). A,B) Representative coronal and maximum intensity projection (MIP) images of 64Cu-NOTA-RMT3–23 immunoPET at different time-points after administration of the radiotracer. C) Quantitative analyses of 64Cu-NOTA-RMT3–23 immunoPET imaging data (n = 3). D) Biodistribution data obtained after the terminal immunoPET imaging (n = 3).
2.4. 64Cu-NOTA-RTG2α ImmunoPET Imaging Following Fractionated Radiation Therapy
To demonstrate the specificity of 64Cu-NOTA-RMT3–23 immunoPET imaging, we conjugated a nonspecific 64Cu-NOTA-RTG2α, and further investigated the diagnostic efficacy of this radiotracer in B16F10 melanoma models after fractionated radiation therapy. As shown in Figure 4, although 64Cu-NOTA-RTG2α immunoPET imaging could outline the tumors on the coronal images, the tumor signal on the maximum intensity projection (MIP) images was not distinct from the adjacent intestinal signal. Moreover, the circulation profiles of 64Cu-NOTA-RTG2α differed significantly from that of the 64Cu-NOTA-RMT3–23. As early as 4 h post-injection, 64Cu-NOTA-RTG2α was largely deposited in the liver, spleen, and kidney (Figure 4A,B). ROI analyses showed uptake of the radiotracer in the aforementioned three organs at the first time-point was 21.63 ± 1.16, 9.23 ± 3.90, and 9.5 ± 2.43%ID g−1 (n = 3), respectively. And, the corresponding value dropped to 10.67 ± 2.37, 3.6 ± 0.79, and 4.47 ± 0.91%ID g−1 (n = 3) two days later (Figure 4C). The accumulation of 64Cu-NOTA-RTG2α in the tumor plateaued at 4 h (4.90 ± 0.70%ID g−1, n = 3) and then decreased during the imaging period (4.70 ± 0.95%ID g−1 at 24 h and 3.77 ± 1.06%ID g−1 at 48 h, respectively). Multiple comparisons showed that tumor uptake of the 64Cu-NOTA-RMT3–23 (either in the nonirradiation group, in the 12 Gy in one fraction irradiation group, or in the 12 Gy in six fractions irradiation group) was significantly higher than that of the 64Cu-NOTA-RTG2α at all the time-points except that at 4 h. Ex vivo biodistribution study confirmed the in vivo immunoPET imaging results, showing a relatively higher uptake of 64Cu-NOTA-RTG2α in several parenchymal organs (i.e., liver, spleen, intestine, and kidney) where the radiolabeled antibody was metabolized or degraded (Figure 4D). The detailed uptake values of 64Cu-NOTA-RMT3–23 and 64Cu-NOTA-RTG2α in the tumor and other major organs (or tissues) based on biodistribution data are presented in Table 1.
Figure 4.
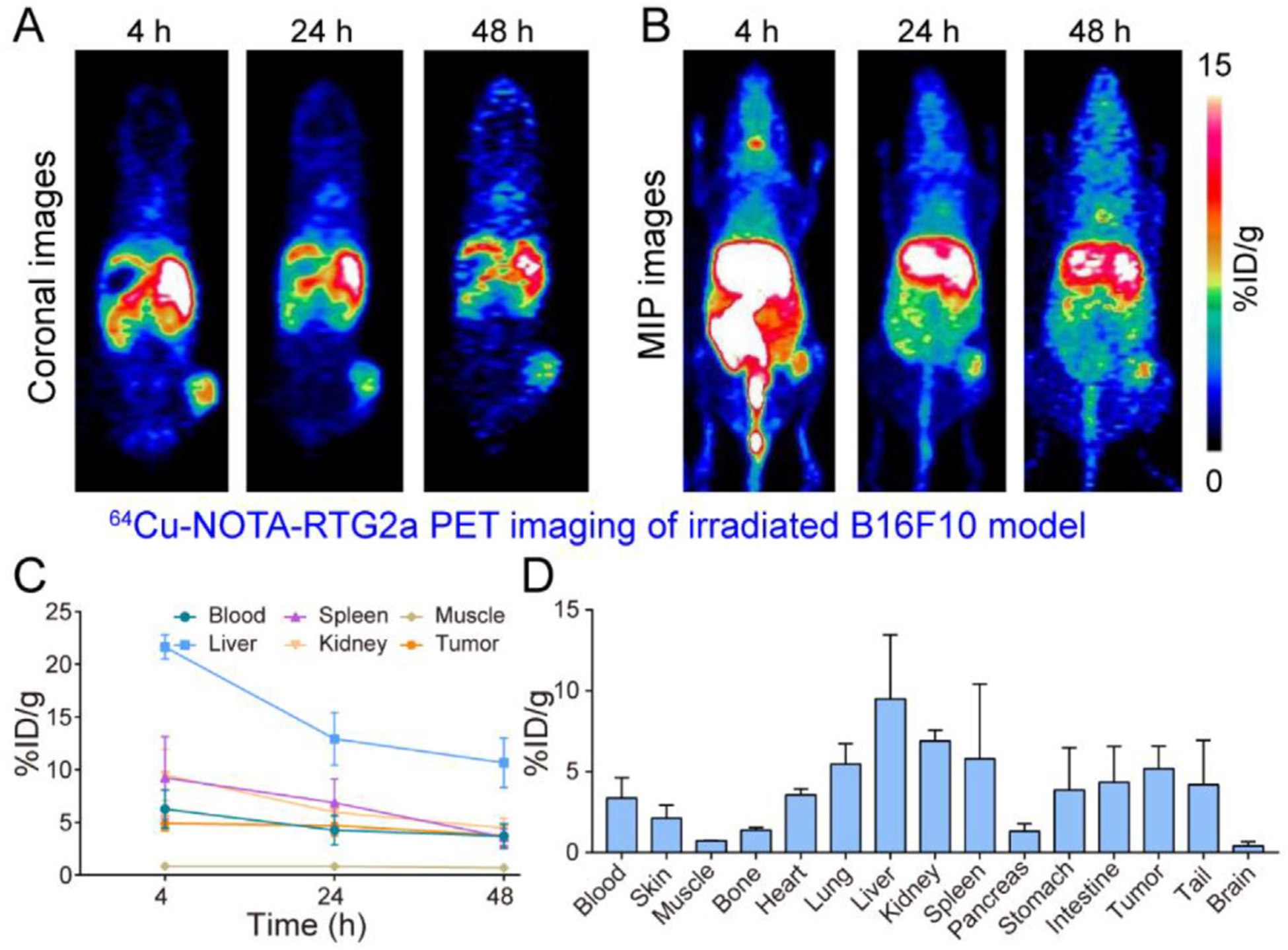
64Cu-NOTA-RTG2α immunoPET imaging of irradiated tumor-bearing C57BL/6N mice (12 Gy in six fractions). A,B) Representative coronal and maximum intensity projection (MIP) images of 64Cu-NOTA-RTG2α immunoPET at different time-points after administration of the radiotracer. C) Quantitative analyses of 64Cu-NOTA-RTG2α immunoPET imaging data (n = 3). D) Biodistribution data obtained after the terminal immunoPET imaging (n = 3).
Table 1.
Uptake of 64Cu-NOTA-RMT3–23 and 64Cu-NOTA-RTG2α in murine melanomas.
| TracerOrgan | 64Cu-NOTA-RMT3–23 (naïve mice, n = 3) | 64Cu-NOTA-RMT3–23 (single-dose irradiation, n = 4) | 64Cu-NOTA-RMT3–23 (fractionated irradiation, n = 3) | 64Cu-NOTA-RTG2α (fractionated irradiation, n = 3) |
|---|---|---|---|---|
| Tumor | 10.3 ± 1.25 | 8.94 ± 2.41 | 7.88 ± 2.36 | 5.18 ± 1.39 |
| Blood | 31.5 ± 3.03 | 22.8 ± 4.19 | 23.0 ± 2.05 | 3.37 ± 1.25 |
| Bone | 4.51 ± 1.18 | 3.69 ± 1.56 | 3.35 ± 0.81 | 1.38 ± 0.18 |
| Heart | 5.47 ± 1.18 | 5.13 ± 0.42 | 4.80 ± 0.51 | 3.55 ± 0.39 |
| Lung | 8.15 ± 1.31 | 8.45 ± 3.11 | 5.95 ± 2.05 | 5.47 ± 1.26 |
| Liver | 6.68 ± 0.63 | 5.05 ± 1.73 | 4.20 ± 0.66 | 9.49 ± 3.97 |
| Kidney | 8.01 ± 0.35 | 6.87 ± 1.43 | 3.24 ± 0.56 | 6.89 ± 0.68 |
| Spleen | 11.9 ± 0.22 | 6.73 ± 1.63 | 4.94 ± 1.65 | 5.80 ± 4.61 |
| Pancreas | 2.8 ± 0.29 | 2.33 ± 1.01 | 1.57 ± 1.00 | 1.30 ± 0.48 |
| Stomach | 7.36 ± 3.07 | 1.78 ± 0.05 | 2.05 ± 0.71 | 3.87 ± 2.62 |
| Intestine | 6.18 ± 1.23 | 5.00 ± 4.22 | 1.65 ± 0.97 | 4.34 ± 2.22 |
| Brain | 0.79 ± 0.22 | 0.73 ± 0.14 | 0.85 ± 0.43 | 0.42 ± 0.25 |
2.5. Immunofluorescent Staining Studies
Immunofluorescent staining of the resected tissues from the nonirradiated mice showed colocalization of TIM-3 and CD45, a leukocyte common antigen, at the tumor edges (Figure 5). However, a scarcity of TIM-3-positive cells was observed inside the tumors despite the infiltration of CD45-positive lymphocytes. Meanwhile, TIM-3 and CD45 double-positive lymphocytes were noted in the spleens, but not in the stained liver sections. No B16F10 tumor cells were stained by TIM-3, affirming that TIM-3 is not a marker for the B16F10 cell line and tumor. Similar immunofluorescent staining patterns were found in the tissues of the irradiated mice (Figure 6). These results were in accordance with the circular uptake of 64Cu-NOTA-RMT3–23 in the tumors and low uptake of the radiotracer in the livers and spleens revealed by immunoPET imaging.
Figure 5.
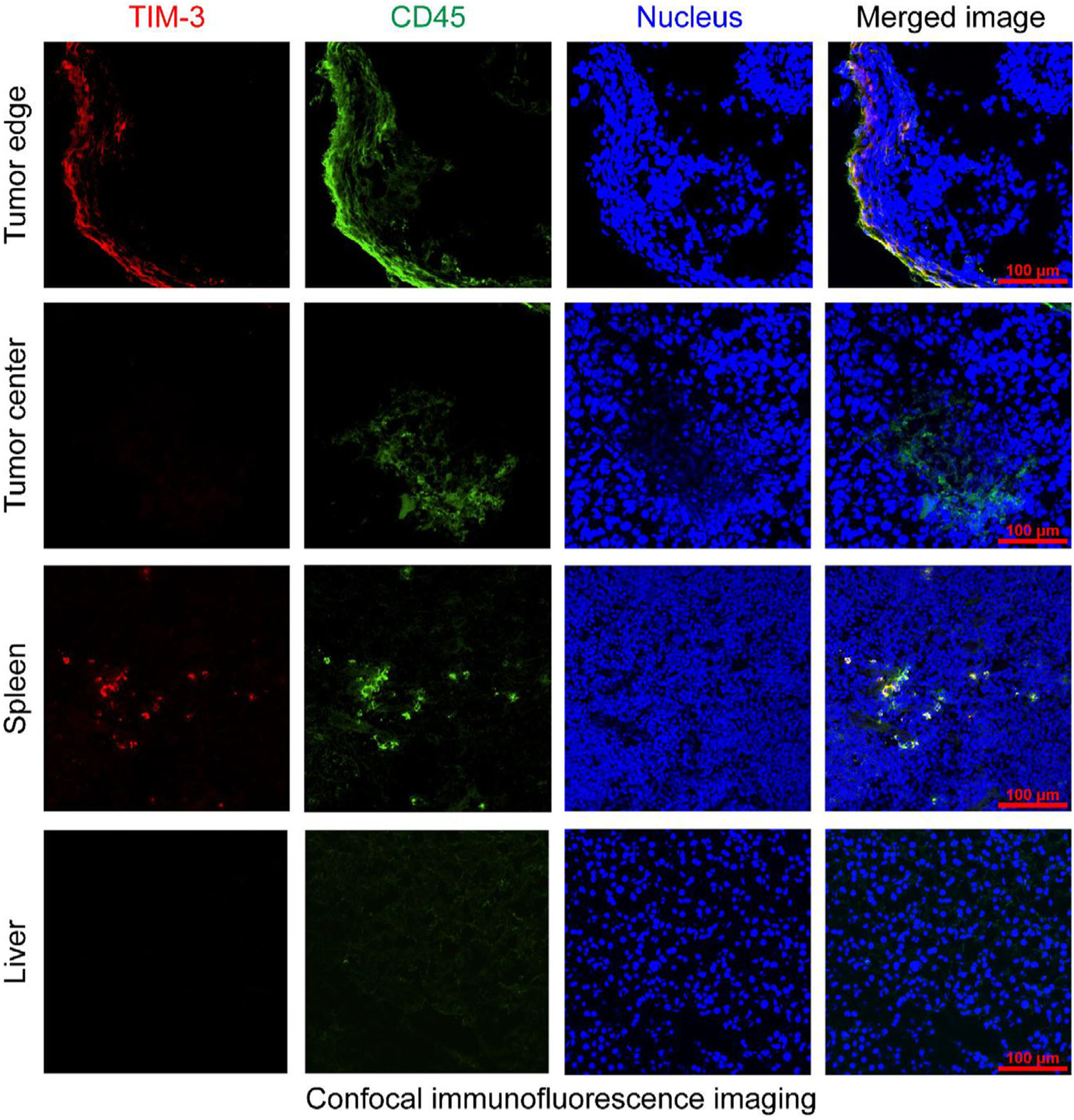
Immunofluorescent staining of the resected tumor tissue, liver, and spleen from the B16F10 tumor-bearing naïve C57BL/6N mice.
Figure 6.
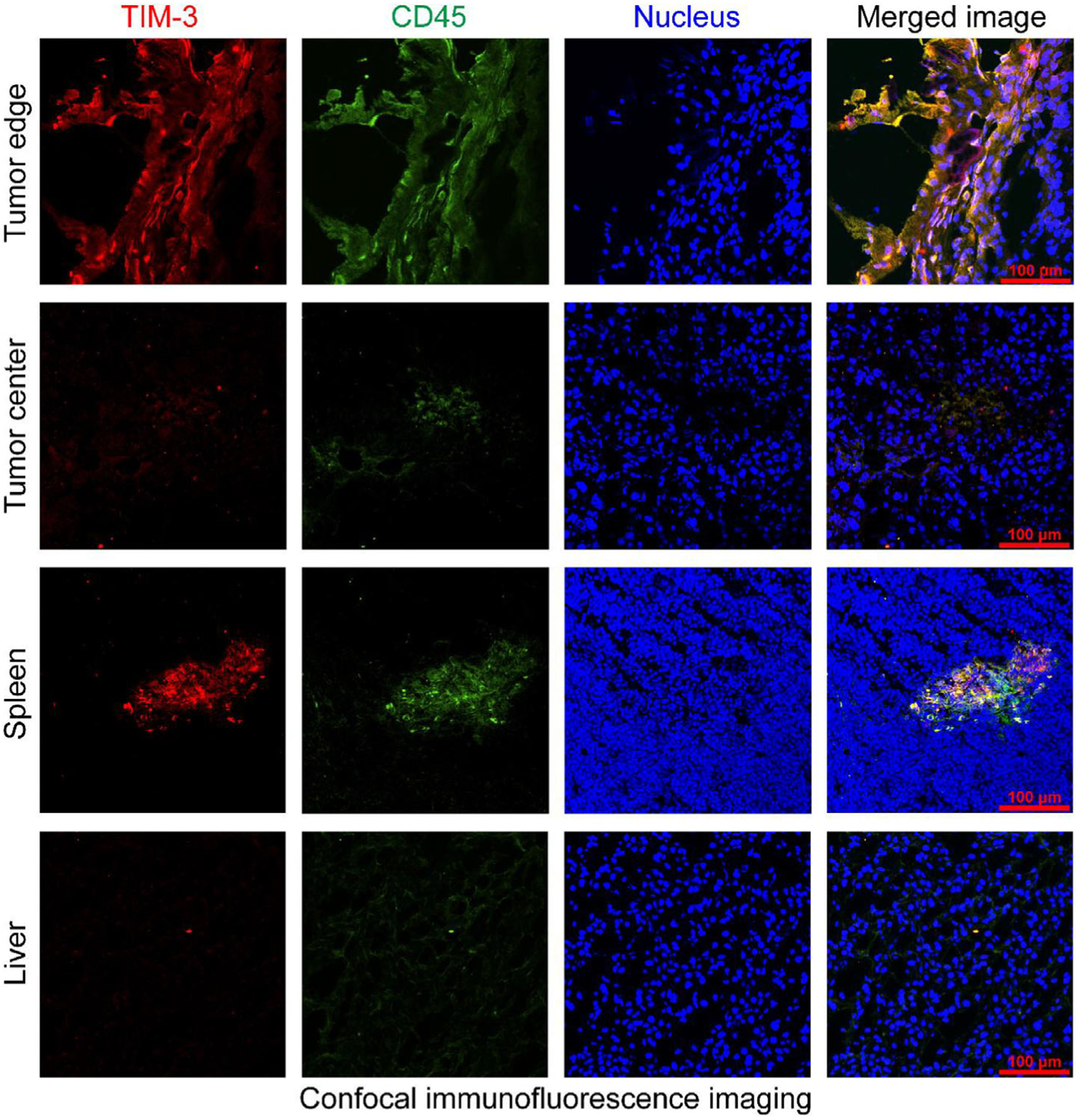
Immunofluorescent staining of the resected tumor tissue, liver, and spleen from the B16F10 tumor-bearing irradiated C57BL/6N mice (12 Gy in six fractions).
3. Discussion
Immune checkpoint inhibition using antibodies blocking either CTLA-4 or PD-1/PD-L1 axis has transformed the care of patients with a broad range of advanced malignancies. Unfortunately, a large proportion of patients have either primary or adaptive resistance to immune checkpoint inhibitors.[26] In the efforts to uncovering the underlying resistance mechanisms, increased expression of TIM-3 has been found to be involved in the adaptive resistance to anti-PD-1 therapies,[24,27,28] and in poorly immunogenic response following radiation and anti-PD-L1 combinatorial therapies.[13] This evidence suggests TIM-3 as a promising immune checkpoint that could be exploited for developing checkpoint-blocking drugs. In addition to its role as a therapeutic target, TIM-3 may also serve as a marker for designing noninvasive diagnostic approaches for patient stratification and therapeutic response assessment. Here, we successfully generated an immunoPET probe for high-resolution in vivo imaging of the immune receptor TIM-3. By harnessing serial immunoPET imaging, we uncovered how the TIM-3-positive immune cells distributed in the tumors and also in other parts of the body in naïve and irradiated melanoma-bearing C57BL/6N mice.
In vivo imaging techniques are being used to visualize the distribution and fate of immune checkpoints and their inhibitors.[17,29] Josefsson et al. reported that PD-L1 was specifically detected by SPECT in tumor and spleen in the immunocompetent mice.[30] Several subsequent studies further demonstrated that PD-1 and PD-L1 targeted immunoPET delineated spleen, lymph nodes, and nonlymphoid organs, such as brown adipose tissue.[31,32] Our study here showed that TIM-3-targeted immunoPET imaging of C57BL/6N mice did not detect signals in secondary lymphoid organs (except the spleen) or in brown adipose tissue. However, we observed prominent immunoPET signals in the peritumoral regions (Figures 1B, 2B, and 3A,B), which were further confirmed by ex vivo immunofluorescent staining of the resected tumor tissues (Figures 5 and 6). Our results are in line with a recent study by Curley et al., which reported that TIM-3 was restricted to peritumoral lymphocytes with scattered positivity in tumor cells in vulvar squamous cell carcinoma.[11] We found low retention of 64Cu-NOTA-RMT3–23 in the livers of irradiated and nonirradiated mice. A previous study also reported the restricted expression of TIM-3 around the portal vein of normal C57BL/6 mice.[22] Our preliminary results reported the unique distribution patterns of TIM-3-positive lymphocytes in murine melanomas and uncovered the role of immunoPET in imaging TIM-3-positive lymphocytes across the body before and after the radiation therapies. However, a rat mAb was used to construct the TIM-3-targeted imaging strategy in our study. Future studies may employ humanized or human antibody vectors to develop translatable immunoPET techniques, which may hopefully optimize the development and use of TIM-3-targeted immunotherapies. Interestingly, the circulation of 64Cu-NOTA-RTG2α, the nonspecific probe developed using a rat IgG2a, κ isotype control, was totally different from that of 64Cu-NOTA-RMT3–23. Indeed, some radiolabeled rat mAbs were rapidly captured by mouse liver and spleen after administration.[33] The underlying reasons for this phenomenon remain to be determined.
Increasing evidence has indicated that anti-CTLA-4 IgG1 antibodies with enhanced fragment crystallizable gamma receptor (FcγR) binding affinity have superior antitumor responses,[34–36] similar to phenomena observed in the antitumor effects of other therapeutic antibodies.[3,37] When it comes to the PD-1/PD-L1 axis, the in vivo antitumor activity of anti-PD-L1 rather than anti-PD-1 antibodies was found to be FcγR-dependent.[38] In contrast, anti-PD-1 antibodies could be rapidly removed from PD-1+ and CD8+ T cells and further transferred to macrophages, which was in part mediated by interactions between Fc domains of the antibodies and FcγRs expressed by the tumor-associated macrophages.[29] In the case of TIM-3, spleen uptake of 64Cu-NOTA-RMT3–23 was quite high at the initial time-point, but it is not known how rat IgG2a interacts with mouse FcγRs. Further investigation and elucidation of these gaps may help optimize the scaffolds of anti-TIM-3 antibodies as diagnostic agents in the first place. Furthermore, optimization of anti-TIM-3 antibodies will mediate the full therapeutic efficacy of TIM-3-targeted therapy either as a single agent or in combination with other immune checkpoint modulators.[14]
Currently, immune checkpoint inhibitors are actively being investigated in synergy with radiation therapy.[39] In head and neck squamous cell carcinomas, a concomitant increase of several T cell-specific surface markers (ICOS, PD-L1, and TIM-3) was detected after radiation plus anti-PD-L1 therapies. More importantly, dual inhibition of PD-L1 and the T cell co-inhibitory receptor TIM-3 in combination with radiation therapy significantly reduced tumor growth accompanied by a prolonged survival.[13] Radiation alone was not able to control the melanoma growth in our study, partially due to the engagement of TIM-3 in the tumor microenvironment. Our results here provided evidence that TIM-3-positive T cells existed in the tumor microenvironment. Therefore, theses subtypes of T cells could be considered as “canaries in the coal mine” or detrimental events indicating less effective therapeutic effect. Based on this observation, future studies could further design combinatorial therapeutic strategies for effective melanoma therapy, because substantial studies have demonstrated that combinatorial immunotherapy and radiation therapy could eradicate tumors.[13,39–41] Currently, we are exploring a combinatorial treatment strategy consisting of local radiation therapy, antibody antagonizing TIM-3, and other immunotherapy regimens for treating both local and distant metastatic melanomas.
4. Conclusion
In this work, we successfully developed a TIM-3-specific immunoPET probe and characterized its diagnostic value in immunocompetent melanoma models. Future studies are needed to develop translatable TIM-3-specific immunoPET that may optimize clinical TIM-3-targeted immunotherapies.
5. Experimental Section
Cell Line and Flow Cytometry:
The B16F10 melanoma cell line was kindly provided by Dr. Ravi Patel (Department of Human Oncology, University of Wisconsin-Madison). The cell line was cultured in Gibco RPMI 1640 medium supplemented with 10% FBS (Gibco) and 1% PenStrep (Invitrogen). The cell line was maintained at 37 °C in a humidified atmosphere with 5% CO2. Following a previously reported protocol,[21] flow cytometry was used to detect TIM-3 expression on the surface of B16F10 cells. Briefly, nonirradiated or irradiated B16F10 cells resuspended in flow cytometry staining buffer (Invitrogen) were incubated with 5 or 10 μg mL−1 of anti-TIM-3 mAb (RMT3–23; rat IgG2a, κ),[22] followed by washing and incubation of 5 μg mL−1 of Alexa Fluor 488-conjugated goat antirat IgG (Jackson ImmunoResearch Laboratories). Cells only group and second antibody only group were stained with no antibody or Alexa Fluor 488-conjugated goat antirat IgG, respectively. All the prepared samples were analyzed with a BD LSR Fortessa flow cytometer (BD Biosciences) and the obtained data were analyzed using the FlowJo analysis software.
Melanoma Models and Radiation Therapies:
All animal experiments were conducted in compliance with the institutional guidelines at the University of Wisconsin-Madison. To establish murine melanoma models for in vivo imaging studies, 5 × 105 or 1 × 106 B16F10 cells per mouse were resuspended in 100 μL of sterile PBS and matrigel matrix (Corning) with a ratio of 1:1 and inoculated subcutaneously (s.c.) into the right posterior flanks of C57BL/6N mice (4–5 weeks, Envigo). Two groups of C57BL/6N mice with different tumor burdens received local tumor radiation. Irradiation was performed using a cabinet orthovoltage X-ray biological irradiator (X-RAD 320; Precision X-Ray, Inc.). Only the tumor areas were irradiated with the rest of the body protected by 3 mm lead shielding. For C57BL/6N mice with relatively large tumor volumes, 12 Gy was delivered in one fraction, whereas mice with small tumor volumes were irradiated with 12 Gy in six fractions on consecutive days. Tumor size was measured every two days with digital calipers and tumor volumes were calculated using the formula (volume = length × width2/2 mm3), where length and width are the longer and shorter diameters of the tumor, respectively.
NOTA Conjugation and 64Cu Radiolabeling:
Two rat mAbs, a TIM-3-specific RMT3–23, and a nonspecific isotype control (RTG2a, rat anti-TNP [Trinitrophenyl] mAb)[22] were used to conjugate immunoPET probes. The detailed procedures for NOTA conjugation and 64Cu-labeling of NOTA-mAbs had been described previously.[42] Briefly, 1 mg of mAb (i.e., RMT3–23 or RTG2a) in PBS was prepared and the pH of the mAb solution was adjusted to 9.0–10.0 by the addition of Na2CO3 (0.1 m). 2-S-(4-Isothiocyanatobenzyl)-1,4,7-triazacyclononane-1,4,7-triacetic acid (p-SCN-Bn-NOTA; Macrocyclics) was freshly dissolved in dimethyl sulfoxide (DMSO) and immediately added to the mAb solution with a chelator/mAb ratio of 10:1. The reaction was incubated at room temperature for 2 h, followed by purification of NOTA-mAb using equilibrated PD-10 desalting columns (GE Healthcare) with PBS as the elution buffer. For 64Cu-labeling of the conjugated mAb, 74 MBq of 64CuCl2 produced in-house was first diluted in 300 μL of sodium acetate buffer (0.1 m, pH 4.5) and then mixed with 200–320 μg of NOTA-mAb. The mixed solution was incubated at 37 °C for 1 h under constant shaking (500 rpm). The final radiopharmaceuticals were purified using equilibrated PD-10 columns and the radiochemical purity of the products was measured using an instant thin-layer chromatography (iTLC; Eckert & Ziegler Radiopharma Inc.).
ImmunoPET Imaging and Data Analysis:
C57BL/6N mice with irradiated or nonirradiated tumors were administered intravenously with either 64Cu-NOTA-RMT3–23 (13.95 ± 1.67 MBq, n = 10) or 64Cu-NOTA-RTG2α (4.87 ± 1.63 MBq, n = 3). The procedures for PET data acquisition, reconstruction, and data analyses had been detailed in the previous work.[21]
Ex Vivo Immunofluorescence Staining:
Tumors, livers, and spleens of two tumor-bearing irradiated mice (7 days after 12 Gy in one fraction treatment) and two tumor-bearing naïve mice were collected and fixed in optimal cutting temperature (OCT) compound (Sakura Finetek USA, Inc.). Frozen tissue sections were sliced and stained with primary antibodies (10 μg mL−1 of RMT3–23 and 10 μg mL−1 of anti-CD45 antibody [Novus Biologicals]), followed by washing and incubation of secondary antibodies (5 μg mL−1 of Cy3-labeled donkey antirat IgG [Jackson ImmunoResearch Laboratories, Inc.] and Alexa Fluor 488-labeled goat antimouse IgG [Invitrogen]). The washed sections were mounted with UltraCruz Hard-set Mounting Medium containing DAPI (4’, 6-diamidino-2-phenylindole) (Santa Cruz Biotechnology) and confocal images were acquired using a Nikon A1R confocal microscope.
Statistical Analysis:
Statistical analyses were performed using the GraphPad software. All data were presented as the mean ± SD. Group data were compared using the two-tailed Student’s t-test, and multiple comparisons of grouped data were calculated using two-way ANOVA (Tukey’s multiple comparisons test). Adjusted p values were considered statistically significant if p < 0.05.
Acknowledgements
W.W. and D.J. contributed equally to this work. This work was supported, in part, by the University of Wisconsin-Madison, the National Institutes of Health (P30CA014520), and the National Natural Science Foundation of China (81771858).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
The ORCID identification number(s) for the author(s) of this article can be found under https://doi.org/10.1002/adtp.202000018
Contributor Information
Weijun Wei, Department of Nuclear Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 1630 Dongfang Rd, Shanghai 200127, China; Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA.
Dawei Jiang, Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA.
Hye Jin Lee, School of Pharmacy University of Wisconsin-Madison, Madison, WI 53705, USA.
Jonathan W. Engle, Department of Medical Physics, University of Wisconsin-Madison, Madison, WI 53705, USA
Hisaya Akiba, Department of Immunology, Juntendo University School of Medicine, Tokyo 113-8421, Japan.
Jianjun Liu, Department of Nuclear Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 1630 Dongfang Rd, Shanghai 200127, China.
Weibo Cai, Department of Radiology, University of Wisconsin-Madison, Madison, WI 53705, USA; School of Pharmacy University of Wisconsin-Madison, Madison, WI 53705, USA; Department of Medical Physics, University of Wisconsin-Madison, Madison, WI 53705, USA; University of Wisconsin Carbone Cancer Center, Madison, WI 53705, USA.
References
- [1].Ribas A, Wolchok JD, Science 2018, 359, 1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Chambers CA, Kuhns MS, Egen JG, Allison JP, Annu. Rev. Immunol 2001, 19, 565. [DOI] [PubMed] [Google Scholar]
- [3].Bulliard Y, Jolicoeur R, Windman M, Rue SM, Ettenberg S, Knee DA, Wilson NS, Dranoff G, Brogdon JL, J. Exp. Med 2013, 210, 1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH, Annu. Rev. Immunol 2016, 34, 539. [DOI] [PubMed] [Google Scholar]
- [5].Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, Smylie M, Dummer R, Hill A, Hogg D, Haanen J, Carlino MS, Bechter O, Maio M, Marquez-Rodas I, Guidoboni M, McArthur G, Lebbe C, Ascierto PA, Long GV, Cebon J, Sosman J, Postow MA, Callahan MK, Walker D, Rollin L, et al. , N. Engl. J. Med 2017, 377, 1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Gide TN, Wilmott JS, Scolyer RA, Long GV, Clin. Cancer Res 2018, 24, 1260. [DOI] [PubMed] [Google Scholar]
- [7].Monney L, Sabatos CA, Gaglia JL, Ryu A, Waldner H, Chernova T, Manning S, Greenfield EA, Coyle AJ, Sobel RA, Freeman GJ, Kuchroo VK, Nature 2002, 415, 536. [DOI] [PubMed] [Google Scholar]
- [8].Das M, Zhu C, Kuchroo VK, Immunol. Rev 2017, 276, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chihara N, Madi A, Kondo T, Zhang H, Acharya N, Singer M, Nyman J, Marjanovic ND, Kowalczyk MS, Wang C, Kurtulus S, Law T, Etminan Y, Nevin J, Buckley CD, Burkett PR, Buenrostro JD, Rozenblatt-Rosen O, Anderson AC, Regev A, Kuchroo VK, Nature 2018, 558, 454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zhou Q, Munger ME, Veenstra RG, Weigel BJ, Hirashima M, Munn DH, Murphy WJ, Azuma M, Anderson AC, Kuchroo VK, Blazar BR, Blood 2011, 117, 4501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Curley J, Conaway MR, Chinn Z, Duska L, Stoler M, Mills AM, Mod. Pathol 2020, 10.1038/s41379-019-0433-3. [DOI] [PubMed]
- [12].Ngiow SF, von Scheidt B, Akiba H, Yagita H, Teng MW, Smyth MJ, Cancer Res 2011, 71, 3540. [DOI] [PubMed] [Google Scholar]
- [13].Oweida A, Hararah MK, Phan A, Binder D, Bhatia S, Lennon S, Bukkapatnam S, Van Court B, Uyanga N, Darragh L, Kim HM, Raben D, Tan AC, Heasley L, Clambey E, Nemenoff R, Karam SD, Clin. Cancer Res 2018, 24, 5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Andrews LP, Yano H, Vignali DAA, Nat. Immunol 2019, 20, 1425. [DOI] [PubMed] [Google Scholar]
- [15].Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, West AN, Carmona M, Kivork C, Seja E, Cherry G, Gutierrez AJ, Grogan TR, Mateus C, Tomasic G, Glaspy JA, Emerson RO, Robins H, Pierce RH, Elashoff DA, Robert C, Ribas A, Nature 2014, 515, 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].England CG, Ehlerding EB, Hernandez R, Rekoske BT, Graves SA, Sun H, Liu G, McNeel DG, Barnhart TE, Cai W, J. Nucl. Med 2017, 58, 162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wei W, Jiang D, Ehlerding EB, Luo Q, Cai W, Trends Cancer 2018, 4, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].England CG, Jiang D, Ehlerding EB, Rekoske BT, Ellison PA, Hernandez R, Barnhart TE, McNeel DG, Huang P, Cai W, Eur. J. Nucl. Med. Mol. Imaging 2018, 45, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ehlerding EB, Lee HJ, Jiang D, Ferreira CA, Zahm CD, Huang P, Engle JW, McNeel DG, Cai W, Am. J. Cancer Res 2019, 9, 53. [PMC free article] [PubMed] [Google Scholar]
- [20].Wei W, Jiang D, Rosenkrans ZT, Barnhart TE, Engle JW, Luo Q, Cai W, Am. J. Cancer Res 2019, 9, 2413. [PMC free article] [PubMed] [Google Scholar]
- [21].Wei W, Jiang D, Ehlerding EB, Barnhart TE, Yang Y, Engle JW, Luo QY, Huang P, Cai W, Adv. Sci 2019, 6, 1801237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Oikawa T, Kamimura Y, Akiba H, Yagita H, Okumura K, Takahashi H, Zeniya M, Tajiri H, Azuma M, J.Immunol 2006, 177, 4281. [DOI] [PubMed] [Google Scholar]
- [23].Wiener Z, Kohalmi B, Pocza P, Jeager J, Tolgyesi G, Toth S, Gorbe E, Papp Z, Falus A, J. Invest. Dermatol 2007, 127, 906. [DOI] [PubMed] [Google Scholar]
- [24].Sakuishi K, Apetoh L, Sullivan JM, Blazar BR, Kuchroo VK, Anderson AC, J. Exp. Med 2010, 207, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dewan MZ, Galloway AE, Kawashima N, Dewyngaert JK, Babb JS, Formenti SC, Demaria S, Clin. Cancer Res 2009, 15, 5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Sharma P, Hu-Lieskovan S, Wargo JA, Ribas A, Cell 2017, 168, 707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shayan G, Srivastava R, Li J, Schmitt N, Kane LP, Ferris RL, OncoImmunology 2017, 6, e1261779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, Gandhi L, Redig AJ, Rodig SJ, Asahina H, Jones RE, Kulkarni MM, Kuraguchi M, Palakurthi S, Fecci PE, Johnson BE, Janne PA, Engelman JA, Gangadharan SP, Costa DB, Freeman GJ, Bueno R, Hodi FS, Dranoff G, Wong KK, Hammerman PS, Nat. Commun 2016, 7, 10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Arlauckas SP, Garris CS, Kohler RH, Kitaoka M, Cuccarese MF, Yang KS, Miller MA, Carlson JC, Freeman GJ, Anthony RM, Weissleder R, Pittet MJ, Sci. Transl. Med 2017, 9, eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Josefsson A, Nedrow JR, Park S, Banerjee SR, Rittenbach A, Jammes F, Tsui B, Sgouros G, Cancer Res 2016, 76, 472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Hettich M, Braun F, Bartholoma MD, Schirmbeck R, Niedermann G, Theranostics 2016, 6, 1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Ingram JR, Dougan M, Rashidian M, Knoll M, Keliher EJ, Garrett S, Garforth S, Blomberg OS, Espinosa C, Bhan A, Almo SC, Weissleder R, Lodish H, Dougan SK, Ploegh HL, Nat. Commun 2017, 8, 647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Bailly C, Gouard S, Lacombe M, Remaud-Le Saec P, Chalopin B, Bourgeois M, Chouin N, Tripier R, Halime Z, Haddad F, Faivre-Chauvet A, Kraeber-Bodere F, Cherel M, Bodet-Milin C, Oncotarget 2018, 9, 9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ingram JR, Blomberg OS, Rashidian M, Ali L, Garforth S, Fedorov E, Fedorov AA, Bonanno JB, Le Gall C, Crowley S, Espinosa C, Biary T, Keliher EJ, Weissleder R, Almo SC, Dougan SK, Ploegh HL, Dougan M, Proc. Natl. Acad. Sci. USA 2018, 115, 3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, Solomon I, Lesko MH, Ruef N, Roddie C, Henry JY, Spain L, Ben Aissa A, Georgiou A, Wong YNS, Smith M, Strauss D, Hayes A, Nicol D, O’Brien T, Martensson L, Ljungars A, Teige I, Frendeus B, TRACERx Melanoma, TRACERx Renal, TRACERx Lung consortia, Pule M, Marafioti T, Gore M, Larkin J, Turajlic S, Swanton C, Peggs KS, Quezada SA, Cancer Cell 2018, 33, 649.29576375 [Google Scholar]
- [36].Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, Korman AJ, Cancer Immunol. Res 2013, 1, 32. [DOI] [PubMed] [Google Scholar]
- [37].Vazquez-Lombardi R, Loetsch C, Zinkl D, Jackson J, Schofield P, Deenick EK, King C, Phan TG, Webster KE, Sprent J, Christ D, Nat. Commun 2017, 8, 15373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV, Cancer Cell 2015, 28, 285. [DOI] [PubMed] [Google Scholar]
- [39].Twyman-Saint Victor C, Rech AJ, Maity A, Rengan R, Pauken KE, Stelekati E, Benci JL, Xu B, Dada H, Odorizzi PM, Herati RS, Mansfield KD, Patsch D, Amaravadi RK, Schuchter LM, Ishwaran H, Mick R, Pryma DA, Xu X, Feldman MD, Gangadhar TC, Hahn SM, Wherry EJ, Vonderheide RH, Minn AJ, Nature 2015, 520, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Morris ZS, Guy EI, Werner LR, Carlson PM, Heinze CM, Kler JS, Busche SM, Jaquish AA, Sriramaneni RN, Carmichael LL, Loibner H, Gillies SD, Korman AJ, Erbe AK, Hank JA, Rakhmilevich AL, Harari PM, Sondel PM, Cancer Immunol. Res 2018, 6, 825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Morris ZS, Guy EI, Francis DM, Gressett MM, Werner LR, Carmichael LL, Yang RK, Armstrong EA, Huang S, Navid F, Gillies SD, Korman A, Hank JA, Rakhmilevich AL, Harari PM, Sondel PM, Cancer Res 2016, 76, 3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yang Y, Hernandez R, Rao J, Yin L, Qu Y, Wu J, England CG, Graves SA, Lewis CM, Wang P, Meyerand ME, Nickles RJ, Bian XW, Cai W, Proc. Natl. Acad. Sci. USA 2015, 112, E6525. [DOI] [PMC free article] [PubMed] [Google Scholar]