

Abstract
We identified an emerging severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variant by viral whole-genome sequencing of 2,172 nasal/nasopharyngeal swab samples from 44 counties in California, a state in the western United States. Named B.1.427/B.1.429 to denote its two lineages, the variant emerged in May 2020 and increased from 0% to >50% of sequenced cases from September 2020 to January 2021, showing 18.6%–24% increased transmissibility relative to wild-type circulating strains. The variant carries three mutations in the spike protein, including an L452R substitution. We found 2-fold increased B.1.427/B.1.429 viral shedding in vivo and increased L452R pseudovirus infection of cell cultures and lung organoids, albeit decreased relative to pseudoviruses carrying the N501Y mutation common to variants B.1.1.7, B.1.351, and P.1. Antibody neutralization assays revealed 4.0- to 6.7-fold and 2.0-fold decreases in neutralizing titers from convalescent patients and vaccine recipients, respectively. The increased prevalence of a more transmissible variant in California exhibiting decreased antibody neutralization warrants further investigation.
Keywords: SARS-CoV-2, COVID-19, viral whole-genome sequencing, molecular dating, genomic epidemiology, spike protein, L452R mutation, variant of concern, B.1.427/B.1.429, 20C/L452R, pseudovirus infectivity studies, antibody neutralization
Graphical abstract
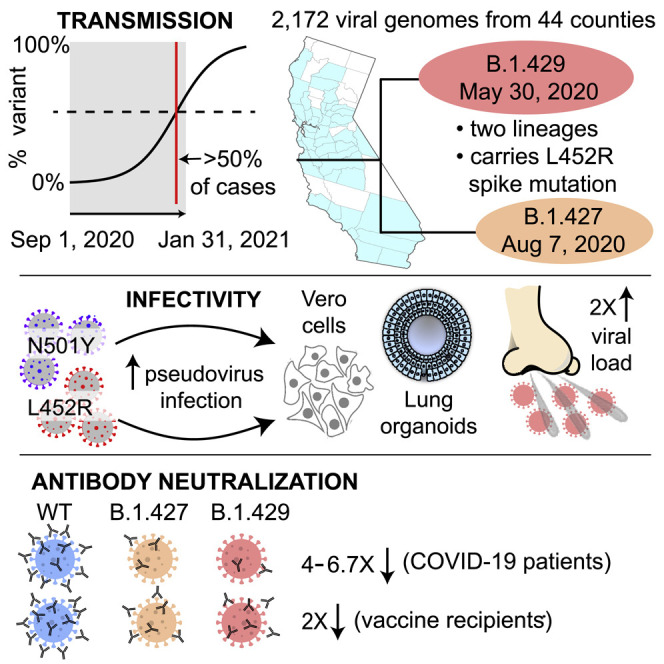
A SARS-CoV-2 variant of concern bearing the L452R spike protein mutation is widely circulating in California, United States, and demonstrates increased transmissibility, infectivity, and avoidance of antibody neutralization.
Introduction
Genetic mutation provides a mechanism for viruses to adapt to a new host and/or evade host immune responses. Although severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has a slow evolutionary rate relative to other RNA viruses (∼0.8 × 10−3 substitutions per site per year) (Day et al., 2020), an unabating coronavirus disease 2019 (COVID-19) pandemic with high viral transmission has enabled the virus to acquire significant genetic diversity since its initial detection in Wuhan, China in December 2019 (Zhu et al., 2020), thereby facilitating the emergence of new variants (Fontanet et al., 2021). Among numerous SARS-CoV-2 variants now circulating globally, those harboring a D614G mutation have predominated since June of 2020 (Korber et al., 2020), possibly due to enhanced viral fitness and transmissibility (Hou et al., 2020; Plante et al., 2021; Zhou et al., 2021).
Emerging variants of SARS-CoV-2 that harbor genome mutations that may impact transmission, virulence, and immunity have been designated “variants of concern” (VOCs). Beginning in the fall of 2020, 3 VOCs have emerged globally, each carrying multiple mutations across the genome, including several in the receptor-binding domain (RBD) of the spike protein. The B.1.1.7 variant, originally detected in the United Kingdom (UK) (Chand et al., 2020), has accumulated 17 lineage-defining mutations, including the spike protein N501Y mutation that confers increased transmissibility over other circulating viruses (Leung et al., 2021; Rambaut et al., 2020b; Volz et al., 2020). Preliminary data suggest that B.1.1.7 may also cause more severe illness (Davies et al., 2021b). As of early 2021, the B.1.1.7 variant has become the predominant lineage throughout the United Kingdom and Europe, with reported cases also rising in the United States (US) (Washington et al., 2021). The other two VOCs, B.1.351 detected in South Africa (Tegally et al., 2020) and P.1 first detected in Brazil (Sabino et al., 2021), carry E484K and K417N/K417T in addition to N501Y mutations. Multiple studies have reported that the E484K mutation in particular may confer resistance to antibody neutralization (Cole et al., 2021; Wang et al., 2021; Wibmer et al., 2021; Wu et al., 2021b; Xie et al., 2021), potentially resulting in decreased effectiveness of currently available vaccines (Liu et al., 2021; Wise, 2021). This phenotype may have also contributed to widespread reinfection by P.1 in an Amazon community that had presumptively achieved herd immunity (Buss et al., 2021; Sabino et al., 2021).
In January 2021, we and others independently reported the emergence of a variant in California carrying an L452R mutation in the RBD of the spike protein (CDPH (California Department of Public Health), 2021a; Zhang et al., 2021). Here, we used viral whole-genome sequencing of nasal/nasopharyngeal (N/NP) swab samples from multiple counties to characterize the emergence and spread of this L452R-carrying variant in California from September 1, 2020, to January 29, 2021. We also combined epidemiologic, clinical, and in vitro laboratory data to investigate transmissibility and susceptibility to antibody neutralization associated with infection by the variant.
Results
Viral genomic surveillance
We sequenced 2,172 viral genomes across 44 California counties from remnant N/NP swab samples testing positive for SARS-CoV-2 (Tables S1 and S2). The counties with proportionally higher representation in the dataset included Santa Clara County (n = 725, 33.4%), Alameda County (n = 228, 10.5%), Los Angeles County (n = 168, 7.7%), and San Francisco County (n = 155, 7.1%) (Figure 1 A). A variant, subsequently named 20C/L452R according to the Nextstrain nomenclature system (Bedford et al., 2021) or B.1.427/B.1.429 according to the Pango system (Rambaut et al., 2020a) (henceforth referred to using the Pango designation to distinguish between the B.1.427 and B.1.429 lineages), was identified in 21.5% (466 of 2,172) of the genomes (Table S1). The frequency of this variant in California increased from 0% at the beginning of September 2020 to >50% of sequenced cases by the end of January 2021, with a similar trajectory to the surge of COVID-19 cases in California from October to December 2020 (Figure S1 ). The rise in the proportion of sequenced cases due to the variant was rapid, with an estimated increase in transmission rate of the B.1.427/B.1.429 variant relative to circulating non-B.1.427/B.1.429 lineages of 20.0% (17.8%–21.1%) and an approximate doubling time of 19.1 days (17.3–21.4 days) (Figure 1B, top panel). The calculated date for when the variant was expected to become predominant (>50% of cases) in California was January 25, 2021, earlier but near in time to the February 5, 2021 date based on additional viral genomic data from samples collected February 1–March 11, 2021 (Figure 1B, bottom panel). Similar epidemic trajectories were observed from multiple counties (Figures 1C–1E and S2 ), despite different sampling approaches used for sequencing. Specifically, genomes from San Francisco County were derived from COVID-19 patients being tested at University of California, San Francisco (UCSF) hospitals and clinics; genomes from Alameda County were derived from community testing; genomes from Santa Clara County were derived from congregate facility, community, and acute care testing; and genomes from Los Angeles County were derived from coroner, community, and inpatient testing.
Figure 1.

Increasing frequency of the B.1.427/B.1.429 variant in California from September 1, 2020 to January 29, 2021
(A) County-level representation of the 2,172 newly sequenced SARS-CoV-2 genomes in the current study. Counties from which at least 1 genome were sequenced are colored in sky blue. The size of the circle is proportionate to the number of genomes sequenced from each county, while points designate counties where fewer than 10 genomes were sequenced.
(B–D) Logistic growth curves fitting the 5-day rolling average of the estimated proportion of B.1.427/B.1.429 variant cases in (B) California, (C) San Francisco County, and (D) Santa Clara County. For each curve, the estimated increase in transmission rate and doubling time are shown, along with their associated 95% confidence intervals. The predicted time when the growth curve crosses 0.5 is indicated by a vertical red line. A vertical black dotted line denotes the transition from 2020 to 2021. (B) Top: the logistic growth curve generated from all 2,172 genomes in the current study. The 95% confidence intervals for the increase in transmission rate and doubling time are shaded in blue and gray, respectively. (B) Bottom: the logistic growth curve with inclusion of an additional 2,737 sequenced genomes from California collected February 1 to March 11, 2021. The increase in transmission rate is defined as the logistic growth rate multiplied by the serial interval (Volz et al., 2020; Washington et al., 2021).
See also Figures S1 and S2 and Tables S1, S2, and S5.
Figure S1.

COVID-19 cases, frequency of the B.1.427/B.1.429 variant, and percentage of sequenced cases in California from April 1, 2020 to April 1, 2021, related to Figure 1
(A) Plot showing the reported COVID-19 cases in California. (B) Plot showing the 784 frequency of sequenced cases corresponding to the B.1.427 or B.1.429 variant. (C) Plot 785 showing the % of COVID-19 cases for which the viral genome is sequenced.
Figure S2.

Increasing frequency of the B.1.427/B.1.429 variant in Los Angeles County and Alameda County from September 2020 to January 2021, related to Figure 1
Logistic growth curves fitting the 5-day rolling average of the estimated proportion of B.1.427/B.1.429 variant cases in Los Angeles County (A) and Alameda County (B). A vertical black dotted line is used to denote the transition from 2020 to 2021.
Phylogenetic and molecular dating analyses
Bayesian phylogenetic analysis of 1,153 genomes subsampled from a 2,519-genome dataset consisting of the 2,172 California genomes sequenced in this study and 347 representative global genomes (Bedford and Neher, 2020) identified two distinct lineages in clade 20C (Nextstrain designation) associated with the variant, B.1.427 and B.1.429 (Figures 2A and 2C). Both lineages share a triad of coding mutations in the spike protein (S13I, W152C, and L452R), one coding mutation in the orf1b protein (D1183Y), and an additional 2 non-coding mutations (Figure 2A). Four additional mutations, one of them a coding mutation in orf1a (I4205V), were specific to B.1.429, while 3 additional mutations, including two coding mutations in orf1a (S3158T) and orf1b (P976L), were specific to B.1.427. A root-to-tip genetic distance plot of the 1,153 subsampled genomes showed no substantial difference between B.1.427/B.1.429 variant and non-variant lineages (Figure 2B).
Figure 2.

Genomic, phylogenetic, and molecular clock analyses of the B.1.427/B.1.429 variant in California
(A) A multiple sequence alignment of 6 representative B.1.427/B.1.429 genomes, 3 from the B.1.427 lineage, and 3 from the B.1.429 lineage, using the prototypical Wuhan Hu-1 genome as a reference. Defining single nucleotide polymorphisms (SNPs) in the B.1.427 and B.1.429 lineages are compared to each other and to other SARS-CoV-2 viruses in Nextstrain clade 20C. The SNPs are color coded as follows: red SNPs are shared between the B.1.427 and B.1.429 lineages, blue SNPs are specific to B.1.427, purple SNPs are specific to B.1.429, brown SNPs are shared with other clade 20C viruses, and gray SNPs are specific to individual viruses.
(B) Root-to-tip divergence plot of number of accumulated mutations by month based on 1,153 genomes subsampled from a complete dataset consisting of the 2,172 genomes recovered in the current study and 347 representative global genomes. The gray highlighted region encompasses the period of sampling for nearly all genomes sequenced in the current study (September 1, 2020 to January 31, 2021), with the exception of the first 2 sequenced B.1.429 genomes from Los Angeles that were reported on July 20, 2020. The orange-red bullseye denotes the first reported genomic sequence of the B.1.429 variant from Los Angeles County from a sample collected July 13, 2020.
(C) Maximum likelihood circular phylogenetic tree of the 1,153 subsampled genomes, denoting the major viral clades. The red asterisk denotes a UK B.1.1.7 variant genome.
(D) Time scaled maximum clade credibility (MCC) tree, showing the median divergence dates and associated 95% highest posterior density (HPD) distributions, or confidence intervals, for the B.1.427/B.1.429 variant (D1), B.1.429 lineage (D2), and B.1.427 lineage (D3), as estimated from TMRCA (time to most recent common ancestor) calculations. The B.1.427 lineage is colored in blue and the B.1.429 lineage in red. The orange-red bullseye denotes the first reported genomic sequence of the B.1.429 variant from Los Angeles County from a sample collected July 13, 2020.
Using a previously reported algorithm to assess divergence time dating (Drummond et al., 2012), we estimated that the most recent common ancestor emerged on May 4, 2020 (95% highest posterior density [HPD] interval: April 10–May 29). The branches giving rise to the B.1.429 and B.1.427 lineages were predicted to have diverged on May 30 (95% HPD: May 10–June 18) and August 7 (95% HPD: June 22–September 18), respectively (Figure 2D).
Increased transmissibility and infectivity
Analysis of available data from 2,126 (97.8%) of the 2,172 sequenced genomes in the current study revealed that the median PCR cycle threshold (Ct) value associated with B.1.427/B.1.429 variant infections was significantly lower overall (p = 4.75 × 10−7) than that associated with non-variant viruses (Figure 3 A). We estimated that in swab samples N/NP viral RNA loads are ∼2-fold higher in B.1.427/B.1.429 than in non-variant viruses (Drew et al., 2020). The differences in cycle threshold were greater during the November and December months relative to January (Figure 3B), although these differences were not statistically significant due to lower sample numbers. There did not appear to be significant differences in cycle threshold between hospitalized patients and outpatients infected with B.1.427/B.1.429 (Figure 3C), nor between B.1.427 and B.1.429 lineages (Figure 3D).
Figure 3.

Higher viral loads in infections from the B.1.427/B.1.429 variant as compared to non-B.1.427/B.1.429 variant lineages
(A–D) Boxen plots of available PCR cycle threshold (Ct) values for B.1.427/B.1.429 variant compared to non-variant identification for (A) all samples sequenced in the current study, (B) samples stratified by month of collection, November 2020–January 2021, (C) samples from hospitalized patients and outpatients at a single tertiary care medical center (University of California, San Francisco), and (D) samples with viruses of B.1.427 or B.1.429 lineage. Note that a Ct difference of 1 represents a 2-fold difference in the virus concentration (Drew et al., 2020). The solid horizontal line in the center box denotes the mean value. ∗∗∗∗p < 0.0001; ∗∗∗p < 0.001; ∗∗p < 0.01; ∗p < 0.05; NS, non-significant. Welch's t-test was used to determine significance.
See also Table S1.
Analysis of the SARS-CoV-2 spike protein complexed to its human ACE2 receptor (Lan et al., 2020) revealed that the L452 residue does not directly contact the receptor. Instead, L452, together with F490 and L492, form a hydrophobic patch on the surface of the spike RBD (Figure 4 A). To understand the effects of L452R RBD mutation on viral entry, pseudoviruses carrying D614G with L452R or W152C, or D614G alone were generated and used for infection of 293T cells stably expressing the ACE2 cell entry receptor and TMPRSS2 cofactor for SARS-CoV-2 (Hoffmann et al., 2020; Khanna et al., 2020). We observed increased entry by pseudoviruses carrying the L452R mutation compared to D614G alone, with a 6.7- to 22.5-fold increase in 293T cells and a 5.8- to 14.7-fold increase in HAOs (Figures 4B and 4C). This increase in pseudovirus infection with the L452R mutation is slightly lower than the increase observed with the N501Y mutation (11.4- to 30.9-fold increase in 293T cells and 23.5- to 37.8-fold increase in HAO relative to D614G alone), which has previously been reported to increase pseudovirus entry (Hu et al., 2021). Pseudoviruses carrying the W152C mutation demonstrated small increases in infection of 293T cells and HAO relative to the D614 control, although these increases were not as pronounced as those observed for the L452R and N501Y pseudoviruses.
Figure 4.

Increased infectivity of L452R-carrying pseudoviruses
(A) Upper panel: ribbon diagram of the SARS-CoV-2 spike RBD in cyan bound to ACE2 receptor in magenta (PDB: 6M0J). The receptor-binding motif of RBD is colored in dark cyan with L452 in solid spheres and F490 and L492 with dotted spheres. Sugars and Zn2+ are shown in gray. The position of N501 in direct contact with the ACE2 receptor is also shown for purposes of comparison. Lower panel: surface representation of the spike RBD showing the hydrophobic patch outlined by L452, F490, and L492.
(B) Levels of infection of SARS-CoV-2 spike pseudoviruses carrying D614G alone or D614G with N501Y, L452R, or W152C mutations in 293T cells stably expressing ACE2 and TMPRSS2. 293T cells were seeded in 96-well plates and infected with high (6 ng, left) or low (3 ng, right) concentrations of the indicated pseudoviruses for 48 h. Two biological replicates were assessed in two independent experiments, with 3 technical replicates per experiment.
(C) Levels of infection in human lung airway organoids (HAO) stably expressing ACE2. HAO were seeded in 24-well plates and infected with high (4 ng, left) or low (2 ng, right) concentrations of the indicated pseudoviruses for 72 h. Pseudovirus cell entry was measured with a luciferase assay. The error bars represent the SD of 3 technical replicates. Dunn’s multiple comparisons test was used to determine significance. Note that each of the N501Y, L452R, and W152G pseudoviruses also carries D614G. NS, not significant.
Reduced susceptibility to neutralizing antibodies from convalescent patients and vaccine recipients
To examine the effect of the L452R mutation on antibody binding, we performed neutralizing antibody assays. We cultured a B.1.429 lineage virus from a patient’s NP swab sample in Vero cells stably expressing TMPRSS2 (Vero-TMPRSS2). We then performed plaque reduction neutralization tests (PRNT) using 21 plasma samples from convalescent patients and vaccine recipients to compare neutralization titers between the B.1.429 isolate and a control isolate USA-WA1/2020 (Figures 5 A and S3 ; Table S3). Twelve samples were collected from individuals after receiving both doses of either the Pfizer BNT16b2 or Moderna mRNA-1273 vaccine, with samples collected 4–28 days after the second dose. Nine samples were convalescent plasma collected from patients who became symptomatic from COVID-19 infection during the June 21–November 11, 2020 time period, during which infection from a VOC in California was highly unlikely. Convalescent samples were collected 18–71 days after symptom onset. Measurable neutralizing antibody responses in the assay range were not observed for 1 convalescent patient and 1 vaccine recipient.
Figure 5.

B.1.427/B.1.429 variant resistance to antibody neutralization in vitro
(A) Antibody neutralization titers from 9 convalescent patients and 12 vaccine recipients against cultured WA1 (control), D614G (control), and B.1.429 viral isolates were assessed using a PRNT assay. Lines connect the individual plasma samples tested pairwise for neutralization (top row). Only a subset of the plasma samples were tested with the WA1 and D614G head-to-head comparisons (top row, right). The dotted lines denote the upper and lower bounds for the PRNT assay (1:100 to 1:3,200). Plasma samples that did not exhibit detectable neutralizing activity at titers above the lower threshold are shown as transparent. Individual PRNT50 measurements are plotted along with error bars denoting the median and SD (bottom row).
(B) Antibody neutralization titers from 10 convalescent patients against cultured WA1 (control), D614G (control), and B.1.427 viral isolates were assessed by 50% CPE endpoint dilution. Lines connect the individual plasma samples tested pairwise for neutralization (top row). Individual TCID50 measurements are plotted along with error bars denoting the median and SD (bottom row). A Wilcoxon matched pairs signed-rank test was used to determine significance. NS, not significant; PRNT, plaque-reduction neutralization test; CPE, cytopathic effect; TCID, tissue culture infective dose.
Figure S3.

Differential neutralization of WA1 and B.1.429 viruses as measured by plaque-reduction neutralization tests, related to Figure 5
Representative 6-well plates arranged in one line showing viral plaques formed after co-culture with plasma samples from a convalescent patient and vaccine recipient. The same negative control well image is shown in line with the respective viral strain for both vaccine and convalescent samples. The plaques from B.1.429 lineage virus are observed to be small and lighter than those from control WA1 virus. The larger plaques for WA1 are likely due to adaptation in Vero E6 cells; these adaptation mutations have been reported not to impact neutralization responses (Klimstra et al., 2020).
We found that in comparison to USA-WA1/2020, 7 of 8 (88%) convalescent patients and 6 of 11 (55%) vaccine recipients showed reduced PRNT50 titers to a B.1.429 lineage virus, with 6.7-fold (p = 0.016) and 2-fold (p = 0.031) median reductions, respectively (Figure 5A). There were no differences in neutralization between WA1 and D614G isolates by convalescent or post-vaccination plasma (Figure 5A, right).
Next, we independently evaluated neutralizing antibody responses against a cultured B.1.427 lineage virus. The TCID50, or median tissue culture infective dose at which 50% of cultures exhibited cytopathic effect (CPE), was determined for 10 different convalescent plasma samples collected from COVID-19 patients from June 19 to August 19, 2020, with samples collected 21–85 days after symptom onset. Nine of 10 (90%) convalescent patients showed reduced TCID50 titers to a B.1.427 lineage virus (Figure 5B), with 5.3-fold (p = 0.0039) and 4.0-fold (p = 0.0039) median reductions compared to USA-WA1/2020) and D614G isolates, respectively.
Discussion
As of early 2021, multiple SARS-CoV-2 variants have emerged in different regions of the world, each rapidly establishing itself as the predominant lineage within a few months after its initial detection (Chand et al., 2020; Faria et al., 2021; Sabino et al., 2021; Tegally et al., 2020). In the current study, we describe the spread of an emerging B.1.427/B.1.429 variant in California carrying a characteristic triad of spike protein mutations (S13I, W152C, and L452R) that is predicted to have emerged in May 2020 and increased in frequency from 0% to >50% of sequenced cases from September 2020 to January 2021. Importantly, this variant was found to comprise 2 separate lineages, B.1.427 and B.1.429, with each lineage rising in parallel in California as well as in multiple other states (Gangavarapu et al., 2020). Potential increased transmissibility of the B.1.427/B.1.429 variant is also supported by findings of an ∼2-fold increase in median viral loads in infected patients and increased infectivity of cultured cells and lung organoids in vitro. We also observed a moderate resistance to neutralization by antibodies elicited by prior infection (4.0- to 6.7-fold) or vaccination (2-fold). These findings indicate that the B.1.427/B.1.429 variant warrants close monitoring and further investigation regarding its potential to cause future surges in COVID-19 cases, accumulate further mutations, and/or decrease vaccine effectiveness.
The results here highlight the urgent need for implementation of a robust genomic surveillance system in the United States and globally to rapidly identify and monitor SARS-CoV-2 variants. Although our findings suggest that the B.1.427/B.1.429 variant emerged as early as May 2020, the first cases of B.1.427 and B.1.429 in the United States were not identified by sequencing until September 28, 2020, and July 13, 2020, respectively. Sparse genomic sequencing of circulating viruses likely contributed to delayed identification of the B.1.427/B.1.429 variant. Furthermore, unlike in countries such as the United Kingdom (COVID-19 Genomics UK (COG-UK), 2020) and South Africa (Msomi et al., 2020), the United States lacks an organized system for real-time analysis and reporting of variants that is tied to actionable public health responses. Public disclosure of the existence of this variant, initiated by us in coordination with local and state public health agencies and the United States Centers for Disease Control and Prevention (US CDC), did not occur until January 17, 2021 (CDPH (California Department of Public Health), 2021a), by which time the variant had already become the dominant lineage in several California counties and spread to multiple other states (Gangavarapu et al., 2020). Earlier identification and monitoring of the variant might have guided focused contact tracing efforts by public health to slow its spread, as well as enabled more timely investigation of its potential significance. Our identification of the B.1.427/B.1.429 variant was made possible by California COVIDNet, a collaborative sequencing network working to track transmission and evolution of SARS-CoV-2 in the state by viral whole-genome sequencing (CDPH (California Department of Public Health), 2021a).
The B.1.427/B.1.429 variant carries 4 coding mutations, including 3 in the spike protein, that are not found in the 3 other SARS-CoV-2 VOCs (B.1.1.7, B.1.351, and P.1) or in other major circulating lineages. The appearance of several new mutations in a variant over a short period of time is not unexpected and may be indicative of sudden increase in the evolutionary rate of a directly ancestral lineage (Rambaut et al., 2020b). Indeed, the B.1.1.7, B.1.351, and P.1 variants exhibit striking genetic divergence, with each carrying over 8 missense mutations in the spike protein (Faria et al., 2021; Rambaut et al., 2020b; Tegally et al., 2020). The evolutionary mechanism underlying these changes remains unexplained but may potentially be due to accelerated viral quasispecies evolution in chronically infected patients (Avanzato et al., 2020; Choi et al., 2020; Kemp et al., 2021). In contrast to VOCs such as B.1.1.7 (Rambaut et al., 2020b), the root-to-tip divergence plot corresponding to the B.1.427/B.1.429 variant is consistent with gradual accumulation of mutations over time. However, we also cannot rule out accelerated evolution of the variant given the absence of sequenced genomes directly ancestral to the B.1.427 and B.1.429 lineages, possibly due to limited genomic sampling (Figure S1), as well as the anomalous position of the first sequenced B.1.429 genome from Los Angeles County in July 2020 on the root-to-tip divergence plot.
Prior studies have suggested that the L452R mutation may stabilize the interaction between the spike protein and its human ACE2 receptor and thereby increase infectivity (Chen et al., 2020; Teng et al., 2021). Our findings of enhanced infection of 293T cells and lung organoids by pseudoviruses carrying L452R confirm these early predictions. Notably, the L452 residue does not directly contact the ACE2 receptor, unlike the N501 residue that is mutated to Y501 in the highly transmissible B.1.1.7, B.1.351, and P.1 variants. However, given that L452 is positioned in a hydrophobic patch of the spike RBD, it is plausible that the L452R mutation causes structural changes in the region that promote the interaction between the spike protein and its ACE2 receptor. Notably, our findings reveal that the infectivity of L452R pseudoviruses was higher than D614G, but slightly reduced compared to that of N501Y pseudoviruses in 293T cells and human airway lung organoids.
Interestingly, we found that the observed differences in viral load were more pronounced during the November and December months, when cases and deaths of COVID-19 in California were surging (CDPH (California Department of Public Health), 2021b), than in December. These findings likely reflect sampling bias, with a possible increased focus on sequencing cases in symptomatic patients and/or associated with outbreaks. Nevertheless, the impact of increased transmissibility associated with B.1.427/B.1.429 on disease severity is a critical question that we are aiming to address in ongoing studies. It is notable that infection by the highly contagious N501Y-carrying B.1.1.7 variant has been shown to be associated with an increased risk of severe disease and death (Challen et al., 2021; Davies et al., 2021c). In addition, whether the L452R-carrying B.1.427/B.1.429 will continue to remain the predominant circulating strain in California, or whether it will eventually be replaced by the B.1.1.7 variant (Washington et al., 2021) remains unclear.
The L452R mutation in the B.1.427/B.1.429 variant has been observed previously in rare, mostly singleton cases, first reported from Denmark on March 17, 2020, and also reported from multiple US states and the United Kingdom prior to September 1, 2020 (Gangavarapu et al., 2020). Given our findings of increased infectivity of L452R pseudoviruses, it is unclear why surges in L452R-carrying lineages have not occurred earlier. We speculate that although these lineages may have been more infective, transmission may not have reached a critical threshold locally or may have been influenced by other factors such as population density and/or public health interventions. An alternative (but not mutually exclusive) possibility is that the additional mutations in B.1.427/B.1.429, especially the W152C and S13I mutations in the spike protein, may contribute to increased infectivity of the variant relative to lineages carrying the L452R mutation alone. Indeed, in the current study we observed smaller but statistically significant increases in infection of 293T cell and lung organoids by pseudoviruses carrying W152C. Studies of pseudoviruses carrying the 3 spike mutations or the full complement of mutations in the B.1.427/B.1.429 variant are needed to address these hypotheses.
Our neutralization findings are consistent with a prior report showing decreased binding of L452R-carrying pseudoviruses by antibodies from previously infected COVID-19 patients and escape from neutralization in 3 of 4 convalescent plasma samples (Liu et al., 2020). We speculate that mutation of the L452 residue in a hydrophobic pocket may induce conformational changes in the RBD that impact neutralizing antibody binding. Of note, a >4-fold decrease in neutralizing antibody titers in convalescent plasma suggests that immune selection pressure from a previously exposed population may be partly driving the emergence of L452R variants. These data also raise questions regarding potential higher risk of re-infection and the therapeutic effectiveness of monoclonal antibodies and convalescent plasma to treat COVID-19 disease from the B.1.427/B.1.429 variant.
Overall, the modest 2-fold decrease in neutralizing antibody titers in vaccine recipients to the B.1.429 variant is an indication of the robust neutralizing antibody responses elicited by mRNA vaccines in the face of variants under immune selection pressure. Indeed, a reduction in neutralization of a similar magnitude associated with the L452R mutation has been reported following mRNA vaccination in studies using pseudotype assays (Garcia-Beltran et al., 2021; Wu et al., 2021a). The use of a B.1.429 isolate in the present study, carrying the full complement of mutations that characterize the lineage, may account for relative fold differences between these two aforementioned studies and ours, and the contribution of epistatic mutations to neutralization phenotypes for SARS-CoV-2 variants merits further study. In addition, because neutralizing antibodies in natural infection have been shown to wane over time (Lau et al., 2021; Seow et al., 2020), longitudinal serologic studies are needed to determine whether these modest decreases will affect the long-term durability of vaccine-elicited immune responses to the B.1.427/B.1.429 variant. Of concern is also the possibility that B.1.427/B.1.429 lineages may accumulate additional mutations in the future that may further enhance the escape phenotype.
Limitations of study
Although in this study, we obtain robust estimates for the emergence and growth of the B.1.427/B.1.429, these estimates may be biased by uneven sampling and limited genomic sampling overall relative to the number of COVID-19 infections in California (Figure S1). The pseudovirus infectivity studies evaluated only the L452R mutation, and the impact of other mutations in the B.1.427/B.1.429 genome in combination needs to be studied experimentally. The neutralization studies included a limited number of convalescent patients (n = 19 in total) and vaccine recipients (n = 12); in addition, some of the vaccine recipients had not yet received the second dose or were sampled prior to 14 days after their second dose. Further investigation of potential antibody neutralization escape associated with the B.1.427/B.1.429 variant in larger cohorts of patients and vaccinees is needed to confirm our results.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| CoV-2/human/USA/CA-UCSF-0001C/2020 | Isolated from patient; Samuel et al., 2020 | N/A |
| Isolate USA-WA1/2020, GenBank: MN985325 | BEI Resources | NR-52281 |
| Isolate hCoV-19/USA/CA-UCSF-UC691-P1/2020 (B.1.429) | Isolated from patient with SARS-CoV-2 genome hCoV-19/USA/CA-UCSF-UC691/2020 | N/A |
| Isolate hCoV-19/USA/CA-CDPH309-P1/2020 (B.1.427) | Isolated from patient with SARS-CoV-2 genome hCoV-19/USA/CA-CDPH309/2020 | N/A |
| Biological samples | ||
| Remnant nasal/nasopharyngeal swab samples in universal transport media | Obtained from patients under IRB-approved biobanking protocol | N/A |
| Peripheral blood plasma | Obtained from patients and vaccinated recipients under IRB-approved biobanking and prospective study protocols | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| DNA/RNA shield | Zymo Research | Cat# R1100-250 |
| Critical commercial assays | ||
| Taqpath 1-Step Multiplex Real-time RT-PCR | ThermoFisher | Cat# A28526 |
| Omega BioTek MagBind Viral DNA/RNA Kit | Omega Biotek | Cat# M6246-03 |
| KingFisherTM Flex Purification System | ThermoFisher | Cat# 5400630 |
| NEBNext Ultra II DNA Library Prep Kit | New England Biolabs | Cat# E7645L |
| NEBNext Multiplex Oligos | New England Biolabs | Cat# E6440L |
| Lenti-X p24 Rapid Titer Kit | Takara | Cat# 632200 |
| Luciferase Assay System | Promega | Cat# E1501 |
| Deposited data | ||
| SARS-CoV-2 genomes in GISAID | Accession numbers in Table S1 | Table S1 |
| SARS-Cov-2 genomes in NIH GenBank | BioProject accession numbers APRJNA722044, PRJNA171119, and PRJNA639591 | Table S1; GenBank: MW972466 - MW974550 |
| FASTA files, XML files, and scripting code used for the SARS-CoV-2 genome assembly and phylogenetic / molecular dating analyses | Chiu and Servellita, 2021 | https://doi.org/10.5281/zenodo.4688394 |
| Experimental models: Cell lines | ||
| Vero E6 | ATCC | CRL-1586 |
| Vero E6 cells stably expressing TMPRSS2 | Case et al., 2020 | N/A |
| A549-ACE2 cells | Peter Jackson lab, Stanford University | N/A |
| Vero-81 | ATCC | CCL-81 |
| 293T cells | ATCC | CRL-3216 |
| HAO-ACE2 | This study | N/A |
| 293T-ACE2-TMPRSS2 | This study | N/A |
| Oligonucleotides | ||
| SARS-CoV-2 primers Version 3 for tiling multiplexed amplicon PCR | Quick et al., 2017 | https://artic.network/ncov-2019 |
| TaqPath COVID-19 primers | Thermo Fisher Scientific | A47814 |
| Recombinant DNA | ||
| pCDNA3.1-SARS-CoV-2-Spike-D614G | This study | N/A |
| pCDNA3.1-SARS-CoV-2-Spike-D614G + N501Y | This study | N/A |
| pCDNA3.1-SARS-CoV-2-Spike-D614G + L452R | This study | N/A |
| pCDNA3.1-SARS-CoV-2-Spike-D614G + W152C | This study | N/A |
| Software and algorithms | ||
| BBTools suite, v38.87 | Bushnell, 2021, https://jgi.doe.gov/data-and-tools/bbtools/ | N/A |
| MAFFT aligner v7.388 | Katoh and Standley, 2013, https://mafft.cbrc.jp/alignment/software/ | N/A |
| Geneious v11.1.5 | Kearse et al., 2012, https://www.geneious.com | N/A |
| Nextstrain/Augur pipeline v3.0.0 | https://github.com/nextstrain/augur | N/A |
| PANGOLIN v.2.3.8 | Rambaut et al., 2020a, https://github.com/cov-lineages/pangolin | N/A |
| GraphPad Prism v9.1.0 (216) | GraphPad Software, https://www.graphpad.com/ | N/A |
| BEAST v2.63 | Bouckaert et al., 2019, https://www.beast2.org/ | N/A |
| R v4.0.3 | https://www.R-project.org/ | N/A |
| Python v3.7.10 | Python Software Foundation, https://www.python.org/ | N/A |
| Adobe Illustrator v23.1.1 | Adobe, https://www.adobe.com/ | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Charles Chiu (charles.chiu@ucsf.edu).
Materials availability
Passaged aliquots of cultured SARS-CoV-2 B.1.427 and B.1.429 viruses, pseudoviruses bearing the D614G, L452R, and/or W152C viruses, and SARS-CoV-2 nasal swab / nasopharyngeal samples and/or RNA extracts are available upon request.
Data and code availability
Assembled SARS-CoV-2 genomes in this study were uploaded to GISAID (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) (accession numbers in Table S1) and can be visualized in Nextstrain. Viral genomes were also submitted to the National Center for Biotechnology Information (NCBI) GenBank database (accession numbers MW972466-MW974550), BioProject accession number PRJNA722044 and umbrella BioProject accession number PRJNA171119, Chiu laboratory at UCSF; umbrella BioProject accession number PRJNA639591, Wyman laboratory at UC Berkeley). FASTA files, XML files, and scripting code used for SARS-CoV-2 genome assembly and phylogenetic / molecular dating analyses are available in a Zenodo data repository (https://doi.org/10.5281/zenodo.4688394) (Chiu and Servellita, 2021).
Experimental model and subject details
Human sample collection and ethics
Remnant nasal/nasopharyngeal (N/NP) swab samples in universal transport media (UTM) or viral transport media (VTM) (Copan Diagnostics, Murrieta, CA, USA) from RT-PCR positive COVID-19 patients were obtained from the University of California, San Francisco (UCSF) Clinical Microbiology Laboratory, the Innovative Genomics Institute (IGI) at University of California, Berkeley, California Department of Public Health, Santa Clara County and Los Angeles County for SARS-CoV-2 genome sequencing. A small fraction of swab samples (< 1%) were obtained from the anterior nares. Clinical samples from state and county public health laboratories were collected and sequenced as part of routine public health surveillance activities. Clinical samples from the IGI were sequenced under a waiver from the UC Berkeley Office for the Protection of Human Subjects. Clinical samples from UCSF were collected for a biorepository and sequenced according to protocols approved by the UCSF Institutional Review Board (protocol number 10-01116, 11-05519). Samples were obtained from pediatric and adult donors of all genders. No analyses based on sex or age were conducted in this study.
Cell culture models
Cells used for this study include Vero E6 cells, Vero-81 cells, Vero cells overexpressing human TMPRSS2 (Vero-TMPRSS2), A549 cells stably expressing ACE2 (A549-ACE2), and 293T cells stably expressing ACE2 and TMPRSS2 (293T-ACE2-TMPRSS2) (Khanna et al., 2020).
. Vero E6 cells were cultured in MEM supplemented with 1x penicillin-streptomycin-glutamine (GIBCO) and 10% fetal calf serum (FCS). Vero-81 cells were cultured with MEM supplemented with 1x penicillin-streptomycin (GIBCO) and glutamine (GIBCO) and 5% FCS (Hyclone). Vero-TMPRSS2 cells were maintained in DMEM supplemented with 1x sodium pyruvate, 1x penicillin-streptomycin-glutamine and 10% FCS. A549-ACE2 cells were cultured in DMEM/F-12 media supplemented with 10% FCS. 293T-ACE2-TMPRSS2 cells were cultured in DMEM supplemented with 10% FCS, 1% penicillin-streptomycin, 10 μg/mL blasticidin and 1 μg/mL puromycin. Cell cultures were maintained in a humidified incubator at 37°C in 5% CO2 in the indicated media and passaged every 3-4 days.
Human airway lung organoids (HAO)
Human airway lung organoids (HAO) were grown from whole-lung lavages from adult donors and cultured as previously reported (Sachs et al., 2019). Briefly, single cells were suspended in 65% reduced growth factor BME2 (Basement Membrane Extract, Type 2). From this mixture, 50 μL drops with 1,000–40,000 cells were seeded in 24-well suspension culture plates to generate three-dimensional organoids representing the 4 major epithelial cell types (basal cells, club cells, goblet cells, and ciliated cells). In order to generate HAO stably expressing ACE2 (HAO-ACE2), organoids were transduced with lentiviruses encoding ACE2 for 6 hours, expanded for 48 hours, and selected with blasticidin (1 μg/ml) for 7 days.
Isolation of SARS-CoV-2 viral strains for neutralization studies
For the B.1.429 neutralization studies, a non-B.1.427/B.1.429 variant SARS-CoV-2/human/USA/CA-UCSF-0001C/2020 clinical isolate carrying the D614G spike mutation was cultured as previously described (Samuel et al., 2020) and passaged in A549-ACE2 expressing cells. For isolation of the B.1.429 lineage virus, 100 μL of a NP swab sample from a COVID-19 patient that was previously sequenced and identified as B.1.429 was mixed 1:1 with serum free DMEM (supplemented with 1x sodium pyruvate and 1x penicillin-streptomycin-glutamine), and two-fold serial dilutions were made of the sample over six wells of a 96-well plate. 100 μL of freshly trypsinized Vero-TMPRSS2 cells resuspended in DMEM (supplemented with 1x sodium pyruvate, 2x penicillin-streptomycin-glutamine, 5 μg/mL amphotericin B and 10% FCS) was added to each well and mixed. The culture was incubated at 37°C in 5% CO2 for 4-6 days and cytopathic effect (CPE) on cells was evaluated daily using a light microscope. The contents of wells positive for CPE were collected and stored at −80°C as a passage 0 stock (P0). P1 stocks were made following infection of four near confluent wells of a 24-well plates with Vero-TMPRSS2 using the P0 stock. Supernatants were harvested 48 hours later after centrifugation at 800 g for 7 minutes. P2 stocks were similarly made after infection of a near confluent T25 plate seeded with Vero E6 cells. All steps for isolation of the B.1.429 lineage virus were done in a Biosafety Level 3 lab using protocols approved by the Institutional Biosafety Committee at UCSF.
For the B.1.427 neutralization studies, B.1.427 and non-B.1.427/B.1.429 variant D614G viruses were cultured from NP swab samples from COVID-19 patients identified by viral whole-genome sequencing as being infected by the B.1.427 or non-B.1.427/B.1.429 variant D614G lineage. Briefly, 100 μL of NP swab sample was diluted 1:5 in PBS supplemented with 0.75% bovine serum albumin (BSA-PBS) and added to confluent Vero-81 cells in a T25 flask. After adsorption for 1 h, additional media was then added, and the flask was incubated at 37°C with 5% CO2 for 3-4 days with daily monitoring for CPE. The contents were collected, clarified by centrifugation and stored at −80C as passage 0 stock. P1 stock was made by inoculation of Vero-81 confluent T150 flasks with 1:10 diluted p0 stock and similarly monitored and harvested to approximately 50% confluency. All steps for isolation of the B.1.427 lineage virus were done in a Biosafety Level 3 lab at the Viral and Rickettisial Disease Laboratory (VRDL) at the California Department of Public Health (CDPH).
For both the B.1.429 and B.1.427 neutralization studies, the SARS-CoV-2 USA-WA1/2020 strain (BEI resources) was passaged in Vero E6 cells or Vero-81 cells and used as a control. All stocks were resequenced and the consensus assembled viral genomes were identical to the genomes derived from the primary NP samples and carried all of the expected mutations.
Method details
SARS-CoV-2 diagnostic testing
Due to variation in results reported by different clinical testing platforms used at UCSF, the Taqpath Multiplex Real-time RT-PCR test, which includes nucleoprotein (N) gene, spike (S) gene, and orf1ab gene targets, was used to determine cycle threshold (Ct) values for PCR-positive samples. The Taqpath assay was also used for determining Ct values for PCR-positive samples from Alameda County that were sequenced by the University of California, Berkeley IGI and from the California Department of Public Health.
SARS-CoV-2 genome sequencing
NP swab samples were prepared using 100 uL of the primary sample in UTM or VTM mixed with 100uL DNA/RNA shield (Zymo Research, #R1100-250). The 1:1 sample mixture was then extracted using the Omega BioTek MagBind Viral DNA/RNA Kit (Omega Biotek, #M6246-03) on KingFisherTM Flex Purification System with a 96 deep-well head (ThermoFisher, 5400630). Extracted RNA was reverse transcribed to complementary DNA and tiling multiplexed amplicon PCR was performed using SARS-CoV-2 primers Version 3 according to a published protocol (Quick et al., 2017). Amplicons were ligated with adapters and incorporated with barcodes using NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, #E7645L). Libraries were barcoded using NEBNext Multiplex Oligos for Illumina (96 unique dual-index primer pairs) (New England Biolabs, #E6440L) and purified with AMPure XP beads (Beckman-Coulter, #63880). Amplicon libraries were then sequenced on either Illumina MiSeq or Novaseq 6000 as 2x150 paired-end reads (300 cycles).
Viral genome assembly and variant calling
Genome assembly of viral reads and variant calling were performed using an in-house developed bioinformatics pipeline as previously described (Deng et al., 2020). In short, Illumina raw paired-end reads were first screened for SARS-CoV-2 sequences using BLASTn (BLAST+ package 2.9.0) alignment against viral reference genome NC_045512, and then processed using the BBTools suite, v38.87 (Bushnell, 2021). Adaptor sequences were trimmed and low-quality reads were removed using BBDuk, and then mapped to the NC_045512 reference genome using BBMap. Variants were called with CallVariants and a depth cutoff of 5 was used to generate the final assembly. A genome coverage breadth of ≥ 70% was required for inclusion in the study. PANGOLIN (Phylogenetic Assignment of Named Global Outbreak LINeages) v.2.3.8 was used to assign SARS-CoV-2 lineages (Rambaut et al., 2020a).
Multiple sequence alignment of 6 B.1.427/B.1.429 genomes and the Wuhan Hu-1 prototypical genome (GISAID ID: EPI_ISL_402125, GenBank: MN908947) was performed using the MAFFT aligner v7.388 (Katoh and Standley, 2013) as implemented in Geneious v11.1.5 (Kearse et al., 2012).
Phylogenetic analysis
High-quality SARS-CoV-2 genomes (n = 2,519, 2,172 generated in the current study and 347 used as representative global genomes) were downloaded from the Global Initiative on Sharing of All Influenza Data (GISAID) database and processed using the Nextstrain bioinformatics pipeline Augur using IQTREE v1.6. Branch locations were estimated using a maximum-likelihood discrete traits model. The resulting tree consisting of 1,153 subsampled genomes was visualized in the Nextstrain web application Auspice (root-to-tip divergence plot in Figure 2B) and in Geneious v11.1.5 (circular phylogenetic tree in Figure 2C) (Kearse et al., 2012).
Molecular dating analysis of SARS-CoV-2 for estimating the TMRCA (time to most recent common ancestor) and divergence dates for the B.1.426/B.1.427 variant was performed using the Markov chain Monte Carlo (MCMC) method implemented by Bayesian Evolutionary Analysis on Sampling Trees (BEAST) software v.2.63 (Bouckaert et al., 2019; Drummond et al., 2012). To decrease computational turnaround time, a representative subset of 490 out of the 1,153 subsampled genomes was identified by combining 225 of the 227 B.1.427/B.1.429 genomes, 100 randomly selected non-B.1.427/B.1.429 variant genomes from California, and all 165 global sequences outside of California. Two B.1.427/B.1.429 genomes (UC1504 and UC464) were found to be outliers that did not map to the B.1.427/B.1.429 phylogenetic cluster due to regions of low genomic coverage and were removed from further analysis. BEAST analysis of the 490 representative genomes was performed using an HKY85 nucleotide substitution model with a strict clock and exponential population growth (Laplace distribution). All models were run using default priors. The chain length was set to 100 million states with a 10% burn-in. Convergence was evaluated using Tracer v1.7.1 (Rambaut et al., 2018). As a single BEAST run resulted in some parameters with effective sample size (ESS) values of < 200, the logged MCMC output of two runs, each consisting of 100 million states, was combined using LogCombiner v.1.10.4 from the BEAST package. The two runs were inspected prior to combining them and were found to yield nearly identical tree topologies. After combining the MCMC chains from both runs, the ESS values for all parameters were > 200, ranging from 265 to 13,484. The resulting maximum clade credibility (MCC) tree was generated using TreeAnnotator v.2.6.3 (Drummond et al., 2012) and visualized using FigTree v.1.4.4 (Rambaut, 2021).
SARS-CoV-2 receptor binding domain mutagenesis and pseudovirus infection assay
SARS-CoV-2 spike mutants (D614G, D614G+W152C, D614G+L452R, and D614G+N501Y) were cloned using standard site-directed mutagenesis and PCR. Pseudoviruses typed with these spike mutants were generated as previously described with modifications (Crawford et al., 2020). Briefly, 293T cells were transfected with plasmid DNA (per 6-well plate: 340 ng of spike mutants, 1 μg CMV-Gag-Pol (pCMV-dΔR8.91), 125 ng pAdvantage (Promega), 1 μg Luciferase reporter) for 48 h. Supernatant containing pseudovirus particles was collected, filtered (0.45 μm), and stored in aliquots at −80°C. Pseudoviruses were quantified with a p24 assay (Takara #632200), and normalized based on titer prior to infection for entry assays.
Human airway organoids (HAO) stably expressing ACE2 (HAO-ACE2) or 293T cells stably expressing ACE2 and TMPRSS2 (293T-ACE2-TMPRSS2) were infected with an equivalent amount of the indicated pseudoviruses in the presence of 5-10 ug/ml of polybrene for 72h. Pseudovirus entry was assayed using a luciferase assay (Promega #E1501) and luminescence was measured in a plate reader (TECAN, Infinite 200 Pro M Plex). Two independent experiments were run for the 293T pseudovirus assays (2 biological replicates), with 3 technical replicates run per experiment. The HAO pseudovirus assays were run as a single experiment with 3 technical replicates.
Plaque reduction neutralization tests using a B.1.429 lineage virus
Conventional PRNT assays were done using P2 stocks of B.1.429 lineage viruses and the USA-WA1/2020 isolate passaged on Vero E6 cells. Patient plasma was heat inactivated at 56°C for 30 minutes, clarified by centrifugation at 10,000 relative centrifugal force (rcf.) for 5 minutes and aliquoted to minimize freeze thaw cycles. Serial 2-fold dilutions were made of plasma in PBS supplemented with 0.75% bovine serum albumin (BSA). Plasma dilutions were mixed with ∼100 plaque forming units (pfu) of viral isolates in serum free MEM in a 1:1 ratio and incubated for 1 hr at 37°C. Final plasma dilutions in plasma-virus mixtures ranged from 1:100 to 1:3200. 250 μL of plasma-virus mixtures were inoculated on a confluent monolayer of Vero E6 cells in 6-well plates, rocked and incubated for 1 h in a humidified incubator at 37°C in 5% CO2. After incubation, 3 mL of a mixture of MEM containing a final concentration of 2% FCS, 1x penicillin-streptomycin-glutamine and 1% melted agarose, maintained at ∼56°C, was added to the wells. After 72 h of culture as above, the wells were fixed with 4% paraformaldehyde for 2 h, agarose plugs were removed, and wells were stained with 0.1% crystal violet solution. Plaques were counted and the PRNT50 values were defined as the serum dilution at which 50% or more of plaques were neutralized. Assays were done in duplicate, and a positive control and negative control were included using plasma with known neutralizing activity (diluted 1:50) and from a SARS-CoV-2 unexposed individual (1:20 dilution), respectively. All steps were done in a Biosafety Level 3 lab using protocols approved by the Institutional Biosafety Committee at UCSF.
CPE endpoint neutralization assays using a B.1.427 lineage virus
CPE endpoint neutralization assays were done following the limiting dilution model (Wang et al., 2005) and using P1 stocks of B.1.427, D614G, and USA-WA1/2020 lineages. Convalescent patient plasma was diluted 1:10 and heat inactivated at 56°C for 30 min. Serial 2-fold dilutions of plasma were made in BSA-PBS. Plasma dilutions were mixed with 100 TCID50 of each virus diluted in BSA-PBS at a 1:1 ratio (220 μL plasma dilution and 220 μL virus input) and incubated for 1 hour at 37C. Final plasma dilutions in plasma-virus mixture ranged from 1:20 to 1:2560. 100 μL of the plasma-virus mixtures were inoculated on confluent monolayer of Vero-81 cells in 96-well plates in quadruplicate and incubated at 37°C with 5% CO2 incubator. After incubation 150 μL of MEM containing 5% FCS was added to the wells and plates were incubated at 37°C with 5% CO2 until consistent CPE was seen in virus control (no neutralizing plasma added) wells. Positive and negative controls were included as well as cell control wells and a viral back titration to verify TCID50 viral input. Individual wells were scored for CPE as having a binary outcome of ‘infection” or ‘no infection’. The TCID50 was calculated as the dose that produced cytopathic effect in > 50% of the inoculated wells. All steps were done in a Biosafety Level 3 lab using approved protocols.
Data visualization
The plots in Figure S1 were generated using graphical visualization tools at outbreak.info (Gangavarapu et al., 2020) and Microsoft Excel v16.47.1 and edited in Adobe Illustrator 23.1.1, using data from GISAID (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) and the California COVID-19 data tracker (CDPH (California Department of Public Health), 2021b). Other figures were generated using R v4.0.3 and Python v3.7.10 and edited in Adobe Illustrator.
Quantification and statistical analysis
The proportion of B.1.427/B.1.429 was estimated by dividing the number of B.1.427/B.1.429 variant cases by the total number of samples sequenced at a given location and collection date. A logistic growth curve fitting to the data points was generated using a non-linear least-squares approach, as implemented by the nls() function in R(version 4.0.3), and using code generated by the laboratory of Dr. Kristian Andersen at the Scripps Institute (https://github.com/andersen-lab/paper_2021_early-b117-usa). We estimated the increase in relative transmission rate of the B.1.427/B.1.429 variant by multiplying the logistic growth rate, defined as the change in the proportion of B.1.427/B.1.429 cases per day, by the serial interval, as previously described (Volz et al., 2020; Washington et al., 2021). The serial interval or generation time was defined as the average time taken for secondary cases to be infected by a primary case. The serial interval was found to be linearly proportional to the calculated transmission rate and did not affect the doubling time (Table S4). For SARS-CoV-2 infection, the serial interval has been estimated at 5 – 5.5 days (Rai et al., 2021), 5.5 days (Davies et al., 2021a; Washington et al., 2021), and 6.5 days (Volz et al., 2020); for the data in Figure 1, we used 5.5 days for the serial interval. Similar to the analyses in Washington et al., the doubling time was approximated using the formula: log (2) / logistic growth rate. Outliers corresponding to rolling average date ranges during which only a single B.1.427/B.1.429 variant genome was sequenced (100% proportion of the variant) were removed prior to curve fitting.
Welch’s t test, as implemented in R (version 4.0.3) using the rstatix_0.7.0 package and Python (version 3.7.9) using scipy package (version 1.5.2), was used to compare the N gene Ct values between B.1.427/B.1.429 variant and non-B.1.427/B.1.429 groups. For the in vitro pseudovirus infectivity studies, a one-way ANOVA test was used to determine significance. For the PRNT studies, a Wilcoxon matched pairs signed rank test was used to determine significance.
Acknowledgments
We acknowledge the help from Delsy Martinez and Tyler Miyasaki at UCSF CAT core facility for genome sequencing efforts using NovaSeq. We acknowledge Maria Salas, Elizabeth Baylis, and the entire COVIDNet team at the Viral and Rickettsial Disease Laboratory of the California Department of Public Health for their support of our community viral whole-genome sequencing efforts. We thank the Whelan lab at the Washington University School of Medicine for the Vero-TMPRSS2 cells. We thank the Jackson lab at Stanford University for the A549-ACE2 cells. We thank Satish Pillai and Hannah S. Sperbert at Vitalant Research Institute (San Francisco, CA) for providing the 293T-ACE2-TMPRSS2 cells used for pseudovirus entry assays. We thank Mehrdad Arjomandi at the San Francisco Veterans Administration Medical Center for providing the clinical samples to generate the human alveolar lung organoids. We thank Dr. Kristian Andersen and his laboratory at the Scripps Research Institute for generating code (available on Github) to calculate logistic growth models of SARS-CoV-2 infection based on positive tests over time. We gratefully acknowledge the authors from the originating laboratories and the submitting laboratories who generated and shared via GISAID genetic sequence data from an additional 2,737 genomes (from samples from California collected February 1 to March 11, 2021) on which this research is based (Table S5). The findings and conclusions in this article are those of the author(s) and do not necessarily represent the views or opinions of the California Department of Public Health or the California Health and Human Services Agency. This work has been funded by US CDC Epidemiology and Laboratory Capacity (ELC) for Infectious Diseases grants (6 NU50CK000539 to CDPH COVIDnet and 6 NU50CK000498 to Los Angeles County Public Health Laboratories), a Laboratory for Genomics Research (LGR) Excellence in Research Award (L.F.L.), a Fast Grant from Emergent Ventures (S.K.W.), the Innovative Genomics Institute (C.Y.C., M.O., L.F.L., S.K.W., P.A.F., and H.S.), the New Frontiers in Research Fund provided by the Canadian Institutes of Health Research (C.Y.C.), the Roddenberry Foundation (M.O.), and NIH (R33-AI129455 to C.Y.C. and 5DP1DA038043 to M.O.).
Author contributions
C.Y.C., M.O., and R.A. conceived and designed the study. C.Y.C., X.D., M.A.G.-K., V.S., C.W., and G.R.K. coordinated the sequencing efforts and laboratory studies. X.D., M.A.G.-K., M.M.K., V.S., C.W., A.S.-G., D.R.G., K.R.R., C.S.S.M., B.S., P.-Y.C., U.S.-G., T.Y.T., J.M.H., C.R.S., S. McMahon, P.V.L., Y.X., and M.K.M. performed experiments. C.Y.C., S.F., and X.D. assembled and curated viral genomes. C.Y.C. performed the phylogenetic and molecular clock analyses. C.Y.C., X.D., M.A.G.-K., V.S., C.W., K.R.R., A.S.-G., N.P.R., J. Balcerek, J.T., J.C., G.R.K., and C.Y.C. analyzed data. A.E., C.K., M.H., J. Bell, J.K.H., C.H., and D.A.W. collected and analyzed samples throughout California. V.S., C.W., A.S.-G., A.S.G., N.P.R., K.R.R., J.A.S., and S. Miller collected and sequenced SARS-CoV-2 samples from UCSF and throughout California. P.H. and N.M.G. collected and sequenced samples from Los Angeles County. C.A. and D.F. collected and sequenced samples from Monterey County. P.A.F., H.S., L.F.L., and S.K.W. collected and sequenced samples from Alameda County. C.Y.C., X.D., M.A.G.-K., V.S., and C.W. wrote the manuscript. C.Y.C., M.A.G.-K., G.R.K., and V.S. prepared the figures. C.Y.C., X.D., M.A.G.-K., V.S., D.A.W., J.K.H., and C.W. edited the manuscript. C.Y.C., X.D., M.A.G.-K., V.S., S.K.W., M.O., and R.A. revised the manuscript. C.Y.C. and V.S. edited and revised the figures. All authors read the manuscript and agree to its contents.
Declaration of interests
C.Y.C. receives support for SARS-CoV-2 research unrelated to this study from Abbott Laboratories and Mammoth Biosciences. The other authors declare no competing interests.
Published: April 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.04.025.
Supplemental information
References
- Avanzato V.A., Matson M.J., Seifert S.N., Pryce R., Williamson B.N., Anzick S.L., Barbian K., Judson S.D., Fischer E.R., Martens C., et al. Case Study: Prolonged Infectious SARS-CoV-2 Shedding from an Asymptomatic Immunocompromised Individual with Cancer. Cell. 2020;183:1901–1912. doi: 10.1016/j.cell.2020.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford T., Neher R. 2020. A Getting Started Guide to the Genomic Epidemiology of SARS-CoV-2. Nextstrain.org.https://nextstrain.github.io/ncov/ [Google Scholar]
- Bedford T., Hodcroft E.B., Neher R.A. 2021. Updated Nextstrain SARS CoV-2 clade naming strategy.http://nextstrain.org/blog/2021-01-06-updated-SARS-CoV-2-clade-naming [Google Scholar]
- Bouckaert R., Vaughan T.G., Barido-Sottani J., Duchêne S., Fourment M., Gavryushkina A., Heled J., Jones G., Kühnert D., De Maio N., et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2019;15:e1006650. doi: 10.1371/journal.pcbi.1006650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B. 2021. BBMap short read aligner, and other bioinformatic tools.https://sourceforge.net/projects/bbmap/ [Google Scholar]
- Buss L.F., Prete C.A., Jr., Abrahim C.M.M., Mendrone A., Jr., Salomon T., de Almeida-Neto C., França R.F.O., Belotti M.C., Carvalho M.P.S.S., Costa A.G., et al. Three-quarters attack rate of SARS-CoV-2 in the Brazilian Amazon during a largely unmitigated epidemic. Science. 2021;371:288–292. doi: 10.1126/science.abe9728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case J.B., Rothlauf P.W., Chen R.E., Liu Z., Zhao H., Kim A.S., Bloyet L.M., Zeng Q., Tahan S., Droit L., et al. Neutralizing Antibody and Soluble ACE2 Inhibition of a Replication-Competent VSV-SARS-CoV-2 and a Clinical Isolate of SARS-CoV-2. Cell Host Microbe. 2020;28:475–485.e5. doi: 10.1016/j.chom.2020.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDPH (California Department of Public Health) 2021. COVID-19 Variant First Found in Other Countries and States Now Seen More Frequently in California. California Department of Public Health.https://www.ucsf.edu/news/2021/01/419656/covid-19-variant-first-found-other-countries-and-states-now-seen-more [Google Scholar]
- CDPH (California Department of Public Health) 2021. Tracking COVID-19 in California. California Department of Public Health.https://covid19.ca.gov/state-dashboard [Google Scholar]
- Challen R., Brooks-Pollock E., Read J.M., Dyson L., Tsaneva-Atanasova K., Danon L. Risk of mortality in patients infected with SARS-CoV-2 variant of concern 202012/1: matched cohort study. BMJ. 2021;372:n579. doi: 10.1136/bmj.n579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chand M., Hopkins S., Dabreara G., Allen H., Lamagni T., Edeghere O., Achison C., Myers R., Barclay W., Ferguson N., et al. 2020. Investigation of novel SARS-COV-2 Variant of Concern.https://www.gov.uk/government/publications/investigation-of-novel-sars-cov-2-variant-variant-of-concern-20201201 [Google Scholar]
- Chen J., Wang R., Wang M., Wei G.W. Mutations Strengthened SARS-CoV-2 Infectivity. J. Mol. Biol. 2020;432:5212–5226. doi: 10.1016/j.jmb.2020.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu C.Y., Servellita V. 2021. Zenodo.https://zenodo.org/ [Google Scholar]
- Choi B., Choudhary M.C., Regan J., Sparks J.A., Padera R.F., Qiu X., Solomon I.H., Kuo H.H., Boucau J., Bowman K., et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020;383:2291–2293. doi: 10.1056/NEJMc2031364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole S., Gazy I., Jackson L., Hwa S.-H., Tegally H., Lustig G., Giandhari J., Pillay S., Wilkinson E., Naidoo Y., et al. 2021. Escape of SARS-CoV-2 501Y.V2 variants from neutralization by convalescent plasma. medRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COVID-19 Genomics UK (COG-UK) An integrated national scale SARS-CoV-2 genomic surveillance network. Lancet Microbe. 2020;1:e99–e100. doi: 10.1016/S2666-5247(20)30054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford K.H.D., Eguia R., Dingens A.S., Loes A.N., Malone K.D., Wolf C.R., Chu H.Y., Tortorici M.A., Veesler D., Murphy M., et al. Protocol and Reagents for Pseudotyping Lentiviral Particles with SARS-CoV-2 Spike Protein for Neutralization Assays. Viruses. 2020;12:E513. doi: 10.3390/v12050513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.G., Abbott S., Barnard R.C., Jarvis C.I., Kucharski A.J., Munday J.D., Pearson C.A.B., Russell T.W., Tully D.C., Washburne A.D., et al. CMMID COVID-19 Working Group. COVID-19 Genomics UK (COG-UK) Consortium Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science. 2021;372:eabg3055. doi: 10.1126/science.abg3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies N.G., Jarvis C.I., Edmunds W.J., Jewell N.P., Diaz-Ordaz K., Keogh R.H. Increased hazard of death in community-tested cases of SARS-CoV-2 Variant of Concern 202012/01. medRxiv. 2021 doi: 10.1101/2021.02.01.21250959. [DOI] [Google Scholar]
- Davies N.G., Jarvis C.I., Edmunds W.J., Jewell N.P., Diaz-Ordaz K., Keogh R.H., CMMID COVID-19 Working Group Increased mortality in community-tested cases of SARS-CoV-2 lineage B.1.1.7. Nature. 2021 doi: 10.1038/s41586-021-03426-1. Published online March 15, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Day T., Gandon S., Lion S., Otto S.P. On the evolutionary epidemiology of SARS-CoV-2. Curr. Biol. 2020;30:R849–R857. doi: 10.1016/j.cub.2020.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X., Gu W., Federman S., du Plessis L., Pybus O.G., Faria N.R., Wang C., Yu G., Bushnell B., Pan C.Y., et al. Genomic surveillance reveals multiple introductions of SARS-CoV-2 into Northern California. Science. 2020;369:582–587. doi: 10.1126/science.abb9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drew R.J., O’Donnell S., LeBlanc D., McMahon M., Natin D. The importance of cycle threshold values in interpreting molecular tests for SARS-CoV-2. Diagn. Microbiol. Infect. Dis. 2020;98:115130. doi: 10.1016/j.diagmicrobio.2020.115130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A.J., Suchard M.A., Xie D., Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012;29:1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbe S., Buckland-Merrett G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017;1:33–46. doi: 10.1002/gch2.1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria N.R., Mellan T.A., Whittaker C., Claro I.M., Candido D.D.S., Mishra S., Crispim M.A.E., Sales F.C., Hawryluk I., McCrone J.T., et al. Genomics and epidemiology of a novel SARS-CoV-2 lineage in Manaus, Brazil. medRxiv. 2021 doi: 10.1101/2021.02.26.21252554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontanet A., Autran B., Lina B., Kieny M.P., Karim S.S.A., Sridhar D. SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet. 2021;397:952–954. doi: 10.1016/S0140-6736(21)00370-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangavarapu K., Alkuzweny M., Cano M., Haag E., Latif A.A., Mullen J.L., Rush B., Tsueng G., Zhou J., Andersen K.G., et al. 2020. outbreak.info.https://outbreak.info/ [Google Scholar]
- Garcia-Beltran W.F., Lam E.C., St Denis K., Nitido A.D., Garcia Z.H., Hauser B.M., Feldman J., Pavlovic M.N., Gregory D.J., Poznansky M.C., et al. Multiple SARS-CoV-2 variants escape neutralization by vaccine-induced humoral immunity. Cell. 2021 doi: 10.1016/j.cell.2021.03.013. S0092-8674(21)00298-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann M., Kleine-Weber H., Schroeder S., Krüger N., Herrler T., Erichsen S., Schiergens T.S., Herrler G., Wu N.H., Nitsche A., et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Y.J., Chiba S., Halfmann P., Ehre C., Kuroda M., Dinnon K.H., 3rd, Leist S.R., Schäfer A., Nakajima N., Takahashi K., et al. SARS-CoV-2 D614G variant exhibits efficient replication ex vivo and transmission in vivo. Science. 2020;370:1464–1468. doi: 10.1126/science.abe8499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J., Peng P., Wang K., Fang L., Luo F.Y., Jin A.S., Liu B.Z., Tang N., Huang A.L. Emerging SARS-CoV-2 variants reduce neutralization sensitivity to convalescent sera and monoclonal antibodies. Cell. Mol. Immunol. 2021;18:1061–1063. doi: 10.1038/s41423-021-00648-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K., Standley D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 2013;30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M., Moir R., Wilson A., Stones-Havas S., Cheung M., Sturrock S., Buxton S., Cooper A., Markowitz S., Duran C., et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012;28:1647–1649. doi: 10.1093/bioinformatics/bts199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemp S.A., Collier D.A., Datir R.P., Ferreira I.A.T.M., Gayed S., Jahun A., Hosmillo M., Rees-Spear C., Mlcochova P., Lumb I.U., et al. CITIID-NIHR BioResource COVID-19 Collaboration. COVID-19 Genomics UK (COG-UK) Consortium SARS-CoV-2 evolution during treatment of chronic infection. Nature. 2021;592:277–282. doi: 10.1038/s41586-021-03291-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna K., Raymond W., Charbit A.R., Jin J., Gitlin I., Tang M., Sperber H.S., Franz S., Pillai S., Simmons G., et al. Binding of SARS-CoV-2 spike protein to ACE2 is disabled by thiol-based drugs; evidence from in vitro SARS-CoV-2 infection studies. bioRxiv. 2020 doi: 10.1101/2020.12.08.415505. [DOI] [Google Scholar]
- Klimstra W.B., Tilston-Lunel N.L., Nambulli S., Boslett J., McMillen C.M., Gilliland T., Dunn M.D., Sun C., Wheeler S.E., Wells A., et al. SARS-CoV-2 growth, furin-cleavage-site adaptation and neutralization using serum from acutely infected hospitalized COVID-19 patients. J. Gen. Virol. 2020;101:1156–1169. doi: 10.1099/jgv.0.001481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korber B., Fischer W.M., Gnanakaran S., Yoon H., Theiler J., Abfalterer W., Hengartner N., Giorgi E.E., Bhattacharya T., Foley B., et al. Sheffield COVID-19 Genomics Group Tracking Changes in SARS-CoV-2 Spike: Evidence that D614G Increases Infectivity of the COVID-19 Virus. Cell. 2020;182:812–827.e19. doi: 10.1016/j.cell.2020.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan J., Ge J., Yu J., Shan S., Zhou H., Fan S., Zhang Q., Shi X., Wang Q., Zhang L., Wang X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220. doi: 10.1038/s41586-020-2180-5. [DOI] [PubMed] [Google Scholar]
- Lau E.H.Y., Tsang O.T.Y., Hui D.S.C., Kwan M.Y.W., Chan W.H., Chiu S.S., Ko R.L.W., Chan K.H., Cheng S.M.S., Perera R.A.P.M., et al. Neutralizing antibody titres in SARS-CoV-2 infections. Nat. Commun. 2021;12:63. doi: 10.1038/s41467-020-20247-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung K., Shum M.H., Leung G.M., Lam T.T., Wu J.T. Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill. 2021;26:2002106. doi: 10.2807/1560-7917.ES.2020.26.1.2002106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z., VanBlargan L.A., Bloyet L.M., Rothlauf P.W., Chen R.E., Stumpf S., Zhao H., Errico J.M., Theel E.S., Liebeskind M.J., et al. Landscape analysis of escape variants identifies SARS-CoV-2 spike mutations that attenuate monoclonal and serum antibody neutralization. bioRxiv. 2020 doi: 10.1101/2020.11.06.372037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Liu J., Xia H., Zhang X., Fontes-Garfias C.R., Swanson K.A., Cai H., Sarkar R., Chen W., Cutler M., et al. Neutralizing Activity of BNT162b2-Elicited Serum - Preliminary Report. N. Engl. J. Med. 2021 doi: 10.1056/NEJMc2102017. Published online February 17, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Msomi N., Mlisana K., de Oliveira T., Network for Genomic Surveillance in South Africa writing group A genomics network established to respond rapidly to public health threats in South Africa. Lancet Microbe. 2020;1:e229–e230. doi: 10.1016/S2666-5247(20)30116-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plante J.A., Liu Y., Liu J., Xia H., Johnson B.A., Lokugamage K.G., Zhang X., Muruato A.E., Zou J., Fontes-Garfias C.R., et al. Spike mutation D614G alters SARS-CoV-2 fitness. Nature. 2021;592:116–121. doi: 10.1038/s41586-020-2895-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J., Grubaugh N.D., Pullan S.T., Claro I.M., Smith A.D., Gangavarapu K., Oliveira G., Robles-Sikisaka R., Rogers T.F., Beutler N.A., et al. Multiplex PCR method for MinION and Illumina sequencing of Zika and other virus genomes directly from clinical samples. Nat. Protoc. 2017;12:1261–1276. doi: 10.1038/nprot.2017.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai B., Shukla A., Dwivedi L.K. Estimates of serial interval for COVID-19: A systematic review and meta-analysis. Clin. Epidemiol. Glob. Health. 2021;9:157–161. doi: 10.1016/j.cegh.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A. 2021. FigTree.http://tree.bio.ed.ac.uk/software/figtree/ [Google Scholar]
- Rambaut A., Drummond A.J., Xie D., Baele G., Suchard M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018;67:901–904. doi: 10.1093/sysbio/syy032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Holmes E.C., O’Toole Á., Hill V., McCrone J.T., Ruis C., du Plessis L., Pybus O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020;5:1403–1407. doi: 10.1038/s41564-020-0770-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rambaut A., Loman N., Pybus O., Barclay W., Barrett J., Carabelli A., Connor T., Peacock T., Robertson D.L., Volz E. 2020. Preliminary genomic characterisation of an emergent SARS-CoV-2 lineage in the UK defined by a novel set of spike mutations.https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 [Google Scholar]
- Sabino E.C., Buss L.F., Carvalho M.P.S., Prete C.A., Jr., Crispim M.A.E., Fraiji N.A., Pereira R.H.M., Parag K.V., da Silva Peixoto P., Kraemer M.U.G., et al. Resurgence of COVID-19 in Manaus, Brazil, despite high seroprevalence. Lancet. 2021;397:452–455. doi: 10.1016/S0140-6736(21)00183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachs N., Papaspyropoulos A., Zomer-van Ommen D.D., Heo I., Böttinger L., Klay D., Weeber F., Huelsz-Prince G., Iakobachvili N., Amatngalim G.D., et al. Long-term expanding human airway organoids for disease modeling. EMBO J. 2019;38:e100300. doi: 10.15252/embj.2018100300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel R.M., Majd H., Richter M.N., Ghazizadeh Z., Zekavat S.M., Navickas A., Ramirez J.T., Asgharian H., Simoneau C.R., Bonser L.R., et al. Androgen Signaling Regulates SARS-CoV-2 Receptor Levels and Is Associated with Severe COVID-19 Symptoms in Men. Cell Stem Cell. 2020;27:876–889.e12. doi: 10.1016/j.stem.2020.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seow J., Graham C., Merrick B., Acors S., Pickering S., Steel K.J.A., Hemmings O., O’Byrne A., Kouphou N., Galao R.P., et al. Longitudinal observation and decline of neutralizing antibody responses in the three months following SARS-CoV-2 infection in humans. Nat. Microbiol. 2020;5:1598–1607. doi: 10.1038/s41564-020-00813-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y., McCauley J. GISAID: Global initiative on sharing all influenza data - from vision to reality. Euro Surveill. 2017;22:30494. doi: 10.2807/1560-7917.ES.2017.22.13.30494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tegally H., Wilkinson E., Giovanetti M., Iranzadeh A., Fonseca V., Giandhari J., Doolabh D., Pillay S., San E.J., Msomi N., et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv. 2020 doi: 10.1101/2020.12.21.20248640. [DOI] [Google Scholar]
- Teng S., Sobitan A., Rhoades R., Liu D., Tang Q. Systemic effects of missense mutations on SARS-CoV-2 spike glycoprotein stability and receptor-binding affinity. Brief. Bioinform. 2021;22:1239–1253. doi: 10.1093/bib/bbaa233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volz E., Mishra S., Chand M., Barrett J.C., Johnson R., Geidelberg L., Hinsley W.R., Laydon D.J., Dabrera G., O’Toole A., et al. Transmission of SARS-CoV-2 Lineage B.1.1.7 in England: Insights from linking epidemiological and genetic data. medRxiv. 2020 doi: 10.1101/2020.12.30.20249034. [DOI] [Google Scholar]
- Wang S., Sakhatskyy P., Chou T.H., Lu S. Assays for the assessment of neutralizing antibody activities against Severe Acute Respiratory Syndrome (SARS) associated coronavirus (SCV) J. Immunol. Methods. 2005;301:21–30. doi: 10.1016/j.jim.2005.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Schmidt F., Weisblum Y., Muecksch F., Barnes C.O., Finkin S., Schaefer-Babajew D., Cipolla M., Gaebler C., Lieberman J.A., et al. mRNA vaccine-elicited antibodies to SARS-CoV-2 and circulating variants. Nature. 2021;592:616–622. doi: 10.1038/s41586-021-03324-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington N.L., Gangavarapu K., Zeller M., Bolze A., Cirulli E.T., Schiabor Barrett K.M., Larsen B.B., Anderson C., White S., Cassens T., et al. Genomic epidemiology identifies emergence and rapid transmission of SARS-CoV-2 B.1.1.7 in the United States. medRxiv. 2021 doi: 10.1101/2021.02.06.21251159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibmer C.K., Ayres F., Hermanus T., Madzivhandila M., Kgagudi P., Oosthuysen B., Lambson B.E., de Oliveira T., Vermeulen M., van der Berg K., et al. SARS-CoV-2 501Y.V2 escapes neutralization by South African COVID-19 donor plasma. Nat. Med. 2021;27:622–625. doi: 10.1038/s41591-021-01285-x. [DOI] [PubMed] [Google Scholar]
- Wise J. Covid-19: The E484K mutation and the risks it poses. BMJ. 2021;372:n359. doi: 10.1136/bmj.n359. [DOI] [PubMed] [Google Scholar]
- Wu K., Werner A.P., Koch M., Choi A., Narayanan E., Stewart-Jones G.B.E., Colpitts T., Bennett H., Boyoglu-Barnum S., Shi W., et al. Serum Neutralizing Activity Elicited by mRNA-1273 Vaccine. N. Engl. J. Med. 2021 doi: 10.1056/NEJMc2102179. Published online March 17, 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K., Werner A.P., Moliva J.I., Koch M., Choi A., Stewart-Jones G.B.E., Bennett H., Boyoglu-Barnum S., Shi W., Graham B.S., et al. mRNA-1273 vaccine induces neutralizing antibodies against spike mutants from global SARS-CoV-2 variants. bioRxiv. 2021 doi: 10.1101/2021.01.25.427948. [DOI] [Google Scholar]
- Xie X., Liu Y., Liu J., Zhang X., Zou J., Fontes-Garfias C.R., Xia H., Swanson K.A., Cutler M., Cooper D., et al. Neutralization of SARS-CoV-2 spike 69/70 deletion, E484K and N501Y variants by BNT162b2 vaccine-elicited sera. Nat. Med. 2021;27:620–621. doi: 10.1038/s41591-021-01270-4. [DOI] [PubMed] [Google Scholar]
- Zhang W., Davis B.D., Chen S.S., Sincuir Martinez J.M., Plummer J.T., Vail E. Emergence of a Novel SARS-CoV-2 Variant in Southern California. JAMA. 2021;325:1324–1326. doi: 10.1001/jama.2021.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B., Thao T.T.N., Hoffmann D., Taddeo A., Ebert N., Labroussaa F., Pohlmann A., King J., Steiner S., Kelly J.N., et al. SARS-CoV-2 spike D614G change enhances replication and transmission. Nature. 2021;592:122–127. doi: 10.1038/s41586-021-03361-1. [DOI] [PubMed] [Google Scholar]
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., et al. China Novel Coronavirus Investigating and Research Team A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020;382:727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Assembled SARS-CoV-2 genomes in this study were uploaded to GISAID (Elbe and Buckland-Merrett, 2017; Shu and McCauley, 2017) (accession numbers in Table S1) and can be visualized in Nextstrain. Viral genomes were also submitted to the National Center for Biotechnology Information (NCBI) GenBank database (accession numbers MW972466-MW974550), BioProject accession number PRJNA722044 and umbrella BioProject accession number PRJNA171119, Chiu laboratory at UCSF; umbrella BioProject accession number PRJNA639591, Wyman laboratory at UC Berkeley). FASTA files, XML files, and scripting code used for SARS-CoV-2 genome assembly and phylogenetic / molecular dating analyses are available in a Zenodo data repository (https://doi.org/10.5281/zenodo.4688394) (Chiu and Servellita, 2021).