

Abstract
Selections of yeast-displayed ligands on mammalian cell monolayers benefit from high target expression and nanomolar affinity, which are not always available. Prior work extending the yeast–protein linker from 40 to 80 amino acids improved yield and enrichment but is hypothesized to be below the optimal length, prompting evaluation of an extended amino acid linker. A 641-residue linker provided enhanced enrichment with a 2-nM affinity fibronectin ligand and 105 epidermal growth factor receptors (EGFR) per cell (14 ± 2 vs. 8 ± 1, P = 0.008) and a >600-nM affinity ligand, 106 EGFR per cell system (23 ± 7 vs. 0.8 ± 0.2, P = 0.004). Enhanced enrichment was also observed with a 310-nM affinity affibody ligand and 104 CD276 per cell, suggesting a generalizable benefit to other scaffolds and targets. Spatial modeling of the linker suggests that improved extracellular accessibility of ligand enables the observed enrichment under conditions not previously possible.
Keywords: ligand, protein engineering, surface display linker, yeast display
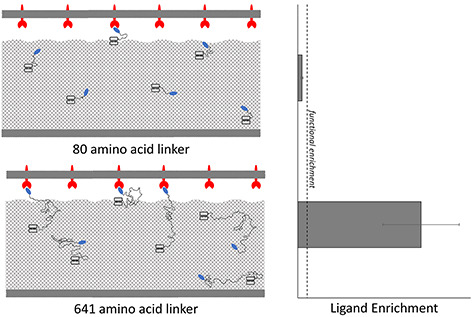
Graphical Abstract
Graphical Abstract.
Introduction
Protein scaffolds have enabled the efficient engineering of specific molecular binding activity while maintaining desirable biophysical properties for both therapeutic and diagnostic applications (Banta et al., 2013; Stern et al., 2013). Often, the discovery of binding ligands to clinically relevant biomarkers is conducted through the generation of large combinatorial libraries (Gera et al., 2011; Plückthun, 2015; Woldring et al., 2015, 2017). To effectively screen these libraries, high-throughput selection strategies including magnetic bead capture (Orlova et al., 2006; Ackerman et al., 2009; Dreier and Plückthun, 2012; Horiya et al., 2017) and fluorescence-activated cell sorting (Boder and Wittrup, 1997; Beerli et al., 2008) have been coupled with yeast (Boder and Wittrup, 1997; Hackel and Wittrup, 2011; Tillotson et al., 2015) and phage (Smith, 1985; Bratkovič, 2010; Even-Desrumeaux and Chames, 2012) display technologies. These strategies often use recombinantly produced, soluble ectodomains as analogs for full-length proteins. While proven successful (Bozovičar and Bratkovič, 2020), these methods may generate ligands that bind strongly to the biomarker analogs yet fail to translate binding to genuine antigen on target-expressing cells (Stern et al., 2019). As a solution, several selection techniques utilizing target-expressing cells have been developed to use full-length, native biomarkers to aid in translatable ligand selection, including biopanning on adherent cell monolayers (Wang and Shusta, 2005; Tillotson et al., 2013; Stern et al., 2016), magnetic bead-immobilized cells (Krohl et al., 2020; Lown and Hackel, 2020) or flow cytometry with suspended cells (Yang et al., 2019). Adherent cell panning has isolated high-affinity, translatable binders in multiple scenarios (Cai and Garen, 1995; Xu et al., 2004; Shen et al., 2007; Wang et al., 2007; Even-Desrumeaux and Chames, 2012; Dangaj et al., 2013; Jones et al., 2014; Williams et al., 2014; Zorniak et al., 2017; Umlauf et al., 2019). Yet studies conducted using epidermal growth factor receptor- (EGFR-) binding fibronectins found that enrichment—the ratio of the yields (fractions of yeast collected) of ligand-displaying and nondisplaying yeast in a sort and a metric for the relative increase in frequency of ligands with desired target-binding relative to nonbinding background—is hindered when using cells with low-to-moderate (≤105 targets/cell) expression or ligands of micromolar or weaker affinity in both adherent and suspension systems (Stern et al., 2016; Lown and Hackel, 2020).
One hypothesis for the inability of these studies to effectively enrich ligands in nonideal systems is due to limited ligand–target binding. The yeast cell wall is ~115 nm thick (Dupres et al., 2010), throughout which the yeast surface display construct is distributed by glycosylphosphatidylinositol (GPI)-mediated anchoring (Klis et al., 2002; Huang et al., 2003). Previous research with similar constructs indicated that cell wall glycans cause steric occlusion of ligand binding, with a 649-amino acid linker providing consistent molecular engagement regardless of target molecular weight (McMahon et al., 2018). Similarly, a 122-nm linker was required for a plasma membrane-tethered yeast surface construct to reliably contact an extracellular molecular probe (Dupres et al., 2010). The linker of the classic pCT yeast surface display construct is 40 amino acids with a maximum end-to-end distance of 15 nm (assuming an average distance of 3.8 Å between alpha-carbons) (Wang et al., 2005), far below either studied limit. This may decrease the number of yeast-displayed ligands able to access the extracellular space and engage with their target, lowering the effective avidity of the system. Further supporting this, a study that appended an additional 40-amino acid flexible linker showed enhanced binding of high- and low-affinity EGFR-binding fibronectins on high-expressing cells (Stern et al., 2016). However, even this 80-amino acid linker is expected to be far under the length for optimal mammalian cell surface engagement, thus motivating the creation of even longer yeast surface display linkers for the purpose of adherent mammalian cell panning.
This study aims to evaluate the effectiveness of a substantially extended linker, 8-fold longer at 641 amino acids, in providing more robust ligand–biomarker interaction in the context of adherent mammalian cell panning in comparison to the previously established 80-amino acid linker. To create a generalizable comparison, parameters including mammalian cell target expression, ligand-binding affinity, ligand–protein scaffold and target biomarker were studied. Sorting mixtures containing a low frequency of yeast expressing pCT-80 or pCT-641 display plasmids, and a majority of plasmidless EBY100 were used to mimic de novo selection from naïve libraries and panned in parallel. The longer linker significantly increased enrichment in both high-affinity, moderate target expression and low-affinity, high target expression EGFR systems. In the latter case, the longer linker provided significant effective enrichment, whereas the shorter linker could not. However, the longer linker did not provide significant enrichment in the low affinity, moderate target expression system, indicating that further modifications to yeast surface display may be needed to enrich low-affinity ligands in nonideal conditions. Significantly increased enrichment was also observed in a low-affinity, low expression CD276 system, indicating this benefit may be generalizable to other protein scaffolds and biomarkers. This increased enrichment was observed despite significantly decreased display levels of the pCT-641 construct on the yeast surface. Modeling the dynamics of the two linkers as stiffness-modified Gaussian chains (Flory, 1969; Zhou, 2004; Wang et al., 2005) uniformly distributed throughout the cell wall indicated that the longer linker spends more time extended into the extracellular environment, supporting the hypothesis that the increased performance is a result of more robust ligand accessibility to the extracellular space. Combined, these results advance understanding of the factors that dictate binding ligand yield and enrichment in adherent mammalian cell panning and provide tools for isolating binding ligands in conditions not previously possible.
Results
Iterative restriction enzyme digestion and destructive ligation (Berges, 2017) were used to exponentially expand the existing pCT-80 linker (Supplementary Table S1) (Stern et al., 2016). In short, the existing vector was digested in parallel by AvrII and BamHI to generate an insert and by NheI and BamHI to create an acceptor vector. While compatible for ligation, the resulting hybrid recognition site of AvrII and NheI cannot be digested by either enzyme, effectively breaking the recognition site while doubling the linker region. To achieve the desired 8-fold increase in linker length, this process was repeated over sufficient iterations to create a 641-amino acid linker (Fig. 1). This new linker, containing a truncated version of the original linker with 18 amino acids of a proline–alanine–serine-rich segment (based on PAS#1) (Schlapschy et al., 2013) and (G4S)3 bridged by alanine and arginine encoded for by the broken restriction site (Supplementary Table S1) was expected to have similar conformational flexibility and secondary structure due to its similar composition. However, detailed characterization was outside the scope of this study.
Fig. 1.
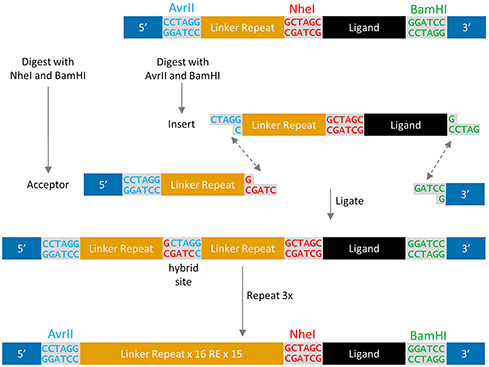
Exponential expansion of a repetitive linker by iterative restriction enzyme digest and destructive ligation. Digesting the initial construct in parallel creates an insert and acceptor with compatible overlaps. Ligation results in a hybrid sequence of NheI and AvrII that is not recognized by either restriction enzyme, preventing further digestion and a doubling of the linker region. Each repetition of the procedure doubles the existing linker region.
Longer linkers increase the yield of displaying yeast and enrichment in low-affinity or moderate target expression systems
To compare the efficacy of the extended linker in enriching binding ligands, adherent cell panning was conducted in parallel with both the 80- and 641-residue linkers. The A431 cell line, which highly overexpresses EGFR (4 ± 1 × 106 EGFR/cell), was the panning target for a mixture of yeast containing plasmidless EBY100 yeast and yeast expressing display plasmids encoding high-affinity (Kd = 2 ± 2 nM) EGFR-binding fibronectin domain E6.2.6′ (Hackel et al., 2010) from either the pCT-80 or pCT-641 construct. Upon selection, the yields of nondisplaying and ligand-displaying yeast were measured via dilution plating, and ligand enrichment was computed. Compared with the pCT-80 construct, pCT-641 showed a higher yield of ligand-displaying yeast (7.5 ± 1.0% vs. 2.8 ± 0.5%, P < 0.0001; Fig. 2B) accompanied with a moderate increase in the yield of nondisplaying yeast (0.15 ± 0.03% vs. 0.10 ± 0.03%, P = 0.05; Fig. 2C), resulting in a nominal, statistically insignificant elevation in enrichment (120 ± 40 vs. 50 ± 10, P = 0.12; Fig. 2A).
Fig. 2.
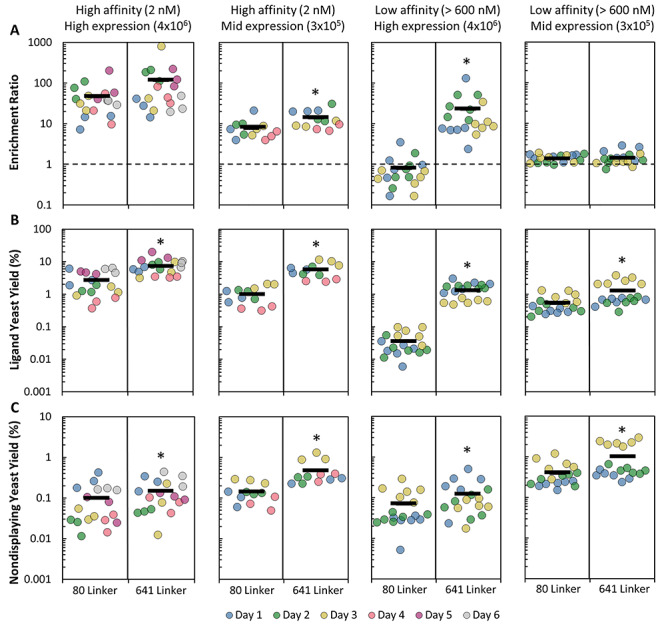
The effect of ligand affinity and target expression on yield and enrichment across 80- and 641-amino acid linkers in an EGFR system. Yeast displaying either pCT-80 or pCT-641 tethered ligands were mixed with nondisplaying yeast and sorted in parallel by adherent cell panning. (A) Enrichment ratio (B) ligand-displaying yeast yield and (C) nondisplaying yeast yield were quantified on both high- (E6.2.6′) and low- (E6.2.6’ AASV) affinity ligands as well as high (A431) and mid (MDA-MB-231) expression cell lines. Dotted lines (A) indicate the limit of functional enrichment. *P < 0.05 relative to the 80-amino acid linker
The lack of a significant increase in enrichment suggested that, while the extended linker may effectively increase avidity, E6.2.6′ is sufficiently high affinity that very little avidity is needed to provide consistent attachment of displaying yeast to the highly expressing mammalian cell surface. Thus, it was decided to move to more challenging systems, which is also where more technological advancement is needed for binder isolation. On mid-expressing MDA-MB-231 cells (2.9 ± 1.7 × 105 EGFR/cell), the longer linker improved enrichment of the high-affinity E6.2.6′ (14 ± 2 vs. 8 ± 1, P = 0.008; Fig. 2A) as well as yield (5.6 ± 0.8% vs. 1.0 ± 0.2%, P < 0.0001; Fig. 2B). The increased stringency of panning with the roughly 14-fold reduction in mammalian cell expression between the mid- and high-expressing system is sufficient to reveal the extended linker’s avidity advantage. Even more striking, for low-affinity ligand E6.2.6’ AASV (Kd > 600 nM for EGFR) (Stern et al., 2016) panned on high-expressing A431 cells, the 641-residue linker increased ligand-displaying yeast enrichment from nonfunctional 0.8 ± 0.2 to highly effective 23 ± 7 (P = 0.004; Fig. 2A). This was driven by an increased yield (1.4 ± 0.2% vs. 0.036 ± 0.007%, P < 0.0001; Fig. 2B).
However, while E6.2.6’ AASV panned on mid-expressing MDA-MB-231 cells provided significantly higher ligand-displaying yeast yield (1.3 ± 0.2% vs. 0.53 ± 0.08%, P < 0.0001; Fig. 2B) for the longer linker, this did not translate to significantly increased enrichment (1.4 ± 0.1 vs. 1.39 ± 0.07, P = 0.73; Fig. 2A), indicating that any potential increase in ligand accessibility by the longer linker is not enough to overcome a combination of low ligand affinity and limited biomarker expression. Thus, enriching weaker affinity ligands on all but the highest expressing cell lines remains challenging in adherent cell panning. Additionally, the moderate but significant increase in nondisplaying yeast yield, seen for the longer linker across all model EGFR systems, somewhat hindered enrichment. The increased accessibility may allow additional nonspecific interactions between the linker and/or ligand with nondisplaying yeast, which could potentially be ameliorated using blocking agents such a milk powder or fetal bovine serum; however, this has not been evaluated.
Improved linker performance is generalizable to CD276 and affibodies
To assess whether the increased performance of the pCT-641 construct was target or ligand dependent, further sorts were conducted with a cell line that has low expression of CD276 (MDA-MB-231: 4 ± 2 × 104 CD276/cell) and a low-affinity affibody ligand: AC2 (Kd = 310 nM; 240–390 nM 68% confidence interval [CI]) (Stern et al., 2019). This system showed similar trends to the EGFR system, with an increase in ligand-displaying yeast yield (0.9 ± 0.1% vs. 0.6 ± 0.1%, P = 0.003; Fig. 3B) and enrichment (7.5 ± 0.8 vs. 4.3 ± 0.4, P = 0.0003; Fig. 3A) of the pCT-641 construct relative to pCT-80. This indicates that the observed benefit of a longer linker is not limited to EGFR and fibronectin domains and may be generalizable to other targets and protein scaffolds. Cell panning is a potential method for screening libraries for ligands of micromolar affinity on cell lines expressing on the order of millions of targets per cell or libraries of ligands of up to 300 nM affinity on cell lines expressing tens-to-hundreds of thousands of targets per cell. Despite this, nonspecific interactions remain a significant issue in cell panning screens and sufficient depletion strategies should be employed when conducting cell panning.
Fig. 3.
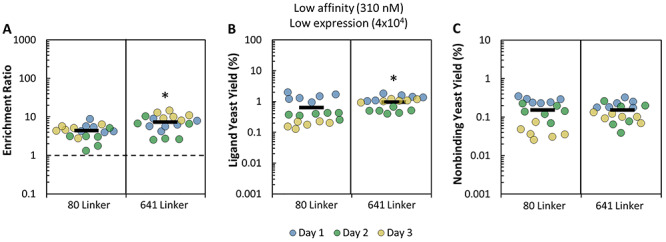
Yield and enrichment of 80- and 641-amino acid linkers in a CD276 system. Yeast displaying either pCT-80 or pCT-641 tethered ligands were mixed with nondisplaying yeast and sorted in parallel by adherent cell panning. (A) Enrichment ratio, (B) ligand-displaying yeast yield and (C) nondisplaying yeast yield were quantified. Ligand: AC2. Cell Line: MDA-MB-231. The dotted line (A) indicates the limit of functional enrichment. *P < 0.05 relative to the 80- amino acid linker
Improved linker performance is observed despite lower yeast cell surface expression
We hypothesize that the increased binding and enrichment for the extended linker results from improved ligand accessibility, which translates to elevated functional valency. Yet it is possible that the extended linker constructs achieved higher valency by simply expressing more abundantly on the yeast surface. To assess this possibility, the amount of surface displayed ligand was quantified using a single aliquot of each population after induction and before sorting (apart from AC2, which used a different induction). Yeast containing the pCT-641 plasmid were found to express significantly fewer ligands per cell relative to yeast containing pCT-80 (25% fewer for E6.2.6′, 10% fewer E6.2.6’ AASV and 34% fewer for AC2) (Fig. 4A). This lower expression indicates that the longer linker allowed the ligand to engage more reliably with its molecular target despite decreased ligand surface expression. Additionally, if a large portion of ligand-negative yeast cells contain plasmid offering antibiotic resistance, a lower percent induction could artificially deflate the displaying yeast yield, and therefore the enrichment ratio, of the assay. However, enrichment benefit was observed despite the percent induction of pCT-641 being comparable or moderately lower compared with the pCT-80 counterparts (Fig. 4B). Additional experiments evaluating per cell expression and percent of cells displaying ligand, while displaying a differing trend of per cell expression, indicated that the induction conditions used to generate data were roughly optimal for both linkers (Supplementary Fig. S1).
Fig. 4.
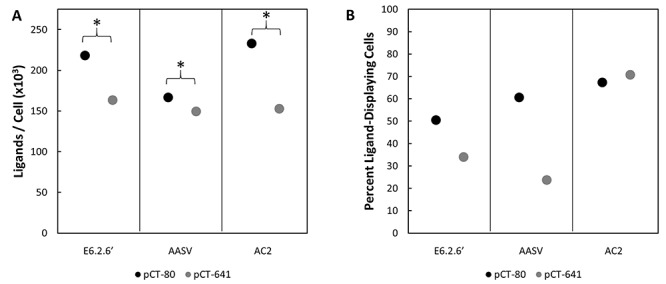
Yeast containing pCT-80 or pCT-641 and encoding for E6.2.6′, E6.2.6’ AASV or AC2 were labeled with an anti-C-Myc antibody and FITC secondary. Their fluorescence was analyzed by flow cytometry and compared to a quantitative bead standard labeled with the same antibody mixture to determine ligand expression. The same induction of E6.2.6′ and E6.2.6’ AASV ligands were used to generate the sorting data in Figure 2. (A) Ligand expression is presented as the mean of >10 000 events from a single culture. 95% CIs were calculated but are <0.8% of the mean and therefore not visible. *P < 0.05 difference in expression levels. (B) The percent of ligand-displaying cells (fluorescein isothiocyanate [FITC] positive) in each sample
Gaussian chain model shows linker stiffness and length drives extracellular accessibility
To further understand the impact of the yeast surface display linkers and the parameters that affect extracellular accessibility, geometric models were constructed and evaluated for the two linker systems. Both GGSGGS and PAS#1, polypeptides of similar composition to the (G4S)3- and PAS#1-based repeats that primarily make up the pCT-80 and pCT-641 linkers, have been previously described as random coil structures (Evers et al., 2006; Breibeck and Skerra, 2018). This guided the selection of the Gaussian chain model to describe both linkers (Flory, 1969; Zhou, 2004; Wang et al., 2005). To account for the nonideal behavior of real polypeptide chains, the model was modified with a characteristic ratio correction factor, using homopolymer values for glycine, alanine and proline (Brant et al., 1967; Schimmel and Flory, 1967). While experimental characterization of the characteristic ratios for the 80- and 641-amino acid linkers is outside the scope of this study, GGSGGS linkers have a characteristic ratio similar to polyglycine (Evers et al., 2006), and PAS#1 has been described as having a stiffness between polyalanine and polyproline (Breibeck and Skerra, 2018). This indicates that, while not experimentally determined, the characteristic ratio of the linkers should fall within the range of values provided, thus allowing for a semi-quantitative framework for comparison of length distributions between both linkers. To account for the GPI-mediated attachment of Aga1p, the model allowed linkers to anchor anywhere within the cell wall (assumed to be 115 nm thick) (Dupres et al., 2010). Lacking any prior knowledge of the distribution of Aga1p in the cell wall, a uniform distribution was chosen for simplicity.
With this model, the probability distribution of ligand distance from the yeast cell surface was calculated, assuming the linker is a homopolymer consisting of either polyglycine, polyalanine or polyproline. This reveals a relatively constant probability within the cell wall, due to the homogenous distribution of the yeast surface display construct, with a sharp decrease across the boundary of the cell wall (Fig. 5A). The fraction of ligands in the extracellular space correlated with both length and stiffness, with pCT-641 having a higher fraction of extracellular ligands compared with pCT-80 regardless of stiffness. Increasing the number of linker amino acids from 80 to 641 provided a 2.8–3.2-fold increase in the fraction of ligands in the extracellular space, supporting our hypothesis that the improved performance of pCT-641 is more consistent ligand–target engagement as a result of increased ligand accessibility to the extracellular space (Fig. 5B).
Fig. 5.
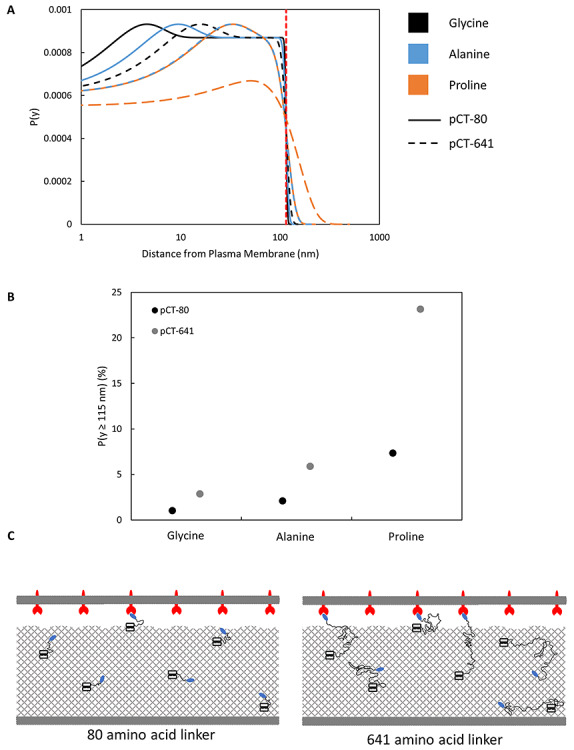
pCT-641 extends farther than pCT-80, on average, regardless of material composition. (A) Probability distribution functions of ligand distance from the cell surface were constructed for pCT-80 and pCT-641 using a characteristic ratio corrected Gaussian chain model. Curves were constructed for the characteristic ratios of glycine, alanine and proline. The vertical dotted line indicates the approximate boundary between the cell wall and the extracellular space. (B) The area under each curve beyond the cell wall boundary was integrated to determine the percentage of total ligand accessible to the extracellular space. (C) Schematic of a yeast cell and mammalian cell in close contact. Aga1p–Aga2p anchor complexes are uniformly distributed throughout the cell wall with ligands tethered through representative linkers to illustrate how increased length relates to extracellular accessibility
While the model does not take into account the observed expression levels or the exact stiffness of the linker, a reasonable approximation for the number of ligands with accessibility to the extracellular space can be made by estimating the fraction of accessible ligands for each linker as a linear combination of the values from Fig. 5B weighted by the fractional composition of glycine, alanine and proline, using expression levels from Figure 4A, and assuming a yeast cell to be an 8 μm diameter sphere. This estimate suggests 24–36 accessible ligands per μm2 for yeast expressing pCT-80 versus 63–69 accessible ligands per μm2 for pCT-641, a moderate increase in spite of the expression difference. Dependent upon cell–cell shape complementarity and target density, these estimates make it reasonable that the longer linker transitions these interactions from monovalent to multivalent.
The model theoretically suggests an infinite length linker to maximize ligand–target engagement but does not account for several factors including genetic efficiency, extracellular shuttling and stability. These factors may restrict the length of realistically functional linkers and reduce ligand expression, which would create an optimum that is not readily extractable from existing data. However, the inability of a 40-, 80- or 641-amino acid flexible linker to effectively enrich the low-affinity linker in low expression conditions (Stern et al., 2016) either suggests that the length needed for effective avidity may be prohibitively long or that other approaches may be needed to enhance the extracellular exposure of ligands. For instance, stiffer linkers provide a higher fraction of accessible ligands relative to flexible linkers with the same number of residues. This could potentially motivate the design of a hybrid linker with a stiff base to allow extracellular access and a flexible distal portion to allow the ligand to sample space. Another suggestion is to pretreat the yeast with Zymolyase to effectively thin the cell wall (Dupres et al., 2010), although this may also result in the removal of ligand attached to the degraded portion of the cell wall.
While the above data highlight the usefulness of extended linkers in yeast surface display systems when conducting cell panning, recommendations for linker length in other display platforms (ribosome, phage and mammalian cell) are less clear. In the case of ribosome and phage display, the small size of the display platform limits steric hindrance between the platform and target cell. In these cases, short linkers are often incorporated more to aid in the stability of the displayed protein fusion. However, it remains possible that the analytical study of linker length in these systems could see a useful improvement. While there is no need to penetrate through a cell wall in mammalian cell display, the size of yeast and mammalian cells remain similar. Thus, a similar level of steric hindrance could be expected, which motivates the switch from existing linkers to one similar in length to those mentioned above and a more thorough investigation of the effect of linker length on cell panning performance in mammalian cell display systems.
Conclusion
A dramatically extended yeast surface display linker provides more robust extracellular engagement with cell surface biomarkers. The 641-amino acid linker provided significantly improved ligand yield and enrichment in adherent cell panning for both high-affinity ligands on cells with moderate target expression and low-affinity ligands on cells with high target expression relative to the existing 80-amino acid linker. This enhanced yield and enrichment was observed despite 10–34% fewer ligands displayed per yeast cell and was observed with multiple scaffolds and targets. In addition, modeling the linkers suggests pCT-641 has a higher fraction of extracellularly accessible ligands, which suggests the observed performance benefits are due to more consistent ligand–target interactions. Ultimately, the findings of this study provide a new yeast surface display construct for use in adherent cell panning while motivating further study into other construct improvements.
Materials and Methods
Cells and cell culture
A431 cells were provided by Professor Daniel Vallera (Department of Therapeutic Radiology, University of Minnesota—Twin Cities). MDA-MB-231 was provided by Prof. Jayanth Panyam (Department of Pharmaceutics, University of Minnesota—Twin Cities). All cell lines were grown with Dulbecco's modified Eagle medium (DMEM) containing 4.5 g/l D-glucose, sodium pyruvate and L-glutamine and supplemented with 10% (v/v) fetal bovine serum and 1% (v/v) 10,000 U/ml penicillin–streptomycin. All cell lines were incubated at 37°C in a humidified atmosphere with 5% CO2.
Yeast surface display was performed largely as previously described (Chen et al., 2013). Expression plasmids, explained below, were transformed into Saccharomyces cerevisiae yeast strain EBY100 by EZ-Yeast Transformation (Zymo Research, Irvine, CA). Yeast harboring expression plasmids were grown in SD-CAA medium (16.8 g/l sodium citrate dihydrate, 3.9 g/l citric acid, 20.0 g/l dextrose, 6.7 g/l yeast nitrogen base and 5.0 g/l casamino acids in deionized H2O) at 30°C with shaking for at least 7 h. Protein expression was induced on the yeast surface by transferring yeast cells in logarithmic phase (OD600nm < 6) into SG-CAA medium (10.2 g/l sodium phosphate dibasic heptahydrate, 8.6 g/l sodium phosphate monobasic monohydrate, 19.0 g/l galactose, 1.0 g/l dextrose, 6.7 g/l yeast nitrogen base and 5.0 g/l casamino acids in deionized H2O) and growing at 20°C for 24–48 h. EBY100 without expression plasmids were grown in YPD medium (20.0 g/l peptone, 20.0 g/l dextrose and 10.0 g/l yeast extract in deionized H2O) at 30°C with shaking.
Expression plasmids
The pCT-80 and pCT-641 plasmids were used as the expression vectors for yeast surface display on the C-terminus of Aga2p. pCT-80 encodes for Aga2p followed by an 80-amino acid linker (composed of a Factor Xa cleavage site, an HA epitope, a proline/alanine/serine peptide based upon the PAS#1 motif [PAS40], a glycine-rich peptide [(G4S)3], and an NheI recognition site), the ligand, a BamHI recognition site, and a C-terminal Myc epitope (Supplementary Table S1). This is, essentially, the classic pCT yeast surface display plasmid with the addition of a 40-amino acid PAS#1-based domain.
To further extend the linker, the latter 38 amino acids of PAS40 (PAS38), the ligand and C-terminal Myc epitope were polymerase chain reaction amplified with the addition of an AvrII recognition site 5′ of PAS38. This construct was inserted into NheI and BamHI digested pCT-80 by HiFi DNA Assembly (New England Biolabs), creating the starting construct for further expansion. The linker was then exponentially expanded through iterative restriction enzyme digestion and destructive ligation, as previously described (Berges, 2017). The construct was digested with AvrII (New England Biolabs) and BamHI (New England Biolabs) to generate an insert containing PAS38, (G4S)3, an NheI recognition site and the ligand. The same plasmid was separately digested with NheI and BamHI to create an acceptor vector. Ligation resulted in the duplication of PAS18-(G4S)3 as well as the destruction of the cut NheI and AvrII sites. This process was repeated three additional times. Each plasmid was digested by AvrII and BamHI before being analyzed by gel electrophoresis to confirm proper duplication of the linker. The longest plasmid constructed was named pCT-641 as it is, essentially, the classic pCT yeast surface display plasmid with the addition of a 601-amino acid linker (Supplementary Table S1).
Receptor expression quantification
Cellular expression of EGFR, CD276 and yeast-displayed ligand were quantified using polystyrene beads with known quantities of immobilized anti-mouse IgG (Bangs Laboratories, Inc., Fishers, IN) to construct a calibration curve. Beads and cells were separately labeled with either mouse anti-EGFR clone ab30 (4 μg/ml) (Abcam), mouse anti-CD276 clone 185504 (4 μg/ml) (Biotechne) or anti-c-Myc clone 9E10 (4 μg/mL) (BioLegend) for 30 min at 4°C. Beads and cells were washed once with phosphate-buffered saline with 0.1% (w/v) bovine serum albumin, 1 mM CaCl2 and 0.5 mM Mg2SO4 (PBSACM) and pelleted at 500 g for 3 min. The beads and cells were then labelled with goat anti-mouse Alexa Fluor 647 conjugate (10 μg/ml) (Life Technologies A-21235) or goat anti-mouse FITC conjugate (10 μg/ml) (Sigma-Aldrich F0257) for 30 min at 4°C, washed once with PBSACM and again pelleted at 500 g for 3 min. Fluorescence was analyzed by flow cytometry using an Accuri C6 Plus (BD Biosciences). Bead fluorescence was used to construct a calibration curve from which the EGFR, CD276 or yeast-displayed ligand expression was quantified.
Adherent mammalian cell panning
Adherent cell panning selections were carried out with minor modifications from previous literature (Stern et al., 2016). Mammalian cells were grown in 12-well plates to ~70–90% confluency. The culture medium was aspirated, and cells washed once with ice-cold PBSACM. Yeast mixtures containing 1 × 106 or 1 × 107 ligand-displaying yeast and 1 × 108 EBY100 were washed in PBSACM and added dropwise to each well in 1 ml of ice-cold PBSACM. Plates were incubated statically for 15 min at 4°C and the unbound yeast were removed. Wells were washed with 1 ml of ice-cold PBSACM four times with 25 gentle tilts and 5 gentle nutations and a fifth time with 10 nutations. Cell monolayers with bound yeast were removed by scraping and resuspended in 1 ml of PBSACM. The yield of plasmid-harboring yeast was quantified by plating mixture dilutions on SD-CAA plates, whereas the yield of total yeast was quantified by plating mixture dilutions on YPD plates. Enrichment ratio was calculated as the yield of plasmid-harboring yeast divided by the yield of plasmidless yeast.
Modeling ligand distribution on the yeast cell surface
Linker length was substantially shorter than yeast cell radius; thus, curvature was neglected and the cell surface was approximated as an impermeable plane. The linker was modeled as a 1-D Gaussian chain model and P(z), the probability distribution function of the linker extending an end-to-end distance z in the direction perpendicular to the cell surface, is given by:
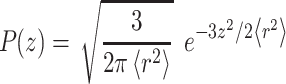 |
where 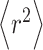 is the mean-square end-to-end length of the peptide chain and is given by:
is the mean-square end-to-end length of the peptide chain and is given by:
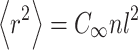 |
where 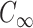 is the length-independent characteristic ratio,
is the length-independent characteristic ratio, 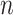 is the number of peptide bonds and
is the number of peptide bonds and 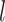 is the average distance between adjacent Cα atoms (3.8 Å) (Wang et al., 2005). The linker was assumed to be uniformly distributed within the cell wall (estimated as 115 nm) (Dupres et al., 2010) and the conditional probability of the linker ending a distance y from the plasma membrane given that the linker starts a distance a from the plasma membrane is given by:
is the average distance between adjacent Cα atoms (3.8 Å) (Wang et al., 2005). The linker was assumed to be uniformly distributed within the cell wall (estimated as 115 nm) (Dupres et al., 2010) and the conditional probability of the linker ending a distance y from the plasma membrane given that the linker starts a distance a from the plasma membrane is given by:
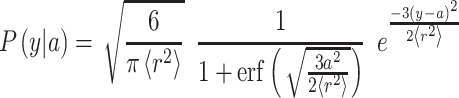 |
This equation was multiplied by the probability distribution of linker in the cell wall and numerically integrated to generate the probability distribution function 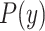 using the law of total probability. The characteristic ratios for polyglycine (2.16), polyalanine (9.27) and polyproline (116) (Brant et al., 1967; Schimmel and Flory, 1967) were used to generate curves for polypeptides of 80 and 641 residues and the fraction of ligands extending beyond the cell wall.
using the law of total probability. The characteristic ratios for polyglycine (2.16), polyalanine (9.27) and polyproline (116) (Brant et al., 1967; Schimmel and Flory, 1967) were used to generate curves for polypeptides of 80 and 641 residues and the fraction of ligands extending beyond the cell wall.
Statistical analysis
All cell panning was binned by day and linker prior to significance testing by two-way analysis of variance with only main effects. Significance testing of yeast surface display levels was conducted by constructing 95% CIs. All statistics are reported as mean ± standard deviation except for the following: yeast surface display levels were reported as 95% CIs and AC2 affinity was reported as a 68% CI.
Funding
The American Cancer Society (130418-RSG-17-110-01-TBG); the National Institutes of Health (R01 EB023339).
Supplementary Material
Edited By: Timothy Whitehead
Contributor Information
Patrick S Lown, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
Jessy J Cai, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
Seth C Ritter, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
Jacob J Otolski, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
Ryan Wong, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
Benjamin J Hackel, Department of Chemical Engineering and Materials Science, University of Minnesota—Twin Cities, Minneapolis, MN 55455, USA.
References
- Ackerman, M., Levary, D., Tobon, G., Hackel, B., Orcutt, K.D. and Wittrup, K.D. (2009) Biotechnol. Prog., 25, 774–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banta, S., Dooley, K. and Shur, O. (2013) Annu. Rev. Biomed. Eng., 15, 93–113. [DOI] [PubMed] [Google Scholar]
- Beerli, R.R., Bauer, M., Buser, R.B., Gwerder, M., Muntwiler, S., Maurer, P., Saudan, P. and Bachmann, M.F. (2008) Proc. Natl. Acad. Sci. U. S. A., 105, 14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berges, S.M. (2017) Engineering a Fluorescent Barcoding System for Highly Multiplexed, Single-Cell Analysis of Biomolecular and Cellular Libraries. University of Delaware,Newark, DE, USA. [Google Scholar]
- Boder, E.T. and Wittrup, K.D. (1997) Nat. Biotechnol., 15, 553–557. [DOI] [PubMed] [Google Scholar]
- Bozovičar, K. and Bratkovič, T. (2019) Int. J. Mol. Sci., 21, 215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brant, D.A., Miller, W.G. and Flory, P.J. (1967) J. Mol. Biol., 23, 47–65. [Google Scholar]
- Bratkovič, T. (2010) Cell. Mol. Life Sci., 67, 749–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breibeck, J. and Skerra, A. (2018) Biopolymers, 109, e23069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, X. and Garen, A. (1995) Proc. Natl. Acad. Sci. U. S. A., 92, 6537–6541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, T.F., De Picciotto, S., Hackel, B.J. and Wittrup, K.D. (2013) Methods Enzymol., 523, 303–326. [DOI] [PubMed] [Google Scholar]
- Dangaj, D., Lanitis, E., Zhao, A.et al. (2013) Cancer Res., 73, 4820–4829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier, B. and Plückthun, A. (2012) Methods Mol. Biol., 805, 261–286. [DOI] [PubMed] [Google Scholar]
- Dupres, V., Dufreěne, Y.F. and Heinisch, J.J. (2010) ACS Nano, 4, 5498–5504. [DOI] [PubMed] [Google Scholar]
- Even-Desrumeaux, K. and Chames, P. (2012) Methods Mol. Biol., 907, 225–235. [DOI] [PubMed] [Google Scholar]
- Evers, T.H., Van Dongen, E.M.W.M., Faesen, A.C., Meijer, E.W. and Merkx, M. (2006) Biochemistry, 45, 13183–13192. [DOI] [PubMed] [Google Scholar]
- Flory, P.J. (1969) Statistical Mechanics of Chain Molecules. Interscience Publishers, New York. [Google Scholar]
- Gera, N., Hussain, M., Wright, R.C. and Rao, B.M. (2011) J. Mol. Biol, 409, 601–616. [DOI] [PubMed] [Google Scholar]
- Hackel, B.J., Ackerman, M.E., Howland, S.W. and Wittrup, K.D. (2010) J. Mol. Biol., 401, 84–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackel, B.J. and Wittrup, D. (2011) Protein Engineering Handbook, Vol. 1 & 2, Wiley, Hoboken, NJ, USA pp. 621–648. [Google Scholar]
- Horiya, S., Bailey, J.K. and Krauss, I.J. (2017) Methods Enzymol., 597, 83–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, G., Zhang, M. and Erdman, S.E. (2003) Eukaryot. Cell. 2, 1099–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, A.R., Stutz, C.C., Zhou, Y., Marks, J.D. and Shusta, E.V. (2014) Biotechnol. J., 9, 664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klis, F.M., Mol, P., Hellingwerf, K. and Brul, S. (2002) FEMS Microbiol. Rev. 26, 239–256 [DOI] [PubMed] [Google Scholar]
- Krohl, P.J., Kim, K.B., Lew, L., Van Dyke, D., Ludwig, S.D. and Spangler, J.B. (2020) AIChE J., 66, e16995. [Google Scholar]
- Lown, P.S. and Hackel, B.J. (2020) ACS Comb. Sci. 22, 274–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon, C., Baier, A.S., Pascolutti, R.et al. (2018) Nat. Struct. Mol. Biol. 25, 289–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova, A., Magnusson, M., Eriksson, T.L.J.et al. (2006) Cancer Res., 66, 4339–4348. [DOI] [PubMed] [Google Scholar]
- Plückthun, A. (2015) Annu. Rev. Pharmacol. Toxicol, 55, 489–511. [DOI] [PubMed] [Google Scholar]
- Schimmel, P.R. and Flory, P.J. (1967) Proc. Natl. Acad. Sci. U. S. A., 58, 52–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlapschy, M., Binder, U., Börger, C., Theobald, I., Wachinger, K., Kisling, S., Haller, D. and Skerra, A. (2013) Protein Eng. Des. Sel., 26, 489–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Y., Yang, X., Dong, N., Xie, X., Bai, X. and Shi, Y. (2007) Cell Res., 17, 650–660. [DOI] [PubMed] [Google Scholar]
- Smith, G.P. (1985) Science, 228, 1315–1317. [DOI] [PubMed] [Google Scholar]
- Stern, L.A., Case, B.A. and Hackel, B.J. (2013) Curr. Opin. Chem. Eng., 2, 425–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, L.A., Lown, P.S., Kobe, A.C., Abou-Elkacem, L., Willmann, J.K. and Hackel, B.J. (2019) ACS Comb. Sci., 21, 207–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern, L.A., Schrack, I.A., Johnson, S.M., Deshpande, A., Bennett, N.R., Harasymiw, L.A., Gardner, M.K. and Hackel, B.J. (2016) Biotechnol. Bioeng., 113, 2328–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson, B.J., Cho, Y.K. and Shusta, E.V. (2013) Methods, 60, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tillotson, B.J., Lajoie, J.M. and Shusta, E.V. (2015) Methods Mol. Biol., 1319, 65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umlauf, B.J., Clark, P.A., Lajoie, J.M., Georgieva, J.V., Bremner, S., Herrin, B.R., Kuo, J.S. and Shusta, E.V. (2019) Sci. Adv., 5, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L., Rivera, E.V., Benavides-Garcia, M.G. and Nall, B.T. (2005) J. Mol. Biol., 353, 719–729. [DOI] [PubMed] [Google Scholar]
- Wang, X.X., Cho, Y.K. and Shusta, E.V. (2007) Nat. Methods, 4, 143–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X.X. and Shusta, E.V. (2005) J. Immunol. Methods, 304, 30–42. [DOI] [PubMed] [Google Scholar]
- Williams, R.M., Hajiran, C.J., Nayeem, S. and Sooter, L.J. (2014) BMC Biotechnol., 14, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldring, D.R., Holec, P.V., Zhou, H. and Hackel, B.J. (2015) PLoS One, 10, e0138956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woldring, D.R., Holec, P.V., Stern, L.A., Du, Y. and Hackel, B.J. (2017) Biochemistry, 56, 1656–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, M.Y., Xu, X.H., Chen, G.Z., Deng, X.L., Li, J., Yu, X.J. and Chen, M.Z. (2004) World J. Gastroenterol., 10, 2619–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z., Wan, Y., Tao, P., Qiang, M., Dong, X., Lin, C.W., Yang, G., Zheng, T. and Lerner, R.A. (2019) Proc. Natl. Acad. Sci. U. S. A., 116, 14971–14978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, H.X. (2004) Biochemistry, 43, 2141–2154. [DOI] [PubMed] [Google Scholar]
- Zorniak, M., Clark, P.A., Umlauf, B.J., Cho, Y., Shusta, E.V. and Kuo, J.S. (2017) Sci. Rep., 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.