

Abstract
We disclose a catalytic method for β-C(sp3)–H functionalization of N-alkylamines for synthesis of enantiomerically enriched β-substituted amines, entities prevalent in pharmaceutical compounds and used to generate different families of chiral catalysts. We demonstrate that a catalyst system comprising of seemingly competitive Lewis acids, B(C6F5)3 and a chiral Mg- or Sc-based complex, promotes the highly enantioselective union of N-alkylamines and α,β-unsaturated compounds. An array of δ-amino carbonyl compounds was synthesized under redox-neutral conditions by enantioselective reaction of a N-alkylamine-derived enamine and an electrophile activated by the chiral Lewis acid co-catalyst. The utility of the approach is highlighted by late-stage β-C–H functionalization of bioactive amines. Investigations in regard to the mechanistic nuances of the catalytic processes are described.
Graphical Abstract
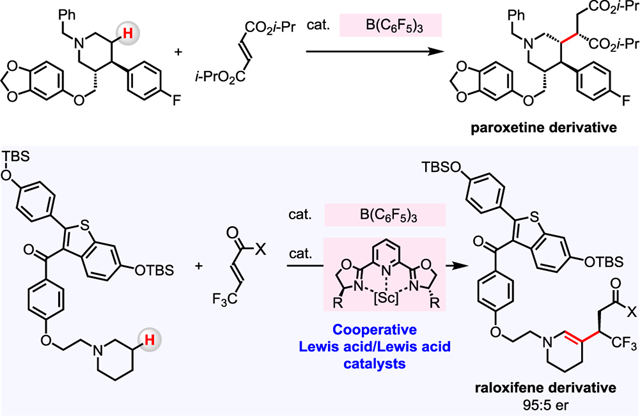
1. INTRODUCTION
Enantioselective synthesis of N-alkylamines through the transformations of amino C(sp3)–H bonds has emerged as a powerful strategy to access key building blocks of N-based natural products, drugs and catalysts for stereoselective synthesis.1–18 A plethora of organometallic catalysts have been introduced that can promote reaction at an α-amino C–H bond by conversion of an amine substrate to appropriately reactive intermediate (e.g., α-amino radical, iminium ion),19–31 metal–carbenoid insertion,32–33 or heteroatom-directed metalation.34–36 More remote γ-amino C–H bonds may be activated by LnPd-catalyzed and N-directed cyclometalation.37–41 However, development of methods for synthesis of enantiomerically enriched amines by β-amino C–H functionalization is a major challenge that remains to be addressed.42–50 α-Amino C–H activation by hydrogen atom or hydride transfer is facilitated by stabilization of the resulting species through hyperconjugation with the nitrogen lone pair. Nonetheless, such processes do not readily occur at the β position of amines. Furthermore, LnPd-catalyzed β-amino C–H activation requires the presence of a strained 4-membered palladacycle (vs a more favorable 5-membered metallacycle formed by γ-amino C–H activation).42–44
There are only a limited number of protocols for preparing enantiomerically enriched β-substituted amines through activation of a β-amino C–H bond.51–54 A notable strategy is Pd/phosphoric acid-catalyzed and IOAc-mediated desymmetrization of gem-dimethyl groups in tetramethyl-morpholinone 1a to give organometallic intermediate I en route to aziridine 2a (Figure 1a).52 Equally noteworthy is Pd/PR3-catalyzed stereospecific cross-coupling of 1,3-oxazinane 1c (prepared by s-BuLi/(+)-sparteine-mediated α-lithiation of 1b and Li/Zn exchange with ZnCl2) and Ph–Br to give 2c via LnPd–enamine II (Figure 1b).54 Still, key shortcomings remain unaddressed. For instance, acid–base complexation often occurs in a mixture that contains a Lewis acidic catalyst (e.g., Pd(OAc)2) and a Lewis basic N-alkylamine substrate and/or product which may also contain other Lewis acid-sensitive functional groups.55–62 Additionally, because these methods require IOAc or s-BuLi, moieties that readily react with oxidants and Lewis bases can be problematic. As a result, substrate scope is confined to highly sterically encumbered and/or Boc-protected amines (1a, 1b and analogues) with minimal electronic and steric affinity towards Lewis acids, Lewis bases or oxidants. Development of a sustainable element-based, highly functional group tolerant and enantioselective catalyst system which allows access to β-substituted N-alkylamines via β-amino C–H functionalization under redox-neutral conditions therefore constitutes a compelling research objective.
Figure 1.
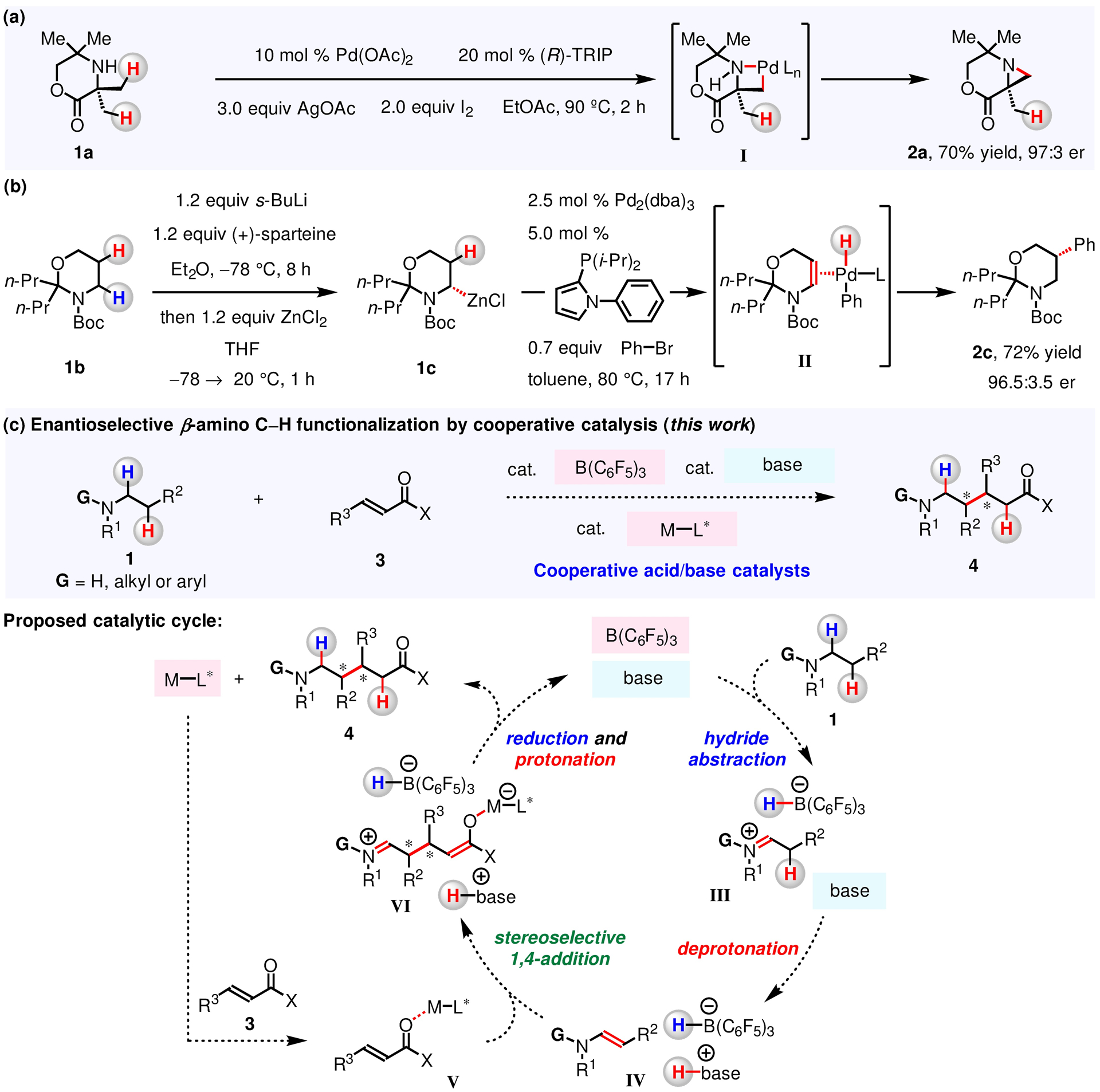
Enantioselective Transformations of β-Amino C–H Bonds
Conversion of β-amino C–H bonds can be achieved nonstereoselectively through in situ generation of enamines from N-alkylamines;63–85 but enamine precursors are largely limited to N,N-dialkylanilines. In the case of more Lewis basic trialkylamines and substrates containing acid- and/or base-sensitive moieties, catalyst deactivation can become problematic.55–62 One strategy to overcome mutual quenching would be to use strongly Lewis acidic and sterically hindered B(C6F5)386–88 to convert trialkylamines into enamines;63–76 this would involve (F5C6)3B-mediated hydride abstraction from the amine to give a borohydride/iminium complex,63–76 which would in turn be deprotonated to yield an enamine (Figure 1c, 1 → III → IV).89–92 Chang and co-workers have shown that B(C6F5)3 is capable of promoting β-silylation of N-arylpiperidines to generate rac-bridged sila-N-heterocycles.64 We have introduced a method for (F5C6)3B-catalyzed hydrogen isotope exchange involving β-amino C–H bonds of various bioactive molecules.65 Still, engagement of the enamine intermediates formed by (F5C6)3B/Brønsted base-catalyzed dehydrogenation of N-alkylamines has not been utilized in an enantioselective transformation.
In contemplating ways to develop a protocol that involves β-C–H alkylation of N-alkylamines (1) through an enantioselective union with α,β-unsaturated compounds (3), we envisioned using a set of B(C6F5)3, a Brønsted base catalyst, and a chiral Lewis acid co-catalyst (M–L*) that might be induced to function cooperatively (Figure 1c).63–76, 93–102 We imagined that B(C6F5)3 being the recipient of a hydride from an amine (1), leading to the formation of a borohydride and an iminium ion (III). A Brønsted base catalyst would subsequently deprotonate III to furnish enamine IV. An ensuing enantio- and diastereo-selective C–C bond formation between enamine IV and α,β-unsaturated compound, activated by the chiral Lewis acid co-catalyst (V), would deliver zwitterionic VI. This would be followed by protonation and reduction to give β-alkylation product 4. A key advantage of using untethered and independently operational catalysts is that efficiency and stereoselectivity may be easily optimized by evaluation of readily accessible Lewis acids, Lewis bases and chiral ligands (vs bifunctional catalysts that require tethering of different catalytic sites). However, B(C6F5)3 and the chiral Lewis acid co-catalyst must be able to perform their independent roles without overlapping functions, as otherwise, B(C6F5)3 could promote the racemic reaction through activation of both substrates, most likely resulting in diminished enantioselectivity. Here, we report that functionally similar B(C6F5)3 and a chiral Lewis acid co-catalyst can operate in concert to engender enantioselective coupling of N-alkylamines and α,β-unsaturated compounds.
2. RESULTS AND DISCUSSION
2.1. Method Development
2.1.1. Identification of Optimal Conditions.
To begin, we set out to investigate if B(C6F5)3 might activate both N,N-dibenzylethanamine (1d) and diisopropyl fumarate (3d), generating 4d (Table 1). Treatment of 1d and 3d with 10 mol % B(C6F5)3 and 10 mol % Et3N, 2,2,6,6-tetramethylpiperidine (TMP) or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded 4d in 50%, 25%, and <5% yield, respectively (entries 1–3). Other than 4d, over alkylation product 5d was also formed (entries 1–6), probably through in situ conjugate addition of an enolate (VI, Figure 1c) to 3d. When the transformation was carried out without a Brønsted base, 4d (73% yield) and 5d (25% yield) were formed more efficiently (entry 4), suggesting that 1d, 4d and/or 5d can deprotonate an iminium to form an enamine (III → IV, Figure 1c). To suppress formation of 5d, the mixture was diluted (entries 5–6); by using 10 mol % of B(C6F5)3 and 0.80 mL of benzene (vs 0.20 mL used in entries 1–4), we were able to obtain 4d (91% yield) within 12 hours (entry 6).103 Moreover, no 4d was formed when less hindered BCl3 or less acidic BPh3 were involved (entries 7–8). Without B(C6F5)3, 4d was not generated (entry 9). These findings support the notion that strongly Lewis acidic B(C6F5)3 in combination with sterically demanding and electron-rich N-alkylamines constitute the most effective catalyst–substrate combination.
Table 1.
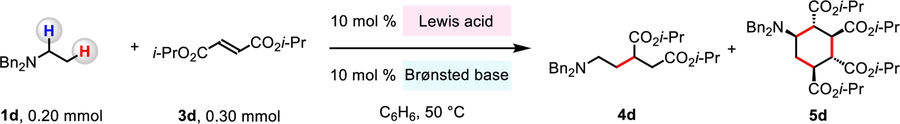 | |||||
|---|---|---|---|---|---|
| entry | Lewis acid | Brønsted base | time (h) | yield of 4d (%) | yield of 5d (%) |
| 1 | B(C6F5)3 | Et3N | 3 | 50 | 23 |
| 2 | B(C6F5)3 | TMP | 3 | 25 | 29 |
| 3 | B(C6F5)3 | DBU | 3 | <5 | <5 |
| 4 | B(C6F5)3 | none | 3 | 73 | 25 |
| 5d | B(C6F5)3 | none | 3 | 54 | 5 |
| 6d | B(C6F5)3 | none | 12 | 91 | 7 |
| 7 | BCI3 | none | 12 | 0 | 0 |
| 8 | BPh3 | none | 12 | 0 | 0 |
| 9 | none | none | 12 | 0 | 0 |
Conditions: N,N-dibenzylethanamine (1d, 0.20 mmol), diisopropyl fumarate (3d, 0.30 mmol), Lewis acid, Brønsted base, C6H6 (0.20 mL), under N2, 50 °C.
Yield was determined by the 1H NMR analysis of unpurified reaction mixtures with m-xylene as the internal standard.
The structure and relative configuration of 5d were established by the nuclear Overhauser effect spectroscopy (NOESY) studies.
0.80 mL of C6H6 was used.
2.1.2. Scope.
Many acyclic and cyclic N-alkylamines may be used in the reaction with diisopropyl fumarate 3d to generate the corresponding β-substituted amines (4d–4o, Figure 2). Reaction with N,N-dibenzylethanamine 1d and 3d afforded 4d in 88% yield. N-Benzhydryl-substituted secondary amines 1e and 1f were suitable substrates, furnishing 4e (93% yield) and 4f (87% yield, 2.1:1 dr), respectively. None of the 1,4-addition product was observed in the case of a secondary amine bearing a more hindered trityl group (e.g., N-tritylethanamine); this may be attributed to rapid substrate decomposition. With less hindered N-benzylethanamine, the formation of (F5C6)3B–amine adduct may compete with Lewis acid-catalyzed hydride abstraction, resulting in minimal formation of the desired product. We then investigated reactions with different chiral N-alkylamines (1g–1i, Figure 2). Accordingly, 4g was produced in 90% yield as a 1.4:1 mixture of easily separable diastereomers, allowing us to secure the amino ester in enantiomerically pure form. As indicated by synthesis of 4h and 4i, chiral pyrrolidines may be used as starting materials. Whereas the reaction with the less hindered 1-(4-methoxy-2,6-dimethylphenyl)piperidine furnished 4j as the only product in 89% yield, with the bulkier 4,4-dimethyl-substituted piperidine substrate, enamine 4k was obtained in 93% yield.104
Figure 2. β-Alkylation of Different N-Alkylamines.
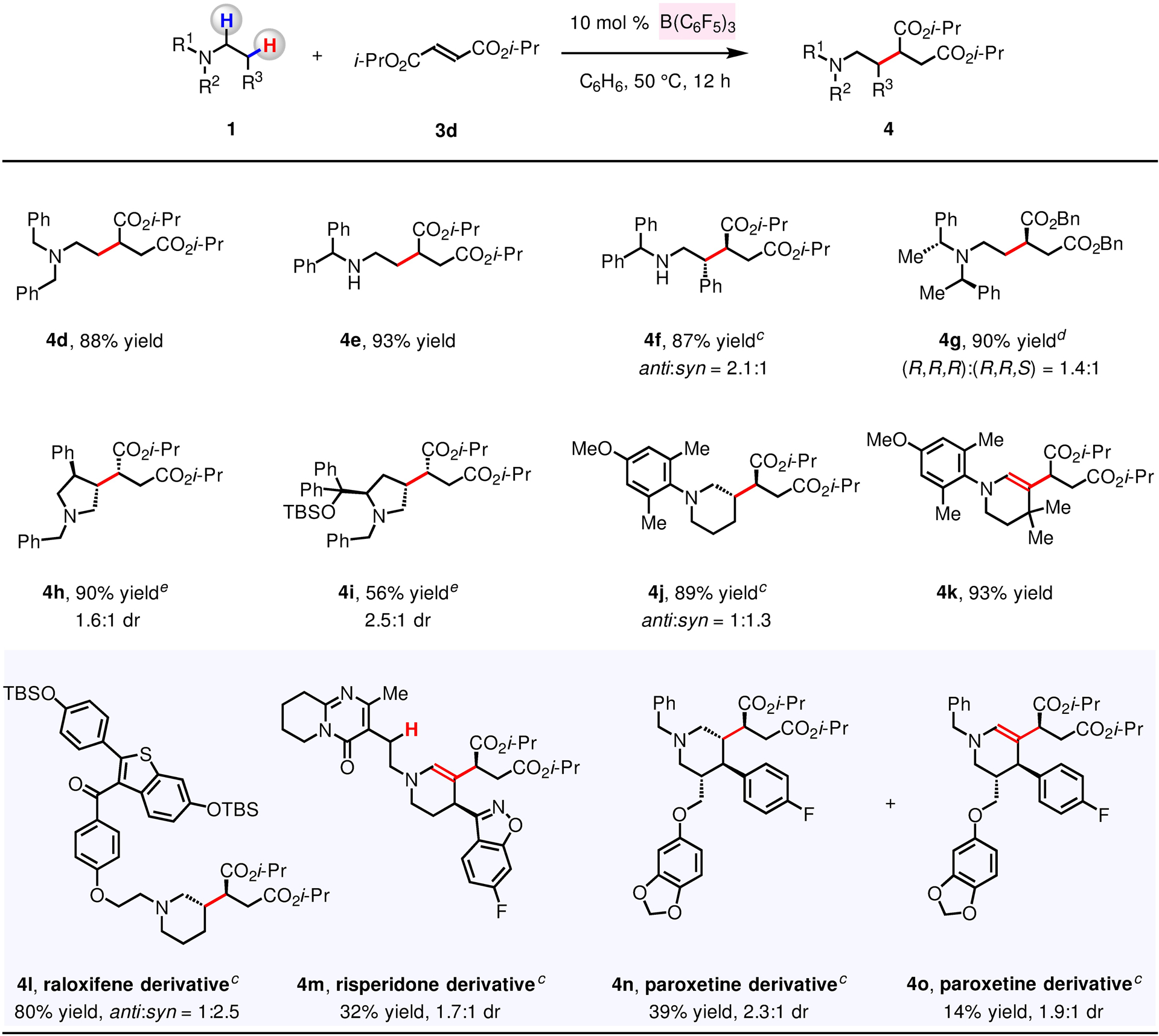
The values correspond to yields of isolated and purified products. a Conditions: N-alkylamine (1, 0.20 mmol), diisopropyl fumarate (3d, 0.30–0.40 mmol), B(C6F5)3 (10 mol %), C6H6, under N2, 50 °C. b Yield of isolated and purified product. The dr values were determined by the 1H NMR analysis of the unpurified reaction mixtures. c The structure and relative configuration of 4f, 4j, 4l, and 4m were assigned in analogy. The stereochemistry of 4n and 4o was assigned in analogy and also by the NOESY studies. d The absolute configuration of 4g was determined based on the specific rotation of the derivative. e The structure and absolute configuration of 4i was established by the X-ray crystallographic analysis. The stereochemistry of 4h was assigned in analogy and also by the NOESY studies. See the Supporting Information for details.
The method is applicable to late-stage modification of N-containing bioactive molecules that possess an array of Lewis acid-sensitive functional groups (1l–1n; Figure 2). In addition to the N-alkylamine moieties of 1l–1n, a ketone (1l), a benzothiophene (1l), an ether (1l, 1n), a pyrimidinone (1m) and a benzoisoxazole (1m) were tolerated. Thus, structures of piperidine-based compounds, such as raloxifene (osteoporosis treatment), risperidone (anti-psychotic) and paroxetine (antidepressant) could be readily altered. Silyl-protected raloxifene 1l reacted efficiently with 3d to afford 4l in 80% yield and 2.5:1 dr. Risperidone 1m, possessing more sterically hindered β-amino C–H bonds (vs. 1l), was merged with 3d, furnishing enamine 4m (32% yield); minimal amounts (<5%) of the product derived from alkylation of acyclic β-amino C–H bond (H labelled in red) could be observed (1H NMR analysis). The process involving N-benzyl paroxetine 1n and 3d afforded 4n and 4o in 39% and 14% yield, respectively.
2.1.3. Diastereo- and Enantio-selective Processes.
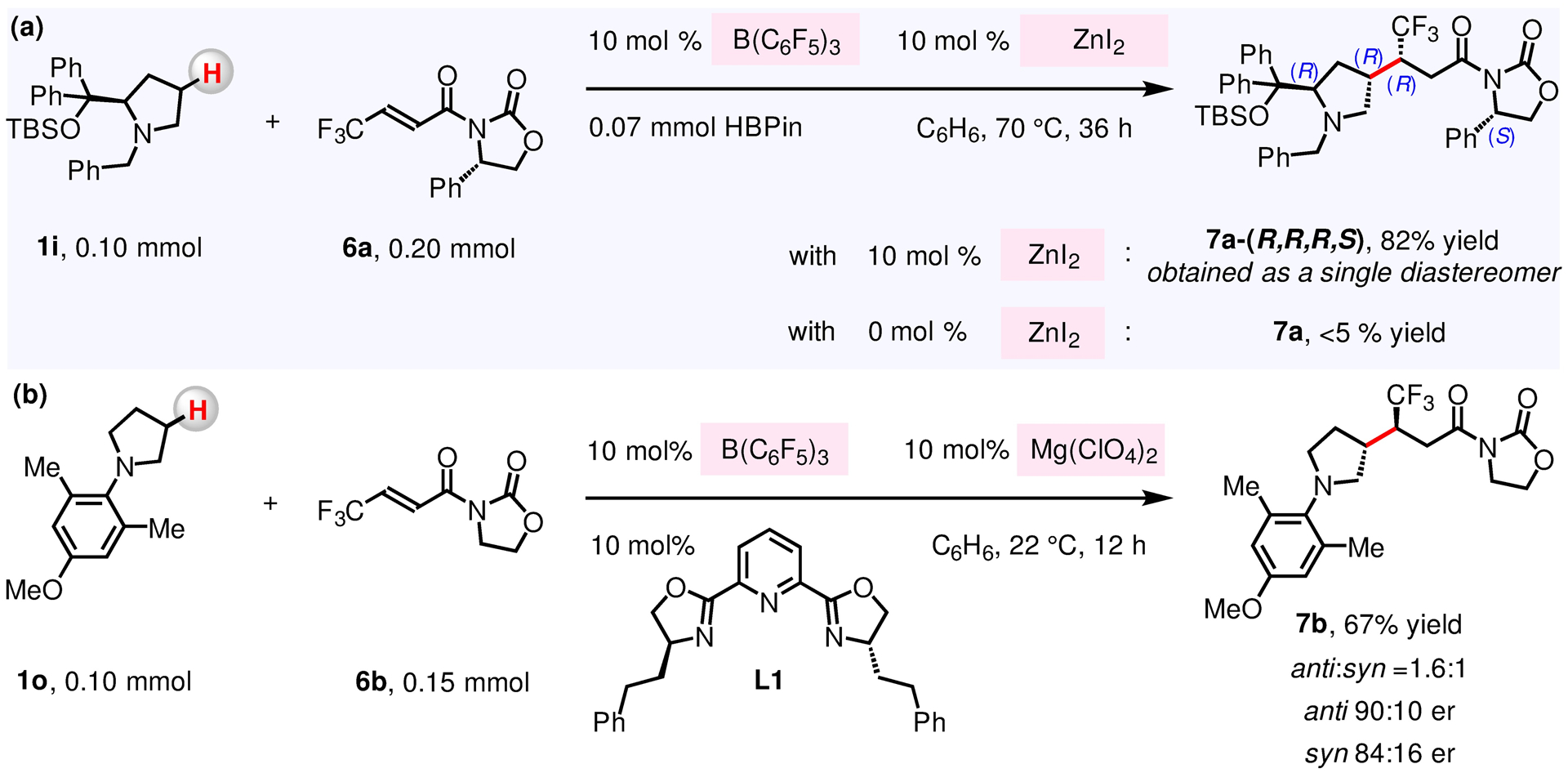
To develop a highly diastereoselective and catalytic β-amino C–H alkylation variant, we chose to utilize the prolinol derivative 1i105–108 and (S,E)-4-phenyl-3-(4,4,4-trifluorobut-2-enoyl)oxazolidin-2-one 6a as model substrates (Figure 3a). In the event, (F5C6)3B-catalyzed reaction of 1i and diisopropyl fumarate 3d gave β-alkylation product 4i in only 56% yield and 2.5:1 dr (Figure 2). To probe whether the use of enantiomerically pure electrophile leads to improved dr, we reacted 1i and 6a in the presence of B(C6F5)3 to find that product 7a was formed in <5% yield. These findings suggest that B(C6F5)3 facilitates the conversion of 1i into an enamine but does not sufficiently activate 6a. We therefore decided to evaluate a blend of B(C6F5)3 and various Lewis acid co-catalysts, latter of which may activate 6a, to establish that, with 10 mol % B(C6F5)3 and ZnI2, 7a-(R,R,R,S) can be obtained in 82% yield and as a single diastereomer (>20:1 dr; see the Supporting Information for details). The presence of HBPin led to enhanced dr, as 7a was isolated in 67% yield and 10:1 dr in its absence (see the Supporting Information for details). We also explored the suitability of developing an enantioselective β-amino C–H alkylation reaction between achiral substrates 1o and 6b, promoted by a combination of B(C6F5)3 and an enantiomerically pure organometallic co-catalyst (Figure 3b). We discovered that, with a complex of PyBOX L1–Mg(ClO4)2, β-amino C–H alkylation of 1o proceeds to afford 7b in 67% yield (1.6:1 dr, up to 90:10 er). However, neither efficiency nor enantioselectivity could be improved by further catalyst optimization (with 6b as electrophile; see the Supporting Information for details).
Figure 3. Stereoselective β-Functionalizations of N-Alkylamines.
The values correspond to yields of isolated and purified products. See the Supporting Information for details.
To improve enantioselectivity, we probed the transformations involving a number of chiral Lewis acid co-catalysts and α,β-unsaturated compounds bearing different auxiliaries.109–112 We found that 1-(4-methoxyphenyl)pyrrolidine 1p reacts efficiently with 2-acryloylpyrazolidinone derivative 6c in the presence of 10 mol % of B(C6F5)3 together with Sc(OTf)3 and various chiral bisoxazoline ligands (Figure 4). We then found that reactions with Ph–BOX, Ph–DBFOX and Ph–PyBOX ligands (e.g., L2–L4) are hardly enantioselective (58:42–50:50 er). The situation improved considerably with alkyl-substituted PyBOX ligands (e.g., L5–L8), and in the presence of (S)-i-Bu–PyBOX (L8), 7c was formed in 72% yield, 2.7:1 dr and up to 98:2 er. Reaction efficiency improved (85% yield) when L9 and its diastereomer L10 were used. The stereochemical course of 1p-derived enamine addition to [L10–Sc(OTf)3]-activated 6c can be rationalized by the models presented in Figure 5.113–115 Models VII and VIII represent the energetically minimized structures of [L10–Sc(OTf)3] docked with 6c.113–115 As shown in model VII, the high level of enantioselectivity observed in the formation of 7c-(R,S) can be explained by selective 1,4-addition of the enamine to the re-face of [L10–Sc(OTf)3]-bound 6c; as depicted in model VIII, the si-face is effectively shielded by a sec-butyl group of the PyBOX ligand.
Figure 4. Evaluation of Chiral Ligands.
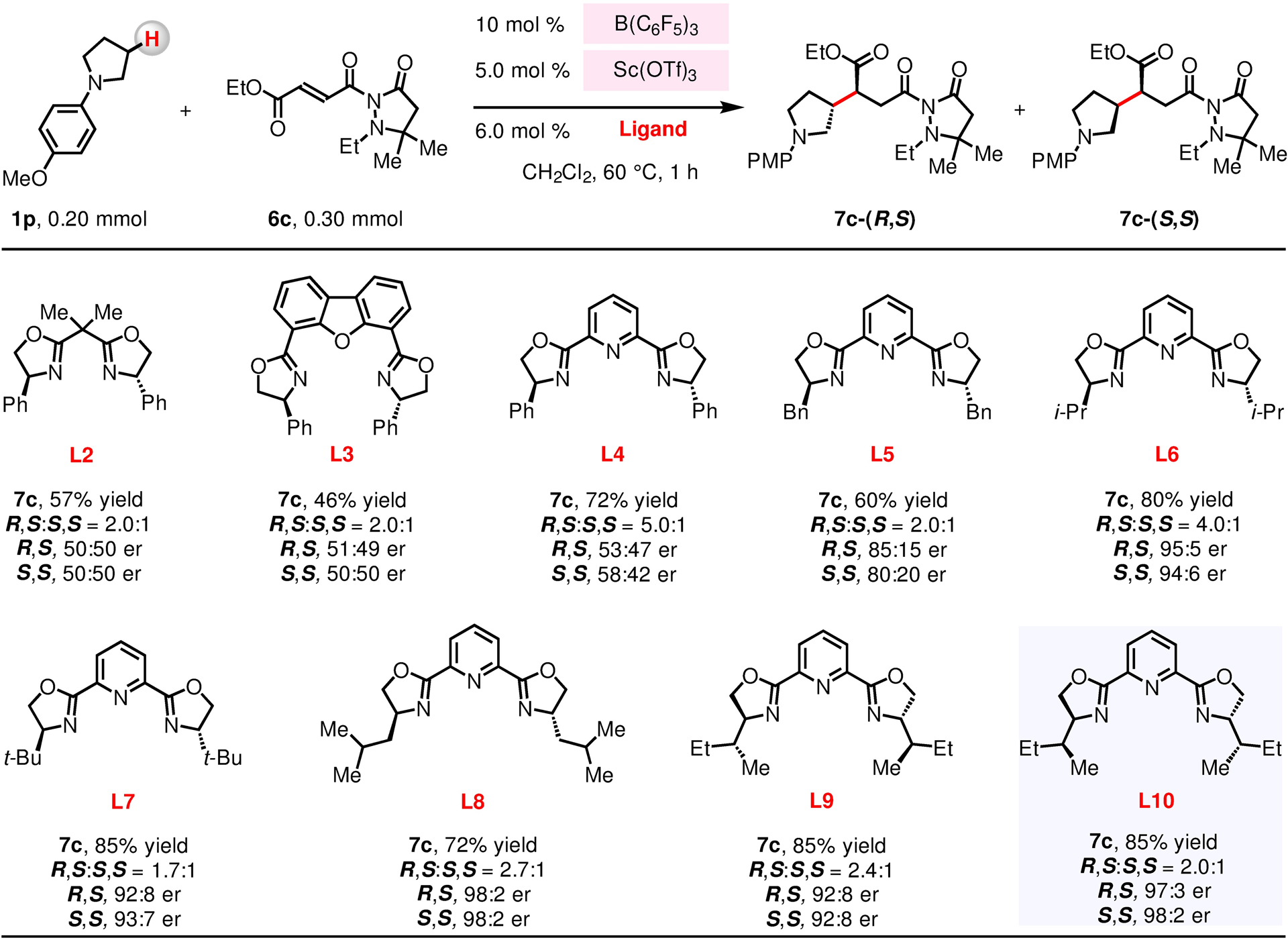
Yield and diastreomeric ratio (dr) values were determined by the 1H NMR analysis of unpurified reaction mixtures with mesitylene as the internal standard. Enantiomeric ratio (er) values were determined by the HPLC analysis of isolated and purified product. Conditions: N-arylpyrrolidine (1p, 0.20 mmol), α,β-unsaturated compound (6c, 0.30 mmol), B(C6F5)3 (10 mol %), Sc(OTf)3 (5.0 mol %), ligand (6.0 mol %), CH2Cl2 (2.0 mL), under N2, 60 °C, 1 h. See the Supporting Information for details.
Figure 5.
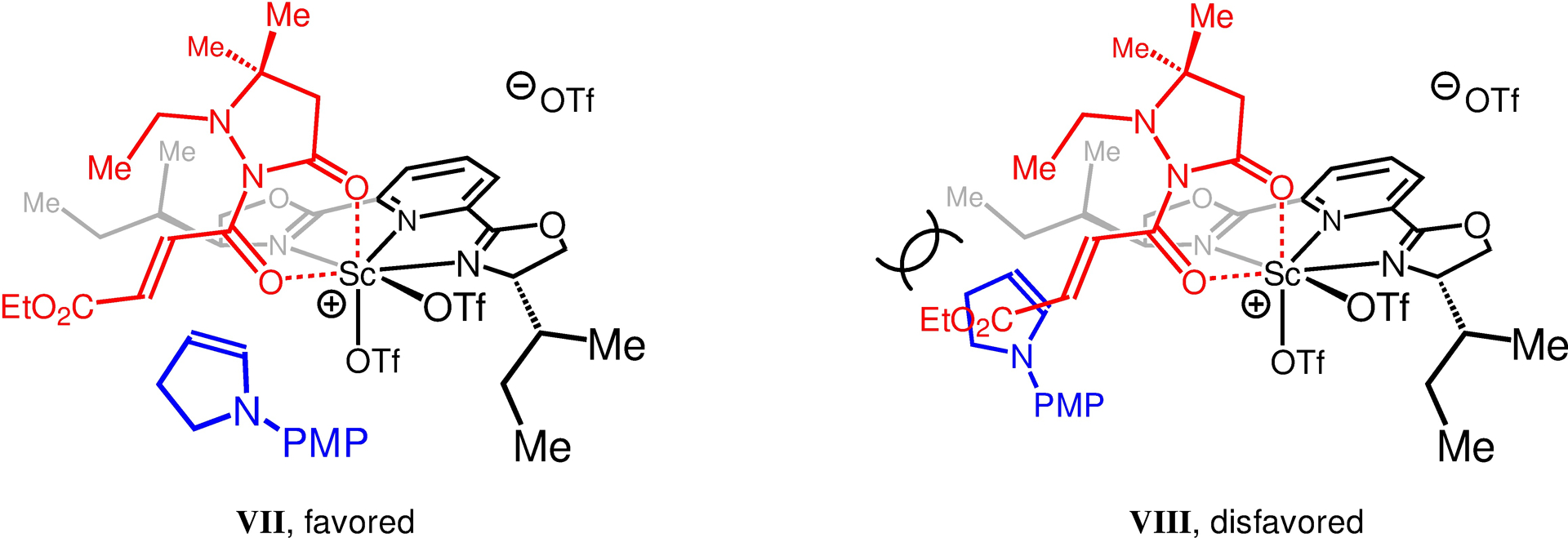
Stereochemical Rationale for Enamine Addition to α,β-Unsaturated Compounds Catalyzed by [L10–Sc(OTf)3].
Enantioselective reactions with an array of N-alkylamines were carried out in the presence of B(C6F5)3, a L–Sc(OTf)3 complex and 6c–6f (Figure 6). β-Alkyl derivatives of 1-(4-methoxyphenyl)pyrrolidine (1p) bearing γ-ester, ketone or CF3 groups (7c–7f) were thus synthesized in 62–83% yield, 2.0:1–6.7:1 dr and 95:5–98:2 er. The reaction of 1p with 6c (R5 = Et) and 6d (R5 = Bn) gave 7c (2.0:1 dr, up to 98:2 er) and 7d (3.0:1, up to 95:5 er), respectively, indicating that N-substituents of the pyrazolidinone unit have influence over both diastereo- and enantio-selectivity. When acyclic N-ethyl-4-methoxy-N,2,6-trimethylaniline 1q was reacted with F3C-substituted 6f (Figure 7a), N-arylpiperidine derivative 7g was produced in 67% yield (>20:1 dr, 95:5 er); generation of 7g probably entails β-amino C–H alkylation of 1q by 6f to afford a zwitterionic intermediate containing an iminium and an enolate (IX), followed by isomerization of the iminium to give X; ensuing intramolecular Mannich-type reaction gives 7g. The unions of N-arylpiperidines (1j–1k) or N-arylazapene (1r) with 6f afforded enamines 7h–7j in 74–95% yield and 94:6–96:4 er. The reaction of N-arylpiperidine-3,3,5,5-d4 1j-d (0.10 mmol) and 6f (0.20 mmol) gave 7h-d (84% yield, 95:5 er) and 8h-d (0.08 mmol; Figure 7b). Spectroscopic analysis of 7h-d and 8h-d revealed that there is deuterium incorporation at their enolizable α-carbonyl units, and that there is D/H exchange at C5 of 7h-d (>95% in 1j-d → 81% in 7h-d).65 These results imply that in situ generated [(F5C6)3B–H]− [base–D]+ reacts with 6f to produce 8h-d to regenerate B(C6F5)3 (vs by releasing H–D). We were able to functionalize the β-Amino C–H bonds of bioactive trialkylamines, including cloperastine (cough suppressant) 1s and raloxifene 1l, to generate enamines 7k and 7l in 53% yield (90:10 er) and 57% yield (95:5 er), respectively.
Figure 6. Stereoselective Processes.
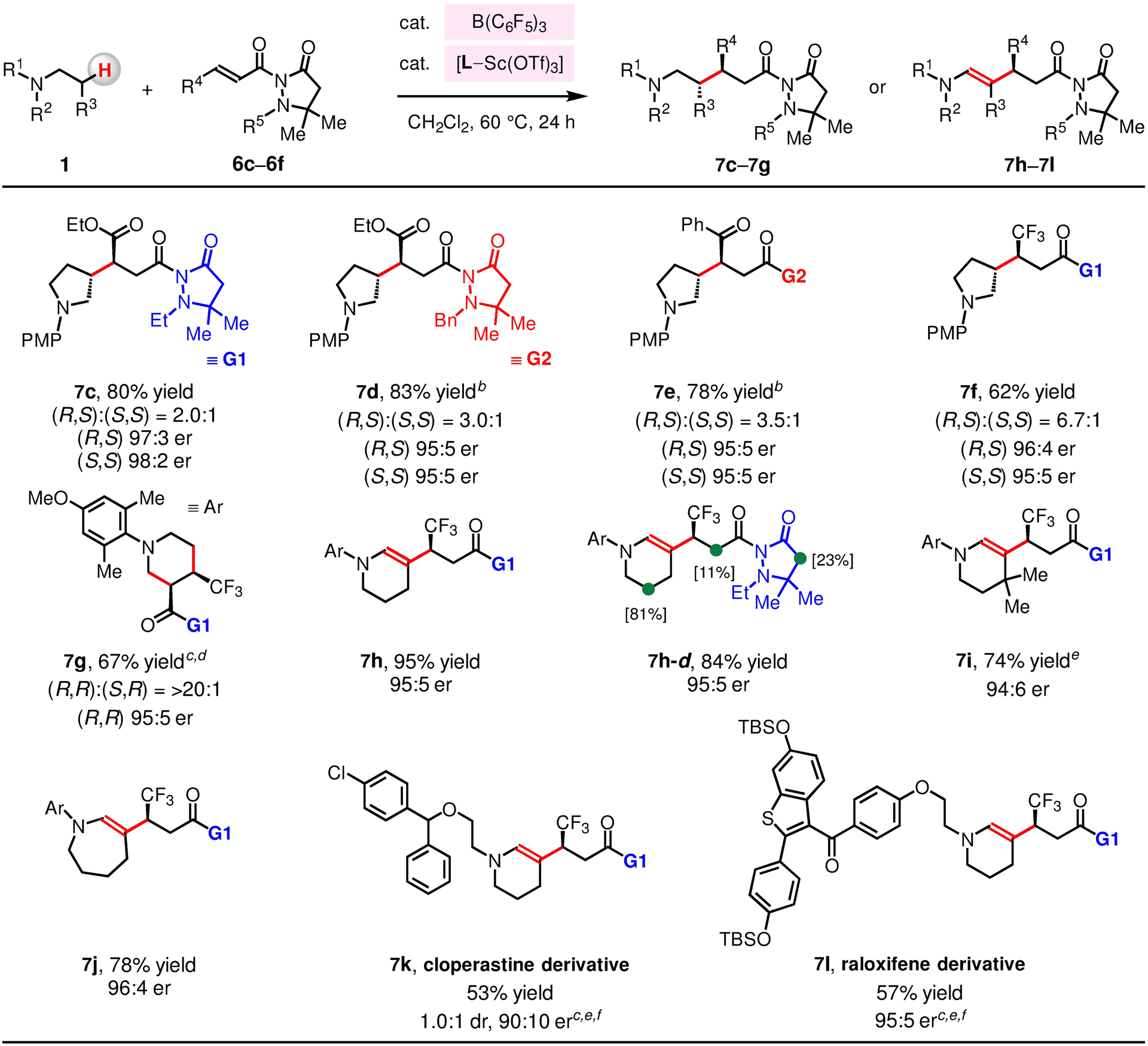
Cooperative functions of B(C6F5)3 and L–Sc(OTf)3 promote stereoselective β-alkylation of N-alkylamines. a Conditions: N-alkylamine (1, 0.10 mmol), α,β-unsaturated compound (6, 0.15–0.20 mmol), B(C6F5)3 (10 mol %), L10–Sc(OTf)3 (10 mol %), CH2Cl2 (1.0 mL), under N2, 60 °C, 1–36 h. b 10 mol % of L6–Sc(OTf)3 was used. c 20 mol % of B(C6F5)3 was used. d 20 mol % of L10–Sc(OTf)3 was used. e The reaction was performed at 80 °C. f 10 mol % of L8–Sc(OTf)3 was used. See the Supporting Information for details.
Figure 7. Additional Data Interpretation.
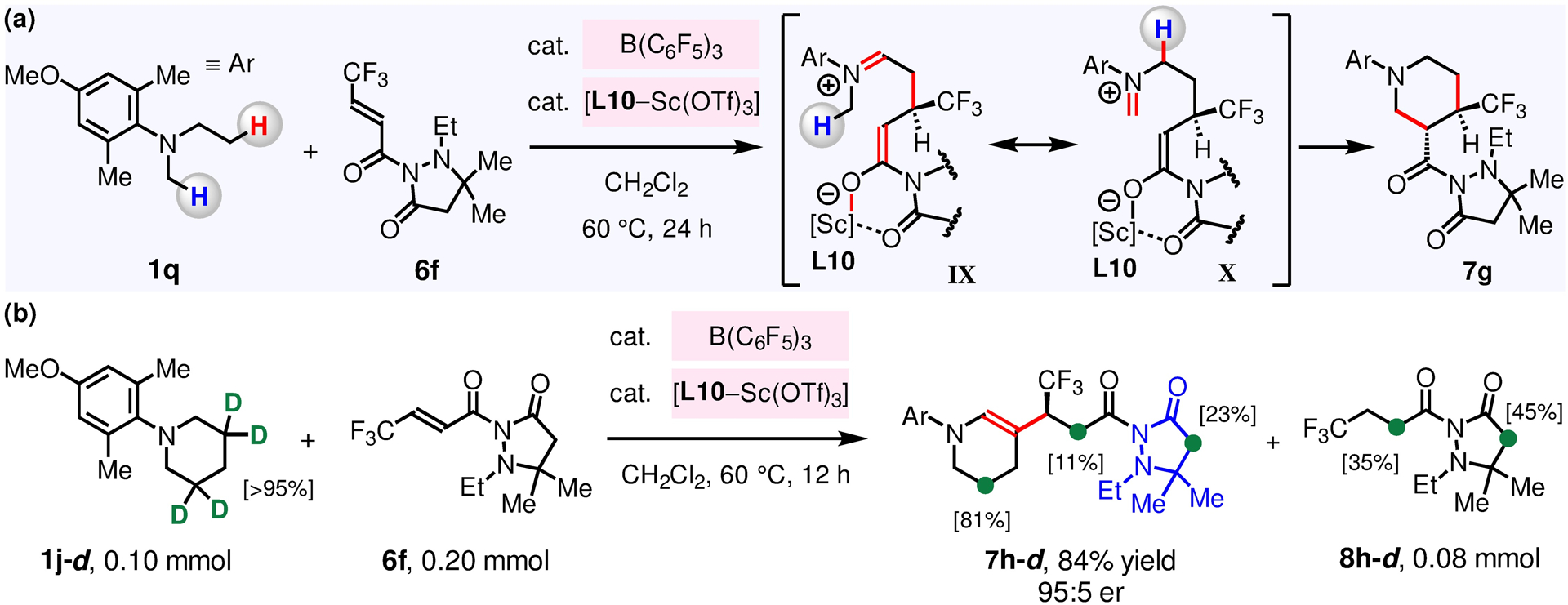
(a) Sequential β-alkylation and Mannich-type reaction gave piperidine derivative 7g. (b) D+ derived from 1j-d was incorporated into enolizable positions of 7h-d and 8h-d.
2.1.4. Scalability and Modifications of β-Functionalized Amines.
The catalytic method is scalable. For example, treatment of 4.0 mmol of N-arylpiperidine 1j and 6f with 10 mol % B(C6F5)3 and 10 mol % L10–Sc(OTf)3, (CH2Cl2, 12 h, 60 °C) afforded 7h in 95% yield (1.9 g; Figure 8a). Treatment of 7f-(R,S) with LiSEt followed by reduction of the resulting thioester with LiAlH4 furnished alcohol 9a-(R,S) in 95% overall yield (Figure 8b). Compound 9a-(R,S) was subsequently converted to its derived carbamate 9b-(R,S) which was subjected to the X-ray crystallographic analysis for determination of absolute configuration (see the Supporting Information for details). Enamines obtained by enantioselective β-alkylation were found to be versatile intermediates (Figure 8c). Hydrogenation of enamine 7h by a chiral Ir-based catalyst afforded 9c in 83% yield and 5.0:1 dr,116 and treatment of 7h with NFSI and TMSCN gave fluorocyanation product 9d in 88% yield and 4.0:1 dr.117
Figure 8. Modification of Products and Scalability.
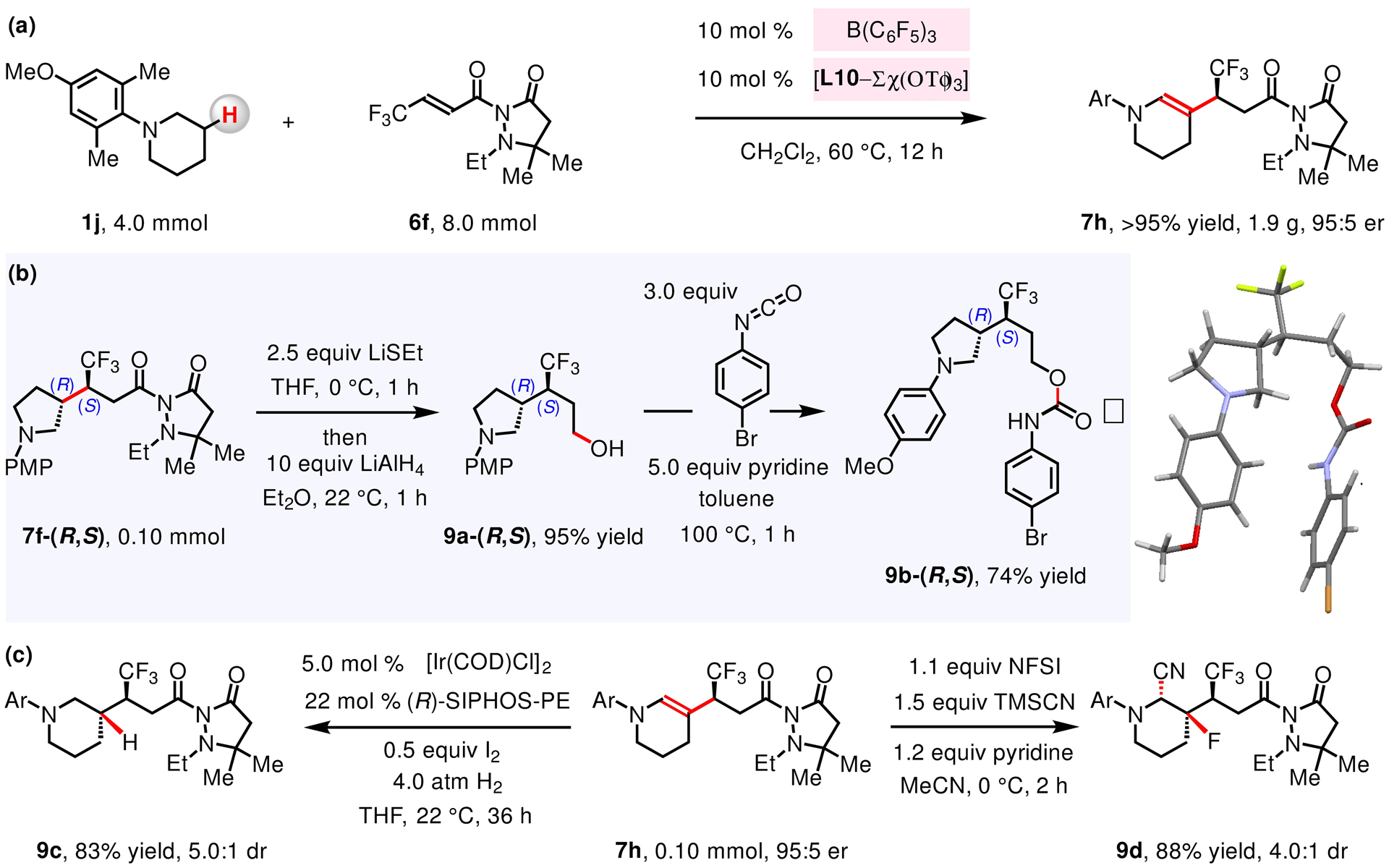
(a) The enantioselective reaction is amenable to gram-scale operations. (b) Transformation of pyrazolidinone auxiliary into thioester, alcohol and then to carbamate was achieved. Absolute configuration of 9b was obtained through X-ray crystallographic analysis. (c) Versatility of enamine 7h was demonstrated through catalytic hydrogenation and fluorocyanation reactions. See the Supporting Information for details.
2.2. Mechanistic Investigations
We designed and performed studies to gain insight regarding the mechanistic nuances of the catalytic process.118 These studies included determining reaction orders, kinetic isotope effects, and Hammett ρ values (Figures 9, 12, and 13, respectively). Additionally, these investigations led to revised pathways for catalytic β-C–H alkylation reaction (Figures 10–11).
Figure 9. Determination of Reaction Orders.
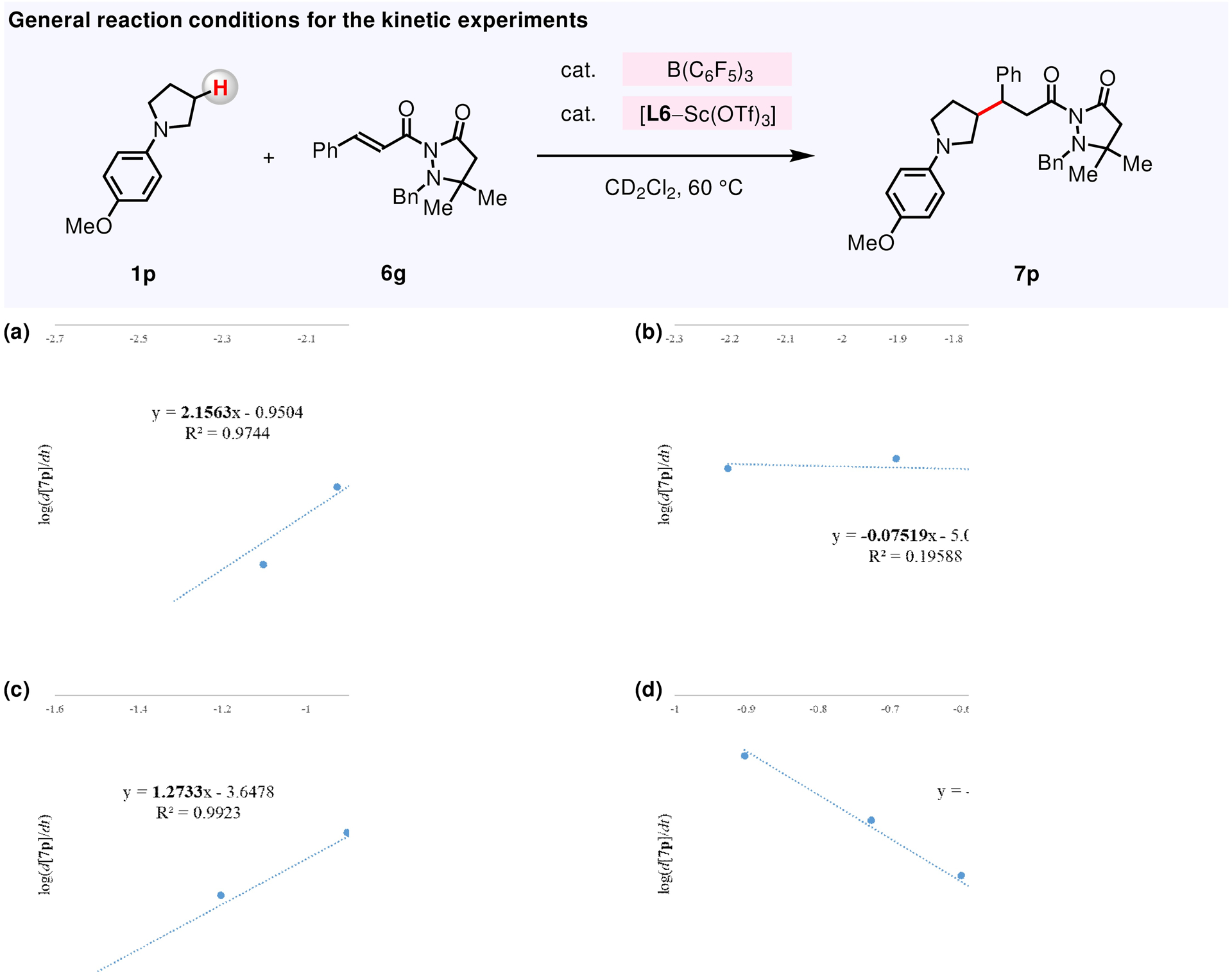
Log(rate) vs log(concentration) plot is employed to determine the reaction order for: (a) B(C6F5)3. (b) L6–Sc(OTf)3. (c) amine 1p. (d) α,β-unsaturated compound 6g. See the Supporting Information for details.
Figure 12. Kinetic Isotope Effect Studies.
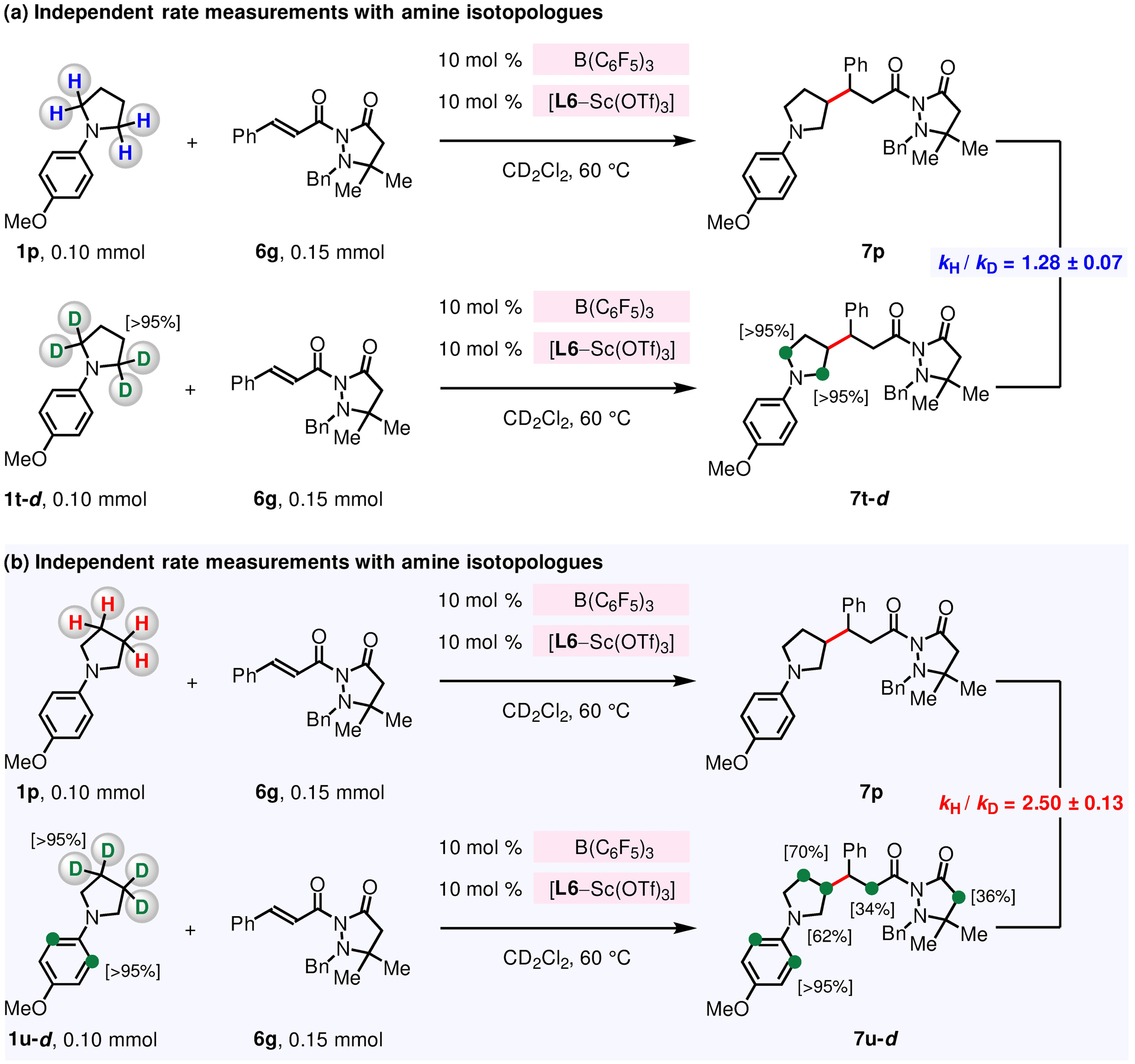
The independent rate measurement studies indicate that intramolecular proton transfer is the turnover-limiting step. See the Supporting Information for details.
Figure 13. Hammett Studies.
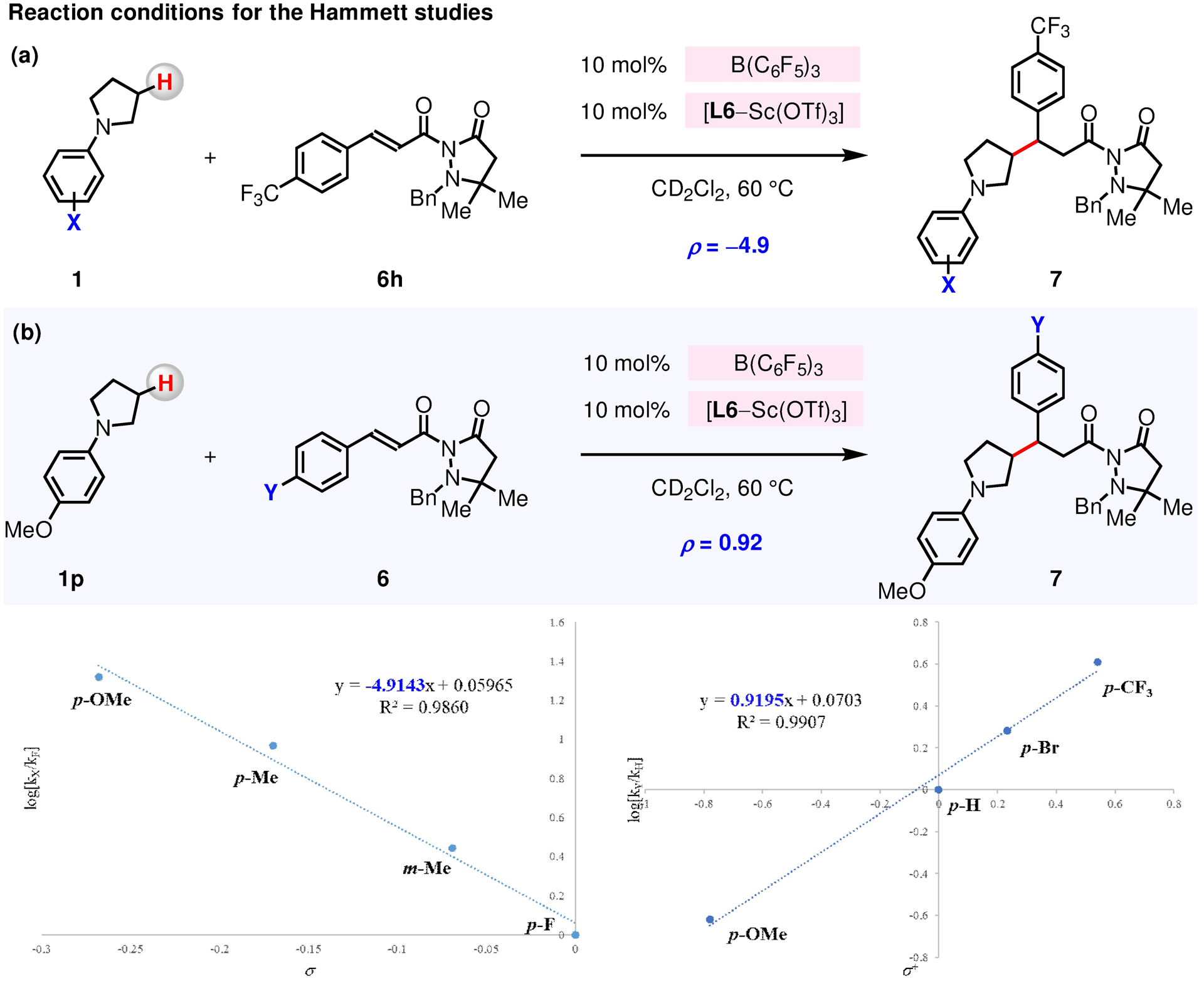
Hammett plots of rates for the reaction of N-aryl substituted pyrrolidines 1 and aryl substituted α,β-unsaturated compounds 6. (a), (c) N-Arylpyrrolidine derivatives 1 that contain electron-donating substituents reacted more rapidly. (b), (d) α,β-Unsaturated compounds 6 that contain electron-withdrawing substituents reacted more rapidly. See the Supporting Information for details.
Figure 10. A Catalytic Cycle Consistent with the Results of Mechanistic Investigations.
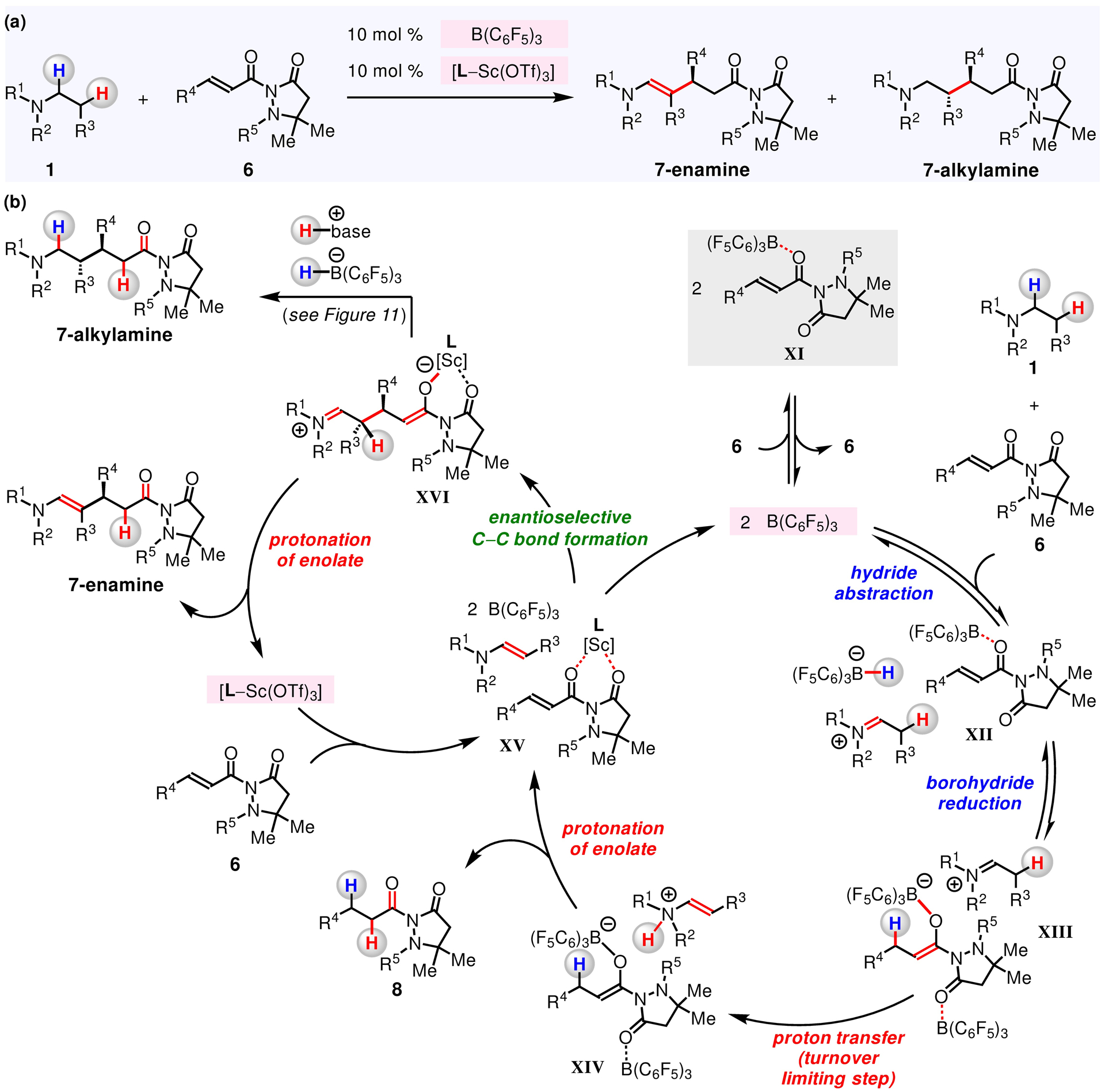
Kinetic and NMR studies indicate that the turnover-limiting step is intramolecular proton transfer step.
Figure 11. Origin of Enamine and N-Alkylamine Products.
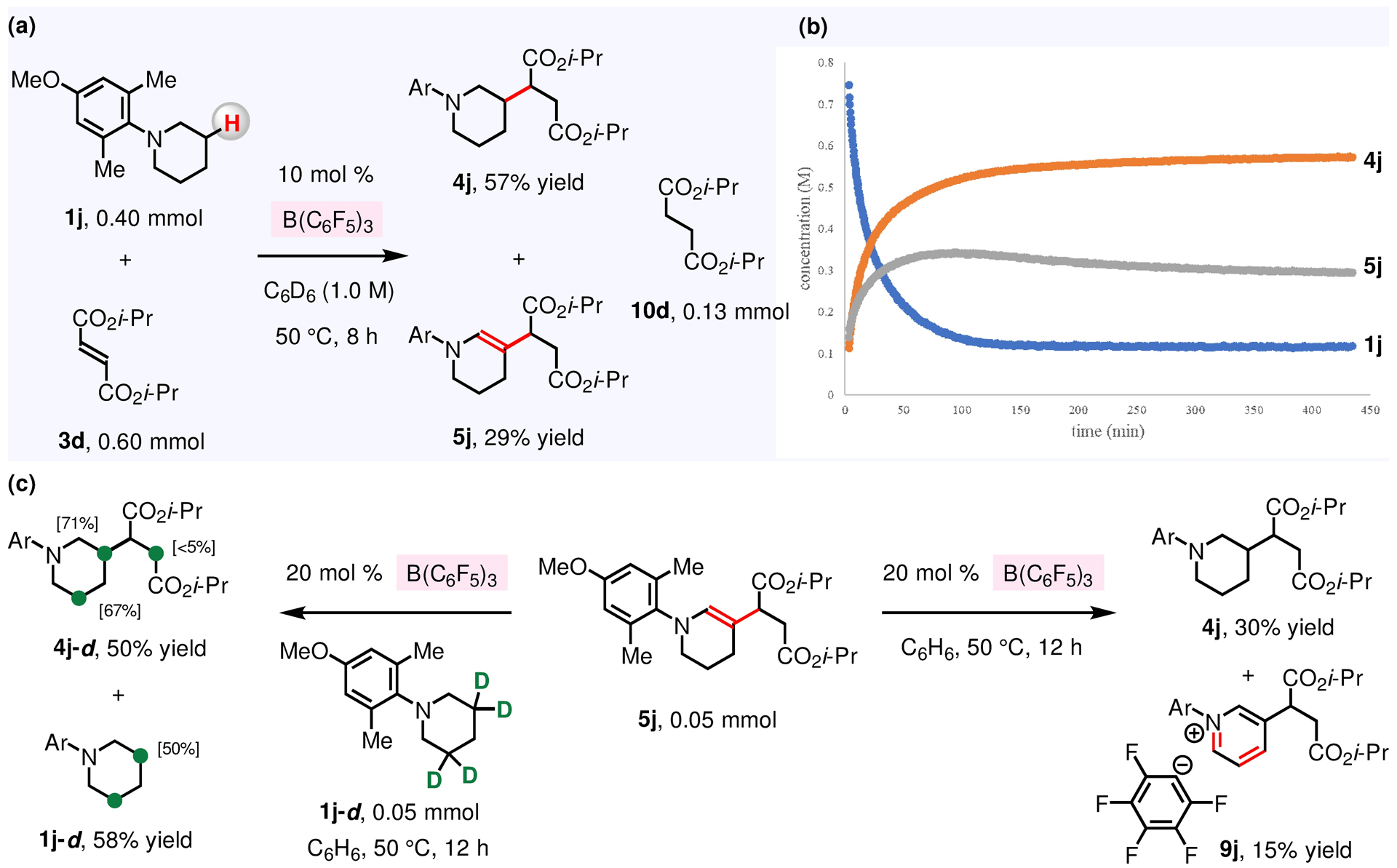
(a), (b) Reaction progress analysis of the coupling of 1j and 3d. (c) 5j and/or 1j-d can serve as H+ or D+ and hydride source in (F5C6)3B-catalyzed transfer hydrogenation of 5j. See the Supporting Information for details.
2.2.1. Determination of Reaction Orders and a Consistent Mechanistic Pathway.
We found that the reaction of N-arylpyrrolidine 1p with α,β-unsaturated compound 6g has a second-order dependence on B(C6F5)3 concentration (Figure 9a) but is not at all impacted by the concentration of L6–Sc(OTf)3 complex (Figure 9b). Furthermore, we found a first-order dependence on amine concentration of 1p (Figure 9c) but, a reverse first-order dependence on the concentration of electrophile 6g (Figure 9d). The independence of the reaction rate on the initial concentration of L6–Sc(OTf)3 suggests that enantioselective C–C bond forming event between in situ generated enamine and [L6–Sc(OTf)3]-activated 6g (Figure 10, XV → XVI) occurs after the turnover-limiting step. Furthermore, the negative first-order dependence on the concentration of 6g implies that the resting state consists of 6g and the Lewis acid. Spectroscopic analysis of the reaction mixture (19F NMR) supports the proposal in regard to formation of [6g–B(C6F5)3] (XI).119
Thus, it is likely that β-C–H alkylation proceeds by the release of B(C6F5)3 from XI, which then abstracts a hydride from 1 to form an iminium/borohydride complex (XII). Borohydride reduction of (F5C6)3B-activated 6 delivers a [(F5C6)3B–enolate]−[iminium]+ complex (XII → XIII).28, 120 Subsequent irreversible isomerization of the iminium into an enammonium (XIII → XIV) is likely the turnover-limiting step (see the following kinetic isotope effect and Hammett studies, as well as the Supporting Information for details).121–124 Protonolysis of [(F5C6)3B–enolate]− in XIV releases B(C6F5)3, 8, and an enamine (XV). Then, enantioselective C–C bond forming reaction between the enamine and [L–Sc(OTf)3]-activated electrophile 6 leads to a zwitterionic intermediate that bears an iminium and an enolate moiety (XV → XVI). Finally, proton transferwithin XVI produces 7-enamine and regenerates L–Sc(OTf)3, thus closing the cycle. Alternatively, borohydride reduction of the iminium and protonation of the resulting enolate in XVI produces an N-alkylamine product (7-alkylamine), as illustrated by the studies described below.
2.2.2. Origins of Enamine and N-Alkylamine Products.
β-Amino C–H alkylation products (Figures 2 and 6) were obtained either as an enamine, an N-alkylamine, or a mixture of the two (e.g., 4n and 4o, Figure 2). We wondered if the N-alkylamine products were formed by transfer hydrogenation of XVI (XVI → 7-alkylamine, Figure 10), or whether they were generated through reduction of enamines (7-enamine → 7-alkylamine). To identify each product’s origins, we studied the progress of (F5C6)3B-catalyzed reaction between N-arylpiperidine 1j and 3d (Figure 11a) which gives a mixture of N-alkylamine 4j (57% yield) and enamine 5j (29% yield) using 0.4 mL of C6D6 (vs the process involving 1.6 mL of C6H6 which selectively gives 4j; Figure 2). We found that there is minimal transformation of 5j into 4j as evidenced by the mostly unchanged concentration of 5j once the β-alkylation reaction completed (2 h; Figure 11b). These results are consistent with the scenario that 7-alkylamine (Figure 10a) is formed by transfer hydrogenation of XVI without the intermediacy of 7-enamine. Nonetheless, the source of proton and hydride remained to be identified.
To probe further if, and under precisely what conditions 5j can be converted to 4j, we investigated the transformation of 5j (isolated and purified by flash silica gel chromatography) in the presence of 20 mol% B(C6F5)3 (C6H6, 12 h; Figure 11c). This led us to observe that there was 30% conversion to 4j; in addition, [pyridinium]+[C6F5]− (9j, 15 %) was also produced. Treatment of 5j with N-arylpiperidine-3,3,5,5-d4 1j-d furnished 4j-d in 50% yield, and not only was there significant deuterium incorporation at C3 and C5 positions of 4j-d (71% and 67%, respectively), recovered 1j-d had also undergone D/H exchange. These data suggest that B(C6F5)3 can catalyze transfer hydrogenation of 5j in the presence of another molecule of 5j and/or 1j-d serving as sources of H+ (or D+) and hydride (Figure 11c). Nevertheless, under the standard conditions for (F5C6)3B-catalyzed β-C–H alkylation reaction (Figures 10 and 11a), in situ generated [(F5C6)3B–H]−[Base–H]+ (derived from the reaction of B(C6F5)3 and N-alkylamine 1) appears to react with either a highly reactive zwitterionic intermediate (XVI → 7-alkylamine) or (F5C6)3B-activated α,β-unsaturated compounds (XII → XIII → XIV → 8). As a consequence, hydrogenation of the relatively unreactive enamine 5j to give 4j may be outcompeted by these more facile processes.
2.2.3. Kinetic Isotope Effect Studies.
To shed light on the hydride abstraction (Figure 10, 1 → XII), and deprotonation steps (XIII → XIV → XV), deuterium-labeled N-arylpyrrolidines 1t-d and 1u-d were prepared, and their reactions with 6g were probed (Figure 12). Based on the aforementioned rate studies (Figure 9), hinting that the turnover-limiting is prior to the stereoselective C–C bond forming process (Figure 10, XV → XVI), the overall rate of the reaction can be affected for reactions involving both α-deuterated 1t-d and β-deuterated 1u-d.125–126 However, independent rate measurements involving 1p and 1t-d (Figure 12a) were found to have kH/kD = 1.28 ± 0.07.127 On the other hand, comparison of the reaction rate between 1p and 1u-d (Figure 12b) revealed that 1p reacts 2.5 times faster than 1u-d (kH/kD = 2.50 ± 0.13).127 These KIE experiments support the notion that the turnover-limiting step is the conversion of iminium into enammonium (Figure 10, XIII → XIV) which entails the cleavage of α-imino C–H or C–D bonds; furthermore, (F5C6)3B-catalyzed hydride abstraction (1 → XII) and the following borohydride reduction (XII → XIII) steps are reversible, thus leading to the equilibrium KIE of 1.28.
2.2.4. Hammett Studies.
Hammett studies revealed a strong dependence of the reaction rate on the electronic properties of the N-alkylamines, with N-arylpyrrolidine derivatives (1) bearing electron-donating substituents reacting more rapidly (ρ = −4.9, Figures 13a and 13c). The large negative ρ value obtained supports the proposed mechanism (Figure 10) in which B(C6F5)3 abstracts a hydride from N-arylpyrrolidine 1 into a N-aryl iminium cation (1 → XII), and its isomerization into an enammonium species (XII → XIII → XIV); these processes take place at or prior to the turnover-limiting step. While the reaction rate was found to be less dependent of the electronic properties of α,β-unsaturated compounds 6, those involving more electron-withdrawing groups reacted with higher efficiency (ρ = 0.92, Figures 13b and 13d). This latter outcome is congruent with the hypothesis that 6 reacts with in situ generated [(F5C6)3B–H]− to afford a boron–enolate intermediate (Figure 10b; XII → XIII), and that this hydride transfer also occurs at or prior to the turnover-limiting step.
3. CONCLUSIONS
In brief, we have developed an efficient catalytic method for functionalization of β-amino C–H bonds to generate enantioenriched δ-amino carbonyl compounds. We find that by using a blend of B(C6F5)3 and a chiral Sc-based complex, it is possible to convert an N-alkylamine into an enamine and then promote its enantio- and diastereo-selective reaction with an α,β-unsaturated compound. The catalyst system is tolerant of a wide variety of Lewis acid-sensitive functional units and therefore applicable to late-stage modification of relatively complex (and bioactive) trialkylamine molecules. Mechanistic investigations reveal that the turnover-limiting step is probably isomerization of N-alkylamine-derived iminium ion into an enammonium intermediate.
The principles outlined above demonstrate that proper combination of an achiral organoborane and an enantiomerically pure organometallic catalyst may be used for chemo- and enantioselective C–H bond functionalization, providing a rational basis for future development of processes for late-stage stereoselective β-functionalization of multifunctional bioactive amines. Studies aimed at achieving these objectives are underway.
Supplementary Material
ACKNOWLEDGEMENTS
Financial support was provided by the NIH (GM-128695), Sloan Foundation and Boston College. Y. C. was supported by Boston College Chemistry Department Graduate Fellowship. We thank Professor Amir H. Hoveyda for helpful discussions and Professor Shih-Yuan Liu for assistance with mechanistic investigations. Dr. Juan del Pozo del Valle and Dr. Filippo Romitti are thanked for their assistance with this project. We are grateful to Dr. Bo Li for the X-ray crystallographic analysis.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- (1).Vardanyan R in Piperidine-Based Drug Discovery; Vardanyan R, Ed.; Elsevier: 2017, p 147. [Google Scholar]
- (2).McGrath NA; Brichacek M; Njardarson JT A graphical journey of innovative organic architectures that have improved our lives. J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]
- (3).Campos KR; Coleman PJ; Alvarez JC; Dreher SD; Garbaccio RM; Terrett NK; Tillyer RD; Truppo MD; Parmee ER The importance of synthetic chemistry in the pharmaceutical industry. Science 2019, 363, eaat0805. [DOI] [PubMed] [Google Scholar]
- (4).Chiral Amine Synthesis: Methods, Developments and Applications; Nugent TC, Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2010, p 15–457. [Google Scholar]
- (5).Weiner B; Szymański W; Janssen DB; Minnaard AJ; Feringa BL Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev 2010, 39, 1656–1691. [DOI] [PubMed] [Google Scholar]
- (6).Dick AR; Sanford MS Transition metal catalyzed oxidative functionalization of carbon-hydrogen bonds. Tetrahedron 2006, 62, 2439–2463. [Google Scholar]
- (7).Davies HM; Manning JR Catalytic C–H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Baudoin O Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds. Chem. Soc. Rev 2011. 40, 4902–4911. [DOI] [PubMed] [Google Scholar]
- (9).Haibach MC; Seidel D C–H Bond functionalization through intramolecular hydride transfer. Angew. Chem., Int. Ed 2014, 53, 5010–5036. [DOI] [PubMed] [Google Scholar]
- (10).Girard SA; Knauber T; Li C-J The cross-dehydrogenative coupling of Csp3–H bonds: A versatile strategy for C–C bond formations. Angew. Chem., Int. Ed 2014, 53, 74–100. [DOI] [PubMed] [Google Scholar]
- (11).Daugulis O; Roane J; Tran LD Bidentate, monoanionic auxiliary-directed functionalization of carbon-hydrogen bonds. Acc. Chem. Res 2015, 48, 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).He J; Wasa M; Chan KSL; Shao Q; Yu J-Q Palladium-catalyzed transformations of alkyl C–H bonds. Chem. Rev 2017, 117, 8754–8786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chu JCK; Rovis T Complementary strategies for directed C(sp3)–H functionalization: A comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbine/nitrene transfer. Angew. Chem., Int. Ed 2018, 57, 62–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Davies HM; Liao K Dirhodium tetracarboxylates as catalysts for selective intermolecular C–H functionalization. Nat. Rev. Chem 2019, 3, 347–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Campos KR Direct sp3 C–H bond activation adjacent to nitrogen in heterocycles. Chem. Soc. Rev 2007, 36, 1069–1084. [DOI] [PubMed] [Google Scholar]
- (16).Mitchell EA; Peschiulli A; Lefevre N; Meerpoel L; Maes BUW Direct α-functionalization of saturated cyclic amines. Chem. Eur. J 2012, 18, 10092–10142. [DOI] [PubMed] [Google Scholar]
- (17).He C; Whitehurst WG; Gaunt MJ Palladium-catalyzed C(sp3)–H bond functionalization of aliphatic amines. Chem 2019, 5, 1031–1058. [Google Scholar]
- (18).Trowbridge A; Walton SM; Gaunt MJ New strategies for the transition-metal catalyzed synthesis of aliphatic amines. Chem. Rev 2020, 120, 2613–2692. [DOI] [PubMed] [Google Scholar]
- (19).Noble A; MacMillan DW Photoredox α-vinylation of α-amino acids and N-aryl amines. J. Am. Chem. Soc 2014, 136, 11602–11605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Osberger TJ; Rogness DC; Kohrt JT; Stepan AF; White MC Oxidative diversification of amino acids and peptides by small-molecule iron catalysis. Nature 2016, 537, 214–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Le C; Liang Y; Evans RW; Li X; MacMillan DWC Selective sp3 C–H alkylation via polarity-match-based cross-coupling. Nature 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).McManus JB; Onuska NPR; Nicewicz DA Generation and alkylation of α-carbamyl radicals via organic photoredox catalysis. J. Am. Chem. Soc 2018, 140, 9056–9060. [DOI] [PubMed] [Google Scholar]
- (23).Ye J; Kalvet I; Schoenebeck F; Rovis T Direct α-alkylation of primary aliphatic amines enabled by CO2 and electrostatics. Nat. Chem 2018, 10, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li Z; Li C-J Catalytic enantioselective alkynylation of prochiral sp3 C–H bonds adjacent to a nitrogen atom. Org. Lett 2004, 6, 4997–4999. [DOI] [PubMed] [Google Scholar]
- (25).Murarka S; Deb I; Zhang C; Seidel D Catalytic enantioselective intramolecular redox reactions: Ring-fused tetrahydroquinolines. J. Am. Chem. Soc 2009, 131, 13226–13227. [DOI] [PubMed] [Google Scholar]
- (26).Mori K; Ehara K; Kurihara K; Akiyama T Selective activation of enantiotopic C(sp3)–hydrogen by means of chiral phosphoric acid: Asymmetric synthesis of tetrahydroquinoline derivatives. J. Am. Chem. Soc 2011, 133, 6166–6169. [DOI] [PubMed] [Google Scholar]
- (27).DiRocco DA; Rovis T Catalytic asymmetric α-acylation of tertiary amines mediated by a dual catalysis mode: N-heterocyclic carbene and photoredox catalysis. J. Am. Chem. Soc 2012, 134, 8094–8097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Shang M; Chan JZ; Cao M; Chang Y; Wang Q; Cook B; Torker S; Wasa M C–H Functionalization of amines via alkene-derived nucleophiles through cooperative action of chiral and achiral Lewis acid catalysts: Applications in enantioselective synthesis. J. Am. Chem. Soc 2018, 140, 10593–10601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chan JZ; Yesilcimen A; Cao M; Zhang Y; Zhang B; Wasa M Direct conversion of N-alkylamines to N-propargylamines through C–H activation promoted by Lewis acid/organocopper catalysis: Application to late-stage functionalization of bioactive molecules. J. Am. Chem. Soc 2020, 142, 16493–16505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen W; Ma L; Paul A; Seidel D Direct α-C–H bond functionalization of unprotected cyclic amines. Nat. Chem 2018, 10, 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Paul A; Seidel D α-Functionalization of cyclic secondary amines: Lewis acid promoted addition of organometallics to transient imines. J. Am. Chem. Soc 2019, 141, 8778–8782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Davies HM; Venkataramani C; Hansen T; Hopper DW New strategic reactions for organic synthesis: catalytic asymmetric C–H activation α to nitrogen as a surrogate for the Mannich reaction. J. Am. Chem. Soc 2003, 125, 6462–6468. [DOI] [PubMed] [Google Scholar]
- (33).Liu W; Babl T; Röther A; Reiser O; Davies HM Functionalization of piperidine derivatives for the site‐selective and stereoselective synthesis of positional analogues of methylphenidate. Chem. Eur. J 2020, 26, 4236–4241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Campos KR; Klapars A; Waldman JH; Dormer PG; Chen CY Enantioselective, palladium-catalyzed α-arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc 2006, 128, 3538–3539. [DOI] [PubMed] [Google Scholar]
- (35).Cordier CJ; Lundgren RJ; Fu GC Enantioconvergent cross-couplings of racemic alkylmetal reagents with unactivated secondary alkylelectrophiles: Catalytic asymmetric Negishi α-alkylations of N-Boc-pyrrolidine. J. Am. Chem. Soc 2013, 135, 10946–10949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Jain P; Verma P; Xia G; Yu JQ Enantioselective amine α-functionalization via palladium-catalysed C–H arylation of thioamides. Nat. Chem 2017, 9, 140–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Topczewski JJ; Cabrera PJ; Saper NI; Sanford MS Palladium-catalysed transannular C–H functionalization of alicyclic amines. Nature 2016, 531, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Xu Y; Young MC; Wang C; Magness DM; Dong G Catalytic C (sp3)–H arylation of free primary amines with an exo directing group generated in situ. Angew. Chem., Int. Ed 2016, 55, 9084–9087. [DOI] [PubMed] [Google Scholar]
- (39).Chan KS; Fu HY; Yu JQ; Palladium(II)-catalyzed highly enantioselective C–H arylation of cyclopropylmethylamines. J. Am. Chem. Soc 2015, 137, 2042–2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Rodrigalvarez J; Nappi M; Azuma H; Floden NJ; Burns ME; Gaunt MJ Catalytic C(sp3)–H bond activation in tertiary alkylamines. Nat. Chem 2020, 12, 76–81. [DOI] [PubMed] [Google Scholar]
- (41).Zhuang Z; Yu JQ Pd (II)-catalyzed enantioselective γ-C(sp3)–H functionalizations of free cyclopropylmethylamines. J. Am. Chem. Soc 2020, 142, 12015–12019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).McNally A; Haffemayer B; Collins BSL; Gaunt MJ Palladium-catalysed C–H activation of aliphatic amines to give strained nitrogen heterocycles. Nature 2014, 510, 129–133. [DOI] [PubMed] [Google Scholar]
- (43).Smalley AP; Gaunt MJ Mechanistic insights into the palladium-catalyzed aziridination of aliphatic amines by C–H activation. J. Am. Chem. Soc 2015, 137, 10632–10641. [DOI] [PubMed] [Google Scholar]
- (44).Willcox D; Chappell BGN; Hogg KF; Calleja J; Smalley AP; Gaunt MJ A general catalytic β-C–H carbonylation of aliphatic amines to β-lactams. Science 2016, 354, 851–857. [DOI] [PubMed] [Google Scholar]
- (45).Huang Z; Wang C; Dong G A hydrazone‐based exo‐directing‐group strategy for β C–H oxidation of aliphatic amines. Angew. Chem., Int. Ed 2016, 55, 5299–5303. [DOI] [PubMed] [Google Scholar]
- (46).Cabrera-Pardo JR; Trowbridge A; Nappi M; Ozaki K; Gaunt MJ Selective palladium(II)-catalyzed carbonylation of methylene β-C–H bonds in aliphatic amines. Angew. Chem., Int. Ed 2017, 56, 11958–11962. [DOI] [PubMed] [Google Scholar]
- (47).Hogg KF; Trowbridge A; Alvarez-Pérez A; Gaunt MJ The α-tertiary amine motif drives remarkable selectivity for Pd-catalyzed carbonylation of β-methylene C–H bonds. Chem. Sci 2017, 8, 8198–8203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Nappi M; He C; Whitehurst WG; Chappell BGN; Gaunt MJ Selective reductive elimination at alkyl palladium(IV) by dissociative ligand ionization: Catalytic C(sp3)–H amination to azetidines. Angew. Chem., Int. Ed 2018, 57, 3178–3182. [DOI] [PubMed] [Google Scholar]
- (49).Millet A; Dailler D; Larini P; Baudoin O Ligand-controlled α- and β-arylation of acyclic N-Boc amines. Angew. Chem., Int. Ed 2014, 53, 2678–2682. [DOI] [PubMed] [Google Scholar]
- (50).He Y; Wang F; Zhang X; Fan X C(sp3)–H Dehydrogenation and C(sp2)–H alkoxy carbonylation of inactivated cyclic amines towards functionalized N-heterocycles. Chem. Commun 2017, 53, 4002–4005. [DOI] [PubMed] [Google Scholar]
- (51).He J; Li S; Deng Y; Fu H; Laforteza BN; Spangler JE; Homs A; Yu J-Q Ligand-controlled C(sp3)–H arylation and olefination in synthesis of unnatural chiral α-amino acids. Science 2014, 343, 1216–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Smalley AP; Cuthbertson JD; Gaunt M,J Palladium-catalyzed enantioselective C–H activation of aliphatic amines using chiral anionic BINOL-phosphoric acid ligands. J. Am. Chem. Soc 2017, 139, 1412–1415. [DOI] [PubMed] [Google Scholar]
- (53).Su B; Lee T; Hartwig JF Iridium-catalyzed, β-selective C(sp3)–H silylation of aliphatic amines to form silapyrrolidines and 1,2-amino alcohols. J. Am. Chem. Soc 2018, 140, 18032–18038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Lin W; Zhang K-F; Baudoin O Regiodivergent enantioselective C–H functionalization of Boc-1,3-oxazinanes for the synthesis of β 2- and β 3-amino acids. Nat. Catal 2019, 2, 882–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yamamoto H; Futatsugi K “Designer Acids”: Combined acid catalysis for asymmetric synthesis. Angew. Chem., Int. Ed 2005, 44, 1924–1942. [DOI] [PubMed] [Google Scholar]
- (56).Paull DH; Abraham CJ; Scerba MT; Alden-Danforth E; Lectka T Bifunctional asymmetric catalysis: Cooperative Lewis acid/base systems. Acc. Chem. Res 2008, 41, 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Kobayashi S; Mori Y; Fossey JS; Salter MM Catalytic enantioselective formation of C–C bonds by addition to imines and hydrazones: A ten-year update. Chem. Rev 2011, 111, 2626–2704. [DOI] [PubMed] [Google Scholar]
- (58).Trost BM; Bartlett MJ ProPhenol-catalyzed asymmetric additions by spontaneously assembled dinuclear main group metal complexes. Acc. Chem. Res 2015, 48, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Shibasaki M; Kumagai N in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Peters R, Eds.; Wiley-VCH: New York, 2015; p 1–24. [Google Scholar]
- (60).Lu X; Deng L in Cooperative Catalysis: Designing Efficient Catalysts for Synthesis, Peters R, Eds.; Wiley-VCH: New York, 2015; p 145–167. [Google Scholar]
- (61).Wang MH; Scheidt KA Cooperative catalysis and activation with N-heterocyclic carbenes. Angew. Chem., Int. Ed 2016, 55, 14912–14922. [DOI] [PubMed] [Google Scholar]
- (62).Romiti F; del Pozo J; Paioti PHS; Gonsales SA; Li X; Hartrampf FWW; Hoveyda AH Different strategies for designing dual-catalytic enantioselective processes: From fully cooperative to non-cooperative systems. J. Am. Chem. Soc 2019, 141, 17952–17961. [DOI] [PubMed] [Google Scholar]
- (63).Millot N; Santini CC; Fenet B; Basset JM Formation and characterization of zwitterionic stereoisomers from the reaction of B(C6F5)3 and NEt2Ph: (E)- and (Z)-[EtPhN+=CHCH2−B−(C6F5)3]. Eur. J. Inorg. Chem 2002, 2002, 3328–3335. [Google Scholar]
- (64).Zhang J; Park S; Chang S Catalytic access to bridged sila-N-heterocycles from piperidines via cascade sp3 and sp2 C–Si bond formation. J. Am. Chem. Soc 2018, 140, 13209–13213. [DOI] [PubMed] [Google Scholar]
- (65).Chang Y; Yesilcimen A; Cao M; Zhang Y; Zhang B; Chan JZ; Wasa M Catalytic deuterium incorporation within metabolically stable β-amino C–H bonds of drug molecules. J. Am. Chem. Soc 2019, 141, 14570–14575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Focante F; Mercandelli P; Sironi A; Resconi L Complexes of tris(pentafluorophenyl)boron with nitrogen-containing compounds: Synthesis, reactivity and metallocene activation. Coord. Chem. Rev 2006, 250, 170–188. [Google Scholar]
- (67).Dureen MA; Brown CC; Stephan DW Addition of enamines or pyrroles and B(C6F5)3 “Frustrated Lewis Pairs” to alkynes. Organometallics 2010, 29, 6422–6432. [Google Scholar]
- (68).Schwendemann S; Fröhlich R; Kehr G; Erker G Intramolecular frustrated N/B Lewis pairs by enamine hydroboration. Chem. Sci 2011, 2, 1842–1849. [Google Scholar]
- (69).Maier AFG; Tussing S; Schneider T; Flörke U; Qu Z-W; Grimme S; Paradies J Frustrated Lewis pair catalyzed dehydrogenative oxidation of indolines and other heterocycles. Angew. Chem., Int. Ed 2016, 55, 12219–12223. [DOI] [PubMed] [Google Scholar]
- (70).Kojima M; Kanai M Tris(pentafluorophenyl)borane-catalyzed acceptorless dehydrogenation of N-heterocycles. Angew. Chem., Int. Ed 2016, 55, 12224–12227. [DOI] [PubMed] [Google Scholar]
- (71).Maier AFG; Tussing S; Zhu H; Wicker G; Tzvetkova P; Flörke U; Daniliuc CG; Grimme S; Paradies J Borane-catalyzed synthesis of quinolines bearing tetrasubstituted stereocenters by hydride abstraction-induced electrocyclization. Chem. Eur. J 2018, 24, 16287–16291. [DOI] [PubMed] [Google Scholar]
- (72).Tian J-J; Zeng N-N; Liu N; Tu X-S; Wang X-C Intramolecular cyclizations of vinyl-substituted N,N-dialkyl arylamines enabled by borane-assisted hydride transfer. ACS Catal, 9, 2019. 295–300. [Google Scholar]
- (73).Li R; Chen Y; Jiang K; Wang F; Lu C; Nie J; Chen Z; Yang G; Chen Y-C; Zhao Y; Ma C B(C6F5)3-Catalyzed redox-neutral β-alkylation of tertiary amines using p-quinone methides via borrowing hydrogen. Chem. Commun 2019, 55, 1217–1220. [DOI] [PubMed] [Google Scholar]
- (74).Chan JZ; Chang Y; Wasa M B(C6F5)3-Catalyzed C–H alkylation of N-alkylamines using silicon enolates without external oxidant. Org. Lett 2019, 21, 984–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Zhang J; Chang S cine-Silylative ring-opening of α-methyl azacycles enabled by the silylium-induced C–N bond cleavage. J. Am. Chem. Soc 2020, 142, 12585–12590. [DOI] [PubMed] [Google Scholar]
- (76).Chen Y; Wan HL; Huang Y; Liu S; Wang F; Lu C; Nie J; Chen Z; Yang G; Ma CB (C6F5)3-Catalyzed β-functionalization of pyrrolidines using isatins via borrowing hydrogen: Divergent access to substituted pyrrolidines and pyrroles. Org. Lett 2020, 22, 7797–7803. [DOI] [PubMed] [Google Scholar]
- (77).Yu P; Zheng S-C; Yang N-Y; Tan B; Liu X-Y Phosphine-catalyzed remote β-C–H functionalization of amines triggered by trifluoromethylation of alkenes: One-pot synthesis of bistrifluoromethylated enamides and oxazoles. Angew. Chem., Int. Ed 2015, 54, 4041–4045. [DOI] [PubMed] [Google Scholar]
- (78).Ma L; Paul A; Breugst M; Seidel D Redox-neutral aromatization of cyclic amines: Mechanistic insights and harnessing of reactive intermediates for amine α- and β-C–H functionalization. Chem. Eur. J 2016, 22, 18179–18189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Xia X-F; Shu X-Z; Ji K-G; Yang Y-F; Shaukat A; Liu X-Y; Liang Y-M Platinum-catalyzed Michael addition and cyclization of tertiary amines with nitroolefins by dehydrogenation of α,β-sp3 C–H bonds. J. Org. Chem 2010, 75, 2893–2902. [DOI] [PubMed] [Google Scholar]
- (80).Sundararaju B; Tang Z; Achard M; Sharma GVM; Toupet L; Bruneau C Ruthenium-catalyzed cascade N- and C(3)-dialkylation of cyclic amines with alcohols involving hydrogen autotransfer processes. Adv. Synth. Catal 2010, 352, 3141–3146. [Google Scholar]
- (81).Sundararaju B; Achard M; Sharma GVM; Bruneau C sp3 C–H Bond activation with ruthenium(II) catalysts and C(3)-alkylation of cyclic amines. J. Am. Chem. Soc 2011, 133, 10340–10343. [DOI] [PubMed] [Google Scholar]
- (82).Yuan K; Jiang F; Sahli Z; Achard M; Roisnel T; Bruneau C Iridium-catalyzed oxidant-free dehydrogenative C–H bond functionalization: Selective preparation of N-arylpiperidines through tandem hydrogen transfers. Angew. Chem., Int. Ed 2012, 51, 8876–8880. [DOI] [PubMed] [Google Scholar]
- (83).Takasu N; Oisaki K; Kanai M Iron-catalyzed oxidative C(3)–H functionalization of amines. Org. Lett 2013, 15, 1918–1921. [DOI] [PubMed] [Google Scholar]
- (84).Zhou M-J; Zhu S-F; Zhou Q-L Copper-catalyzed Mannich-type oxidative β-functionalization of tertiary amines. Chem. Commun 2017, 53, 8770–8773. [DOI] [PubMed] [Google Scholar]
- (85).Shi X; Chen X; Wang M; Zhang X; Fan X Regioselective synthesis of acylated N-heterocycles via the cascade reactions of saturated cyclic amines with 2-oxo-2-arylacetic acids. J. Org. Chem 2018, 83, 6524–6533. [DOI] [PubMed] [Google Scholar]
- (86).Ishihara K; Hananki N; Yamamoto H Tris(pentafluorophenyl) boron as a new efficient, air stable, and water tolerant catalyst in the aldol-type and Michael reactions. Synlett 1993, 1993, 577–579. [Google Scholar]
- (87).Ishihara K; Funahashi M; Hanaki N; Miyata M; Yamamoto H Tris(pentafluorophenyl) boron as an efficient catalyst in the aldol-type reaction of ketene silyl acetals with imines. Synlett 1994, 1994, 963–964. [Google Scholar]
- (88).Ishihara K; Hanaki N; Yamamoto H Tris(pentafluorophenyl)boron as an efficient catalyst in the stereoselective rearrangement of epoxides. Synlett 1995, 1995, 721–722. [Google Scholar]
- (89).Wagner J; Lerner RA; Barbas CF; Efficient aldolase catalytic antibodies that use the enamine mechanism of natural enzymes. Science 1995, 270, 1797–1800. [DOI] [PubMed] [Google Scholar]
- (90).Bock DA; Lehmann CW; List B Crystal structures of proline-derived enamines. PNAS 2010, 107, 20636–20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Nielsen M; Worgull D; Zweifel T; Gschwend B; Bertelsen S; Jørgensen KA Mechanisms in aminocatalysis. Chem. Commun 2011, 47, 632–649. [DOI] [PubMed] [Google Scholar]
- (92).Mukherjee S; Yang JW; Hoffmann S; List B Asymmetric enamine catalysis. Chem. Rev 2007, 107, 5471–5569. [DOI] [PubMed] [Google Scholar]
- (93).Phipps RJ; Hamilton GL; Toste FD The progression of chiral anions from concepts to application in asymmetric catalysis. Nat. Chem 2012, 4, 603–614. [DOI] [PubMed] [Google Scholar]
- (94).Brak K; Jacobsen EN Asymmetric ion-paring catalysis. Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Frustrated Lewis Pairs I: Uncovering and Understanding; Stephan DW; Erker G Eds.; Springer: Berlin, 2013; Vol. 332, p 1–289. [Google Scholar]
- (96).Frustrated Lewis Pairs II: Expanding the Scope; Erker G; Stephan DW Eds.; Springer: Berlin, 2013; Vol. 334, p 1–311. [Google Scholar]
- (97).Ashley AE; O’Hare D FLP-mediated activations and reductions of CO2 and CO. Top. Curr. Chem 2013, 334, 191–218. [DOI] [PubMed] [Google Scholar]
- (98).Feng X; Du H Metal-free asymmetric hydrogenation and hydrosilylation catalyzed by frustrated Lewis pairs. Tetrahedron Lett 2014, 55, 6959–6964. [Google Scholar]
- (99).Stephan DW; Erker G Frustrated Lewis pair chemistry: Development and perspectives. Angew. Chem., Int. Ed 2015, 54, 6400–6441. [DOI] [PubMed] [Google Scholar]
- (100).Stephan DW Frustrated Lewis pairs. J. Am. Chem. Soc 2015, 137, 10018–10032. [DOI] [PubMed] [Google Scholar]
- (101).Stephan DW The broadening reach of frustrated Lewis pair chemistry. Science 2016, 354, aaf7229. [DOI] [PubMed] [Google Scholar]
- (102).Stephan DW Catalysis, FLPs, and beyond. Chem 2020, 6, 1520–1526. [Google Scholar]
- (103). β-Alkylated amine 4d may undergo (F5C6)3B/base-catalyzed dehydrogenation to afford a di-substituted enamine; however, no such product was obtained. This could be because the di-substituted enamine readily undergoes sequential protonation and borohydride reduction to afford 4d (vs processes involving N-alkylamine substrates that afford more stable tri-substituted enamines; see Figures 2 and 6 for details).
- (104). To synthesize 4j and 4k more selectively, different reaction conditions were used. See the Supporting Information for details.
- (105).Gotoh H; Masui R; Ogino H; Shoji M; Hayashi Y Enantioselective ene reaction of cyclopentadiene and α,β-enals catalyzed by a diphenylprolinol silyl ether. Angew. Chem., Int. Ed 2006, 45, 6853–6856. [DOI] [PubMed] [Google Scholar]
- (106).Hayashi Y; Gotoh H; Masui R; Ishikawa H Diphenylprolinol silyl ether as a catalyst in an enantioselective, catalytic, formal aza [3+3] cycloaddition reaction for the formation of enantioenriched piperidines. Angew. Chem., Int. Ed 2008, 47, 4012–4015. [DOI] [PubMed] [Google Scholar]
- (107).Dell’Amico L; Albrecht Ł; Naicker T; Poulsen PH; Jørgensen KA Beyond classical reactivity patterns: Shifting from 1,4- to 1,6- additions in regio- and enantioselective organocatalyzed vinylogous reactions of olefinic lactones with enals and 2,4-dienals. J. Am. Chem. Soc 2013, 135, 8063–8070. [DOI] [PubMed] [Google Scholar]
- (108).McLeod D; Izzo JA; Jørgensen DKB; Lauridsen RF; Jørgensen KA Development and investigation of an organocatalytic enantioselective [10 + 2] cycloaddition. ACS Catal 2020, 10, 10784–10793. [Google Scholar]
- (109).Evans DA; Chapman KT; Bisaha J Asymmetric Diels-Alder cycloaddition reactions with chiral α,β-unsaturated N-acyloxazolidinones. J. Am. Chem. Soc 1988, 110, 1238–1256. [Google Scholar]
- (110).Sibi MP; Ji J Practical and efficient enantioselective conjugate radical additions. J. Org. Chem 1997, 62, 3800–3801. [Google Scholar]
- (111).Adachi S; Takeda N; Sibi MP Evaluation of achiral templates with fluxional Brønsted basic substituents in enantioselective conjugate additions. Org. Lett 2014, 16, 6440–6443. [DOI] [PubMed] [Google Scholar]
- (112).Espelt LR; McPherson IS; Wiensch EM; Yoon TP Enantioselective conjugate additions of α-amino radicals via cooperative photoredox and Lewis acid catalysis. J. Am. Chem. Soc 2015, 137, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (113).Evans DA; Masse CE; Wu J C2-Symmetric Sc(III)-Complexes as Chiral Lewis Acids. Catalytic Enantioselective Aldol Additions to Glyoxylate Esters. Org. Lett 2002, 4, 3375–3378. [DOI] [PubMed] [Google Scholar]
- (114).Evans DA; Fandrick KR; Song H-J; Scheidt KA; Xu R Enantioselective Friedel– Crafts Alkylations Catalyzed by Bis(oxazolinyl)pyridine–Scandium(III) triflate complexes. J. Am. Chem. Soc 2007, 129, 10029–10041. [DOI] [PubMed] [Google Scholar]
- (115).Sibi MP; Itoh K; Jasperse CP Chiral Lewis Acid Catalysis in Nitrile Oxide Cycloadditions. J. Am. Chem. Soc 2004, 126, 5366–5367. [DOI] [PubMed] [Google Scholar]
- (116).Hou GH; Xie JH; Yan PC; Zhou QL Iridium-catalyzed asymmetric hydrogenation of cyclic enamines. J. Am. Chem. Soc 2009, 131, 1366–1367. [DOI] [PubMed] [Google Scholar]
- (117).Dilman AD; Belyakov PA; Struchkova MI; Arkhipov DE; Korlyukov AA; Tartakovsky VA Fluorocyanation of enamines. J. Org. Chem, 2010, 75, 5367–5370. [DOI] [PubMed] [Google Scholar]
- (118). For the purpose of investigating the rate order, KIE and Hammett ρ values, aryl-substituted α,β-unsaturated compounds were used as electrophiles (e.g., 6g and their related compounds). To probe whether the β-alkylation may proceed through different reaction mechanism when a different electrophile is used, we have performed a series of studies with ester-substituted 6c. See the Supporting Information for details.
- (119).Parks DJ; Piers WE Tris(pentafluorophenyl)boron-catalyzed hydrosilation of aromatic aldehydes, ketones, and esters. J. Am. Chem. Soc 1996, 118, 9440–9441. [Google Scholar]
- (120).Chen GQ; Kehr G; Daniliuc CG; Bursch M; Grimme S; Erker G Intermolecular redox‐neutral amine C–H functionalization induced by the strong boron Lewis acid B(C6F5)3 in the frustrated Lewis pair regime. Chem. Eur. J 2017, 23, 4723–4729. [DOI] [PubMed] [Google Scholar]
- (121).Sorgi KL; Maryanoff CA; McComsey DF; Graden DW; Maryanoff BE Asymmetric induction in an enammonium-iminium rearrangement. Mechanistic insight via NMR, deuterium labeling, and reaction rate studies. Application to the stereoselective synthesis of pyrroloisoquinoline antidepressants. J. Am. Chem. Soc 1990, 112, 3567–3579. [Google Scholar]
- (122).Han J; Lu Z; Flach AL; Paton RS; Hammond GB; Xu B Role of hydrogen-bonding acceptors in organo-enamine catalysis. Chem. Eur. J 2015, 21, 11687–11691. [DOI] [PubMed] [Google Scholar]
- (123).Ashley MA; Hirschi JS; Izzo JA; Vetticatt MJ Isotope effects reveal the mechanism of enamine formation in L‑proline-catalyzed α‑amination of aldehydes. J. Am. Chem. Soc 2016, 138, 1756–1759. [DOI] [PubMed] [Google Scholar]
- (124). The proton transfer process involving the conversion of the iminium to enammonium intermediates (XIII → XIV, Figure 10) may occur through deprotonation of XIII by an appropriate Brønsted base (e.g., H2O), followed by protonation of the resulting enamine species.
- (125).Simmons EM; Hartwig JF On the interpretation of deuterium kinetic isotope effects in C–H bond functionalizations by transition-metal complexes. Angew. Chem., Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- (126).Blackmond DG Kinetic profiling of catalytic organic reactions as a mechanistic tool. J. Am. Chem. Soc 2015, 137, 10852–10866. [DOI] [PubMed] [Google Scholar]
- (127). Intermolecular competition KIE experiments were also conducted, providing the results that are consistent with those of the independent KIE experiments. See the Supporting Information for details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.