

Abstract
The botanical natural product goldenseal can precipitate clinical drug interactions by inhibiting cytochrome P450 (CYP) 3A and CYP2D6. Besides P-glycoprotein, effects of goldenseal on other clinically relevant transporters remain unknown. Established transporter-expressing cell systems were used to determine the inhibitory effects of a goldenseal extract, standardized to the major alkaloid berberine, on transporter activity. Using recommended basic models, the extract was predicted to inhibit the efflux transporter breast cancer resistance protein (BCRP) and uptake transporters organic anion transporting polypeptide (OATP) 1B1/3. Using a cocktail approach, effects of the goldenseal product on BCRP, OATP1B1/3, organic anion transporters (OATs), organic cation transporters (OCTs), multidrug and toxin extrusion (MATE) proteins, and CYP3A were next evaluated in 16 healthy volunteers. As expected, goldenseal increased the area under the plasma concentration-time curve (AUC0-inf) of midazolam (CYP3A; positive control), with a geometric mean ratio (GMR) [90% confidence interval] of 1.43 [1.35–1.53]. However, goldenseal had no effects on the pharmacokinetics of rosuvastatin (BCRP, OATP1B1/3) and furosemide (OAT1/3); decreased metformin AUC0-inf (OCT1/2, MATE1/2-K; GMR, 0.77 [0.71–0.83]); and had no effect on metformin half-life and renal clearance. Results indicated that goldenseal altered intestinal permeability, transport, and/or other processes involved in metformin absorption, which may have unfavorable effects on glucose control. Inconsistencies between model predictions and pharmacokinetic outcomes prompt further refinement of current basic models to include differential transporter expression in relevant organs and intestinal degradation/metabolism of the precipitant(s). Such refinement should improve in vitro-in vivo prediction accuracy, contributing to a standard approach for studying transporter-mediated natural product-drug interactions.
Keywords: drug interaction, goldenseal, metformin, natural product, probe cocktail, transporters, breast cancer resistance protein (BCRP), organic anion transporting polypeptide (OATP), organic anion transporter (OAT), organic cation transporter (OCT), multidrug and toxin extrusion protein (MATE), in vitro-in vivo extrapolation
Introduction
Botanical natural products (NPs), including herbal supplements, constitute a multibillion-dollar industry that has more than doubled in market sales since passage of the Dietary and Supplement Health Education Act in 1994.1 Supplements containing goldenseal [Hydrastis canadensis L. (Ranunculaceae)], a perennial herb native to North America, have consistently ranked among the top 20 highest selling NPs during the last decade.2 Goldenseal products are marketed as licensed natural health products in Canada and as dietary supplements in the United States.3 NPs made from dried roots of the goldenseal plant are purported to have therapeutic value and are used to self-treat a range of medical complications, including the common cold, allergic rhinitis, and digestive disorders such as diarrhea and constipation.4,5 As more patients continue to seek goldenseal and other NPs to self-treat their medical conditions, there is an increasing need to characterize their safety profiles, especially when co-consumed with prescribed medications, which can lead to adverse NP-drug interactions (NPDIs).
The potential for goldenseal to precipitate clinically relevant pharmacokinetic NPDIs have long been suspected.6,7 Mechanisms underlying these interactions have been attributed to inhibition of the cytochromes P450 (CYPs).8 Clinical studies conducted by Gurley and colleagues have since confirmed that a goldenseal extract (900 mg) administered thrice daily for 28 days inhibited CYP2D6 and CYP3A activity by approximately 40%.9–11 Regarding the effects of goldenseal on transporters, only one clinical study has been reported, which showed minimal effects on the pharmacokinetics of the P-glycoprotein (P-gp) probe drug digoxin after administration of a goldenseal extract (1.3 g) to healthy volunteers thrice daily for 14 days.12 These observations contrast with an earlier in vitro study demonstrating that a major alkaloid in goldenseal, berberine, upregulated P-gp function in the human intestinal cell line Caco-213 and with a later in vitro study demonstrating goldenseal extracts and berberine to increase P-gp ATPase activity in a baculovirus system expressing P-gp.14 The effects of goldenseal on the function of other clinically relevant transporters have not been evaluated. Likewise, the effects of berberine and the abundant alkaloids (−)-β-hydrastine and hydrastinine, all of which are time-dependent inhibitors of CYP3A and/or CYP2D6,15 have not been tested on transporter function.
Numerous validated probe drug cocktails for CYPs are widely used to assess drug interaction potential of new chemical entities. Validated cocktails covering a range of clinically relevant transporters that contain probe drug substrates for multiple uptake [organic anion transporters (OATs), organic anion transporting polypeptides (OATPs), organic cation transporter 1 (OCT1)] and efflux [breast cancer resistance protein (BCRP), multidrug and toxin extrusion proteins (MATEs), P-gp] transporters are emerging (Table S1). Selection of a given transporter cocktail, ideally guided by in vitro data, allows for rapid and robust clinical assessment of potential xenobiotic-drug interactions.
Based on the knowledge gaps in transporter-mediated NPDIs, the objective of this work was to assess the effects of a model NP, goldenseal, on clinically relevant transporters using an in vitro-in vivo extrapolation approach. The aims were to (1) test goldenseal alkaloids (berberine, (−)-β-hydrastine, and hydrastinine) and an extract prepared from a well-characterized goldenseal product as inhibitors of multiple transporters using established in vitro systems, (2) use recommended basic models to predict potential transporter-mediated interactions involving the goldenseal product, and (3) evaluate the effects of the goldenseal product on the pharmacokinetics of the probe substrates furosemide [OAT1/3], metformin [OCT1/2, MATE1/2-K], rosuvastatin [OATP1B1/3, BCRP], and midazolam [CYP3A; positive control] administered as a cocktail to healthy volunteers. Results from this comprehensive study provide a foundation to improve in vitro-in vivo prediction accuracy of transporter-mediated NPDIs.
Materials and Methods
Chemicals and reagents
Goldenseal product selection and preparation.
The goldenseal product (500 mg root extract capsules; Now Foods, Bloomingdale, IL) was chosen from a set of 35 commercial goldenseal supplements that were selected based on their popularity among US consumers (determined from Amazon sales data). The number of products selected was consistent with recommended approaches for research studies with botanical natural products.16 One representative sample for the clinical study was selected from among the 35 products based on extensive analysis as described.17 LC-MS metabolomics profiling ensured that the specific product chosen (arbitrarily coded as GS-16) was authentic Hydrastis canadensis.18 Composite scores analysis was used to compare the chemical composition of all 35 commercial H. canadensis samples, and GS-16 (dried Hydrastis canadensis root material) was chosen based on its similarity to reference standards and other commercially available goldenseal supplements.19 LC-MS data comparing GS-16 to other commercial goldenseal products are freely accessible in the Center of Excellence for Natural Product Drug Interaction Research data repository.20
To prepare the extract from the goldenseal product, 200 mg of the goldenseal root material was removed from the capsule and combined with 20 mL methanol in a 20 mL scintillation vial. Mixtures were shaken overnight at room temperature, filtered, dried under a stream of nitrogen, and the resulting extract was stored at room temperature until analysis. Based on the calculated concentration of berberine (0.1926 ± 0.0060 μmol/mg) (Table S2), a 10 mM standard stock was prepared by resuspending the extract (51.9 mg/mL) in DMSO. This diluted goldenseal extract containing a known concentration of berberine is referred to as extract “standardized to berberine”. Berberine served as a marker for assessing potential transporter-mediated drug interactions of the select goldenseal product because the identity of the constituent(s) in the goldenseal extract responsible for transporter inhibition was unknown.
Alkaloid identification and quantification.
Standards of berberine chloride (CAS number 633–65-8) and hydrastinine chloride monohydrate (CAS number 65945–18-8) were purchased from Sigma-Aldrich (Saint Louis, MO). (1R,9S)-(−)-β-Hydrastine (CAS number 118081) was purchased from Cayman Chemical (Ann Arbor, MI). For quantitative analysis by LC-MS, the alkaloid standards were prepared in spectrometric-grade MeOH and diluted in a two-fold dilution series ranging from 0.1–200 μg/mL before injection and analysis using previously established methods.17 Alkaloid concentrations were determined by 1/x2 weighted least-squares linear regression (Table S2).
Clinical study probe substrates.
Furosemide (10 mg/mL solution; West-ward Pharmaceuticals Corp., Eatontown, NJ), metformin (500 mg/5 mL solution; Ranbaxy Pharmaceuticals, Inc., Jacksonville, FL), rosuvastatin (10 mg tablet; Camber Pharmaceuticals Inc., Piscataway, NJ), and midazolam (2 mg/mL syrup; West-ward Pharmaceuticals Corp.) were obtained from a local pharmacy.
In vitro transporter inhibition assays
Solubility of the test articles in buffer.
The solubility of each test article (berberine, (−)-β-hydrastine, hydrastinine, and goldenseal extract) in relevant buffer (transporter, Krebs-Henseleit, or Hank’s Balanced Salt Solution) was assessed to determine the appropriate concentration range for conducting the inhibition assays. In brief, a stock solution of each test article (extract standardized to berberine) was prepared in dimethyl sulfoxide and diluted in a 5-step, 2-fold dilution series to achieve five individual concentrations. Each test article was mixed with appropriate buffer in a 96-well plate to achieve the desired final concentrations (Table S3) and incubated at 37°C (or 32°C for BCRP and P-gp due to temperature compatibility) for 10–15 minutes. Final organic solvent concentration in the incubation mixture did not exceed 1.5% (v/v). Each solution was evaluated by optical microscopy to verify appropriate solubility necessary for transport inhibition assays.
Test articles as inhibitors of transporters.
Inside-out membrane vesicles prepared from HEK293 cells overexpressing a single transporter (vesicular transport inhibition assay) or cell lines (HEK293, MDCKII) stably expressing a single transporter (uptake transporter inhibition) were used to determine the inhibitory effects of each test article on the designated transporter as described21–23 using established probe substrates and inhibitors (Table S4). Each alkaloid was tested at 10 and 100 μM, whereas the goldenseal extract, standardized to berberine concentrations (detailed above), was tested at 1.75 μM and 17.5 μM (based on solubility limits). Inhibition by each test article was then compared against reference inhibitors and negative control cell lines. If ≥50% inhibition was observed, the IC50 was determined using varying concentrations for each goldenseal constituent/transporter combination (Table S5) and recovered using Prism 7.0 (GraphPad, San Diego, CA). Each concentration of test article, reference inhibitor, and solvent control for a given transporter were tested in triplicate.
In vitro-in vivo prediction of goldenseal-drug interactions
Whether co-administration of the goldenseal product with the transporter probe cocktail would result in a clinical NPDI was predicted using basic models described per FDA guidance.24 The models were populated with IC50 values obtained from the goldenseal extract, which was standardized to berberine (detailed above), and estimated maximum berberine concentrations at relevant sites (i.e., intestinal lumen, inlet to the liver, and systemic plasma).
Clinical study
Clinical protocol and study participants.
The clinical protocol was approved by the Washington State University (WSU) Institutional Review Board (IRB #16620), and the study was conducted at the WSU Clinical Research Unit on the Health Sciences Campus in accordance with the Code of Federal Regulations on the Protection of Human Subjects (45 CFR Part 46). The study is registered with the ClinicalTrials.gov database (NCT03772262). Healthy adult volunteers underwent screening that consisted of history, physical exam, and clinical laboratory testing (complete blood count with differentials and platelets, complete metabolic panel, and urinalysis) to determine eligibility; all women underwent serum pregnancy testing prior to enrollment and inpatient study visits. Participation eligibility was based on the screening evaluation and inclusion/exclusion criteria (Table S6). Written informed consent and Health Insurance Portability and Accountability Act authorization were obtained from all participants prior to screening.
Study design and procedures.
Eight men and eight nonpregnant, non-lactating women participated in an open label, 2-arm, fixed sequence crossover study (Figure 1). During Arm 1 (baseline), participants were administered the oral transporter probe cocktail consisting of 1 mg furosemide, 50 mg metformin, 10 mg rosuvastatin, and 2.5 mg midazolam. During Arm 2 (goldenseal exposure), participants were administered 1 g goldenseal thrice daily, evenly spaced during the 8-hour work interval (approximately 8:00 am, 12:00 pm, and 4:00 pm), for 5 consecutive days. On the morning of day 6, subjects were administered the oral probe cocktail with 1 g goldenseal; an additional two doses of goldenseal (1 g) followed, in 4-hour intervals. Blood (8 mL) was collected into BD K3 EDTA-containing vacutainer collection tubes (Fisher Scientific Co., Pittsburgh, PA) from an arm vein via an indwelling venous catheter before and 0.33–12 hours post-cocktail administration. Participants returned for single venous blood draws at 24, 48, 72, and 96 hours after cocktail administration. Urine was collected from 0–12 hours. Upon discharge, subjects were instructed to collect their urine until returning for the 24-hour blood draw; Vital signs (blood pressure, oxygen saturation, pulse) were recorded prior to oral cocktail administration and during every outpatient visit.
Figure 1.
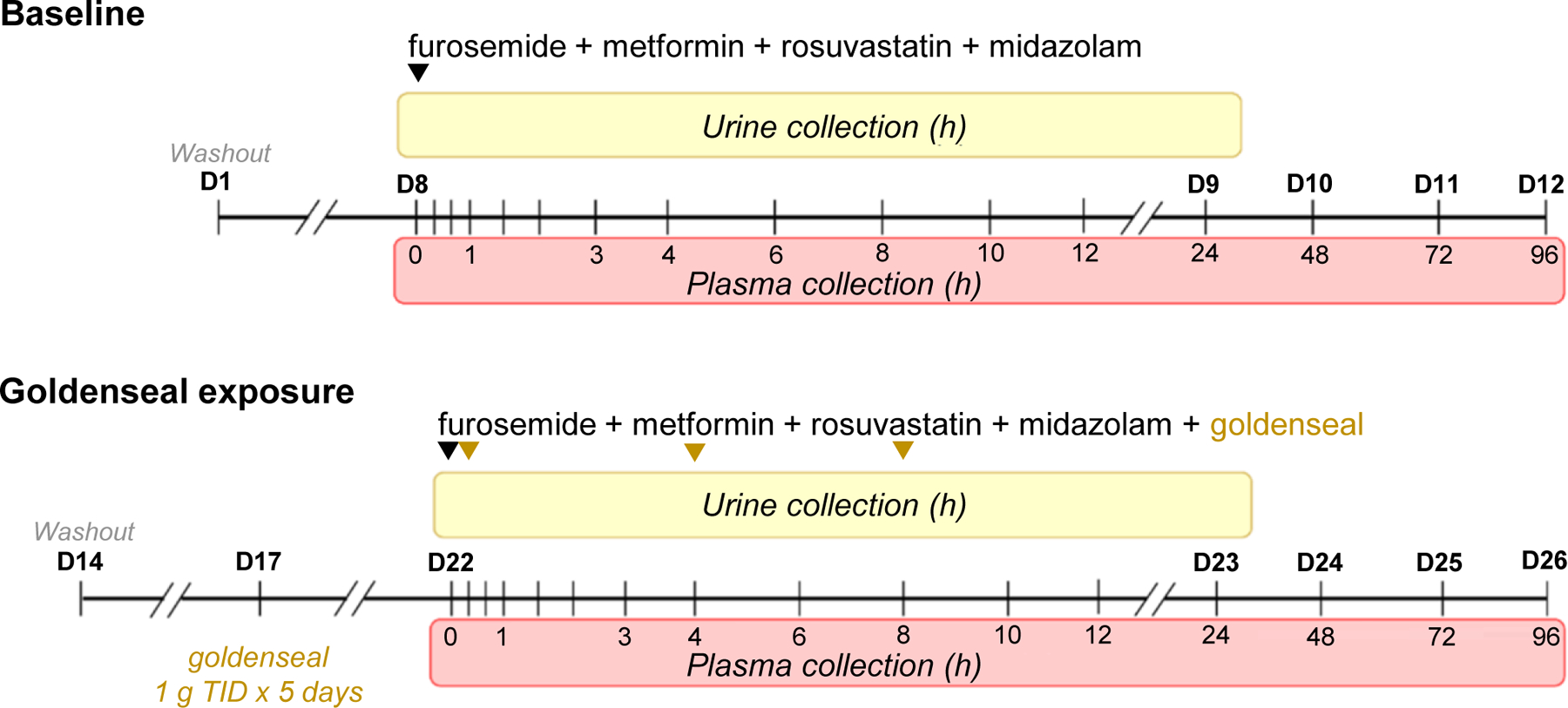
Healthy adult volunteers were enrolled in a two-arm, open-label, fixed sequence, crossover study. Arm 1 (baseline) entailed administration of a transporter probe cocktail containing 1 mg furosemide, 50 mg metformin, 10 mg rosuvastatin, and 2.5 mg midazolam (black inverted triangle). A minimum of 9 days of washout ensured no residual analytes. Arm 2 (goldenseal exposure) entailed administration of 1 g goldenseal thrice daily for 5 consecutive days to ensure berberine reached steady state (t1/2 28 h).49 On day 6 of Arm 2, participants were administered the oral probe cocktail with 1 g goldenseal; two additional doses of goldenseal (1 g) were administered in 4-hour increments (gold inverted triangles). During both arms, plasma was collected at designated times (shaded in red) up to 96 hours post-cocktail administration; urine was collected in 12-hour intervals (0–12 and 12–24; shaded in yellow) immediately following cocktail administration.
Bioanalytical methods.
Plasma was isolated from whole blood via centrifugation (1600 g × 10 min), and 0.5 mL was transferred to a cryovial containing ammonium acetate (1 M, pH 5, 25 μL) to stabilize rosuvastatin lactone; the remaining plasma was transferred to a second cryovial. Samples were stored at −80°C pending analysis for furosemide, metformin, rosuvastatin acid, rosuvastatin lactone, and midazolam by UHPLC-MS/MS (Table S7). Total urine volume and weight from 0–12 and 12–24 hours were recorded, and two aliquots (~5 mL) for each period were stored in 5-mL conical tubes at −80°C pending UHPLC-MS/MS analysis, allowing for robust calculation of analytes excreted in the urine.
Pharmacokinetic analysis.
The pharmacokinetics of each analyte were determined via noncompartmental analysis using Phoenix WinNonlin (version 7.0, Certara, Princeton, NJ). Maximum concentration (Cmax) and time to Cmax (tmax) were recovered directly from the plasma concentration-time profile. Terminal slope (λz) was extrapolated via linear regression of at least three of the last data points. Terminal half-life (t1/2) was calculated as 0.693/λz when the following criteria were satisfied: the plasma concentration time curve spanned at least three t1/2, and the regression about λz (r2) was ≥85%. Area under the plasma concentration-time curve (AUC) from time zero to the time of the last measurable concentration (Clast) was determined using the trapezoidal method with linear up/log down interpolation. Total AUC (AUC0-inf) was calculated as the sum of AUC0-last and Clast/λz. Oral clearance (Cl/F) was calculated as the ratio of the administered dose to AUC0-inf. Renal clearance (ClR) was calculated as the ratio of the mass of analyte recovered in the urine from time zero to the time of Clast to AUC0-last.
Statistical and power analyses.
The sample size of 16 evaluable subjects was based on an 80% power to detect a 20% change in the primary endpoint [geometric mean ratio (GMR) of midazolam AUC0-inf in the presence to absence of goldenseal] with a Type I error of 0.05, assuming a 25% intra-individual variability in midazolam AUC.25 As recommended by FDA guidance,26 two one-sided testing procedure was used to determine whether a pharmacokinetic interaction was evident, with a pre-defined no effect range of 0.80–1.25. Secondary pharmacokinetic endpoints included AUC0-last, t1/2, Cmax, and ClR for midazolam and, when recoverable, AUC0-inf, AUC0-last, t1/2, Cmax, and ClR for the remaining probe drugs. Secondary endpoints were evaluated using a paired Student’s t-test or Wilcoxon signed-rank test when appropriate; a p-value <0.05 was considered statistically significant. Statistical Analysis System STAT (version 14.3, SAS, Cary, NC) was used for all statistical analysis procedures.
Results
Inhibition assays and in vitro-in vivo predictions
The efflux transporters MATE1, MATE2-K, and BCRP were the most sensitive to inhibition by the goldenseal extract, with IC50 values <1 μM (Table 1). The uptake transporters OAT1, OAT3, OCT1, OCT2, and OATP1B1/3 were the next most sensitive, with IC50 values of 1–10 μM. MATE1 and MATE2-K were sensitive to inhibition by berberine (IC50 <1 μM), and OCT1 was sensitive to inhibition by β-hydrastine (IC50 <10 μM), whereas hydrastinine had negligible inhibitory effects (IC50 ≥80 μM) against all transporters tested. Using current FDA-recommended basic models24 the goldenseal product was predicted to inhibit the intestinal efflux transporter BCRP, and the hepatic uptake transporters OATP1B1 and OATP1B3 (Table 2).
Table 1.
Inhibitory effects of key goldenseal alkaloids and goldenseal extract against selected transporters.
| Transporter | Berberine | (−)-β-Hydrastine | Hydrastinine | Goldenseal Extracta |
|---|---|---|---|---|
| IC50 (μM) | ||||
| NTCP | -- | -- | -- | -- |
| OAT1 | -- | 7.3 [1.9–28] | ||
| OAT3 | 79 [47–134] | 203 [169–243] | -- | 4.6 [2.6–8.0] |
| OATP1B1 | -- | 120 [85–165] | -- | 8.0 [6.8–9.5] |
| OATP1B3 | -- | -- | 1.3 [0.5–3.7] | |
| OATP2B1 | -- | -- | -- | |
| OCT1 | 19 [11–31] | 6.6 [5.3–8.1] | 2.6 [1.4–4.7] | |
| OCT2 | 45 [17–117] | 90 [38–214] | 95 [70–129] | 12 [5.6–25] |
| BSEP | -- | -- | -- | -- |
| BCRP | 180 [53–624] | 86.2 [19–391] | -- | 0.59 [0.43–0.78] |
| MATE1 | 0.73 [0.37–1.44] | 110 [90–131] | 82 [44–154] | 0.45 [0.27–0.76] |
| MATE2-K | 0.77 [0.41–1.48] | 0.47 [0.24–0.92] | ||
| MRP2 | -- | -- | -- | -- |
| MRP3 | -- | -- | -- | -- |
| P-gp | -- | -- | -- | -- |
Based on the concentration of berberine quantified in the extract. NTCP, sodium/taurocholate cotransporting polypeptide; OAT, organic anion transporter; OATP, organic anion transporting polypeptide; OCT, organic cation transporter; BSEP, bile salt export pump; BCRP, breast cancer resistant protein; MATE, multidrug and toxin extrusion protein; MRP, multidrug resistance-associated protein. P-gp, P-glycoprotein.
The goldenseal alkaloids berberine, (−)-β-hydrastine, and hydrastinine were screened as inhibitors of each transporters at 10 and 100 μM; the extract was tested at 1.75 and 17.5 μM (based on berberine concentration and solubility limit). --, inhibition not observed; ⬛, 20–50% inhibition; ⬛, 50–70% inhibition; or ⬛, >70% inhibition at the highest concentration tested. Subsequently, IC50 values were determined for test articles demonstrating ≥50% inhibition. Values denote mean IC50 [95% confidence interval] of three technical replicates.
Table 2.
In vitro-in vivo predictions of transporter-mediated goldenseal-drug interactions using established basic models for clinically relevant transporters per FDA guidance.
| Transporter | Basic modela | Goldenseal Extract R-value |
Predicted Effect |
|---|---|---|---|
| BCRP | 601 | ↑ rosuvastatin AUC | |
| -- | No interaction | ||
| OAT1 | <0.001 | No interaction | |
| <0.001 | No interaction | ||
| <0.001 | No interaction | ||
| .002 | No interaction | ||
| 0.002 | No interaction | ||
| OATP1B1 OATP1B3 |
1.5 | ↑ rosuvastatin AUC | |
| 4.2 | ↑ rosuvastatin AUC |
--, No interaction predicted based on low inhibition potential in transporter assay (IC50 > 100 μM).
IC50 values for the goldenseal extract, depicted in Table 1, were used in basic models.
Igut, intestinal luminal concentration of inhibitor, estimated as the ratio of the mass of berberine (29.8 mg) in a 1-gram capsule of goldenseal to 250 mL. fu,p, fraction unbound of inhibitor (berberine) in plasma, estimated to be 0.75 using GastroPlus version 9.7 (Simulations Plus, Lancaster, CA). Imax,u, maximum unbound plasma concentration of berberine at steady state, estimated as the product of fu,p and the maximum reported plasma concentration of berberine (0.44 ng/mL),49 adjusted to dose (0.033 ng/mL), and scaling to 0.20 ng/mL (0.59 nM) based on an accumulation ratio of 5.95, calculated using the equation 1/(1-e−k*T), where k and T are the elimination rate constant32 (0.023 h−1) and dosing interval (8 h), respectively. For simplicity, a dosing interval of 8 h was used. Iin,max, estimated maximum plasma concentration of inhibitor (berberine) at the inlet of the liver, calculated as Imax + (Fa × Fg, × ka × dose)/(Qh/RB), where Fa is the fraction of berberine absorbed after oral administration of a 1-gram capsule of goldenseal, Fg is the fraction of berberine that escapes intestinal metabolism, ka is the first-order absorption rate constant for berberine, Qh is hepatic blood flow (97 L/hr),50 and Rb is the blood-to-plasma concentration ratio. Because the data were not available, Fa, Fg, and Rb were set to 1, and ka was set to 0.1 min−1 per FDA guidance.24
Clinical study
Participants, safety, and tolerability.
Of the 29 subjects screened, 19 were enrolled in the study, and 16 completed both arms. One man was discontinued due to poor venous access and was replaced with another eligible man. Participants who completed the study self-identified as white (7 men, 6 women) or Asian (1 man, two women). The median (range) age was 27 (23–42) and 26 (23–35) for men and women, respectively. None of the participants reported taking concomitant medications or natural products known to modulate the metabolism of midazolam and disposition of the remaining probe drugs. The goldenseal product and probe drugs were well tolerated. No subject experienced a severe adverse event. Three subjects experienced an adverse event during at least one arm (abdominal cramping or intravenous site bruising) that did not result in study discontinuation.
Pharmacokinetics.
As anticipated, midazolam systemic exposure increased in the presence of goldenseal (Figure 2A). The GMR [90% confidence interval] of the primary endpoint, midazolam AUC0-inf, lay outside the pre-defined no effect range in the presence of goldenseal (1.43 [1.35–1.53]; Figure 3, Table 3). The geometric mean Cmax increased by 31%; except for renal clearance (ClR), a minor elimination pathway for midazolam, all other pharmacokinetic endpoints were unchanged in the presence of goldenseal. Plasma concentrations of furosemide were quantifiable for up to 2–6 hours post-cocktail administration in the absence and presence of goldenseal. Because the descending concentrations did not meet the pre-defined criteria to obtain a robust estimate of λz for more than half of the subjects, AUC0-inf, Cl/F, and terminal t1/2 for furosemide could not be determined. Goldenseal had no effect on the remaining evaluable pharmacokinetic endpoints (Figure 2B). The geometric mean AUCinf and Cmax of metformin decreased by 23% and 27%, respectively, in the presence of goldenseal, whereas ClR and t1/2 were unaffected (Figure 2C). The AUC0-last and ClR of both rosuvastatin acid and rosuvastatin lactone were unaffected by goldenseal (Figure 2D). The AUCinf, Cl/F, and t1/2 for both rosuvastatin acid and rosuvastatin lactone could not be determined based on the inability to obtain a robust λz.
Figure 2.
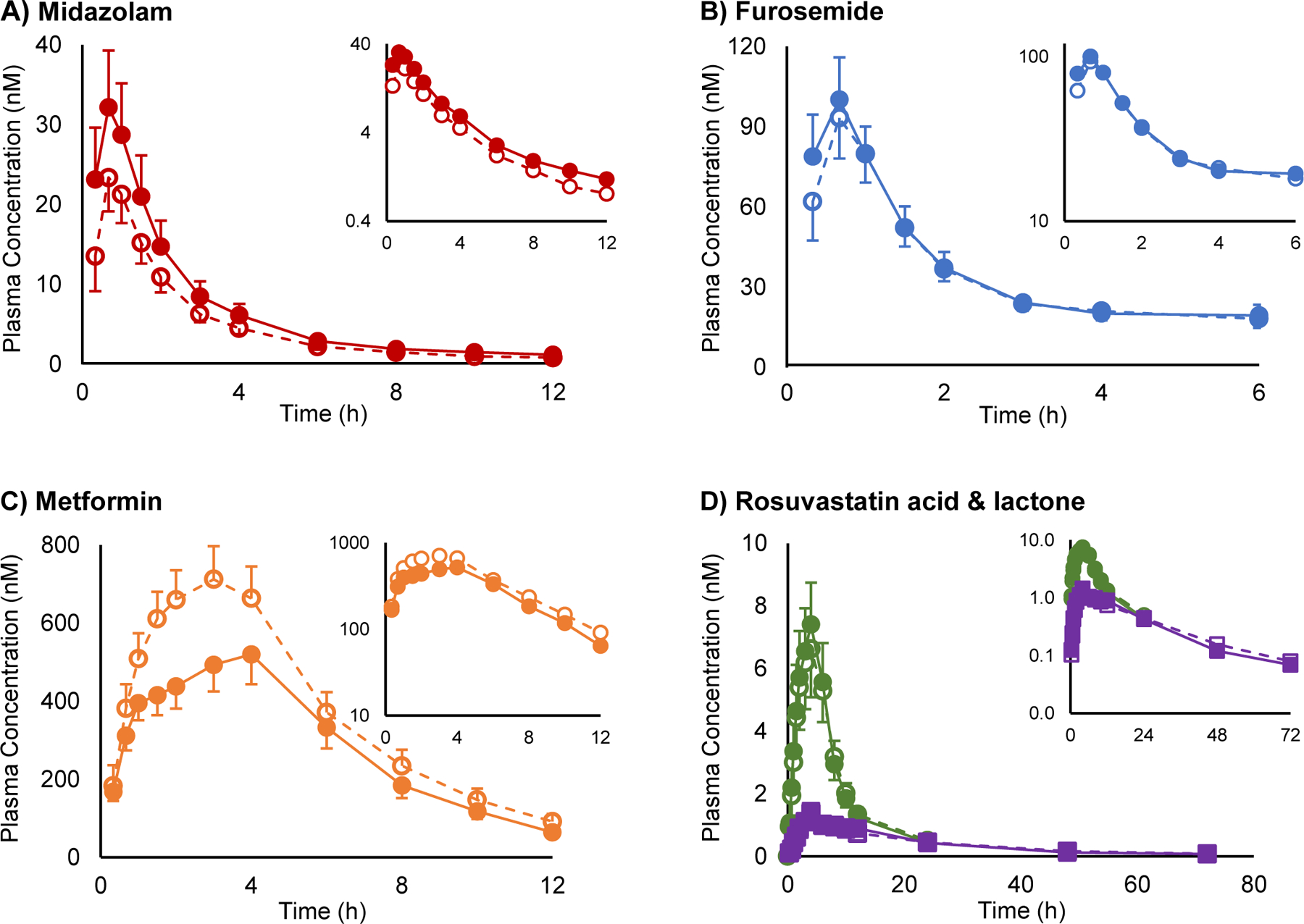
Plasma concentration vs. time profiles for (A) midazolam, (B) furosemide, (C) metformin, and (D) rosuvastatin acid (●) and rosuvastatin lactone (■) following oral administration to 16 healthy volunteers alone (open symbols) or upon five-day exposure to oral goldenseal (solid symbols). Symbols and error bars denote geometric means and 90% confidence intervals, respectively.
Figure 3.
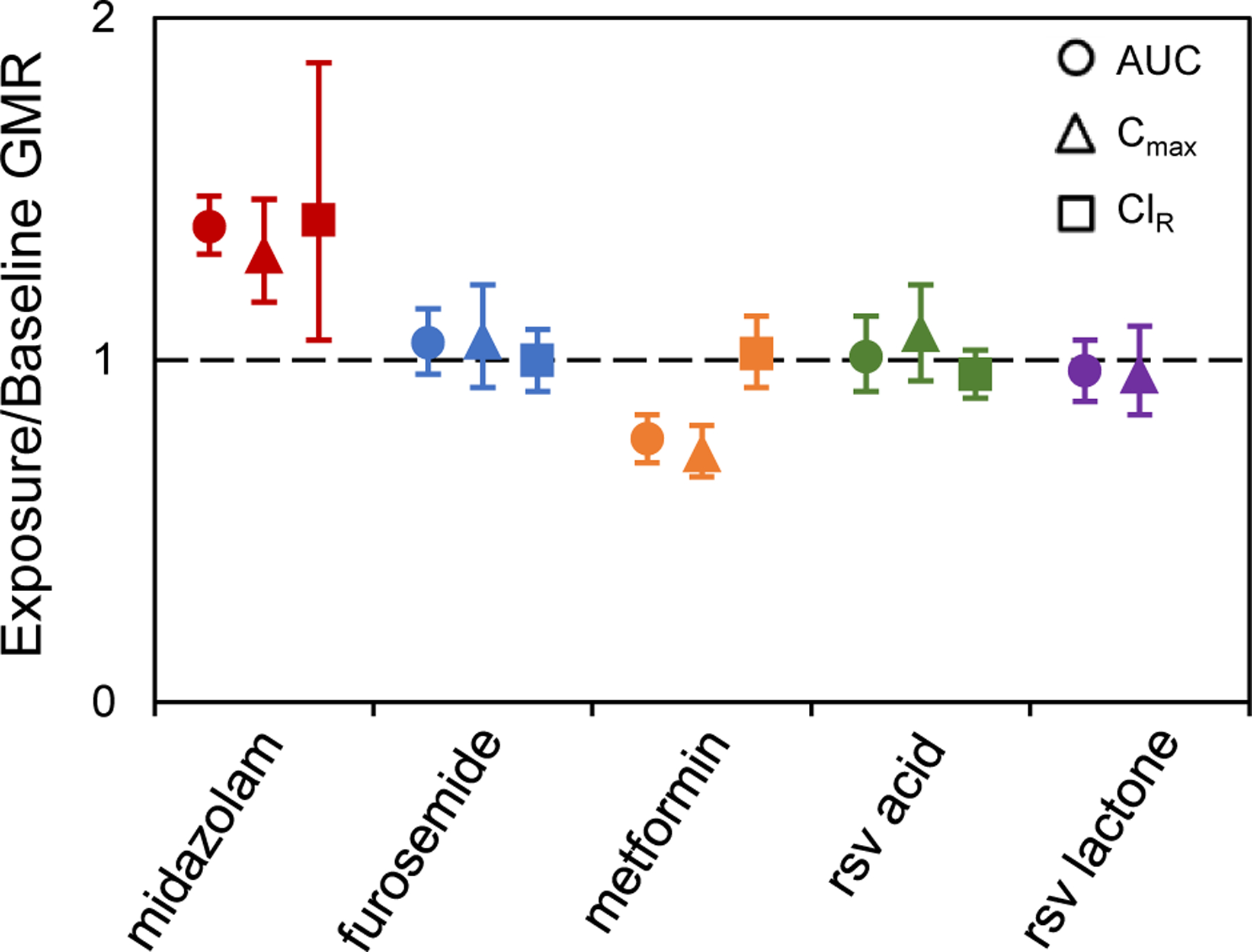
Goldenseal exposure-to-baseline geometric mean ratios (GMRs), with 90% confidence intervals, of the area under the plasma concentration-time curve (AUC), maximum concentration (Cmax), and renal clearance (ClR) for each probe substrate. The AUC ratios for furosemide, rosuvastatin (rsv) acid, and rsv lactone were calculated using plasma concentration-time curves up to the last quantifiable time point (AUC0-last), whereas AUC ratios for midazolam and metformin were calculated using plasma concentration-time curves to time infinity (AUC0-inf). ■ – midazolam; ■ – furosemide; ■ – metformin; ■ – rsv acid; ■ – rsv-lactone.
Table 3.
Pharmacokinetics of furosemide, metformin, rosuvastatin acid, rosuvastatin lactone, and midazolam (n=16 subjects) in the absence and presence of goldenseal co-administration.
| Geometric mean [90% confidence interval] | |||
|---|---|---|---|
| Baseline | Goldenseal exposure | Goldenseal exposure / baseline ratio | |
| Furosemide | |||
| AUC0-inf (μM*h)) | NC | NC | NC |
| AUC0–6 (μM*h) | 0.17 [0.14–0.20] | 0.18 [0.15–0.20] | 1.05 [0.96–1.15] |
| t1/2 (h) | NC | NC | NC |
| Cmax (μM) | 0.10 [0.09–0.12] | 0.11 [0.10–0.12] | 1.06 [0.92–1.22] |
| ClR (L/h) | 7.3 [6.3–8.6] | 7.3 [6.4–8.3] | 1.00 [0.91–1.09] |
| Metformin | |||
| AUC0-inf (μM*h) | 5.0 [4.5–5.6] | 3.8 [3.3–4.4]a | 0.77 [0.71–0.83] |
| AUC0–24h (μM*h) | 4.7 [4.2–5.3] | 3.6 [3.1–4.2]a | 0.77 [0.70–0.84] |
| t1/2 (h) | 3.2 [2.9–3.5] | 3.0 [2.6–3.5] | 0.94 [0.83–1.06] |
| Cmax (μM) | 0.75 [0.67–0.82] | 0.54 [0.48–0.62]a | 0.73 [0.66–0.81] |
| ClR (L/h) | 41 [36–47] | 42 [38–47] | 1.02 [0.92–1.13] |
| Rosuvastatin acid | |||
| AUC0-inf (nM*h) | NC | NC | NC |
| AUC0–24 (nM*h) | 59 [48–72] | 60 [51–70] | 1.01 [0.91–1.13] |
| t1/2 (h) | NC | NC | NC |
| Cmax (nM) | 7.4 [5.9–9.1] | 7.9 [6.7–9.3] | 1.08 [0.94–1.22] |
| ClR (L/h) | 15.4 [13.8–17.2] | 14.8 [13.6–16.0] | 0.96 [0.89–1.03] |
| Rosuvastatin lactone | |||
| AUC0-inf (nM*h) | NC | NC | NC |
| AUC0–72 (nM*h) | 28 [24–32] | 27 [23–32] | 0.97 [0.88–1.06] |
| t1/2 (h) | NC | NC | NC |
| Cmax (nM) | 1.5 [1.3–1.8] | 1.5 [1.3–1.7] | 0.96 [0.84–1.10] |
| ClR (L/h) | NC | NC | NC |
| Midazolam | |||
| AUC0-inf (nM*h) | 67 [55–82] | 96 [78–119]a | 1.43 [1.35–1.53] |
| AUC0–12 (nM*h) | 62 [51–74] | 86 [70–105]a | 1.39 [1.31–1.48] |
| t1/2 (h) | 3.9 [3.4–4.6] | 4.8 [4.2–5.6]a | 1.23 [1.07–1.42] |
| Cmax (nM) | 26 [22–31] | 35 [28–42]a | 1.31 [1.17–1.47] |
| ClR (L/h) | 0.05 [0.03–0.07] | 0.07 [0.05–0.09]a | 1.47 [1.14–1.88] |
AUC0-inf, area under the plasma-concentration time curve from time zero to infinity; AUC0-last, area under the plasma-concentration time curve from time zero to the time of the last measured concentration; t1/2, terminal half-life; Cmax, maximum plasma concentration; ClR, renal clearance. NC, not calculable based on inability to obtain robust estimate of terminal slope.
p < 0.05 compared to baseline.
Discussion
This work represents the first comprehensive in vitro-in vivo assessment of a botanical NP to precipitate transporter-mediated pharmacokinetic drug interactions. Using goldenseal as a model NP, the following three-step approach was taken: (i) the alkaloids berberine, (−)-β-hydrastine, and hydrastinine and an extract prepared from a well- characterized product17 were tested as inhibitors of clinically relevant transporters using established in vitro systems, (ii) basic models were populated with the resulting extract IC50 values and other relevant parameters to predict the likelihood of a clinical interaction, and (iii) model predictions were evaluated via a powered clinical study involving a transporter probe cocktail.
Consistent with previous clinical observations, goldenseal increased systemic exposure to the positive control probe substrate midazolam by ~40%.10 The negligible change in terminal t1/2 further indicated that goldenseal inhibited CYP3A primarily in the intestine. Goldenseal constituents, including the extensively studied major alkaloids berberine and (−)-β-hydrastine, are known time-dependent inhibitors of CYP3A,15 suggesting goldenseal could have prolonged inhibitory effects in vivo similar to grapefruit juice.
Prior to in vitro testing, the goldenseal extract was standardized to contain a fixed concentration of berberine based on typical high abundance in commercial goldenseal products,17 including the product used in the current work (Table S2), and documented modulatory, albeit inconsistent, effects on transporter activity (i.e., P-gp).13,14 Compared to berberine and the other tested alkaloids, (−)-β-hydrastine and hydrastinine, the extract showed the strongest inhibition towards all transporters examined (Table 1), indicating that other uncharacterized constituents in the goldenseal product contributed to the observed effects of the extract or that one or more constituents act synergistically. A systematic analysis using traditional bioassay-guided fractionation or biochemometric approaches of inhibitory activity of goldenseal extracts, fractions, and isolated constituents are needed to identify these constituents.27,28 Nevertheless, with appropriate assumptions (detailed below), the goldenseal extract IC50 values were used for the in vitro to in vivo predictions of potential berberine-mediated goldenseal-drug interactions.
The goldenseal extract showed relatively potent inhibition of the uptake transporters OAT1 and OAT3 (4 < IC50 < 10 μM; Table 1), which are expressed primarily on the basolateral membrane of kidney proximal tubule cells.29,30 As such, precipitant constituents must overcome both intestinal and hepatic first-pass processes before entering the systemic circulation and reaching the target site. These processes contributed to the low anticipated berberine Cmax (~0.59 nM).31–33 By using the extract IC50 that is standardized to berberine, the assumption is that all other alkaloids and uncharacterized constituents in the goldenseal product have no bioavailability concerns, thus contributing to the increased inhibition potential of the extract compared to single alkaloids. Regardless, a berberine-mediated clinical interaction between the goldenseal product and the OAT1/3 probe furosemide was unlikely, as predicted by the basic model, due to the low systemic concentration of berberine (Table 2). Results from the clinical study confirmed these in vitro-in vivo predictions, as goldenseal co-administration had no effect on furosemide renal clearance and systemic exposure (Table 3, Figure 3).
The goldenseal extract showed potent in vitro inhibition (IC50 ~0.6 μM; Table 1) of the apical efflux transporter BCRP, which was unlikely caused by any of the tested alkaloids (IC50 >80 μM). The basic model predicted a berberine-mediated interaction between intestinal BCRP, located on the luminal membrane of enterocytes, and the goldenseal product (increased rosuvastatin AUC predicted; Table 2) because the estimated concentration of berberine in the gut (~350 μM) greatly exceeded the observed IC50. Although BCRP is expressed in other tissues, the intestinal lumen represents the first target and the highest risk for a BCRP-mediated interaction involving orally administered xenobiotics. In addition, the IC50 was obtained using inside-out BCRP-expressing vesicles in compatible assay solution, whereas the berberine gut luminal concentration was calculated using the administered dose and intestinal fluid volume, the latter of which assumes complete dissolution of all goldenseal constituents in intestinal fluids. However, in the clinical study, the pharmacokinetics of rosuvastatin acid and lactone were unchanged in the presence of goldenseal. Two potential mechanisms could explain this in vitro-in vivo disconnect: 1) incomplete dissolution led to insufficient luminal concentrations, and/or 2) if enterocyte concentrations drive the interaction, low permeability or extensive enterocyte metabolism or efflux would lead to a discordance between luminal and enterocyte concentrations (current basic model incorporates luminal concentration).
Once absorbed into the portal circulation, rosuvastatin is taken up into hepatocytes primarily by OATP1B1 and NTCP and to a minor extent by OATP1B3.34,35 The goldenseal extract inhibited OATP1B1 (IC50, 8.0 μM) and OATP1B3 (IC50, 1.3 μM) but not NTCP in vitro. Similar to BCRP, the inhibitory effects on OATP1B1/3 were unlikely due to any of the tested alkaloids (IC50 ≥ 120 μM). Despite the relatively high IC50 for OATP1B1, interactions between both transporters and the probe substrate rosuvastatin were predicted (Table 2) because theoretically, the precipitants could reach the target, basolateral membrane of hepatocytes, in high concentrations immediately following intestinal first-pass processes. As with BCRP, these predictions contradicted in vivo results, as the pharmacokinetics of both rosuvastatin acid and lactone were unaltered by goldenseal co-administration. This misprediction can be attributed to an overestimated hepatic inlet berberine concentration, which could reflect incomplete or impaired intestinal absorption and/or extensive metabolism in the gut.
The goldenseal extract showed the most potent inhibition against the apical efflux transporters MATE1 and MATE2-K (IC50 ~0.5 μM), major mediators of metformin renal excretion, with berberine representing a potential key precipitant (IC50, 0.73 μM). However, based on the low anticipated berberine Cmax, the basic model predicted no clinical interaction with these transporters in the kidney (Table 2). The goldenseal extract was also a modest inhibitor of the basolateral uptake transporter OCT2 (IC50, 10 μM), localized in kidney proximal tubules,36 which was unlikely caused by any of the tested alkaloids. As with MATE1/2-K, the basic model predicted no interaction with OCT2, again due to the low anticipated berberine Cmax. The clinical study demonstrated successful in vitro-in vivo predictions, as co-administration of the goldenseal extract had no effect on metformin ClR.
Although metformin ClR was unaffected by the goldenseal product, metformin AUCinf and Cmax were significantly reduced by 23–27% (Table 3). These observations, coupled with no change in half-life, suggested that goldenseal decreased metformin oral bioavailability by altering intestinal permeability, transport, and/or other processes involved in metformin absorption. Multiple uptake transporters contribute to the intestinal absorption of metformin, including OCT1/3, thiamine transporter 2 (ThTR-2), plasma membrane monoamine transporter (PMAT), and serotonin reuptake transporter (SERT) (Figure 4).37–39 OCT3, ThTR-2, PMAT, and SERT are expressed on the luminal membrane, whereas the reported localization of OCT1 is conflicting.40–42 An interaction with one or more of these transporters could explain the observed decrease in metformin systemic exposure, and follow-up studies are ongoing to address these potential mechanisms.
Figure 4.
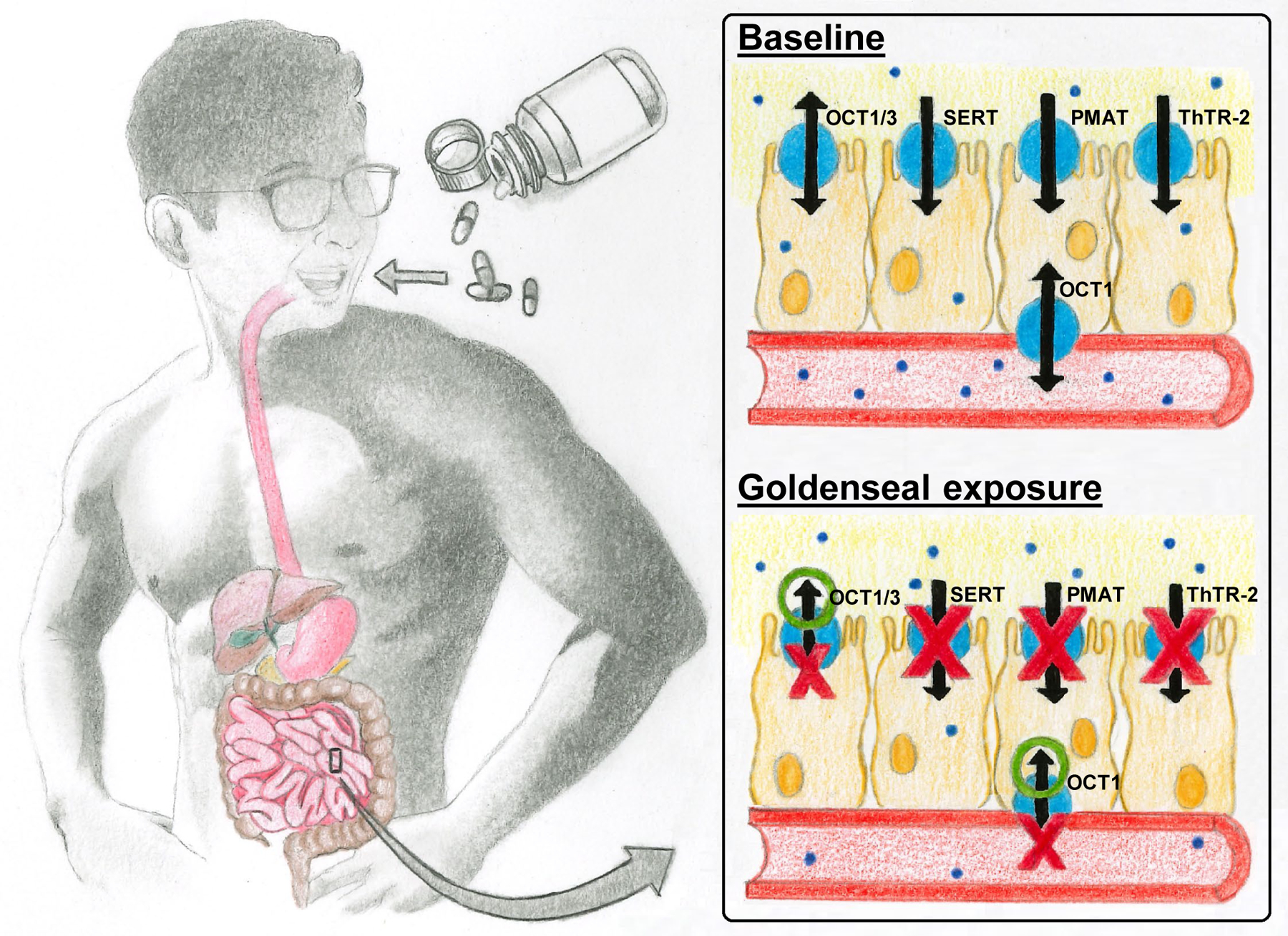
Proposed transporter-mediated mechanism underlying the decreased metformin systemic exposure upon goldenseal exposure. The indicated transporters have been reported to facilitate intestinal absorption of metformin.37–42 Arrows represent uptake or efflux; double-headed arrows represent bidirectional transport. Red X’s represent inhibition of indicated transporters by goldenseal; green O’s represent induction of indicated transporters by goldenseal. Solid blue circles represent metformin molecules. OCT, organic cation transporter; PMAT, plasma membrane monoamine transporter; SERT, serotonin reuptake transporter; ThTR-2, thiamine transporter 2.
The goldenseal extract was a relatively potent inhibitor of OCT1 (IC50 2.6 μM). Because there is currently no recommended basic model for predicting OCT1-mediated drug interactions, a standard in vitro-in vivo prediction was not conducted. However, if OCT1 is localized on the luminal membrane of enterocytes, inhibition of uptake could explain the observed goldenseal-metformin interaction, as the estimated luminal concentration of berberine was ~100x higher than the IC50. This theory is further supported by applying the current basic model for other gut luminal membrane transporters (i.e., BCRP and P-gp) to OCT1, which would predict an interaction (R-value, ~125). In contrast to BCRP, the basic model would successfully predict an interaction for OCT1, suggesting that intestinal luminal concentrations drive apical uptake, whereas enterocyte concentrations drive apical/basolateral efflux. Based on the multiple dose design of this clinical study and the bidirectionality of OCTs localized in enterocytes, other potential mechanisms that could explain the decreased metformin exposure include induction of OCT1/3 efflux on the luminal membrane, induction of OCT1-mediated uptake on the basolateral membrane, or inhibition of OCT1-mediated efflux on the basolateral membrane (Figure 4). However, because goldenseal was administered orally, the high luminal to basolateral concentration gradient would favor modulation of net uptake, suggesting that the observed clinical results (decreased AUC with no change in half-life) were largely due to inhibition of luminal uptake. Additional studies are needed to determine the precise mechanism underlying the observed pharmacokinetic effects.
The current work demonstrated a comprehensive in vitro-in vivo approach involving established transporter inhibition assays, recommended basic models, and clinical pharmacokinetic evaluation to elucidate transporter-mediated goldenseal-drug interactions. Although several nuances regarding the basic models have been addressed, there are additional limitations to consider. First, as mentioned earlier, a confounding factor with the in vitro assays was that the IC50 of a single constituent was not always predictive of the goldenseal extract, suggesting that an uncharacterized constituent(s) in the extract contributed to the inhibitory effects. Second, disconnects between basic model predictions and pharmacokinetic outcomes for some, but not all, of the tested transporters were observed. As noted in a recent publication, refining the relevant models to include potential degradation and metabolism of the NP constituent in the gut could improve prediction accuracy.43 Such refinement may distinguish the false positive prediction for BCRP from the successful prediction for P-gp (i.e., no interaction based on an R value <10 and lack of clinical interaction with digoxin12). Physiologically-based pharmacokinetic (PBPK) models could further improve prediction accuracy by accounting for the variable expression of different transporters in relevant tissues. Third, the lack of pharmacokinetic changes for some transporter probes do not rule out the potential for interactions with competing pathways given that goldenseal was administered for multiple consecutive days. For example, hepatic OATP1B1/3 induction could have counteracted intestinal BCRP inhibition, resulting in the lack of change in systemic exposure to rosuvastatin. However, induction of transporters other than P-gp has become a topic of debate that requires further investigation.44 Finally, multiple transporters mediate the disposition of current clinical probe substrates. Endogenous transporter biomarkers are emerging, including coproporphyrins for OATP1Bs, which could help pinpoint specific transporters mediating a given interaction.45,46
There are potential clinical implications with the experimental observations. Although the decrease in metformin systemic exposure caused by goldenseal co-administration was modest, such a change could impact glucose control in type 2 diabetic patients. Given that metformin is the most prescribed anti-diabetic medication,47 there are widespread concerns because this large patient population has been reported to self-treat their medical condition with goldenseal, as well as berberine.48 Accordingly, supplementing a metformin-based pharmacotherapeutic regimen with goldenseal is not recommended for type 2 diabetic patients.
In summary, the current work represents the first comprehensive translational approach to assess potential transporter-mediated NPDIs. An appropriately designed probe cocktail enabled rapid clinical assessment of the effects of a model NP on multiple transporters. Refinement of current recommended basic models, development of PBPK models, and incorporation of endogenous transporter probes will help establish a standard approach for elucidating transporter-mediated NPDIs. Results will provide additional guidance for consumers and healthcare providers regarding the drug interaction risk, or lack thereof, of goldenseal co-administered with certain drugs.
Supplementary Material
Study Highlights.
What is the current knowledge on the topic?
The natural product (NP) goldenseal is a clinically relevant inhibitor of CYP2D6 and CYP3A, raising concerns for NP-drug interactions (NPDIs). Except P-glycoprotein, the effects of goldenseal on other clinically relevant transporters remain unknown.
What question did this study address?
Do current recommended basic models successfully predict transporter-mediated NPDIs? What are the effects of goldenseal supplementation on the pharmacokinetics of transporter probe substrates?
What does this study add to our knowledge?
The current models successfully predicted some, but not all, NPDIs. An oral transporter probe cocktail helped elucidate potential transporter-mediated NPDIs. Goldenseal unexpectedly decreased metformin systemic exposure. The observed pharmacokinetic changes suggested goldenseal altered processes involved in metformin intestinal absorption.
How might this change clinical pharmacology or translational science?
Results from this translational study prompt refinement of current models to improve in vitro-in vivo prediction accuracy of transporter-mediated NPDIs, which will contribute to establishing a standard approach for studying these complex interactions. The observed pharmacokinetic changes raise concern regarding goldenseal supplementation with standard antidiabetic pharmacotherapies, thus co-consuming goldenseal with metformin is not recommended.
Acknowledgments
The authors thank Ms. Judy Griffin for her expert nursing skills and Ms. Vi Tran for her excellent artistic skills (Figure 4). M.F.P. dedicates this article to Dr. David P. Paine.
Funding: This work was supported by the National Institutes of Health National Center for Complementary and Integrative Health and Office of Dietary Supplements, specifically the Center of Excellence for Natural Product Drug Interaction Research [U54 AT008909].
Footnotes
Conflict of Interest: The authors declared no competing interest for this work.
References
- 1.Natural Products Association Members Reflect on DSHEA at 25 Years. Natural Products Association. (2019). <https://www.npanational.org/news/25th-anniversary-of-dshea-npa-board/> Accessed 20 March 2020. [Google Scholar]
- 2.Smith T, Gillespie M, Eckl V, Knepper J and Reynolds CM Herbal supplement sales in US increase by 9.4% in 2018. Herbalgram. 123, 62–73 (2019). [Google Scholar]
- 3.Goldenseal - Hydrastis Canadensis. Health Canada. (2019). <http://webprod.hc-sc.gc.ca/nhpid-bdipsn/monosReq.do%3Flang=eng> Accessed 20 March 2020. [Google Scholar]
- 4.Hussain MS Patient counseling about herbal-drug interactions. African J. Tradit. Complement. Altern. Med 8, 152–163 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldenseal. National Center for Complementary and Integrative Health. (2016). <https://www.nccih.nih.gov/health/goldenseal> Accessed 15 February 2020. [Google Scholar]
- 6.Wu X, Li Q, Xin H, Yu A and Zhong M Effects of berberine on the blood concentration of cyclosporin A in renal transplanted recipients: Clinical and pharmacokinetic study. Eur. J. Clin. Pharmacol 61, 562–572 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Budzinski JW, Foster BC, Vandenhoek S and Arnason JT An in vitro evaluation of human cytochrome P450 3A4 inhibition by selected commercial herbal extracts and tinctures. Phytomedicine. 7, 273–282 (2000). [DOI] [PubMed] [Google Scholar]
- 8.Guo Y, Chen Y, Tan ZR, Klaassen CD and Zhou HH Repeated administration of berberine inhibits cytochromes P450 in humans. Eur. J. Clin. Pharmacol 68, 213–217 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurley BJ et al. In vivo effects of goldenseal, kava kava, black cohosh, and valerian on human cytochrome P450 1A2, 2D6, 2E1, and 3A4/5 phenotypes. Clin. Pharmacol. Ther 77, 415–426 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gurley BJ et al. Supplementation with goldenseal (Hydrastis canadensis), but not kava kava (Piper methysticum), inhibits human CYP3A activity in vivo. Clin. Pharmacol. Ther 83, 61–69 (2008). [DOI] [PubMed] [Google Scholar]
- 11.Gurley BJ et al. Clinical assessment of CYP2D6-mediated herb-drug interactions in humans: Effects of milk thistle, black cohosh, goldenseal, kava kava, St. John’s wort, and Echinacea. Mol. Nutr. Food Res 52, 755–763 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gurley BJ et al. Effect of goldenseal (Hydrastis canadensis) and kava kava (Piper methysticum) supplementation on digoxin pharmacokinetics in humans. Drug Metab. Dispos 35, 240–245 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeng HJ et al. P-glycoprotein-mediated transport of berberine across Caco-2 cell monolayers. J. Pharm. Sci 91, 2614–2621 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Etheridge AS, Black SR, Patel PR, So J and Mathews JM An in vitro evaluation of cytochrome P450 inhibition and P-glycoprotein interaction with goldenseal, Ginkgo biloba, grape seed, milk thistle, and ginseng extracts and their constituents. Planta Med. 73, 731–741 (2007). [DOI] [PubMed] [Google Scholar]
- 15.McDonald MG, Tian D-D, Thummel KE, Paine MF and Rettie AE Modulation of Major Human Liver Microsomal Cytochromes P450 by Component Alkaloids of Goldenseal: Time-Dependent Inhibition and Allosteric Effects. Drug Metab. Dispos 48, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kellogg JJ, Paine MF, McCune JS, Oberlies NH and Cech NB Selection and characterization of botanical natural products for research studies: A NaPDI center recommended approach. Nat. Prod. Rep 36, 1196–1221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wallace ED, Oberlies NH, Cech NB and Kellogg JJ Detection of adulteration in Hydrastis canadensis (goldenseal) dietary supplements via untargeted mass spectrometry-based metabolomics. Food Chem. Toxicol 120, 439–447 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wallace ED, Todd DA, Harnly JM, Cech NB and Kellogg JJ Identification of adulteration in botanical samples with untargeted metabolomics. Anal. Bioanal. Chem 412, 4273–4286 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kellogg JJ, Kvalheim OM and Cech NB Composite score analysis for unsupervised comparison and network visualization of metabolomics data. Anal. Chim. Acta 1095, 38–47 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Birer-Williams C et al. A New Data Repository for Pharmacokinetic Natural Product-Drug Interactions: from Chemical Characterization to Clinical Studies. Drug Metab. Dispos 48, 1104–1112 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heredi-Szabo K, Kis E and Krajcsi P The Vesicular Transport Assay: Validated In Vitro Methods to Study Drug-Mediated Inhibition of Canalicular Efflux Transporters ABCB11/BSEP and ABCC2/MRP2. Curr. Protoc. Toxicol 23, 1–16 (2012). [DOI] [PubMed] [Google Scholar]
- 22.Li CY et al. Major glucuronide metabolites of testosterone are primarily transported by MRP2 and MRP3 in human liver, intestine, and kidney. J. Steroid Biochem. Mol. Biol 191, ePublication (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doki K, Apati S, Sakata T and Hmma M Involvement of Renal Efflux Transporter MATE1 in Renal Excretion of Flecainide. Biol. Pharm. Bull 42, 1226–1229 (2019). [DOI] [PubMed] [Google Scholar]
- 24.Guidance for Industry: In Vitro Drug Interaction Studies — Cytochrome P450 Enzyme- and Transporter-Mediated Drug Interactions Guidance. FDA Guid. Doc (2020). [Google Scholar]
- 25.Kharasch ED, Jubert C, Senn T, Bowdle TA and Thummel KE Intraindividual Variability in Male Hepatic CYP3A4 Activity Assessed by Alfentanil and Midazolam Clearance. J. Clin. Pharmacol 39, 664–669 (1999). [DOI] [PubMed] [Google Scholar]
- 26.Guidance for Industry: Establishing Bioequivalence Guidance for Industry Statistical Approaches to Establishing Bioequivalence. FDA Guid. Doc (2001). [Google Scholar]
- 27.Tian DD et al. Identification of intestinal UDP-Glucuronosyltransferase inhibitors in green tea (Camellia sinensis) using a biochemometric approach: Application to raloxifene as a test drug via in vitro to in vivo extrapolation. Drug Metab. Dispos 46, 552–560 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee D et al. Aromatase inhibitors from Broussonetia papyrifera. J. Nat. Prod 64, 1286–1293 (2001). [DOI] [PubMed] [Google Scholar]
- 29.Nakajima N et al. Developmental changes in multispecific organic anion transporter 1 expression in the rat kidney. Kidney Int. 57, 1608–1616 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Cha SH et al. Identification and characterization of human organic anion transporter 3 expressing predominantly in the kidney. Mol. Pharmacol 59, 1277–1286 (2001). [DOI] [PubMed] [Google Scholar]
- 31.Gupta P, Gurley B, Barone G and Hendrickson H Clinical Pharmacokinetics and Metabolism of Berberine and Hydrastine Following an Oral Dose of Goldenseal Supplement. Planta Med. 76, 110 (2010). [Google Scholar]
- 32.Li G, Zhao M, Qiu F, Sun Y and Zhao L Pharmacokinetic interactions and tolerability of berberine chloride with simvastatin and fenofibrate: An open-label, randomized, parallel study in healthy chinese subjects. Drug Des. Devel. Ther 13, 129–139 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Spinozzi S et al. Berberine and its metabolites: Relationship between physicochemical properties and plasma levels after administration to human subjects. J. Nat. Prod 77, 766–772 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Ho RH et al. Drug and Bile Acid Transporters in Rosuvastatin Hepatic Uptake: Function, Expression, and Pharmacogenetics. Gastroenterology. 130, 1793–1806 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Zhang Y et al. Dissecting the Contribution of OATP1B1 to Hepatic Uptake of Statins Using the OATP1B1 Selective Inhibitor Estropipate. Mol. Pharm 16, 2342–2353 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Motohashi H et al. Gene expression levels and immunolocalization of organic ion transporters in the human kidney. J. Am. Soc. Nephrol 13, 886–874 (2002). [DOI] [PubMed] [Google Scholar]
- 37.Dawed AY et al. Variation in the plasma membrane monoamine transporter (PMAT) (encoded by SLC29A4) and organic cation transporter 1 (OCT1) (encoded by SLC22A1) and gastrointestinal intolerance to metformin in type 2 diabetes: An IMI direct study. Diabetes Care. 42, 1027–1033 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Han T et al. Four cation-selective transporters contribute to apical uptake and accumulation of metformin in Caco-2 cell monolayerss. J. Pharmacol. Exp. Ther 352, 519–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liang X and Giacomini KM Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci 106, 2245–2250 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Han T et al. Organic cation transporter 1 (OCT1/mOct1) is localized in the apical membrane of Caco-2 cell monolayers and enterocytes. Mol. Pharmacol 84, 182–189 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jonker JW and Schinkel AH Pharmacological and Physiological Functions of the Polyspecific Organic Cation Transporters: OCT1, 2, and 3 (SLC22A1–3). J. Pharmacol. Exp. Ther 308, 2–9 (2004). [DOI] [PubMed] [Google Scholar]
- 42.Liang X et al. Metformin Is a Substrate and Inhibitor of the Human Thiamine Transporter, THTR-2 (SLC19A3). Mol. Pharm 12, 4301–4310 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu J and Ragueneau-Majlessi I In Vitro‐to‐In Vivo Extrapolation of Transporter Inhibition Data for Drugs Approved by the US Food and Drug Administration in 2018. Clin. Transl. Sci 13, 693–699 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamek-Gliszczynski MJ et al. Intestinal P-gp and Putative Hepatic OATP1B Induction: International Transporter Consortium Perspective on Drug Development Implications. Clin. Pharmacol. Ther (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoshikado T et al. PBPK Modeling of Coproporphyrin I as an Endogenous Biomarker for Drug Interactions Involving Inhibition of Hepatic OATP1B1 and OATP1B3. CPT Pharmacometrics Syst. Pharmacol 7, 739–747 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barnett S et al. Comprehensive evaluation of the utility of 20 endogenous molecules as biomarkers of OATP1B inhibition compared with rosuvastatin and coproporphyrin I. J. Pharmacol. Exp. Ther 368, 125–135 (2019). [DOI] [PubMed] [Google Scholar]
- 47.Agarwal A, Jadhav P and Deshmukh Y Prescribing pattern and efficacy of anti-diabetic drugs in maintaining optimal glycemic levels in diabetic patients. J. Basic Clin. Pharm 5, 79–83 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yin J, Xing H and Ye J Efficacy of berberine in patients with type 2 diabetes mellitus. Metabolism. 57, 712–717 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hua W et al. Determination of berberine in human plasma by liquid chromatography-electrospray ionization-mass spectrometry. J. Pharm. Biomed. Anal 44, 931–937 (2007). [DOI] [PubMed] [Google Scholar]
- 50.Yang J, Jamei M, Yeo K, Tucker G and Rostami-Hodjegan A Prediction of Intestinal First-Pass Drug Metabolism. Curr. Drug Metab 8, 676–684 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.