

Abstract
Tumor necrosis factor alpha (TNF) has sleep regulatory and brain development roles. TNF promotes sleep in vivo and in vitro while TNF inhibition diminishes sleep. Transmembrane (tm) TNF and the tmTNF receptors (Rs), are cleaved by tumor necrosis factor alpha convertase to produce soluble (s) TNF and sTNFRs. Reverse signaling occurs in cells expressing tmTNF upon sTNFR binding. sTNFR administration in vivo inhibits sleep, thus we hypothesized that a wake-like state in vitro would be induced by sTNFR-tmTNF reverse signaling. Somatosensory cortical neuron/glia co-cultures derived from male and female mice lacking both TNFRs (TNFRKO), or lacking TNF (TNFKO) and wildtype (WT) mice were plated onto six-well multi-electrode arrays. Daily one-hour electrophysiological recordings were taken on culture days 4 through 14. sTNFR1 (0.0, 0.3, 3, 30, 60, and 120 ng/µL) was administered on day 14. A final one-hour recording was taken on day 15. Four measures were characterized that are also used to define sleep in vivo: action potentials (APs), burstiness index (BI), synchronization of electrical activity (SYN), and slow wave power (SWP; 0.25–3.75 Hz). Development rates of these emergent electrophysiological properties increased in cells from mice lacking TNF or both TNFRs compared to cells from WT mice. Decreased SWP, after the three lowest doses (0.3, 3 and 30 ng/µL) of the sTNFR1, indicate a wake-like state in cells from TNFRKO mice. A wake-like state was also induced after 3 ng/µl sTNFR1 treatment in cells from TNFKO mice, which express the TNFR1 ligand, lymphotoxin alpha. Cells from WT mice showed no treatment effects. Results are consistent with prior studies demonstrating involvement of TNF in brain development, TNF reverse signaling, and sleep regulation in vivo. Further, the current demonstration of sTNFR1 induction of a wake-like state in vitro is consistent with the idea that small neuronal/glial circuits manifest sleep- and wake-like states analogous to those occurring in vivo. Finally, that sTNF forward signaling enhances sleep while sTNFR1 reverse signaling enhances a wake-like state is consistent with other sTNF/tmTNF/sTNFR1 brain actions having opposing activities.
Keywords: Reverse signaling, Tumor necrosis factor, TNF, TNF receptor, Sleep, Wake, Brain development, Local sleep, Cytokine, Tissue culture, In vitro sleep, Interleukin-1
1. Introduction
Substantial evidence indicates that tumor necrosis factor alpha (TNF) is involved in sleep regulation in animals and humans (Rockstrom et al., 2017). Thus, injection of the 17 kD soluble form of TNF into rabbits (Kapas et al., 1992; Kapas and Krueger, 1992), rats (De Sarro et al., 1997; Kubota et al., 2002; Nistico, 1992), or mice (Fang et al., 1997) induces excess non-rapid eye movement sleep (NREMS). Mice lacking one or both TNF receptors (Rs) sleep less than wild type (WT) mice (Fang et al., 1997; Kapas et al., 2008). Brain or blood levels of TNF or TNF mRNA vary with sleep propensity, e.g. after sleep deprivation, TNF increases in brain (Taishi P, 1999) or blood (Haack et al., 2004). TNF protein concentrations are about 10-fold higher in the cerebral cortex and hippocampus, at the onset of light hours, the beginning of the rat sleep phase, compared to levels at the onset of dark hours (Floyd and Krueger, 1997). TNF is produced by neurons (Jewett et al., 2015) and glia (Frei et al., 1987; Ingiosi et al., 2013) and its production in neurons is neuron activity-dependent. Thus, afferent activity into the somatosensory cortex enhances TNF expression (Churchill et al., 2005) and in vitro optogenetic stimulation enhances neuronal expression of TNF immunoreactivity (Jewett et al., 2015). Conditions or substances that induce TNF production also are often associated with periods of excessive sleep, e.g. infection (Kapas et al., 2008), or injection of endotoxin (Layé et al., 1994; Takahashi et al., 1996a). TNF can act on sleep regulatory circuits to promote sleep, e. g. the anterior hypothalamus (Kubota et al., 2002). Systemic injections of somnogenic amounts of TNF promote brain production of TNF mRNA (Zielinski et al., 2013). TNF can also act on local neuronal/glial networks to intensify local sleep states. Thus, injection of TNF unilaterally onto the surface of the cortex enhances electroencephalogram (EEG) slow wave power (SWP; 0.5 – 4 Hz) ipsilaterally, suggesting a more intense sleep state. In contrast, inhibition of TNF by unilateral injection of TNF siRNAs reduces EEG SWP ipsilaterally and TNF immunoreactivity in cortical pyramidal neurons (Taishi et al., 2007). Further, if TNF is applied to individual cortical columns, the columns express a sleep-like state in that evoked response potentials (ERPs) induced by facial whisker stimulation are higher in amplitude (Churchill et al., 2008). Cortical ERPs are greater in magnitude during sleep than during waking states (Rector et al., 2005). Similarly, in vitro cultures of neurons/glia respond to low doses of TNF by exhibiting greater SWP values suggesting a deeper sleep-like state (Jewett et al., 2015).
TNF is initially produced as a 26 kD transmembrane (tm) protein. A soluble 17 kD TNF (sTNF) is cleaved from the 26 kD TNF by TNF converting enzyme (TACE) releasing it into the extracellular space (Black et al., 1997). Within the cerebral cortex, tmTNF is the dominate form present (Churchill et al., 2008). There are two TNFRs, a 55 kD TNFR and a 75 kD TNFR. Both are involved in sleep regulation (Fang et al., 1997; Kapas et al., 2008; Taishi P, 1999). The 55kD TNFR is found in most cells while the 75kD TNF form is in brain found on microglia and endothelial cells (Mccoy and Tansey, 2008; Probert, 2015). Like the TNF ligand, the TNFRs are initially produced as a transmembrane protein and the extracellular moieties of both Rs can be cleaved by TACE (Edwards et al., 2008; Peschon et al., 1998a; Reddy et al., 2000). TACE can be upregulated in a cell-specific manner by various chemokines, cytokines and stimuli, including: TNF (Bzowska et al., 2004; Ge and Vujanovic, 2017), interleukin (IL)1β (Tachida et al., 2008), IL10 (Brennan et al., 2008), hypoxic inducible factor 1 (Charbonneau et al., 2007; Li et al., 2015), oxygen-glucose deprivation (Hurtado et al., 2001), and ischemic preconditioning (Bigdeli et al., 2009; Cárdenas et al., 2002). Soluble (s) Rs are released into the extracellular space (Dostert et al., 2019). Although both membrane bound and soluble TNFRs are active as trimers (Aggarwal, 2003; Banner et al., 1993; Chan et al., 2000), soluble 55 kD TNFR (sTNFR1) forms dimer complexes called pre-ligand assembly domains (PLADs) which, upon ligand binding, undergo conformational changes which may act as a molecular switch for a particular response (Lo et al., 2020; Naismith et al., 1995). The exact natures of tmTNFRs and sTNFRs signaling are dependent upon the intracellular recruitment of adapter proteins. The specific TNFR, adapter protein, and spatial orientation of R-ligand complexes determine target responses, which can range from induction of cell protection to cell death (Dostert et al., 2019; Ledgerwood et al., 1999; Lo et al., 2020; Mccoy and Tansey, 2008). The sTNFRs have biological activities. Thus, sTNFRs can bind extracellular 17kD TNF preventing TNF from binding to the tmTNFRs. In contrast, the sTNFRs can also bind to tmTNF which in turn initiates cell signaling within the cell producing the tmTNF (Mccoy and Tansey, 2008; Watts et al., 1999). This latter action is called reverse signaling. Herein, we describe reverse signaling of sTNFR1 in mixed mature neuronal/glial cultures obtained from WT, TNF knockout (KO), and TNFR1/TNFR2KO (TNFRKO) mice.
Over two decades ago, the sTNFR1 derived from the 55kD TNFR and a synthetic fragment of it containing the TNF binding site were shown to inhibit TNF cytotoxicity on mouse L-M cells and rabbit sleep (Takahashi et al., 1995). Thus, both substances inhibited rabbit spontaneous NREMS after intracerebroventricular injections. Similar experiments in rabbits showed that the soluble synthetic TNFR fragment also inhibited sleep rebound after sleep deprivation (Takahashi et al., 1996a, 1996b) and the increases in sleep that accompany mild increases in ambient temperatures (Takahashi and Krueger, 1997). Finally, the peptide TNFR fragment also inhibited IL1β-induced sleep, but not fever, responses (Takahashi et al., 1999). At the time of those experiments, TNF reverse signaling was unknown and results were interpreted within the context of the soluble receptor binding extracellular TNF thus lowering its effective concentration. Within the past few years TNF reverse signaling has been demonstrated within the larger TNF family of molecules (reviewed (Lee et al., 2019)). Although the TNF reverse signaling literature dealing with brain is relatively small, TNF reverse signaling has been shown to promote axon growth (Kisiswa et al., 2013). More important for the studies described herein, blocking sTNF protected mice against clinical symptoms in an autoimmune encephalomyelitis murine model (Taoufik et al., 2011). Yet if both sTNF and tmTNF were blocked no protection was provided suggesting that sTNF and tmTNF had opposing actions. There is also a hint that tmTNF may play a role in sleep/wake regulation. For example, the neuropeptide, substance P, enhances expression of tmTNF without increasing release of sTNF (Zhou et al., 2010). Interestingly, substance P also increases waking bouts and latency and reduces sleep efficiency in mice (Andersen et al., 2006). Herein, we provide evidence that TNF reverse signaling enhances a wake-like state in vitro.
Previously, an in vitro neuronal/glial cell culture model of sleep was developed (Corner, 2013; Hinard et al., 2012; Jewett et al., 2015; Krueger et al., 2019; Saberi-Moghadam et al., 2018). In such cultures the spontaneous default state is sleep-like in that neuron action potentials (APs) exhibit a burst-pause firing pattern by day four in vitro (Kamioka et al., 1996) that is analogous to the neuronal firing patterns observed during sleep in vivo (Corner, 2013; Hinard et al., 2012; Jewett et al., 2015; Timofeev et al., 2001), suggesting that sleep is the default state of cultured neurons. TNF treatment of these cultures induces a deeper sleep-like state in that SWP (0.5 – 3.5 Hz) is enhanced (Jewett et al., 2015). This measure is also used in vivo to characterize sleep and sleep intensity (Davis et al., 2011). Such cultures also demonstrate sleep homeostasis in that treatments that enhance AP burstiness or SWP, are followed by periods of enhanced sleep-like states (Jewett et al., 2015; Saberi-Moghadam et al., 2018). Further, optogenetic neuronal stimulation of these cultures enhances TNF expression (Jewett et al., 2015). Herein we use similar cultures of cells from wild type (WT), TNFKO, and TNFRKO mice. The lack of TNF or the TNFRs affects culture development rate. Further, treating TNFKO cells or TNFRKO cells in culture with a sTNFR inhibits SWP suggesting an induction of a wake-like state in culture. These findings help our understanding of some of the conflicting TNF-sleep reports, e.g. sTNF induces sleep yet inhibition of TNF by thalidomide induces drowsiness (Rockstrom et al., 2017).
2. Materials and methods
2.1. Primary culture preparation and maintenance
Wildtype mice (WT; C57BL/6J), TNFKO mice (B6.129S-Tnftm1Gkl (Pasparakis et al., 1996)) and TNFRKO mice (B6.129S-Tnfrsf1atm1Imx Tnfrsf1btm1Imx/J (Peschon et al., 1998b)), were purchased from The Jackson Laboratory (JAX stock numbers: 000664, 005540, and 0003243, Bar Harbor, ME, USA). The TNFKO mice lacked TNF alpha while the TNFRKO mice lacked both the p55 (TNFR1) and p75 (TNFR2) receptors. The Washington State University (WSU) Institutional Animal Care and Use Committee (IACUC) approved animal procedures that complied with National Institutes of Health (NIH) guidelines. Homozygous mice were bred for two to six generations in the WSU vivarium in a separate breeding room maintained at 24 ° C ± 2 °C, 40% ± 10% humidity on a 14 h light/10 h dark schedule with lights on at Zeitgeber time zero (ZT0). Somatosensory cortical tissue samples were obtained from one- to two-day old male and female mouse pups at ZT3 ± 1 h. However, mouse pups were differentiated only by strain as sex is difficult to determine at this early stage of development. As previously described (Jewett et al., 2015; Nguyen et al., 2019b), cells were dispersed and seeded onto 6-well multi-electrode arrays (MEAs; Multi-Channel Systems, Reutlingen, Germany). Briefly, immediately after harvesting, tissue was placed in ice-cold Hibernate-E (HE, BrainBits, Springfield, IL), then digested for 20 min at 37 °C with papain (2 mg/mL; LS003119, Worthington Biomedical, Lakewood, NJ); triturated in HE through a syringe twice with a 20-gauge needle and once with a 22-gauge needle; and passed through a 40 µm strainer before centrifugation at 200g for 10 min at room temperature. A warm media solution comprised of Dulbecco’s Modified Eagle Serum (DMEM; D6429, Sigma, St. Louis, MO) with 10% fetal bovine serum (FBS), 1% GlutaMAX ™, and 2% penicillin/streptomycin (pen-strep) dissolved the cell precipitate.
After counting, cells were plated at a concentration of 2 × 105 cells/20 µL onto individual MEA wells previously coated with poly-D-lysine (P6407, Sigma). MEAs with cells were placed in a 5% CO2 for 50 min at 37 °C before adding 50 µL/well of warm DMEM/FBS/Glutamax/pen-strep media and allowed to rest for another 4 h in the incubator. After 4 h, 350 µL media solution of NbActiv4 (Nb4–100, BrainBits), 2% pen-strep, 0.2% Gentamycin (15710–064, Invitrogen) was added to each MEA well. To prevent evaporation, Teflon membranes (ALA Scientific, Farmingdale, NY) covered the MEAs throughout the experiments. Approximately every three to four days, approximately half of the media was removed from the wells and replaced with fresh NbActiv4/pen-strep/Gentamycin media.
2.2. MEA recording
Electrophysiological cell recordings were taken in a dry incubator (37 ° C; 5% CO2) with a MEA600BC system (Multi Channel Systems). Each MEA (MEA1060-Inv-BC) contained 6 independent wells each with 9 titanium nitride recording electrodes spaced 200 µm apart in a 3x3 grid (Fig. 1A) and 1 silicon nitride substrate-integrated reference electrode. Relative to its respective ground electrode, each electrode’s voltage was recorded at a sampling rate of 10 kHz. Consecutive daily one-hour recordings were taken beginning on the fourth day after plating (Jewett et al., 2015; Kamioka et al., 1996; Nguyen et al., 2018, 2019b) and continuing through day (D) 15. All raw data were stored on the recording computer and copied to an external hard drive for later analyses of developmental and sTNFR1-induced changes.
Fig. 1.
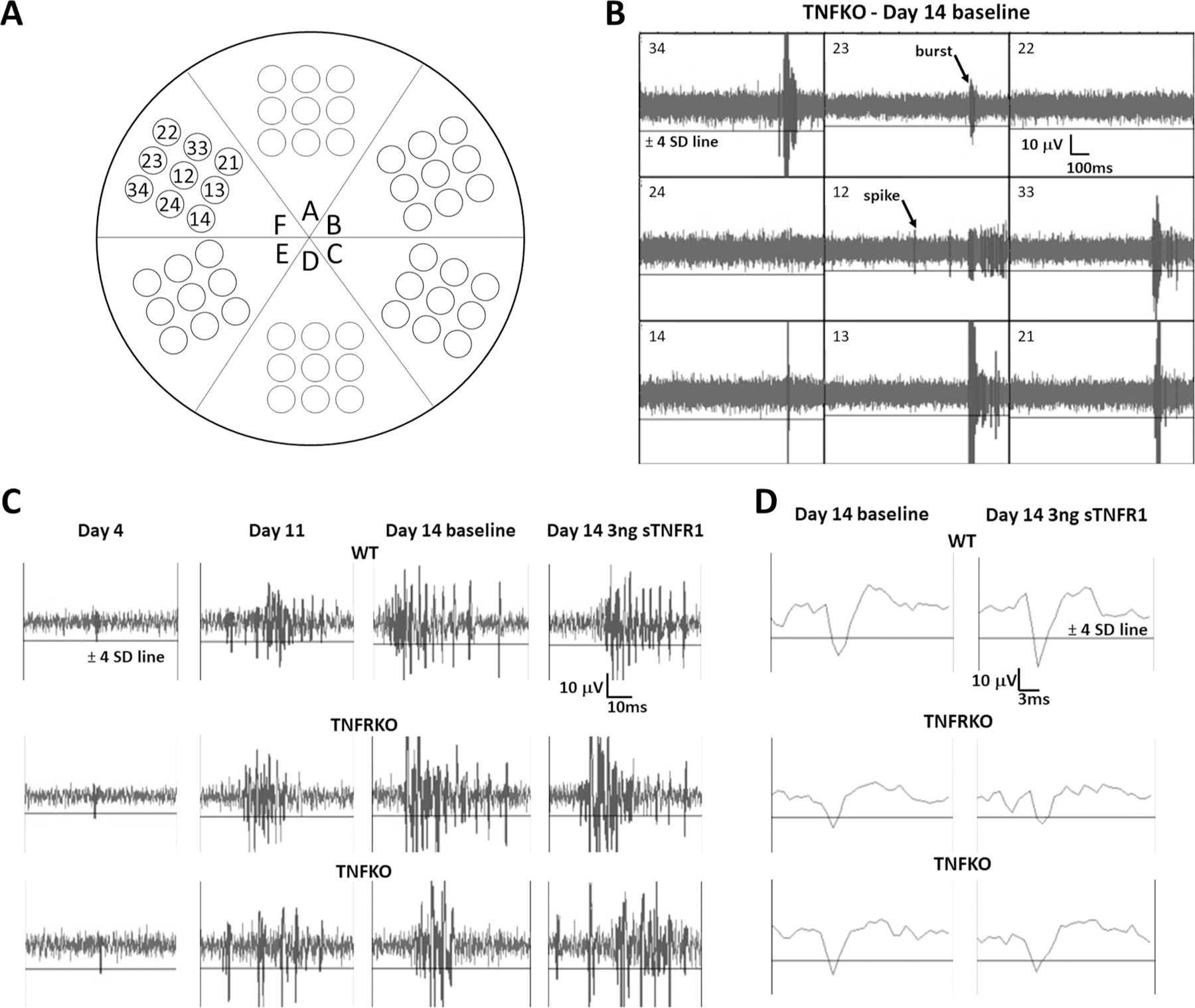
Multi-electrode array (MEA) analyses of spontaneous electrical activity in neuronal/glial co-cultures. A. Configuration cartoon of electrodes in 6-well MEAs. Electrodes are numerically identified for single well, F. B. Representative single-channel raw baseline data for all nine electrodes in well F (refer to (A) for configuration) using a 200-Hz high-pass filter on day (D)14 for cells from mice lacking tumor necrosis factor (TNFKO). Electrodes are identified in the top left corners of the raw data windows and each electrode’s data was analyzed individually. The AP threshold line of −4 standard deviations (SDs) from the baseline average signal is identified only for electrode 34 but shown in all electrode windows. A representative burst is identified on electrode 23 and a representative spike is identified on electrode 12. It is emphasized that electrical activity recorded from each electrode within the well is unique and, therefore, considered as an independent sample. C. Representative single-channel raw data for cells of all strains (wildtype (WT), top row; TNF double receptor knockout (TNFRKO), middle row; TNFKO, bottom row) on development D4 (far left column), development D11, D14 pre-treatment baseline and D14 after treatment with 3 ng soluble TNF receptor 1 (sTNFR1; far right column). D. Representative single action potentials for all three strains (WT, top; TNFRKO, middle; and TNFKO, bottom) for D14 pre-treatment baseline (left column) and D14 post-treatment with 3 ng sTNFR1 (right column). The horizontal line in all windows is the AP threshold of −4 SD from baseline average signal.
2.3. MEA data analyses
MEA data were analyzed to determine several output measures that were chosen because they have been previously described in the literature (Corner, 2008; Hinard et al., 2012; Jewett et al., 2015; Jimbo et al., 1998; Nguyen et al., 2019b; Wagenaar et al., 2005) and are analogous to measures that are used to identify sleep in vivo. These measures include: action potentials per second (APs/sec), an AP burstiness index (BI), synchronization (SYN) of electrical activity between electrodes, and fast Fourier transformation (FFT) of the slow wave component (0.25–3.75 Hz) to obtain slow wave power (SWP).
Output measures on D4 through D15 and after sTNFR1 treatments were analyzed using MC_Rack software (©MultiChannel Systems, Germany) and MATLAB® as previously described (Jewett et al., 2015; Nguyen et al., 2019b). Briefly, the raw signal was filtered using a 200 Hz-high pass second order Butterworth filter with a set limit of ± 4 standard deviations (SDs;(Jimbo et al., 2000) from the mean voltage for each independent electrode to calculate APs. An AP was registered when filtered electrical activity passed the ± 4 SD threshold. See Fig. 1A–C for representative raw data spikes and individual action potentials. We previously determined that APs/sec exponentially decrease during the first half hour of recording (Jewett et al., 2015), likely due to disturbances in the MEA associated with its movement to the recording chamber. To exclude these disturbances, we analyzed only the last 5 min of the hour-long baseline recordings. Those 5 min periods were used to determine baseline values for each electrode. When cells were treated on D14 (see Experiment (Expt.) 2 below), the sTNFR1 was administered to the wells immediately after the baseline recording and then an additional full 1-hour recording was taken and analyzed.
A burst was defined by: (a) at least 4 APs in the first 10 msec, (b)a maximum of 10 msec between APs throughout the burst, (c) at least 10 msec between individual bursts, and (d) a minimum burst duration of 20 msec. The BI (over 5 min periods) was calculated first by dividing the 5 min period into 300 one-sec epochs and summing the number of APs in the top 15% of the epochs containing the most APs (Wagenaar et al., 2005). This number of APs was then divided by the total number of APs in that same 5 min period (Nguyen et al., 2019b; Wagenaar et al., 2005). Thus, BI reflects the portion of APs in bursts and provides a more refined illustration of the structure of AP bursts where higher BI values indicate greater burstiness and lower BI values indicate more random firing.
To determine exclusion criteria for AP and BI developmental data analyses (D4 to D14; Expt. 1), we first used the filtered signal data that previously passed the ± 4 SD threshold to determine the average APs/sec of electrodes in MEA wells that contained media but no cells. Recordings from those cell-less MEAs had an average of 0.21 APs/sec ± one SD (0.15 – 0.27 APs/sec). Thus, electrodes with<0.27 APs/sec on culture D14 and, any electrodes displaying both a decreasing trend in APs/sec from D10 through D14 and with less than one burst were considered to have poor development and were subsequently excluded from further analyses of electrical activity from MEA wells containing cells. Further, values from individual electrode recordings containing obvious artifacts were eliminated; artifacts were determined by reviewing the recorded data and from notes written during the recordings. Additionally, after applying the exclusion criteria, any MEA well with less than half of its total electrodes remaining was also excluded. For analyses of sTNFR1 treatment effects (Expt. 2), only electrodes with 0.25 APs/sec or more during D14 baseline recordings were included. All post-sTNFR1 treatment data analyses on D15 were performed using the same electrodes as the D14 analyses.
SW activity data were extracted using low pass/high pass (100 Hz/0.1 Hz) second order Butterworth filters with 100 Hz data downsampling (10 msec bins). SYN of SW activity between electrodes was calculated as follows. First, pairwise correlation coefficients were calculated for each pair of separate adjacent electrodes in an MEA well using the formula , where V1(t), V2(t) represent voltages at time t from the two electrodes; , , represent sample voltage means ; and σv1, σv2, represent sample SDs. The SW SYN value was then computed by averaging these pairwise correlation coefficients, for all pairs of adjacent electrodes in the MEA well. It is noted that the SW SYN ranges between −1 and + 1. High SYN values indicate greater synchrony between electrodes. Electrodes used for SW analyses were the same as those used for AP/BI analyses.
After filtration and retention of SW activity, the data were subjected to FFT to obtain spectra from randomly sampled active network nodes using a custom MATLAB® program with a cosine tapered window over the 5 min baseline period (D4 to D14) or across pre- and post-sTNFR1 treatment periods (D14 and D15). FFT was calculated for the full 60-min period immediately following TNFR1 treatment on D14 (except for the time course experiments shown in Fig. 6 where data for successive 5 min time blocks were used. For SWP determinations, we binned the range of 0.25–30.25 Hz spectral content into 0.5 Hz frequency bands and then the delta frequency was derived by adding the µV2 values from the 7 individual bins in the range of 0.25–3.75 Hz.
Fig. 6.
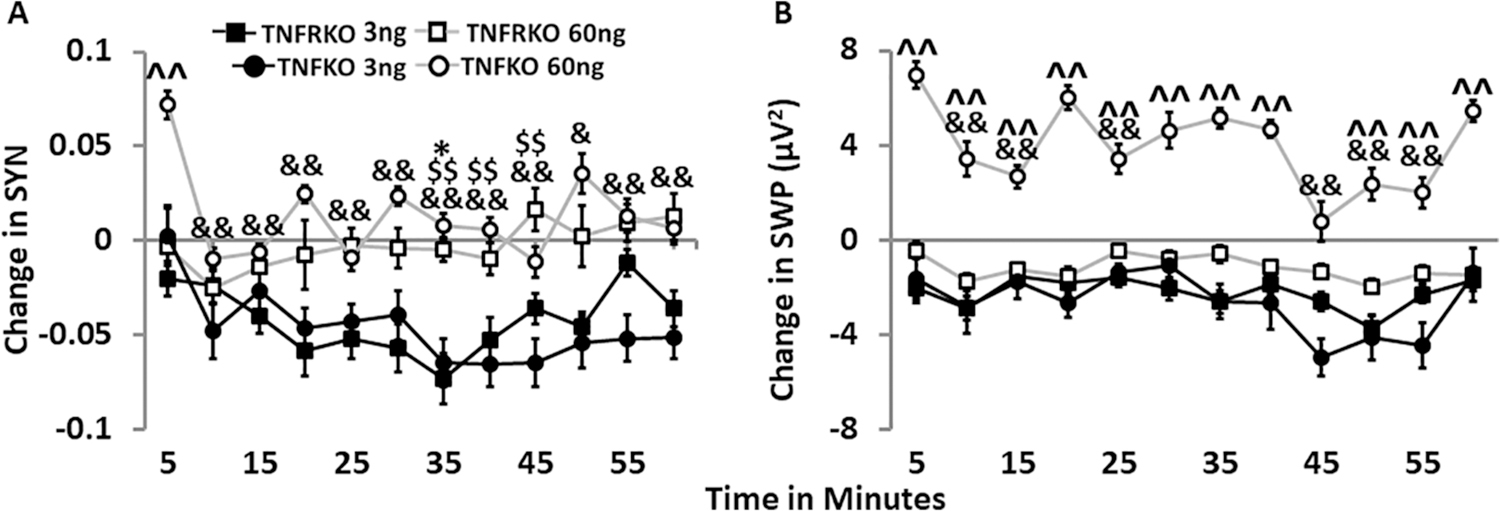
Time course of SYN and SWP after the soluble tumor necrosis factor receptor 1 (TNFR1) treatment (3 ng/µL; 60 ng/µL) in cells from mice lacking both TNFRs (TNFRKO) and cells from mice lacking TNF (TNFKO) on day 14. For each strain, sTNFR1 treatment dose, and parameter, graphically depicted values are means ± standard errors (SEs) of changes in synchrony (SYN) and slow wave delta power (SWP) every five minutes over the course of one hour. Numbers of preparations, MEAs, wells, and electrodes are presented in Table 1. A. Time course of SYN changes after treatment with either 3 ng/µL or 60 ng/µL sTNFR1 in TNFKO and TNFRKO cells. B. Time course of SWP (µV 2, 0.25–3.75 Hz) changes after treatment with either 3 ng/µL or 60 ng/µL sTNFR1 in TNFKO and TNFRKO cells. Changes in each measure were compared first to their day 14 baseline values and then to the sTNFR1 control treatment change from baseline mean for each 5 min time period individually within the same MEA and strain as described in the methods section. Significance of time-induced changes (p < 0.05) was determined by one-way ANOVA (time) followed by post-hoc Tukey’s HSD tests when significant main effects were found within each strain, dose, and parameter. Significant effects of time, strain and time by strain interactions were determined by two-way ANOVA (time, strain, and time × strain) and, where indicated, post-hoc Tukey’s HSD tests within each dose for each parameter. Significant strain differences were found for 60 ng/µL dose only: TNFRKO vs TNFKO: ^^p < 0.01. Within strain and dose, significant time differences vs 5 min: TNFRKO 3 ng, *p < 0.05; TNFKO 3 ng, $ $p < 0.01; TNFKO 60 ng, &p < 0.05, &&p < 0.01. Within strain and dose, significant time differences are reported in the results section.
2.4. Experiment 1: Development of spontaneous electrical activity in neuronalglial co-cultures.
Somatosensory neuron/glia co-cultures from three mouse strains were plated separately onto 6-well MEAs (Table 1). Using microscopy, individual MEA wells were observed 24 h after plating and then repeatedly every 2–3 days after media changes to ensure cell growth and development. Each individual electrode within each MEA well was considered an independent sample (n) since varied cell spatial distribution throughout the developmental period gave the electrodes unique patterns of electrical activity (see Fig. 1B for representative single-well individual electrode raw data). Daily one-hour recordings of electrical activity were taken from each MEA beginning on D4 and continuing through D14. Because electrophysiological changes occurred in response to the addition of fresh media, values obtained on the day following media changes were excluded from analyses. Developmental changes were determined by comparing each day’s parameter for each electrode to the first recording on D4. The average plus/minus standard error (SE) across all MEAs are reported and graphically presented for each measure, strain, and days in vitro. Statistical significances (p < 0.05) between days within each strain were determined by using one-way ANOVA (day) followed by post-hoc Tukey’s honestly significant differences (HSD) tests when significant main effects were found. Significant effects of day, strain, and day by strain interactions were also determined by two-way ANOVA (day and strain) and, where indicated, post-hoc Tukey’s HSD tests were used. Statistical analyses reported for this and all other experiments were performed using RStudio, version 1.3.959 (Team, 2020).
Table 1.
Numbers of preparations, MEAs, wells and electrodes for each strains and experiment.
| Experiment | Cell Strains | Preparations | MEAs | Wells | Electrodes |
|---|---|---|---|---|---|
| Experiment 1: | TNFRKO | 8 | 10 | 51 | 326 |
| TNFKO | 8 | 16 | 73 | 449 | |
| WT | 10 | 19 | 71 | 432 | |
| Experiment 2: sTNFR1 D14 | TNFRKO | 4 | 8 | 32 | 146 |
| WT | 3 | 8 | 34 | 124 | |
| Experiment 2: sTNFR1 D15 | TNFRKO | 4 | 8 | 33 | 147 |
| WT | 3 | 8 | 34 | 126 | |
| Experiment 3: sTNFR1 D14 | TNFKO | 1 | 4 | 23 | 177 |
| Experiment 3: sTNFR1 D15 | TNFKO | 1 | 4 | 23 | 175 |
| Experiment 3: sTNFR1 D14 time course | TNFRKO | 4 | 8 | 20 | 94 |
| TNFKO | 1 | 4 | 13 | 103 |
Abbreviations: TNFRKO – tumor necrosis factor double receptor knockout; TNFKO – tumor necrosis factor alpha knockout; WT – wildtype; sTNFR1 – soluble tumor necrosis factor receptor 1; D – day; MEA – multielectrode array.
2.5. Experiment 2: Addition of the sTNFR1 to WT and TNFRKO cells
2.5.1. Effects of sTNFR1 on electrical activity on developmental day 14
Data from cells derived from TNFRKO and WT mice (Table 1) were used to examine the effects of the sTNFR1 on APs/sec, BI, SYN and SWP to characterize network activity. Various treatment doses (0.0 ng/µL (control), 0.3 ng/µL, 3 ng/µL, 30 ng/µL, 60 ng/µL and, 120 ng/µL (5.71 pM, final high dose concentration) were prepared by dissolving and diluting the sTNFR1 (425-R1–050/CF, R&D Systems, Minneapolis, MN) in phosphate-buffered saline. Hour-long baseline recordings (only the last 5 min was analyzed) were taken on D14 as described in Expt. 1 prior to adding one of the various treatment doses of sTNFR1 in a volume of one µL to individual MEA wells. Immediately after treatment, recording resumed for an additional hour.
APs/sec and BI were calculated as fold change values. Briefly, each electrode value within each treatment dose, including control, sTNFR1, was first divided by its own baseline electrode value for baseline-normalization. All the baseline-normalized control treatment electrode values were then averaged within the same MEA well and strain. Each baseline-normalized non-control treatment electrode value within the same MEA well and strain was divided by its own average baseline-normalized control treatment electrode value to get the fold change values for each parameter. Finally, the average fold change values across all MEAs with the same dose and strain were reported and graphically presented ± SE separately for AP and BI.
SYN and SWP were calculated as change values; SYN values were correlation coefficients and SWP values were µV2. Briefly, for all treatment doses, including control, absolute differences from baseline were calculated by first subtracting each electrode’s baseline value from its own treatment dose value. Then, the difference from baseline values were averaged for control treatment electrodes within the same MEA well and strain. This control average of difference value was then subtracted from each non-control treatment electrode’s difference from baseline value within its own MEA and strain. Finally, the change values across all MEAs with the same dose and strain were averaged and graphically presented ± SE separately for SYN and SWP.
Significance of the sTNFR1 treatment–induced changes (p < 0.05) was determined by one-way ANOVA (dose) followed by post-hoc Tukey’s HSD tests when significant main effects were found within each strain. Significant effects of dose, strain, and dose by strain interactions were also determined by two-way ANOVA (strain and dose) and, where indicated, post-hoc Tukey’s HSD tests were used.
2.5.2. Effects of the sTNFR1 on electrical activity on developmental day 15
To determine if the actions of the sTNFR1 treatment were long- or short-term, a one-hour recording was taken 24 h after treatment and all four output measures were calculated. Data from cells derived from TNFRKO and WT mice cells were used (Table 1). The last 5 min of the hour-long recordings were analyzed respectively for D14 and D15 and then they were compared. The D15 data were determined in a similar manner to that described for D14 experiment above but with changes in D15 data calculated with respect to D14 baseline data. The average values across all MEAs were reported and graphically presented ± SE for each measure, strain, and dose. Significance of dose-induced changes (p < 0.05) was determined by one-way ANOVA (dose) followed by post-hoc Tukey’s HSD tests when significant main effects were found within each strain. Significant effects of dose, strain, and dose by strain interactions were also determined by two-way ANOVA (strain and dose) and, where indicated, post-hoc Tukey’s HSD tests.
2.6. Experiment 3: Effects of the sTNFR1 on TNFKO cells on D14 and D15
2.6.1. Effects of the sTNFR1 on electrical activity in TNFKO cells on developmental days 14 and 15
Data from cells derived from TNFKO mice (were used to analyze the effects of the sTNFR1 after treatment (Table 1). TNFKO cells were treated with various doses of the sTNFR1 (control, 3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL), and were recorded and analyzed on D14 and D15 as described in Expt. 2. Average values across all MEAs were reported and graphically presented ± SE for each measure, dose and post-sTNFR1 treatment time (1 h and 24 h). Significance of dose-induced changes (p < 0.05) was determined by one-way ANOVA (dose) followed by post-hoc Tukey’s HSD tests when significant main effects were found within each post-sTNFR1 treatment time and parameter.
2.6.2. Time course of the sTNFR1 effects on SYN and SWP in TNFRKO and TNFKO cells on day 14
Data from cells derived from TNFRKO and TNFKO mice (Table 1) were used to analyze the effects of the sTNFR1 on SYN and SWP in 5 min time blocks for 1 h after treatment with 3 ng/µL and 60 ng/µL sTNFR1. Results were graphically depicted as average values across all MEAs with the same dose, time, and strain ± SE for each measure. Methods for calculating SYN and SWP changes were like Expt. 2 methods reported above except that changes were calculated for each 5 min time period individually.
For change from baseline values for all treatment doses, including control sTNFR1, each baseline electrode value was first subtracted from its own treatment dose electrode value from each 5 min time period. All the control treatment change from baseline electrode values from the same 5 min period were then averaged within the same MEA and strain. Lastly, this averaged value was then subtracted from each control treatment electrode’s change from baseline value from the same 5 min time-period within its own MEA and strain. Finally, the change values across all MEAs with the same dose, time, and strain were averaged and graphically presented ± SE separately for SYN and SWP. Significance of time-induced changes (p < 0.05) was determined by one-way ANOVA (time) followed by post-hoc Tukey’s HSD tests when significant main effects were found within each strain, dose, and parameter. Significant effects of time, strain and time by strain interactions were determined by two-way ANOVA (time and strain) and, where indicated, post-hoc Tukey’s HSD tests within each dose and time for each parameter.
3. Results
3.1. Experiment 1: Characterization of emergent spontaneous electrical activity
The development of electrophysiological properties within neuronal/glial co-cultures during D4-D14 were characterized. Cells from WT, TNFRKO mice and TNFKO mice were used to determine whether the absence of TNF or its receptors, TNFR1 and TNFR2, alter the timing of emergent spontaneous electrical activity. Strain-specific numbers of preparations, MEAs, wells and electrodes are presented in Table 1. Strain-specific daily numbers of electrodes and spontaneous electrical activity means ± SE values are presented in Supplementary Table 1.We previously reported that APs/sec increase over time from D4 through D14 with increases beginning around D8 or D9 in developing co-cultures of neurons and glia from somatosensory cells obtained from WT mice (Jewett et al., 2015; Nguyen et al., 2019b). The current research confirms these reports. In all three strains, APs/sec increased across D4-D14 (Fig. 2A, Supplementary Table 1). Within strain one-way ANOVA analyses indicated effects during D5 through D14 compared to D4 in all three strains (FWT(10,2562) = 30.47, p < 2E-16; FTNFRKO(10,2213) = 33.64, p < 2E-16; FTNFKO(10,3374) = 103.30, p < 2E-16). Two-way ANOVA analyses used to determine day and strain differences revealed main effects of strain (F(2,8149) = 64.91, p < 2E-16), day (F(10,8149) = 144.90, p < 2E-16), and interaction of day and strain (day × strain; F(20,8149) = 6.07, p < 2E-16). TNFKO cells were the fastest to develop spontaneous electrical activity, with higher APs/sec on D8 (p < 0.05). TNFRKO cells were somewhat slower to develop APs, showing more APs/sec (p < 0.01) on D9. WT cells were slowest to develop, not showing increases in APs/sec until D11 (p < 0.01). Although significantly different from WT cells only on D10 and D12 through D14 (p < 0.01), TNFKO cells had more APs/sec than cells from the other two strains beginning on D5 and continuing throughout the developmental period, suggesting a role for TNF in moderating brain development.
Fig. 2.
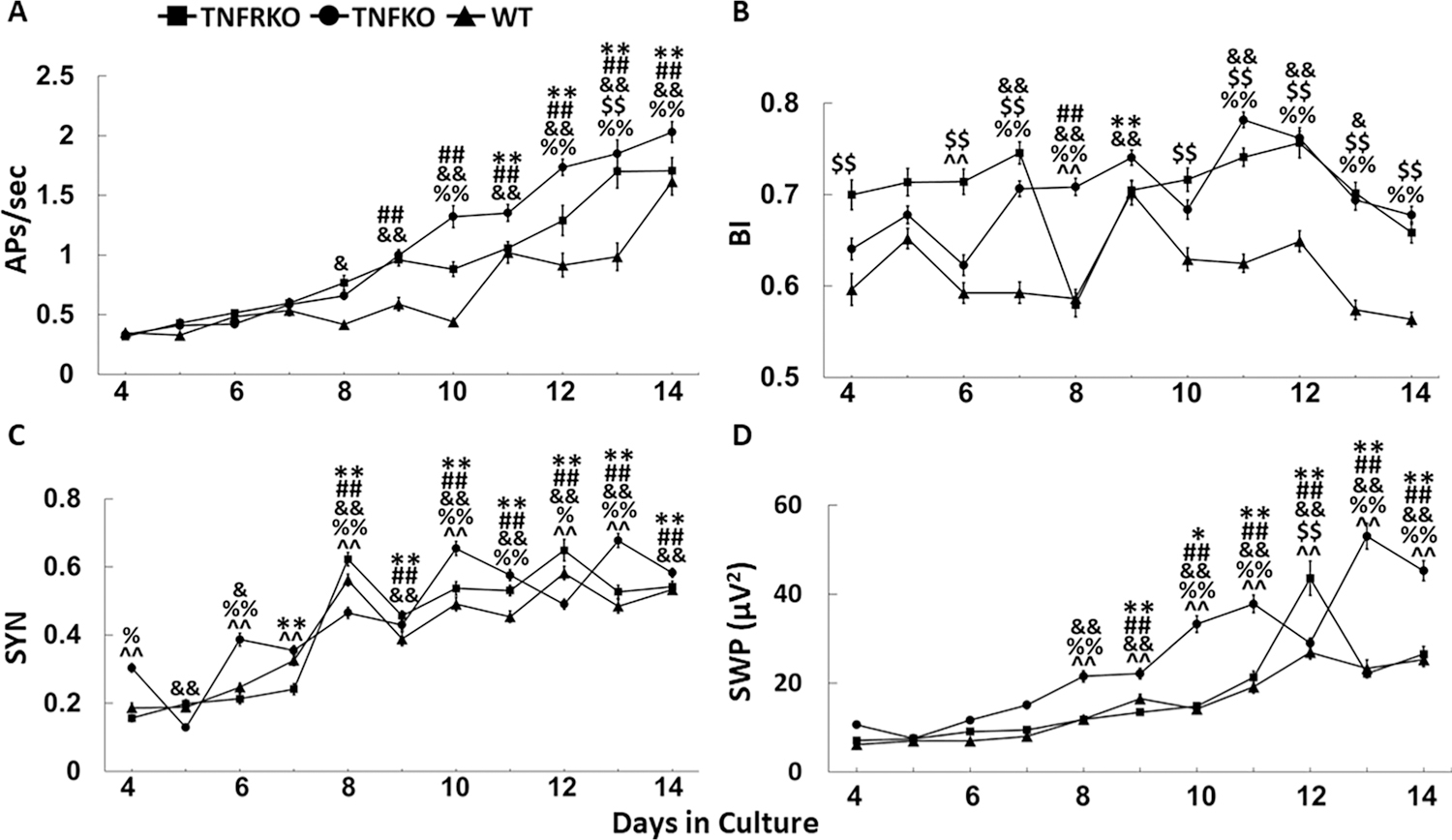
Development of spontaneous electrical activity in somatosensory cortex neuronalglial co-cultures. Cultures were derived from wildtype (WT), double tumor necrosis factor receptor (TNFR1, /TNFR2) knockout mice (TNFRKO), and TNFKO mice. Numbers of preparations, MEAs, wells and electrodes are presented in Table 1. For each strain and parameter, mean values ± standard errors from day (D)4-D14 in cell culture are presented in the figure. In some cases, SE values are so small they are imperceptibly confined within the data points. A. Average values for action potentials per second (APs/sec) are shown. B. Mean burstiness indices (BI) are shown. C. Average synchronization (SYN) values between adjacent electrodes are shown. D. Mean fast Fourier transformation values (slow wave power (SWP; µV2) within the 0.25–3.75 Hz frequency band values are shown. To determine development changes of values on D5-D14, each day’s values were compared to D4 values within each strain for each sleep parameter. One-way ANOVA analyses were followed by post-hoc Tukey’s HSD tests if significant (p < 0.05) main effects were found: WT, *p < 0.05, **p < 0.01; TNFRKO, ##p < 0.01; TNFKO, &p < 0.05, &&p < 0.01; To determine day, strain and day by strain interaction differences for each sleep parameter, two-way ANOVA analyses were used, followed by post-hoc Tukey’s HSD tests if significant (p < 0.05) effects were found: WT vs TNFRKO, $ $p < 0.01; WT vs TNFKO, %p < 0.05, %%p < 0.01; TNFRKO vs TNFKO, ^^p < 0.01.
BI values varied across development days (FWT(10,2562) = 13.78, p < 2E-16; FTNFRKO(10,2213) = 11.60, p < 2E-16; FTNFKO(10,3374) = 23.79, p < 2E-16; Fig. 2B, Supplementary Table 1), confirming our previous report of a pattern of increased and decreased BI in WT cells over development days (Jewett et al., 2015). Two-way ANOVA analyses showed main effects of strain (F(2,8149) = 226.79, p = 2E-16), day (F(10,8149) = 32.90,p < 2E-16) and day × strain (F(20,8149) = 10.09, p < 2E-16). BI values were lowest in WT cells compared to both TNFRKO cells (Ds 4, 6, 7 and 10–14; p < 0.01) and TNFKO cells (Ds 7, 8 and 11–14; p < 0.01). However, BI was only higher in TNFRKO cells compared to TNFKO cells on two days, D6 and D8 (p < 0.01). In TNFRKO cells and WT cells, BI values were only increased on one day compared to D4 (TNFRKO, D8, p < 0.01; WT, D9, p < 0.01). However, in TNFKO cells, BI was increased from D4 on 6 days (D7, D8, D9, D11, D12, p < 01; D13, p < 0.05). In all three strains, BI values did not differ significantly from D4 values. It is possible that, although APs/sec increased through D14 for all three strains, the lack of change to BI values by that day suggest a subtle reorganization of the cellular networks.
Confirming previous reports from WT cells (Jewett et al., 2015; Nguyen et al., 2019b), SYN values in the current study increased from D4 to D14 for all strains (Fig. 2C; Supplementary Table 1). Within strain one-way ANOVA analyses showed effects for SYN development changes on D5 through D14 compared to D4 in all three strains (FWT(10,4239) = 76.56, p < 2E-16; FTNFRKO(10,3834) = 87.83, p < 2E-16; FTNFKO(10,5825) = 103.00, p < 2E-16). Two-way ANOVA analyses showed main effects of strain (F(2,13898) = 21.66, p = 4.05E-10), day (F(10,13898) = 233.11,p < 2E-16) and day × strain (F(20,13898) = 15.46, p < 2E-16). In comparison to D4, D5 SYN values were lower for TNFKO cells (p < 0.01) but were higher on D6 (p < 0.05). This waxing and waning of SYN values continued for TNFKO cells through D14, although, except for D5, were higher than D4 values (D6, p < 0.05; D8-D14, p < 0.01). SYN waxing and waning also occurred in WT and TNFRKO cells with similarly pronounced peaks on D8, when both strains first showed higher SYN values compared with D4 (p < 0.01), and again on D12. Interestingly, although SYN values for TNFKO cells on D4 were higher than those for both WT (p < 0.05) and TNFRKO (p < 0.01) cells, values were similar for all strains by D14.
SWP in intact animals is a defining parameter of NREMS (Davis et al., 2011). In our current study, we substantiated our previous findings that SWP is slow to develop over the first few days in vitro (Jewett et al., 2015; Nguyen et al., 2019b). This was true for all three strains. Within strain one-way ANOVA analyses showed effects for development changes of D5 through D14 compared to day 4 in all three strains (FWT(10,2562) = 39.74, p < 2E-16; FTNFRKO(10,2213) = 56.10, p < 2E-16; FTNFKO(10,3374) = 103.30, p < 2E-16). Two-way ANOVA analyses showed main effects of strain (F(2,8149) = 221.66, p = 2E-19), day (F(10,8149) = 148.75, p < 2E-16) and day × strain (F(20,8149) = 17.77, p < 2E-16). Following the trend for APs/sec, when compared to D4, SWP was faster to develop in TNFKO cells, beginning on D8 (p < 0.01; Fig. 2D, Supplementary Table 1), whereas both WT and TNFRKO cells had higher SWP beginning on D9 (WT and TNFRKO, p < 0.01). Throughout the development period, SWP was highest in TNFKO cells, but barely distinguishable between WT cells and TNFRKO cells. TNFKO SWP was higher than WT and TNFRKO starting on D8 (p < 0.01). This difference continued through D14 for TNFRKO but changed for WT on D9 and D12 when SWP decreased in TNFKO cells and increased in WT cells. In contrast, SWP differed between TNFRKO and WT cells only on D12 when SWP increased in TNFRKO (p < 0.01) compared to WT cells.
Overall, Experiment 1 development data suggest a maturation of both wake-like (more APs/sec occurring within bursts) and sleep-like (greater SYN and SWP) states. Data are consistent with our prior reports (Jewett et al., 2015; Nguyen et al., 2019b). Further, current results clearly indicate a role for the TNF family in brain development as previously described (Merrill, 1992).
3.2. Experiment 2: Effect of the sTNFR1 on spontaneous electrical activity of mature TNFRKO and WT cells
3.2.1. sTNFR1 effects on developmental day 14
To determine the effect of the sTNFR1 on APs/sec, SWP, SYN, and BI, various treatment doses of the sTNFR1 (control, 0.3 ng/µL, 3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) were added directly to MEA wells containing TNFRKO or WT somatosensory cortex neuron-glia co-cultures on D14. sTNFR1 treatment altered the in vitro emergent parameters in dose- and/or strain-specific ways, with the most drastic effects observed in the changes to SYN and SWP.
Changes in APs/sec in TNFRKO cells showed a dose effect (F(5,140) = 3.68, p = 3.68E-3). Decreased APs/sec occurred in TNFRKO cells after the sTNFR1 treatment with 3 ng/µL (p < 0.05) and 30 ng/µL doses compared to the control dose (p < 0.01; Fig. 3A). In WT cells, none of the sTNFR1 doses had significant effects on APs/sec (Fig. 3A)
Fig. 3.
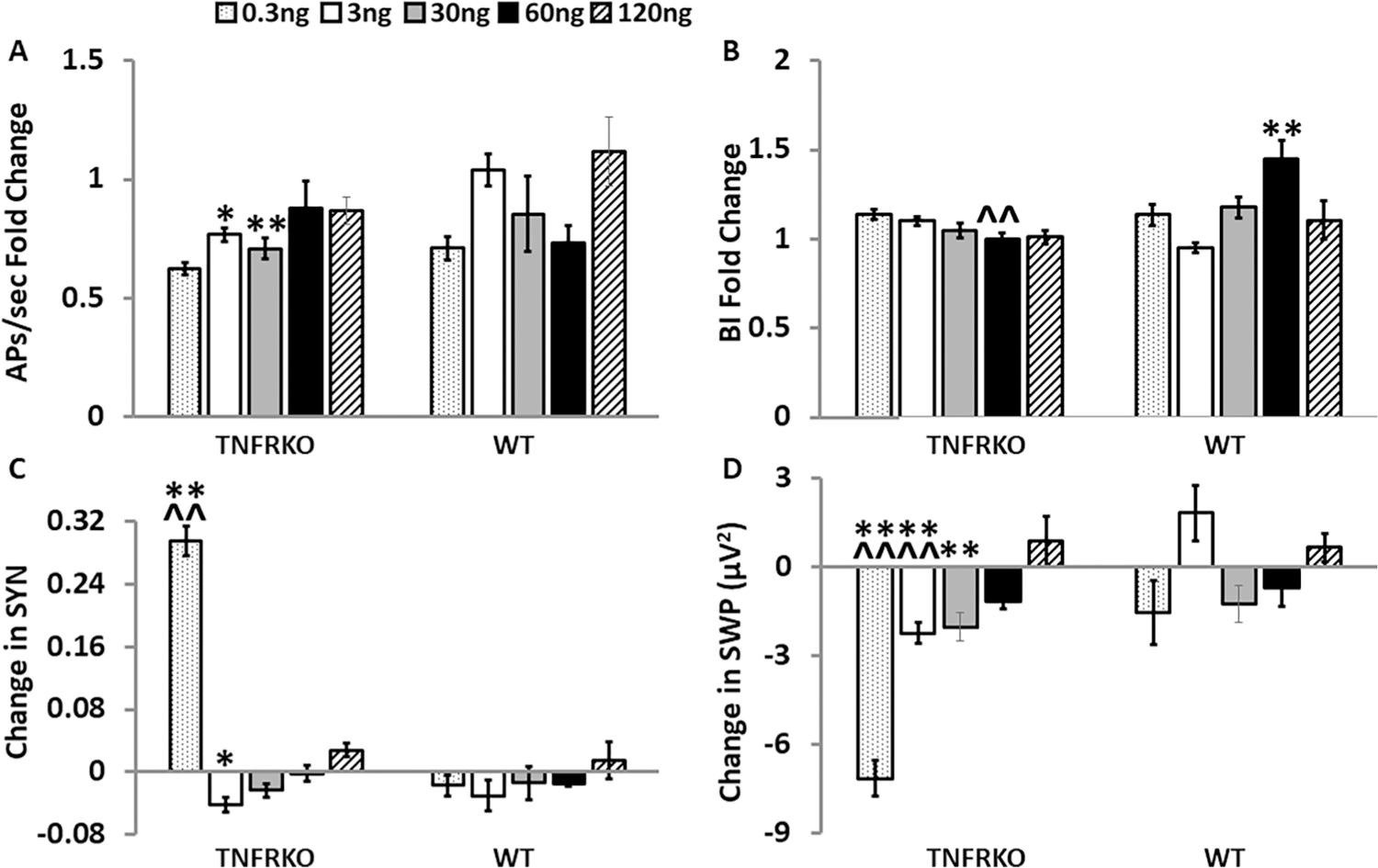
Addition of the soluble tumor necrosis factor receptor 1 (TNFR1) on day 14 altered electrophysiological parameters in a dose- and strain-dependent manner in cells from mice lacking both TNFRs (TNFRKO) mice and cells from wildtype (WT) mice. For each sTNFR1 treatment dose (0.3 ng/µL, 3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) within each strain and parameter, graphically presented values are means ± standard errors of normalized fold changes (APs/sec and BI) and differences (SYN and SWP) relative to the control treatment dose. Changes in each measure were compared first to their respective day 14 baseline values and then to the sTNFR1 control treatment change from baseline mean within the same MEA and strain as described in the methods section. Numbers of preparations, MEAs, wells and electrodes are presented in Table 1. A. Normalized fold change values of action potentials per second (APs/sec). B. Normalized fold change of burstiness index (BI). C. Change in synchrony (SYN) between electrodes. D. Change in slow wave power (SWP) (µV2, 0.25–3.75 Hz). Within strain, significance of dose-induced changes (p < 0.05) was determined by one-way ANOVA (dose) followed by post-hoc Tukey’s honestly significant differences (HSD) tests when significant main effects were found. Significant effects of dose, strain and dose by strain interactions were determined with two-way ANOVA (dose, strain, dose × strain), followed by post-hoc Tukey’s HSD tests when significant (p < 0.05) main effects were found. Within strain significant dose-induced changes compared with the control treatment: *p < 0.05, **p < 0.01. Within dose significant strain differences : ^^p < 0.01. Within strain significant differences between dose-induced changes (0.3 ng/µL, 3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) are not shown in this figure but are reported in the results section.
Effects of strain (F(1,206) = 4.85, p = 0.029) and dose × strain (F(4,206) = 8.16, p = 3.96E-6) were observed for BI changes. Treatment with the 60 ng/µL sTNFR1 dose lowered BI (p < 0.01) in TNFRKO cells compared to WT cells. In contrast, the WT cells increased BI (p < 0.01) after the 60 ng sTNFR1 dose compared to the control dose (Fig. 3B). There was a dose response for BI changes within WT cells (F(5,118) = 5.78, p = 8.26E-5) with higher BI after the 60 ng/µL sTNFR1 dose than each of the other doses (0.3 ng/µL, p < 0.01; 3 ng/µL, p < 0.05, 30 ng/µL, p < 0.01; 120 ng/µL, p < 0.05; significance not shown in Fig. 3B).
Changes in SYN were significant for dose (F(4,197) = 38.95, p < 2.00E-16), strain (F(1,197) = 15.04, p = 1.43E-4) and dose:strain (F(4,197) = 28.59, p < 2.00E-16). SYN increased in TNFRKO cells after treatment with the 0.3 ng/µL sTNFR1 dose compared to the control dose (p < 0.01) and compared to WT cells (p < 0.01; Fig. 3C). There were dose effects on SYN in TNFRKO cells (F(5,177) = 74.66, p < 2.00E-16). SYN increased after the 0.3 ng/µL sTNFR1 dose compared to each of the other doses (3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL; all, p < 0.01). Additionally, treatment with 3 ng/µL sTNFR1 decreased SYN compared to the control (p < 0.01), 60 ng/µL (p < 0.05), and 120 ng/µL (p < 0.01) doses (significance not shown in Fig. 3C).
Changes to SWP had effects of dose (F(4,206) = 6.89, p = 3.19E-5), strain (F(1,206) = 23.21, p = 2.81E-6), and dose:strain (F(4,206) = 5.85, p = 1.77E-4). SWP decreased in TNFRKO cells after treatment with the 0.3 ng/µL, 3 ng/µL and 30 ng/µL sTNFR1 doses compared to the control sTNFR1 dose (all, p < 0.01; Fig. 3D). Additionally, the SWP response after treatment with 0.3 ng/µ L and 3 ng/µL, was decreased compared to WT cells (p < 0.01, Fig. 3D). Dose effects occurred in both strains (TNFRKO, F(5,140) = 17.26, p < 2.73E-13; WT (F(5,18) = 3.30, p = 7.98E-3). In TNFRKO cells, the 0.3 ng/µL dose decreased power compared to all other doses (3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL, all p < 0.01). Additionally, the SWP observed after the 120 ng/µL treatment dose differed from the decreased SWP observed after treatment with 3 ng/µL (p < 0.01), 30 ng/µL (p < 0.01) and 60 ng/µL (p < 0.05) sTNFR1. In WT cells, the SWP after treatment with the 3 ng/µL sTNFR1 dose differed from the SWP observed after treatment with 0.3 ng/µL and with 30 ng/µL (both p < 0.05; significance not shown in Fig. 3D).
3.2.2. sTNFR1 residual effects on developmental day 15
To determine the prolonged effects of the sTNFR1 on APs/sec, SWP, SYN, and BI, electrical activity was recorded from mature WT and TNFRKO cells on D15, 24 h after treatment with the various treatment doses of the sTNFR1.
On D15 compared to D14, there were no significant changes to APs/sec in cells derived from either strain (Fig. 4A). Similarly, there were few changes to BI on D15. However, in TNFRKO cells there was a dose response (F(5,141) = 2.73, p = 0.22); BI increased 24 h after 120 ng/µL sTNFR1 compared the control dose (p < 0.05; Fig. 4B).
Fig. 4.
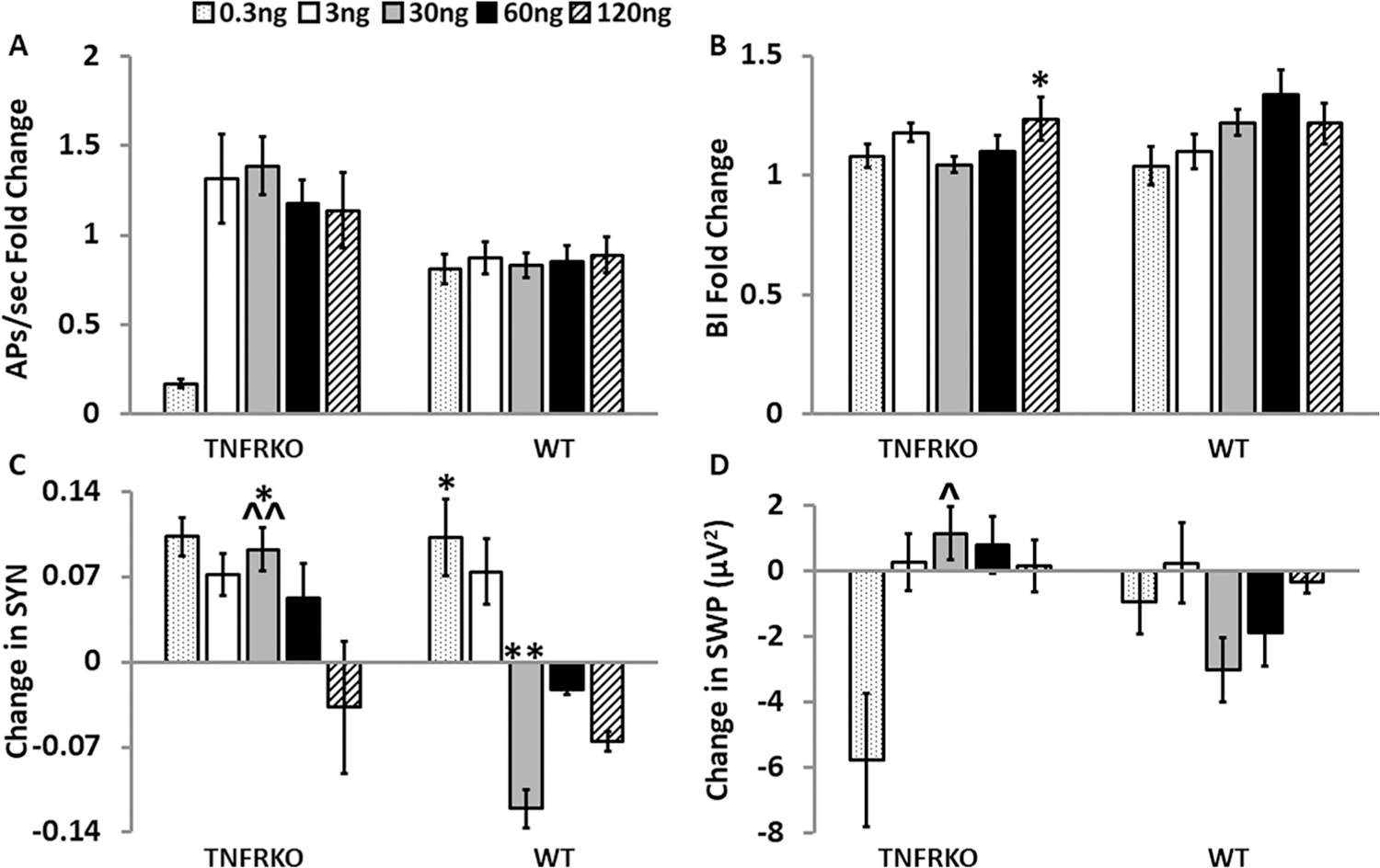
Effects of the soluble tumor necrosis factor receptor 1 (TNFR1) on electrophysiological parameters 24 h after treatment in cells from mice lacking both TNFRs (TNFRKO) mice and cells from wildtype (WT) mice. For each sTNFR1 treatment dose (0.3, 3, 30, 60 and 120 ng/µL) within each strain and parameter, graphically presented values are means ± standard errors of normalized fold changes (APs/sec and BI) and differences (SYN and SWP) relative to the control treatment dose. Changes in each measure were compared first to their day 14 baseline values and then to the sTNFR1 control treatment change from baseline mean within the same MEA and strain as described in the methods section. Numbers of preparations, MEAs, wells and electrodes are presented in Table 1. A. Normalized fold change of action potentials per second (APs/sec). B. Normalized fold change of burstiness index (BI). C. Change in synchrony (SYN) between electrodes. D. Change in slow wave power (SWP) (µV2, 0.25–3.75 Hz). Within strain significance of dose-induced changes (p < 0.05) was determined by one-way ANOVA (dose) followed by post-hoc Tukey’s honestly significant differences (HSD) tests when significant main effects were found. Significant effects of dose, strain and dose by strain interactions were determined with two-way ANOVA (dose, strain, dose × strain) followed by post-hoc Tukey’s HSD tests when significant (p < 0.05) main effects were found. Within strain, significant dose-induced changes compared with the control treatment: *p < 0.05, **p < 0.01. Within dose significant strain differences: ^p < 0.05, ^^p < 0.01. Within strain, significant differences between dose-induced changes (0.3 ng/µL, 3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) are not shown in this figure but are reported in the results section.
The actions of the sTNFR1 were more wide spread for SYN; there were effects of dose (F(4,198) = 4.61, p = 1.40E-3), strain (F(1,198) = 13.34, p = 3.32E-4) and dose:strain (F(4,198) = 6.09, p = 1.21E-4) on D15 compared to D14. In TNFRKO cells, SYN was elevated 24 h after treatment with the 30 ng/µL sTNFR1 dose (p < 0.05) compared to the control dose (p < 0.05; Fig. 4C). In WT cells, on D15 SYN increased after the 0.3 ng/µL dose (p < 0.05) yet decreased after the 30 ng/µL (p < 0.01) dose. The WT cell response after 30 ng/µL also differed from the TNFRKO cell response (p < 0.01; Fig. 4C). In TNFRKO cells, dose effects for SYN occurred (F(5,177) = 3.80, p = 2.73E-3); SYN decreased after the 120 ng/µL sTNFR1 dose compared to the elevated SYN after treatment with 30 ng/µL (p < 0.05). In WT cells, SYN also exhibited dose effects (F(5,75) = 12.83, p = 5.08E-9). SYN decreased 24 h after the 30 ng/µL sTNFR1 dose. However, SYN in WT cells increased after the 0.3 ng/µL dose (both, p < 0.01). Additionally, the increased SYN 24 h after the 0.3 ng/µL sTNFR1 dose differed (p < 0.05) from the decreased SYN 24 h after the 120 ng/µL dose (significance not shown in Fig. 4C).
The differences in SWP between D14 and D15 had effects of dose: strain (F(4,208) = 3.91,p = 4.40E-3). The increased SWP observed in TNFRKO cells 24 h after treatment with the 30 ng/µL sTNFR1 dose differed from WT cells (p < 0.05; Fig. 4D). Additionally, within strain dose effects occurred in TNFRKO cells (F(5,141) = 2.46, p = 0.036) with a decrease in SWP 24 h after the 0.3 ng/µL sTNFR1 dose that differed from the increased power observed 24 h after treatment with the 3 ng/µL, 30 ng/µL and 60 ng/µL sTNFR1 doses (all, p < 0.05).
To summarize, administration of the 0.3 ng/µL, 3 ng/µL and 30 ng/µL sTNFR1 doses on D14 decreased SWP in TNFRKO cells, indicative of a clear enhancement of the wake-like state. This was not a long-lasting effect, having returned to baseline by 24 h after treatment (Compare Fig. 3D and 4D). In contrast, treatment effects in WT were minimal on either D14 or D15.
3.3. Experiment 3: Effect of the sTNFR1 on spontaneous electrical activity of mature TNFKO cells
TNFKO mice, while deficient in TNF, still produce lymphotoxin-alpha (LTα; formerly called TNFβ), a ligand for the sTNFR1. Thus, because TNFβ is somnogenic (Kapas and Krueger, 1992), it was of interest to determine if the sTNFR1 had activity in TNFKO cells.
3.3.1. The sTNFR1 effects on developmental day 14 in TNFKO cells
In TNFKO cells after sTNFR1 treatment, there were dose-dependent effects on APs/sec (F(4,172) = 5.76, p = 2.25E-4) on D14. Dose-dependent effects on APs/sec compared to control were insignificant (Fig. 5A, left bars). However, responses in APs/sec after administration of 3 ng/µL and 30 ng/µL sTNFR1 doses differed from the responses observed after treatment with the 60 ng/µL sTNFR1 dose (vs 3 ng/µL and vs 30 ng/µL, p < 0.05) and 120 ng/µL (vs 3 ng/µL, p < 0.05; vs 30 ng/µL, p < 0.01 doses of sTNFR1).
Fig. 5.
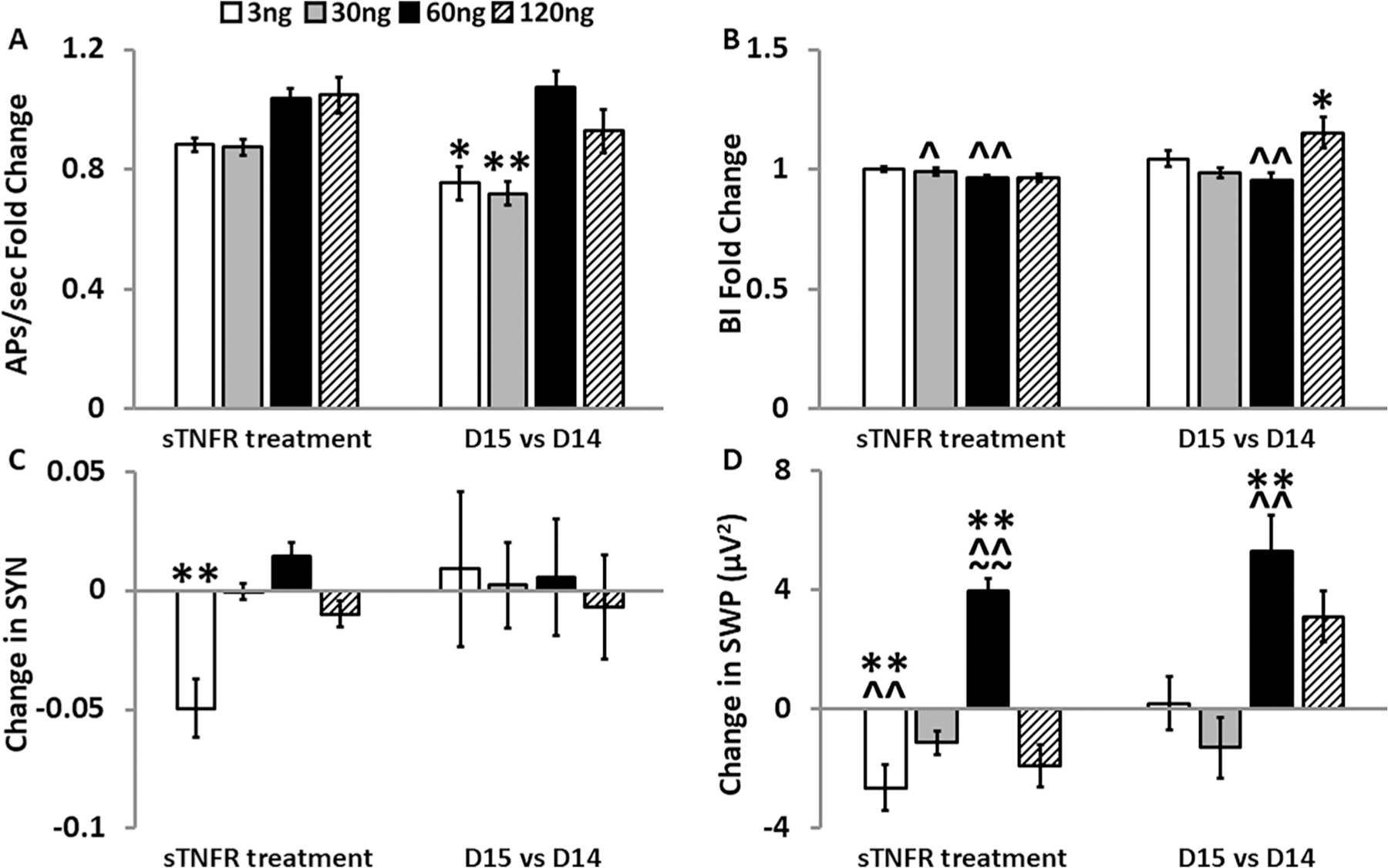
Effects of the soluble tumor necrosis factor receptor 1 (TNFR1) on electrophysiological parameters in cells from mice lacking TNF (TNFKO). For each sTNFR1 treatment dose (3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) within each strain and parameter, graphically presented values are means ± standard errors of normalized fold changes (APs/sec and BI) and differences (SYN and SWP) relative to the control treatment dose. One hour post-sTNFR1 treatment on day (D) 14 values for each measure (left hand bars) were first compared to their D14 baseline values and then to the sTNFR1 control treatment change from baseline mean within the same MEA and strain as described in the methods section. 24 h post-sTNFR1 treatment values for each measure (right hand bars) were first compared to their D14 baseline values and then to the D15 sTNFR1 control treatment change from D14 baseline mean within the same MEA and strain as described in the methods section. Numbers of preparations, MEAs, wells, and electrodes are presented in Table 1. A. Normalized fold change of action potentials per second (APs/sec). B. Normalized fold change of burstiness index (BI). C. Change in synchrony (SYN) between electrodes in TNFKO cells. D. Change in slow wave power (SWP; µV2, 0.25–3.75 Hz). For each day and parameter, a one-way ANOVA determined significant dose effects and was followed by post-hoc Tukey’s HSD tests where indicated and as described in the methods section: Significant dose-induced changes compared with the control treatment: *p < 0.05, **p < 0.01. Significant strain differences vs WT: ^p < 0.05, ^^p < 0.01; vs TNFRKO: ~~p < 0.01. Significant differences between dose-induced changes (3 ng/µL, 30 ng/µL, 60 ng/µL, and 120 ng/µL) are not shown in this figure but are reported in the results section.
Although there were no dose effects on BI changes in TNFKO cells (Fig. 5B, left bars), there were effects of strain (F(2,318) = 14.99, p = 5.99E-7) and dose:strain (F(6,318) = 9.04, p = 3.95E-9). BI in TNFKO cells (Fig. 5B, left bars) decreased compared to WT cells (Fig. 3B) after the 30 ng/µL (p < 0.05) and the 60 ng/µL (p < 0.01) sTNFR1 doses.
Changes to SYN showed dose effects in TNFKO cells (F(4,327) = 11.50, p = 9.62E-9). SYN decreased after treatment with the 3 ng/µL sTNFR1 dose compared to the control dose (p < 0.01; Fig. 5C, left bars) and compared to all other treatment doses (30 ng/µL, 60 ng/µL and 120 ng/µL; all p < 0.01).
Dose effects were also observed in SWP (F(4,172) = 20.72, p = 5.93E-14). SWP decreased after addition of the 3 ng/µL sTNFR1 dose (p < 0.01) yet increased after treatment with the 60 ng/µL sTNFR1 dose (p < 0.01). Further, the increased SWP after the 60 ng/µL sTNFR1 dose varied from the responses observed after treatment with all other sTNFR1 doses (3 ng/µL, 30 ng/µL, and 120 ng/µL, all p < 0.01; Fig. 5D, left bars). SWP also had effects of strain (F(2,318) = 8.45, p = 2.66E-4) and dose:strain (F(6,318) = 12.12, p = 2.82E-12). SWP in TNFKO cells decreased after the 3 ng/µL sTNFR1 dose (Fig. 5D, left bars) compared to WT (Fig. 3D; p < 0.01) and increased after the 60 ng/µL sTNFR1 dose (Fig. 5D, left bars) compared to both TNFRKO and WT (Fig. 3D; both, p < 0.01).
3.3.2. STNFR1 effects on developmental day (D) 15 compared to D14 in TNFKO cells
The changes in APs/sec on D15 compared to D14 had effects of dose (F(4,170) = 7.84, p = 8.00E-6) in TNFKO cells and, compared to the control dose, decreased after the 3 ng/µL (p < 0.05) and after the 30 ng/µL (p < 0.01) sTNFR1 doses (Fig. 5A, right bars). APs/sec increased after the 60 ng/µL sTNFR1 dose compared with the response after the 3 ng/µL (p < 0.01) and after the 30 ng/µL (p < 0.01) doses. Additionally, APs/sec were higher after the 120 ng/µL treatment compared to the 30 ng/µL treatment (p < 0.05).
Similarly, the changes in BI had effects of dose (F(4,170) = 4.47, p = 1.84E-3) in TNFKO cells. BI increased after 120 ng/µL sTNFR1 compared to the control dose (p < 0.05; Fig. 5B, right bars) and compared to BI after the 30 ng/µL and 60 ng/µL doses (both, p < 0.01). Additionally, the increase in BI observed after the 60 ng/µL dose differed from WT (Fstrain (2,318) = 9.34, p = 1.15E-4, Tukey HSD p < 0.01; compare Fig. 5B and 4B, both right bars)
Interestingly, there were no significant changes to SYN on D15 in TNFKO cells (Fig. 5C, right bars). However, SWP showed a dose effect (F(4,170) = 7.99, p = 6.28E-6) with increases in power after administration of the 60 ng/µL sTNFR1 dose compared to the control dose (p < 0.01; Fig. 5D, right bars) and compared to the responses after doses of 3 ng/µ L and 30 ng/µL sTNFR1 (all, p < 0.01). This increased power (Fig. 5D, right bars) also differed from the response in WT cells (Fig. 4D) after the 60 ng/µL sTNFR1 dose (p < 0.01). Additionally, the change in power increased after the 120 ng/µL sTNFR1 dose compared to the response after the 30 ng/µL sTNFR1 dose (p < 0.01).
To summarize, the 3 ng/µL sTNFR1 decreased SWP in TNFKO mice on D14, indicative of a wake-like state. This was a short-lived effect that was abolished 24 h later. In contrast, the 60 ng/µL sTNFR1 dose increased SWP on D14, indicating a deeper sleep-like state that was prolonged and still evident on D15.
3.3.3. Time courses of the sTNFR1 effects on SYN and SWP on day 14 in TNFKO and TNFRKO cells (Experiments 2 and 3)
To further investigate the responses to sTNFR1 after the 3 ng/µL and 60 ng/µ L doses in both TNFKO and TNFRKO cells, we compared the time courses of these treatment responses (Fig. 6) To reiterate, decreases in SYN occurred after the 3 ng/µL sTNFR1 dose compared to the control dose in both TNFKO cells (p < 0.01; Fig. 5C, left bars) and TNFRKO cells (p < 0.05; Fig. 3C). Additionally, after the 60 ng/µL sTNFR1 treatment, the increased SWP observed in TNFKO (Fig. 5D, left bars) cells differed (p < 0.01) from TNFRKO cells (Fig. 3D).
Changes to SYN showed effects of time (F(11,2480) = 5.68, p = 4.34E-9), strain (F(1,2480) = 5.23, p = 0.022), dose (F(1,2480) = 5.68, p = 4.34E-9), and interactions of time:strain (F(11,2480) = 3.14, p = 3.12E-4), time: dose (F(11,2480) = 2.01, p = 0.024), and strain:dose (F(1,2480) = 7.71, p5.53E-3). Within TNFRKO cells, SYN had a time effect after the 3 ng/µL sTNFR1 dose (F(11,672) = 3.00, p = 6.59E-4) and effects of time (F(11,1020) = 2.43, p = 5.56E-3) and dose (F(11,1020) = 62.68, p = 6.32E-15) between the 3 ng/µL and the 60 ng/µL sTNFR1 doses. Within TNFKO cells, SYN showed effects of time after both doses of the sTNFR1 (3 ng/µL, (F(11,836) = 2.31, p = 8.72E-3); 60 ng/µL sTNFR1 (F(11,624) = 11.30, p < 2.30E-16)) and effects of time (F(11,1460) = 6.25, p = 3.89E-10) and dose (F(11,1460) = 173.95, p < 2.00E-16) between the two doses. Compared to the value at 5 min after the 3 ng/µL sTNFR1 dose, SYN decreased at 35 min in both strains (TNFRKO, p < 0.05; TNFKO, p < 0.01; Fig. 6A). Unlike TNFRKO cells, however, SYN in TNFKO cells remained lower for the next 10 min in TNFKO cells (p < 0.01; Fig. 6A). In TNFKO cells, the SYN response after 3 ng/µL sTNFR1 was lower than those after 60 ng/µL sTNFR1 across time at 5 min, 20 min, 30 min, 35 min, 40 min, 50 min, 55 min, (all, p < 0.01) and at 60 min (p < 0.05). However, in TNFRKO cells, this difference between dose responses occurred only at 35 min when SYN was lower after 3 ng/µL compared to 60 ng/µL (p < 0.05). Strain effects occurred only after the 60 ng/µL sTNFR1 dose when SYN was elevated at 5 min in TNFKO cells compared to TNFRKO cells (p < 0.01; Fig. 6A). SYN then decreased in TNFKO cells by 10 min post-treatment and remained lower than the 5 min value for the remainder of the hour-long recording (p < 0.01 for all times except 50 min, p < 0.05; Fig. 6A). Interestingly, there was an effect of time (F(11,624) = 11.30, p < 2.00E-16) in TNFKO cells after the 60 ng/µL sTNFR1 dose, with an increasing and decreasing pattern of SYN values (Fig. 6A).
Changes in SWP effects occurred for time (F(11,1522) = 8.20,p = 4.33E-14), strain (F(1,1522) = 168.27, p < 2.00E-16), dose (F(1,1522) = 455.55, p < 2.00E-16), and interactions of time:strain (F(11,1522) = 3.64, p = 4.07E-5), and strain:dose (F(1,1522) = 241.34, p = 4.33E-14). Within TNFRKO cells, SWP had effects of time after 3 ng/µL sTNFR1 (F(11,480) = 2.11, p = 0.019). Within TNFKO cells, SWP effects of time occurred after the 60 ng/µL sTNFR1 dose (F(11,348) = 9.00, p = 3.78E-14). SWP comparisons between the 3 ng/ul and 60 ng/µL sTNFR1 doses had effects of time (F(11,742) = 6.80, p = 5.92E-11) and dose (F(1,742) = 443.41,p < 2.00E-16). Compared to the SWP at 5 min, the lowest SWP value in TNFKO cells after 3 ng/µL sTNFR1 occurred at 45 min after treatment (p < 0.05), but SWP was also decreased at 15 min and 25 min (both, p < 0.01). Compared to the response at 5 min after the 60 ng/µL treatment in TNFKO cells, SWP was decreased at 10 min, 15 min, 25 min, 45 min, 50 min and 55 min (all, p < 0.01), with the lowest SWP value at 45 min (Fig. 6B). After the initial decreases in SWP from 5 min through 15 min, SWP increased at 20 min (vs 15 min, p < 0.01). After remaining fairly steady between 25 min and 40 min, SWP then dropped to its lowest value at 45 min (vs 40 min, p < 0.01), increasing slightly at 50 min was finally higher again by 60 min (vs 45 min and vs 55 min, p < 0.01). Additionally, SWP responses in TNFKO cells after 60 ng/µL were higher than those after the 3 ng/µL sTNFR1 dose across all time points (all p < 0.01, except 15 min, p < 0.05). Like SYN, changes to SWP had effects of strain only after the 60 ng/µL sTNFR1 dose, with higher SWP in TNFKO cells than in TNFRKO cells at every timepoint (p < 0.01) except at 45 min (Fig. 6B), when the value, as previously mentioned, was at its lowest in TNFKO cells.
In summary, the 60 ng/µL sTNFR1 dose had a more pronounced effect on SYN and SWP in TNFKO cells compared to the 3 ng/µL dose and compared to SYN and SWP values obtained from TNFRKO cells with the same doses. In contrast, both doses of sTNFR1 in TNFRKO cells induced a more wake-like state.
4. Discussion
The TNF family of molecules is large, complex, and plays multiple diverse roles in the central nervous system including sleep regulation (Rockstrom et al., 2017) and development (Merrill, 1992). During development, TNF is upregulated in a region-specific manner in mammalian brain (Dziegielewska et al., 2000; Merrill, 1992). Nevertheless, in many ways TNFKO mice and TNFRKO mice appear to develop normally (Bruce et al., 1996; Fang et al., 1997; Marino et al., 1997). However, gene expression related to neurogenesis differs in TNF deficient mice, which have decreased TNFR2 expression and which develop increased numbers of neurons and decreased numbers of microglia (Yli-Karjanmaa et al., 2019b). Additionally, memory impairments develop in mice lacking TNF and TNFR1 and mice lacking TNF, TNFR1 or TNFR2 have learning disabilities (Camara et al., 2013). Finally, whereas TNF is a well characterized sleep regulatory substance, mice deficient in TNFR1 do not sleep more after treatment with sTNF (Fang et al., 1997). Herein, we show that TNF and TNFRs have a role in the development of neuronal/glial networks in vitro to the extent that rates of emergent electrophysiological properties develop faster in both TNFKO and TNFRKO in somatosensory cortical neuron/glia co-cultures compared to cultured WT cells. Although we did not investigate the glia:neuron ratio, the faster development of APs/sec and SWP could be the result of increased neuronal firings, untempered by glia, in the TNFKO and TNFRKO cultures. Previously, we showed that APs decreased in cultures of WT somatosensory cells enriched in astrocytes (Jewett et al., 2012). Regardless of such speculation, our results confirm that the TNF family of molecules plays a role in brain development.
The major new finding in the current study is that the sTNFR1 enhances the wake-like state as evidenced by decreases in SYN and SWP after the 3 ng dose in cells derived from both TNFRKO and TNFKO mice (Figs. 3 and 5). We previously showed that in WT somatosensory cells in vitro sTNF induces a deeper sleep-like state, as evidenced by increases in SWP (Jewett et al., 2015). Further, electrical stimulation of WT cells in culture reduces SWP values, suggesting a more wake-like state which is followed the next day by a rebound in SWP values, indicating sleep homeostasis can occur in vitro (Jewett et al., 2015). In vivo, EEG SWP is often used to define NREMS and higher values indicate a deeper NREMS (Borbely et al., 1984; Hajnik et al., 2013). Previously, we had also used a sTNFR1, or fragment of it, in vivo to inhibit sleep and EEG SWP but were unaware of tmTNF and TNF reverse signaling at the time (Kubota et al., 2002, 2001; Takahashi et al., 1996a). In the current study, we hypothesized that addition of sTNFR1 would induce a more wake-like state in vitro. The inhibition of the sleep-like state by the sTNFR1 is similar to results reported by Jewett et al after electrical stimulation (Jewett et al., 2015). Thus, these data suggest a reverse signal in the absence of tmTNFRs that induces a wake-like state through binding of sTNFR1 to cells that present tmTNF.
We previously showed that LTα is somnogenic (Kapas and Krueger, 1992). TNFKO mice are null for the TNF gene but still produce LTα (Jackson Labs website, overview for B6.129S-TNFtm1Gkl/J, https://www.jax.org/strain/005540). TNF and LTα are homologous members of the TNF superfamily and TNFR1 is a high affinity receptor for both ligands (Banner et al., 1993; Hohmann et al., 1990; Loetscher et al., 1991; Medvedev et al., 1996; Nedwin et al., 1985; Pennica et al., 1984; Schoenfeld et al., 1991; Smith et al., 1994). TNFR1 form dimers and trimers that bind trimeric and monomeric TNF and LTα, resulting in varied cellular responses through conformational changes in the receptor upon ligand binding (Aggarwal, 2003; Lo et al., 2020; Naismith et al., 1995; Schoenfeld et al., 1991). Thus, whereas TNF is not playing a role in the sleep-like parameter changes observed in TNFKO mice, it is possible that they are the result of LTα:sTNFR binding. Future studies to characterize the mechanisms of TNF:sTNFR1 induced reverse signaling should, therefore, utilize an animal model that silences both TNF and LTα genes.
Although TNFRKO cells exhibit clear sTNFR1 dose SYN- and SWP-responses (Fig. 3), the largest effects occurred with the lowest 0.3 ng sTNFR1 dose. This is not entirely without precedent as we have previously shown that, with other pro-inflammatory cytokines, i.e. low doses of IL1 in vivo promote sleep while high doses inhibit sleep (Opp et al., 1991). In the current study, the direction of the SYN effects were opposite at the 0.3 ng dose compared to the 3 ng sTNFR1 dose. It is not known what is responsible for these effects although it seems possible that receptor dimers and trimers interacting with ligand (sTNF, tmTNF, LTα) monomers and trimers are somehow involved. As the sTNFR1 dose is increased to 3 ng, cells from both TNFRKO and TNFKO mice respond similarly with decreases in SYN and SWP suggesting a more wake-like state in both cell type cultures. However, as the dose increases even more, the response profiles of the two cell types differ from each other and at the higher dose are not different from WT cell responses. Nevertheless, responses of both KO cell types are distinct from WT cell responses suggesting a role for the TNF family of molecules in these responses although it would be speculative to suggest a specific mechanism. We previously showed a sTNFR fragment induces increases in intracellular calcium responses. This effect decreased in a dose-dependent manner from 10 ng/mL to 1000 ng/mL, but then rose again at 10000 ng/mL. In contrast, TNFα (10 ng/mL) increased calcium (De et al., 2003). Additionally, we also previously showed that, in similar WT somatosensory cell cultures used herein, low doses of sTNF enhanced SYN and SWP while higher doses greatly inhibited APs/sec (Jewett et al., 2015). Because TNF is well-characterized for its involvement in synaptic scaling (Turrigiano, 2008) we interpreted those results within the context of glutamate AMPA receptor trafficking. Although speculative, it seems likely that altered TNF-induced AMPA mechanisms are occurring in the TNFRKO and TNFKO cells that in turn manifest in distinct dose response relationships.
To our knowledge, reverse signaling, induced by a soluble receptor binding a transmembrane ligand, has not previously been described in cortical brain tissue with respect to sleep. However, increased levels of sTNFR1 are correlated with increased arousal in obstructive sleep apnea patients (Yue et al., 2009). It is possible that these arousal states are induced through sTNFR1/tmTNF-induced reverse signaling. Reverse signaling of TNF occurs in many cell types including sympathetic neurons from the superior cervical ganglion, where axon growth and tissue innervation is promoted by TNFR1 binding to axonal tmTNF (Kisiswa et al., 2013). Although uninvestigated in these studies, this phenomenon reportedly occurs via dephosphorylation of tmTNF upon binding by sTNFR and is due to a casein kinase I motif in the TNF cytoplasmic domain of TNF (Watts et al., 1999). Future studies are necessary to determine if this is also the case in reverse signal-induced waking.
Several studies demonstrate that the two forms of TNF have opposing roles in the central nervous system; neuroprotection is afforded by tmTNF, but not sTNF. In an in vivo model of multiple sclerosis, inflammation was induced through sTNF signaling whereas tmTNF signaling was neuroprotective (Taoufik et al., 2011). In models of ischemic stroke in vivo and in vitro, tmTNF:TNFR1 but not sTNF:TNFR1 signaling is neuroprotective (Madsen et al., 2016; Taoufik et al., 2007; Yli-Karjanmaa et al., 2019a). Further, anti-TNF agents, infliximab, etanercept and adalimumab, have been used successfully to treat chronic inflammatory diseases by binding and neutralizing sTNF, yet these agents can also bind tmTNF to effect stronger inhibition (Horiuchi et al., 2010). With regards to the sleep effects of these anti-TNF agents, they appear to be better anti-tmTNF agents than anti-sTNF agents because they reduce sleep (Rockstrom et al., 2017). Although we investigated reverse signaling induced by soluble receptor-transmembrane ligand binding, cell to cell contact can also result in reverse signal modulation of physiological processes, including inflammation (Horiuchi et al., 2010) and chronic pain (Zhou et al., 2010), and the current study provides a rationale for future research to investigate the role of tmTNF/sTNFR cell-to-cell reverse signaling in sleep state regulation.
It is not uncommon in the myriad of molecules that work to maintain homeostatic balance, to find a complexity of roles being played within the same protein family. In this vein, the TNF superfamily may be compared the IL1 family, with its multiple anti- and pro-inflammatory members that provide stability to the physiological and pathological process which they regulate. Whereas IL1β, its receptor (IL1R1) and receptor accessory protein (IL1RAP) are pro-inflammatory, the sIL1R, neuron-specific IL1RAP and the receptor antagonist (IL1RA) are anti-inflammatory (Clinton et al., 2011; Nguyen et al., 2019a; Obal and Krueger, 2003; Zielinski and Krueger, 2012). Additionally, IL33, has two forms like TNF, which have opposing anti- and pro-inflammatory properties (reviewed, (Dinarello, 2018)). Further, it is suggested that the sIL1R may regulate inflammation through reverse signaling mechanisms (Sporri et al., 2001). Finally, the TNF and IL1 families are closely interconnected, as both IL1β and TNF self-amplify and increase each other’s mRNA expression in brain (Churchill et al., 2006). Such amplification systems are useful to rapidly amplify responses to inflammatory/infectious challenges. However, if damping systems are impaired, they lead to cytokine storms leading to enhanced morbidity and mortality, e.g. in COVID-19. Regardless, the current study reinforces the concept of sleep being a fundamental process of small local brain cellular circuits to the extent that in vitro the sTNFR1 induces the wake-like state of neuronal/glial cultures in a fashion similar to sTNFR1-enhanced waking in vivo. Further, several sleep regulatory substances, e.g. TNF, IL1, NO, prostaglandins, and adenosine are all produced within local cell circuits in response to cell use. They all alter local cell activity-dependent processes such as local sleep/wake states, cerebral blood flow, metabolism, and neuronal plasticity (Krueger, 2020; Krueger et al., 2019). These molecules are part of biomolecular cascades affecting each other’s expression and regulating the local processes all of which can interact with other homologous processes in adjacent local circuits to manifest as emergent properties at higher levels of tissue organization affecting whole animal behavior/physiology, e.g. sleep, memory, and immunity. Understanding the compartmentalization of these local cell-use driven processes and their emergent properties would greatly enable our understanding of neurobiology.
Supplementary Material
5. Statement of Funding
This work was supported in part by grants from the National Institutes of Health (NS025378 and NS096259) and by the W. M. Keck Foundation.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Aggarwal BB, 2003. Signalling pathways of the TNF superfamily: a double-edged sword. Nat. Rev. Immunol 3, 745–756. [DOI] [PubMed] [Google Scholar]
- Andersen ML, Nascimento DC, Machado RB, Roizenblatt S, Moldofsky H, Tufik S, 2006. Sleep disturbance induced by substance P in mice. Behav. Brain Res 167, 212–218. [DOI] [PubMed] [Google Scholar]
- Banner DW, D’Arcy A, Janes W, Gentz R, Schoenfeld HJ, Broger C, Loetscher H, Lesslauer W, 1993. Crystal structure of the soluble human 55 kd TNF receptor-human TNF beta complex: implications for TNF receptor activation. Cell 73, 431–445. [DOI] [PubMed] [Google Scholar]
- Bigdeli MR, Rahnema M, Khoshbaten A, 2009. Preconditioning with Sublethal Ischemia or Intermittent Normobaric Hyperoxia Up-regulates Glutamate Transporters and Tumor Necrosis Factor-α Converting Enzyme in the Rat Brain. J. Stroke Cerebrovasc. Dis 18, 336–342. [DOI] [PubMed] [Google Scholar]
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP, 1997. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature 385, 729–733. [DOI] [PubMed] [Google Scholar]
- Borbely AA, Tobler I, Hanagasioglu M, 1984. Effect of sleep deprivation on sleep and EEG power spectra in the rat. Behav. Brain Res 14, 171–182. [DOI] [PubMed] [Google Scholar]
- Brennan FM, Green P, Amjadi P, Robertshaw HJ, Alvarez-Iglesias M, Takata M, 2008. Interleukin-10 regulates TNF-alpha-converting enzyme (TACE/ADAM-17) involving a TIMP-3 dependent and independent mechanism. Eur. J. Immunol 38, 1106–1117. [DOI] [PubMed] [Google Scholar]
- Bruce AJ, Boling W, Kindy MS, Peschon J, Kraemer PJ, Carpenter MK, Holtsberg FW, Mattson MP, 1996. Altered neuronal and microglial responses to excitotoxic and ischemic brain injury in mice lacking TNF receptors. Nat. Med 2, 788–794. [DOI] [PubMed] [Google Scholar]
- Bzowska M, Jura N, Lassak A, Black RA, Bereta J, 2004. Tumour necrosis factor-alpha stimulates expression of TNF-alpha converting enzyme in endothelial cells. Eur. J. Biochem 271, 2808–2820. [DOI] [PubMed] [Google Scholar]
- Camara M, Corrigan F, Jaehne E, Jawahar M, Anscomb H, Koerner H, Baune B, 2013. TNF-[alpha] and its receptors modulate complex behaviours and neurotrophins in transgenic mice. Psychoneuroendocrinology 38, 3102. [DOI] [PubMed] [Google Scholar]
- Chan FK, Chun HJ, Zheng L, Siegel RM, Bui KL, Lenardo MJ, 2000. A domain in TNF receptors that mediates ligand-independent receptor assembly and signaling. Science 288, 2351–2354. [DOI] [PubMed] [Google Scholar]
- Charbonneau M, Harper K, Grondin F, Pelmus M, McDonald PP, Dubois CM, 2007. Hypoxia-inducible factor mediates hypoxic and tumor necrosis factor alpha-induced increases in tumor necrosis factor-alpha converting enzyme/ADAM17 expression by synovial cells. J. Biol. Chem 282, 33714–33724. [DOI] [PubMed] [Google Scholar]
- Churchill L, Rector DM, Yasuda K, Fix C, Rojas MJ, Yasuda T, Krueger JM, 2008. Tumor necrosis factor alpha: activity dependent expression and promotion of cortical column sleep in rats. Neuroscience 156, 71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churchill L, Taishi P, Wang M, Brandt J, Cearley C, Rehman A, Krueger JM, 2006. Brain distribution of cytokine mRNA induced by systemic administration of interleukin-1beta or tumor necrosis factor alpha. Brain Res 1120, 64–73. [DOI] [PubMed] [Google Scholar]
- Churchill L, Yasuda K, Yasuda T, Blindheim KA, Falter M, Garcia-Garcia F, Krueger JM, 2005. Unilateral cortical application of tumor necrosis factor alpha induces asymmetry in Fos- and interleukin-1beta-immunoreactive cells within the corticothalamic projection. Brain Res 1055, 15–24. [DOI] [PubMed] [Google Scholar]
- Clinton JM, Davis CJ, Zielinski MR, Jewett KA, Krueger JM, 2011. Biochemical regulation of sleep and sleep biomarkers. J. Clin. Sleep Med 7, S38–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corner MA, 2008. Spontaneous neuronal burst discharges as dependent and independent variables in the maturation of cerebral cortex tissue cultured in vitro: a review of activity-dependent studies in live ‘model’ systems for the development of intrinsically generated bioelectric slow-wave sleep patterns. Brain Res. Rev 59, 221–244. [DOI] [PubMed] [Google Scholar]
- Corner MA, 2013. From neural plate to cortical arousal-a neuronal network theory of sleep derived from in vitro “model” systems for primordial patterns of spontaneous bioelectric activity in the vertebrate central nervous system. Brain Sci 3, 800–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cárdenas A, Moro MA, Leza JC, O’Shea E, Dávalos A, Castillo J, Lorenzo P, Lizasoain I, 2002. Upregulation of TACE/ADAM17 after ischemic preconditioning is involved in brain tolerance. J. Cereb. Blood Flow Metab 22, 1297–1302. [DOI] [PubMed] [Google Scholar]
- Davis CJ, Clinton JM, Jewett KA, Zielinski MR, Krueger JM, 2011. Delta wave power: an independent sleep phenotype or epiphenomenon? J. Clin. Sleep Med 7, S16–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM, 2003. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res 981, 133–142. [DOI] [PubMed] [Google Scholar]
- De Sarro G, Gareri P, Sinopoli VA, David E, Rotiroti D, 1997. Comparative, behavioural and electrocortical effects of tumor necrosis factor-alpha and interleukin-1 microinjected into the locus coeruleus of rat. Life Sci 60, 555–564. [DOI] [PubMed] [Google Scholar]
- Dinarello CA, 2018. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev 281, 8–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostert C, Grusdat M, Letellier E, Brenner D, 2019. The TNF Family of Ligands and Receptors: Communication Modules in the Immune System and Beyond. Physiol. Rev 99, 115–160. [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Møller JE, Potter AM, Ek J, Lane MA, Saunders NR, 2000. Acute-phase cytokines IL-1beta and TNF-alpha in brain development. Cell Tissue Res 299, 335–345. [DOI] [PubMed] [Google Scholar]
- Edwards DR, Handsley MM, Pennington CJ, 2008. The ADAM metalloproteinases. Mol. Aspects Med 29, 258–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Wang Y, Krueger JM, 1997. Mice lacking the TNF 55 kDa receptor fail to sleep more after TNFalpha treatment. J. Neurosci 17, 5949–5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd RA, Krueger JM, 1997. Diurnal variation of TNF alpha in the rat brain. NeuroReport 8, 915–918. [DOI] [PubMed] [Google Scholar]
- Frei K, Siepl C, Groscurth P, Bodmer S, Schwerdel C, Fontana A, 1987. Antigen presentation and tumor cytotoxicity by interferon-γ-treated microglial cells. Eur. J. Immunol 17, 1271–1278. [DOI] [PubMed] [Google Scholar]
- Ge L, Vujanovic NL, 2017. Soluble TNF Regulates TACE via AP-2α Transcription Factor in Mouse Dendritic Cells. J. Immunol 198, 417–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haack M, Pollmacher T, Mullington JM, 2004. Diurnal and sleep-wake dependent variations of soluble TNF- and IL-2 receptors in healthy volunteers. Brain Behav. Immun 18, 361–367. [DOI] [PubMed] [Google Scholar]
- Hajnik T, Toth A, Detari L, 2013. Characteristic changes in the slow cortical waves after a 6-h sleep deprivation in rat. Brain Res 1501, 1–11. [DOI] [PubMed] [Google Scholar]
- Hinard V, Mikhail C, Pradervand S, Curie T, Houtkooper RH, Auwerx J, Franken P, Tafti M, 2012. Key electrophysiological, molecular, and metabolic signatures of sleep and wakefulness revealed in primary cortical cultures. J. Neurosci 32, 12506–12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann HP, Remy R, Poschl B, van Loon AP, 1990. Tumor necrosis factors-alpha and -beta bind to the same two types of tumor necrosis factor receptors and maximally activate the transcription factor NF-kappa B at low receptor occupancy and within minutes after receptor binding. J. Biol. Chem 265, 15183–15188. [PubMed] [Google Scholar]
- Horiuchi T, Mitoma H, Harashima S, Tsukamoto H, Shimoda T, 2010. Transmembrane TNF-alpha: structure, function and interaction with anti-TNF agents. Rheumatology (Oxford) 49, 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado O, Cárdenas A, Lizasoain I, Boscá L, Leza JC, Lorenzo P, Moro MA, 2001. Up-regulation of TNF-alpha convertase (TACE/ADAM17) after oxygen-glucose deprivation in rat forebrain slices. Neuropharmacology 40, 1094–1102. [DOI] [PubMed] [Google Scholar]
- Ingiosi AM, Opp MR, Krueger JM, 2013. Sleep and immune function: glial contributions and consequences of aging. Curr. Opin. Neurobiol 23, 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jewett K, Sengupta P, Davis CJ, Krueger JM, 2012. Altering Neuronal Firing by Changing Astrocyte-to-Neuron Ratio in Vitro. Sleep 35, A25–A25. [Google Scholar]
- Jewett KA, Taishi P, Sengupta P, Roy S, Davis CJ, Krueger JM, 2015. Tumor necrosis factor enhances the sleep-like state and electrical stimulation induces a wake-like state in co-cultures of neurons and glia. Eur. J. Neurosci 42, 2078–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimbo Y, Kawana A, Parodi P, Torre V, 2000. The dynamics of a neuronal culture of dissociated cortical neurons of neonatal rats. Biol. Cybern 83, 1–20. [DOI] [PubMed] [Google Scholar]
- Jimbo Y, Robinson HP, Kawana A, 1998. Strengthening of synchronized activity by tetanic stimulation in cortical cultures: application of planar electrode arrays. IEEE Trans. Biomed. Eng 45, 1297–1304. [DOI] [PubMed] [Google Scholar]
- Kamioka H, Maeda E, Jimbo Y, Robinson HPC, Kawana A, 1996. Spontaneous periodic synchronized bursting during formation of mature patterns of connections in cortical cultures. Neurosci. Lett 206, 109–112. [DOI] [PubMed] [Google Scholar]
- Kapas L, Bohnet SG, Traynor TR, Majde JA, Szentirmai E, Magrath P, Taishi P, Krueger JM, 2008. Spontaneous and influenza virus-induced sleep are altered in TNF-alpha double-receptor deficient mice. J. Appl. Physiol 1985 (105), 1187–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapas L, Hong L, Cady AB, Opp MR, Postlethwaite AE, Seyer JM, Krueger JM, 1992. Somnogenic, pyrogenic, and anorectic activities of tumor necrosis factor-alpha and TNF-alpha fragments. Am. J. Physiol 263, R708–715. [DOI] [PubMed] [Google Scholar]
- Kapas L, Krueger JM, 1992. Tumor necrosis factor-beta induces sleep, fever, and anorexia. Am. J. Physiol 263, R703–707. [DOI] [PubMed] [Google Scholar]
- Kisiswa L, Osorio C, Erice C, Vizard T, Wyatt S, Davies AM, 2013. TNF alpha reverse signaling promotes sympathetic axon growth and target innervation. Nat. Neurosci 16, 865–U264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, 2020. Sleep and circadian rhythms: Evolutionary entanglement and local regulation. Neurobiol. Sleep Circadian Rhythms 9, 100052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger JM, Nguyen JT, Dykstra-Aiello CJ, Taishi P, 2019. Local sleep. Sleep Med. Rev 43, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota T, Li N, Guan Z, Brown RA, Krueger JM, 2002. Intrapreoptic microinjection of TNF-alpha enhances non-REM sleep in rats. Brain Res 932, 37–44. [DOI] [PubMed] [Google Scholar]
- Kubota T, Majde JA, Brown RA, Krueger JM, 2001. Tumor necrosis factor receptor fragment attenuates interferon-gamma-induced non-REM sleep in rabbits. J. Neuroimmunol 119, 192–198. [DOI] [PubMed] [Google Scholar]
- Layé S, Parnet P, Goujon E, Dantzer R, 1994. Peripheral administration of lipopolysaccharide induces the expression of cytokine transcripts in the brain and pituitary of mice. Brain Res. Mol. Brain Res 27, 157–162. [DOI] [PubMed] [Google Scholar]
- Ledgerwood EC, Pober JS, Bradley JR, 1999. Recent advances in the molecular basis of TNF signal transduction. Lab. Invest 79, 1041–1050. [PubMed] [Google Scholar]
- Lee WH, Seo D, Lim SG, Suk K, 2019. Reverse Signaling of Tumor Necrosis Factor Superfamily Proteins in Macrophages and Microglia: Superfamily Portrait in the Neuroimmune Interface. Front. Immunol 10, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Uttarwar L, Gao B, Charbonneau M, Shi Y, Chan JS, Dubois CM, Krepinsky JC, 2015. High Glucose Up-regulates ADAM17 through HIF-1α in Mesangial Cells. J. Biol. Chem 290, 21603–21614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo CH, Huber EC, Sachs JN, 2020. Conformational states of TNFR1 as a molecular switch for receptor function. Protein Sci 29, 1401–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loetscher H, Gentz R, Zulauf M, Lustig A, Tabuchi H, Schlaeger EJ, Brockhaus M, Gallati H, Manneberg M, Lesslauer W, 1991. Recombinant 55-kDa tumor necrosis factor (TNF) receptor. Stoichiometry of binding to TNF alpha and TNF beta and inhibition of TNF activity. J. Biol. Chem 266, 18324–18329. [PubMed] [Google Scholar]
- Madsen PM, Clausen BH, Degn M, Thyssen S, Kristensen LK, Svensson M, Ditzel N, Finsen B, Deierborg T, Brambilla R, Lambertsen KL, 2016. Genetic ablation of soluble tumor necrosis factor with preservation of membrane tumor necrosis factor is associated with neuroprotection after focal cerebral ischemia. J. Cereb. Blood Flow Metab 36, 1553–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MW, Dunn A, Grail D, Inglese M, Noguchi Y, Richards E, Jungbluth A, Wada H, Moore M, Williamson B, Basu S, Old LJ, 1997. Characterization of tumor necrosis factor-deficient mice. Proc. Natl. Acad. Sci. U.S.A 94, 8093–8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mccoy MK, Tansey MG, 2008. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J. Neuroinflammat 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medvedev AE, Espevik T, Ranges G, Sundan A, 1996. Distinct roles of the two tumor necrosis factor (TNF) receptors in modulating TNF and lymphotoxin alpha effects. J. Biol. Chem 271, 9778–9784. [DOI] [PubMed] [Google Scholar]
- Merrill JE, 1992. Tumor necrosis factor alpha, interleukin 1 and related cytokines in brain development: normal and pathological. Dev. Neurosci 14, 1–10. [DOI] [PubMed] [Google Scholar]
- Naismith JH, Devine TQ, Brandhuber BJ, Sprang SR, 1995. Crystallographic evidence for dimerization of unliganded tumor necrosis factor receptor. J. Biol. Chem 270, 13303–13307. [DOI] [PubMed] [Google Scholar]
- Nedwin GE, Naylor SL, Sakaguchi AY, Smith D, Jarrett-Nedwin J, Pennica D, Goeddel DV, Gray PW, 1985. Human lymphotoxin and tumor necrosis factor genes: structure, homology and chromosomal localization. Nucleic Acids Res 13, 6361–6373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J, Gibbons CM, Dykstra-Aiello C, Ellingsen R, Koh KMS, Taishi P, Krueger JM, 2019a. Interleukin-1 receptor accessory proteins are required for normal homeostatic responses to sleep deprivation. J. Appl. Physiol 1985 (127), 770–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen J, Taishi P, Jewett K, Sandip R, Xue MR, Krueger JM, 2018. Neuron-specific interleukin-1 receptor accessory protein is required for the maturation of small network emergent sleep-like electrophysiological properties. Sleep Res. J
- Nguyen JT, Sahabandu D, Taishi P, Xue M, Jewett K, Dykstra-Aiello C, Roy S, Krueger JM, 2019b. The neuron-specific interleukin-1 receptor accessory protein alters emergent network state properties in Vitro. Neurobiol Sleep Circadian Rhythms 6, 35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nistico GD, Rotiroti D, 1992. Behavioral and electrocortical spectum power changes of interleukins and tumor necrosis factor after microinjection into different areas of the brain. In: Smirne SFM; Ferini-Stambi L; Zucconi M (Eds.), Sleep, Hormones and Immunological System Mason, Milan, pp. 11–22. [Google Scholar]
- Obal F Jr., Krueger JM, 2003. Biochemical regulation of non-rapid-eye-movement sleep. Front. Biosci 8, d520–550. [DOI] [PubMed] [Google Scholar]
- Opp MR, Obal F Jr., Krueger JM, 1991. Interleukin 1 alters rat sleep: temporal and dose-related effects. Am. J. Physiol 260, R52–58. [DOI] [PubMed] [Google Scholar]
- Pasparakis M, Alexopoulou L, Episkopou V, Kollias G, 1996. Immune and inflammatory responses in TNF alpha-deficient mice: a critical requirement for TNF alpha in the formation of primary B cell follicles, follicular dendritic cell networks and germinal centers, and in the maturation of the humoral immune response. J. Exp. Med 184, 1397–1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennica D, Nedwin GE, Hayflick JS, Seeburg PH, Derynck R, Palladino MA, Kohr WJ, Aggarwal BB, Goeddel DV, 1984. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature 312, 724–729. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA, 1998a. An essential role for ectodomain shedding in mammalian development. Science 282, 1281–1284. [DOI] [PubMed] [Google Scholar]
- Peschon JJ, Torrance DS, Stocking KL, Glaccum MB, Otten C, Willis CR, Charrier K, Morrissey PJ, Ware CB, Mohler KM, 1998b. TNF receptor-deficient mice reveal divergent roles for p55 and p75 in several models of inflammation. J. Immunol 160, 943–952. [PubMed] [Google Scholar]
- Probert L, 2015. TNF and its receptors in the CNS: The essential, the desirable and the deleterious effects. Neuroscience 302, 2–22. [DOI] [PubMed] [Google Scholar]
- Rector DM, Topchiy IA, Carter KM, Rojas MJ, 2005. Local functional state differences between rat cortical columns. Brain Res 1047, 45–55. [DOI] [PubMed] [Google Scholar]
- Reddy P, Slack JL, Davis R, Cerretti DP, Kozlosky CJ, Blanton RA, Shows D, Peschon JJ, Black RA, 2000. Functional analysis of the domain structure of tumor necrosis factor-alpha converting enzyme. J. Biol. Chem 275, 14608. [DOI] [PubMed] [Google Scholar]
- Rockstrom MD, Chen L, Taishi P, Nguyen JT, Gibbons CM, Veasey SC, Krueger JM, 2017. Tumor necrosis factor alpha in sleep regulation. Sleep Med. Rev [DOI] [PMC free article] [PubMed]
- Saberi-Moghadam S, Simi A, Setareh H, Mikhail C, Tafti M, 2018. In vitro cortical network firing is homeostatically regulated: A model for sleep regulation. Sci. Rep 8, 6297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenfeld HJ, Poeschl B, Frey JR, Loetscher H, Hunziker W, Lustig A, Zulauf M, 1991. Efficient purification of recombinant human tumor necrosis factor beta from Escherichia coli yields biologically active protein with a trimeric structure that binds to both tumor necrosis factor receptors. J. Biol. Chem 266, 3863–3869. [PubMed] [Google Scholar]
- Smith CA, Farrah T, Goodwin RG, 1994. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death pp. 959–962. [DOI] [PubMed]
- Sporri B, Bickel M, Dobbelaere D, Machado J, Lottaz D, 2001. Soluble interleukin-1 receptor–reverse signaling in innate immunoregulation. Cytokine Growth Factor Rev 12, 27–32. [DOI] [PubMed] [Google Scholar]
- Tachida Y, Nakagawa K, Saito T, Saido TC, Honda T, Saito Y, Murayama S, Endo T, Sakaguchi G, Kato A, Kitazume S, Hashimoto Y, 2008. Interleukin-1 beta up-regulates TACE to enhance alpha-cleavage of APP in neurons: resulting decrease in Abeta production. J. Neurochem 104, 1387–1393. [DOI] [PubMed] [Google Scholar]
- Taishi P, Churchill L, Wang M, Kay D, Davis CJ, Guan X, De A, Yasuda T, Liao F, Krueger JM, 2007. TNFalpha siRNA reduces brain TNF and EEG delta wave activity in rats. Brain Res 1156, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taishi P, Chen GJ, Fang Z, Krueger J, 1999. Sleep deprivation increases the expression of TNF alpha mRNA and TNF 55kD receptor mRNA in rat brain. The Physiologist 42, A4. [Google Scholar]
- Takahashi S, Kapas L, Fang J, Krueger JM, 1999. Somnogenic relationships between tumor necrosis factor and interleukin-1. Am. J. Physiol 276, R1132–1140. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kapas L, Krueger JM, 1996a. A tumor necrosis factor (TNF) receptor fragment attenuates TNF-alpha- and muramyl dipeptide-induced sleep and fever in rabbits. J. Sleep Res 5, 106–114. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Kapas L, Seyer JM, Wang Y, Krueger JM, 1996b. Inhibition of tumor necrosis factor attenuates physiological sleep in rabbits. NeuroReport 7, 642–646. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Krueger JM, 1997. Inhibition of tumor necrosis factor prevents warming-induced sleep responses in rabbits. Am. J. Physiol 272, R1325–1329. [DOI] [PubMed] [Google Scholar]
- Takahashi S, Tooley DD, Kapas L, Fang J, Seyer JM, Krueger JM, 1995. Inhibition of tumor necrosis factor in the brain suppresses rabbit sleep. Pflugers Arch 431, 155–160. [DOI] [PubMed] [Google Scholar]
- Taoufik E, Tseveleki V, Chu SY, Tselios T, Karin M, Lassmann H, Szymkowski DE, Probert L, 2011. Transmembrane tumour necrosis factor is neuroprotective and regulates experimental autoimmune encephalomyelitis via neuronal nuclear factor-kappaB. Brain 134, 2722–2735. [DOI] [PubMed] [Google Scholar]
- Taoufik E, Valable S, Müller GJ, Roberts ML, Divoux D, Tinel A, Voulgari-Kokota A, Tseveleki V, Altruda F, Lassmann H, Petit E, Probert L, 2007. FLIP (L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J. Neurosci 27, 6633–6646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Team R, 2020. RStudio: Integrated Development Environment for R RStudio, PBC, Boston, MA. [Google Scholar]
- Timofeev I, Grenier F, Steriade M, 2001. Disfacilitation and active inhibition in the neocortex during the natural sleep-wake cycle: an intracellular study. Proc. Natl. Acad. Sci. U.S.A 98, 1924–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, 2008. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell 135, 422–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagenaar DA, Madhavan R, Pine J, Potter SM, 2005. Controlling bursting in cortical cultures with closed-loop multi-electrode stimulation. J. Neurosci 25, 680–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts AD, Hunt NH, Wanigasekara Y, Bloomfield G, Wallach D, Roufogalis BD, Chaudhri G, 1999. A casein kinase I motif present in the cytoplasmic domain of members of the tumour necrosis factor ligand family is implicated in ‘reverse signalling’. EMBO J 18, 2119–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yli-Karjanmaa M, Clausen BH, Degn M, Novrup HG, Ellman DG, Toft-Jensen P, Szymkowski DE, Stensballe A, Meyer M, Brambilla R, Lambertsen KL, 2019a. Topical Administration of a Soluble TNF Inhibitor Reduces Infarct Volume After Focal Cerebral Ischemia in Mice. Front. Neurosci 13, 781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yli-Karjanmaa M, Larsen KS, Fenger CD, Kristensen LK, Martin NA, Jensen PT, Breton A, Nathanson L, Nielsen PV, Lund MC, Carlsen SL, Gramsbergen JB, Finsen B, Stubbe J, Frich LH, Stolp H, Brambilla R, Anthony DC, Meyer M, Lambertsen KL, 2019b. TNF deficiency causes alterations in the spatial organization of neurogenic zones and alters the number of microglia and neurons in the cerebral cortex. Brain Behav. Immun 82, 279–297. [DOI] [PubMed] [Google Scholar]
- Yue HJ, Mills PJ, Ancoli-Israel S, Loredo JS, Ziegler MG, Dimsdale JE, 2009. The roles of TNF-alpha and the soluble TNF receptor I on sleep architecture in OSA. Sleep Breath 13, 263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Hagshenas J, Insolera R, Fink DJ, Mata M, 2010. A novel cell-cell signaling by microglial transmembrane TNFa with implications for neuropathic pain. Pain 151, 296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Dunbrasky DL, Taishi P, Souza G, Krueger JM, 2013. Vagotomy attenuates brain cytokines and sleep induced by peripherally administered tumor necrosis factor-alpha and lipopolysaccharide in mice. Sleep 36 (1227–1238), 1238A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielinski MR, Krueger J, 2012. Inflammation and Sleep. In: Barkoukis TJ, Matheson JK, Ferber R, Doghramji K (Eds.), Therapy in Sleep Medicine Elsevier, pp. 607–616. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.