

SPEG (Striated Preferentially Expressed Protein Kinase), a member of the myosin light chain kinase family, is critical for cardiac and skeletal muscle function involved in excitation-contraction coupling. Genetic variants in SPEG are associated with centronuclear myopathy with or without dilated cardiomyopathy (DCM).1,2 Here, we report 3 individuals from 2 families who presented with nonsyndromic DCM, carrying a homozygous in-frame deletion in SPEG (NM_005876.5; c.9028_9030delGAG, p.Glu3010del). This study was approved by an institutional review committee (IRB00010471), and informed consent from the patients or their parents was obtained. The data that support the findings of this study are available from the corresponding author upon reasonable request.
Patient 1, born at full term to consanguineous parents and currently 5 years old, presented at 2 years of age with poor feeding, breathing difficulties, and metabolic acidosis. Electrocardiogram showed normal sinus rhythm and no history of arrhythmia. Echocardiogram revealed severely dilated left ventricle (LV), depressed LV ejection fraction (EF) of 13%, and moderate mitral valve regurgitation (MVR). He was started on anti-failure medications with some improvement. Last evaluation at 5 years of age showed dilated left atrium and LV, moderate MVR, and estimated LVEF of 30.3 % with fractional shortening of 14.3%. Growth and development were normal with normal tone and muscle strength. At age of 4 years, he started to have unexplained episodes of hypoglycemia of unknown cause despite extensive investigation. He is currently on carvedilol in addition to furosemide, lisinopril, and digoxin. Hypoglycemia secondary to carvedilol, a nonselective both beta- and alpha-adrenergic receptor blocker, cannot be completely excluded.
Patient 2 is the younger sister who also had DCM with moderate LV dilatation, severely impaired LV function (LVEF data unavailable), moderate MVR, and mild tricuspid regurgitation. At 5 months of age, she was noted to have some developmental delay (not rolling, smiling only) and mild generalized hypotonia. She died at 8 months of age, likely due to heart failure.
Exome sequencing for both siblings revealed a homozygous in-frame deletion in SPEG (c.9028_9030delGAG, p.Glu3010del), parents being carriers for the variant. This variant is absent from the Genome Aggregation Database, and the glutamate 3010 residue is highly conserved in vertebrates and invertebrates. No other variants in DCM-related genes were reported.
Patient 3 is a 2-year-old girl born to consanguineous parents, who was hospitalized at 5 months of age with DCM requiring inotropic support. Electrocardiogram showed normal sinus rhythm with T-wave inversion in lateral leads and there was no history of arrhythmia. Echocardiogram showed severely dilated left atrium and LV, depressed LVEF of 22%, mild MVR, and moderate tricuspid valve repair. Muscle tone and motor development were normal. Latest echocardiogram at 24 months of age showed moderately dilated left atrium and LV, mild MVR, and estimated LVEF of 26.9 % with fractional shortening of 12.3%. Developmentally she achieved all her gross and fine motor and cognitive milestones at appropriate age and has normal muscle tone and strength.
The father, currently 30-year-old, has history of near syncope due to complete heart block needing permanent cardiac pacemaker. Echocardiogram showed normal LV size and function with EF 50% to 55%, grade 2 LV diastolic dysfunction, normal right ventricle size with mild right ventricle systolic dysfunction. Twenty-four–hour Holter monitoring showed minimum heart rate of 31 bpm and average 61 bpm with high-grade complete heart block. The mother and the healthy brother did not have cardiac evaluation yet. The same SPEG variant was identified in the proband on quad exome sequencing, both parents being carriers and a healthy brother homozygous for the normal allele. No other variants were reported in the father.
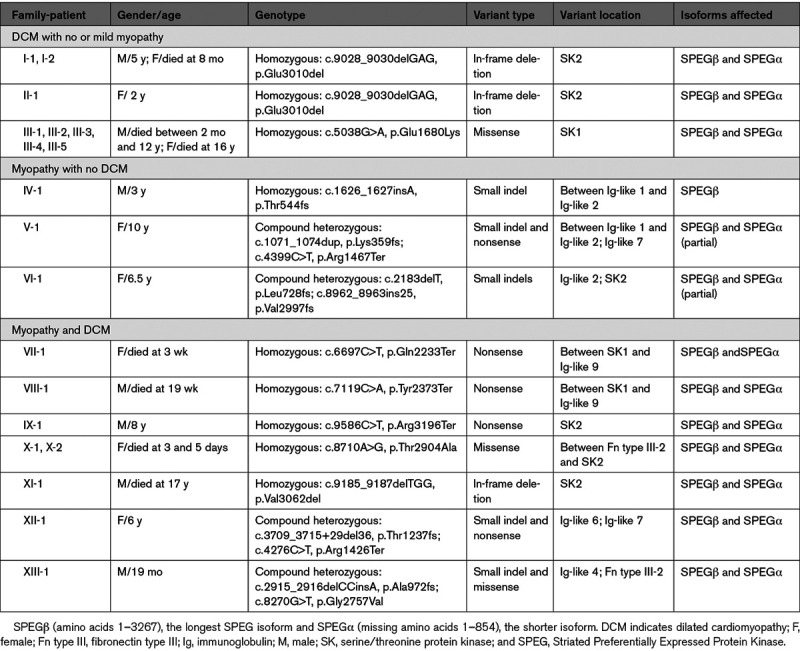
In summary, we report 3 patients with SPEG-associated DCM homozygous for an in-frame deletion (p.Glu3010del). None of them have history of ophthalmoplegia or ptosis, although formal neurological evaluation or electromyography/muscle biopsy were not performed. In the Table, we summarize the clinical and molecular findings of all 19 patients with SPEG mutations.1–3 Although majority of the patients had myopathy with or without DCM, isolated DCM without myopathy was recently described in a family of five members carrying homozygous missense mutation (c.5038G>A, p.Glu1680Lys) in the SK (serine/threonine protein kinase)-1 domain of SPEG.3 Here, we identified 3 patients with a homozygous in-frame deletion in the SK2 domain of SPEG. Structure-based alignment indicated that both residues (glutamate) are highly conserved and may function as nucleotide-binding sites. The SK1 domain of SPEG has been shown to phosphorylate junctophilin-2 in cardiac muscle, whereas SK2 phosphorylates sarco-endoplasmic reticulum ATPase-2a.4,5 Further investigations are needed to understand how these glutamate residue mutations may specifically affect the role of SPEG in the cardiac muscle.
Table.
Clinical and Molecular Findings in Individuals Carrying SPEG Mutations
This study expands the phenotypic heterogeneity associated with SPEG mutations with the identification of isolated DCM without myopathy, and further describes genotype-phenotype correlations to guide appropriate clinical diagnosis and management. A better understanding of the underlying mechanism of DCM associated with unique SPEG variants could unravel potential therapeutic avenues for the patients.
Acknowledgments
We thank the study participants and their families for their support and enrollment.
Sources of Funding
P.B. Agrawal is supported by R01 AR068429 from National Institute of Arthritis and Musculoskeletal and Skin Diseases of National Institute of Health (NIH).
Disclosures:
None.
Footnotes
For Sources of Funding and Disclosures, see page 257.
Contributor Information
Shiyu Luo, Email: shiyu.luo@childrens.harvard.edu.
Eissa Faqeih, Email: efaqeih@kfmc.med.sa.
Fuad Al Mutairi, Email: almutairifu@ngha.med.sa.
Qifei Li, Email: qifei.li@childrens.harvard.edu.
References
- 1.Agrawal PB, Pierson CR, Joshi M, Liu X, Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC, Haliloğlu G, et al. SPEG interacts with myotubularin, and its deficiency causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum Genet. 2014; 95:218–226. doi: 10.1016/j.ajhg.2014.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tang J, Ma W, Chen Y, Jiang R, Zeng Q, Tan J, Jiang H, Li Q, Zhang VW, Wang J, et al. Novel SPEG variant cause centronuclear myopathy in China. J Clin Lab Anal. 2020; 34:e23054. doi: 10.1002/jcla.23054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Levitas A, Muhammad E, Zhang Y, Perea Gil I, Serrano R, Diaz N, Arafat M, Gavidia AA, Kapiloff MS, Mercola M, et al. A novel recessive mutation in SPEG causes early onset dilated cardiomyopathy. PLoS Genet. 2020; 16:e1009000. doi: 10.1371/journal.pgen.1009000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Quick AP, Wang Q, Philippen LE, Barreto-Torres G, Chiang DY, Beavers D, Wang G, Khalid M, Reynolds JO, Campbell HM, et al. SPEG (striated muscle preferentially expressed protein kinase) is essential for cardiac function by regulating junctional membrane complex activity. Circ Res. 2017; 120:110–119. doi: 10.1161/CIRCRESAHA.116.309977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quan C, Li M, Du Q, Chen Q, Wang H, Campbell D, Fang L, Xue B, MacKintosh C, Gao X, et al. SPEG controls calcium reuptake into the sarcoplasmic reticulum through regulating SERCA2a by its second kinase-domain. Circ Res. 2019; 124:712–726. doi: 10.1161/CIRCRESAHA.118.313916 [DOI] [PubMed] [Google Scholar]