

Abstract
G-protein-biased agonists with reduced β-arrestin-2 activation are being investigated as safer alternatives to clinically-used opioids. β-arrestin-2 has been implicated in the mechanism of opioid-induced antinociceptive tolerance. Opioid-induced analgesic tolerance is classically considered as centrally-mediated, but recent reports implicate nociceptive dorsal root ganglia neurons as critical mediators in this process. Here, we investigated the role of β-arrestin-2 in the mechanism of opioid tolerance in dorsal root ganglia nociceptive neurons using β-arrestin-2 knockout mice and the G-protein-biased μ-opioid receptor agonist, TRV130. Whole-cell current-clamp electrophysiology experiments revealed that 15-18-h overnight exposure to 10 μM morphine in vitro induced acute tolerance in β-arrestin-2 wild-type but not knockout neurons. Furthermore, in wild-type neurons circumventing β-arrestin-2 activation by overnight treatment with 200 nM TRV130 attenuated tolerance. Similarly, acute morphine tolerance in vivo in β-arrestin-2 knockout mice was prevented in the warm-water tail-withdrawal assay. Treatment with 30 mg/kg TRV130 s.c. also inhibited acute antinociceptive tolerance in vivo in wild-type mice. Alternately, in β-arrestin-2 knockout neurons tolerance induced by 7-day in vivo exposure to 50 mg morphine pellet was conserved. Likewise, β-arrestin-2 deletion did not mitigate in vivo antinociceptive tolerance induced by 7-day exposure to 25 mg or 50 mg morphine pellet in both female or male mice, respectively. Consequently, these results indicated that β-arrestin-2 mediates acute but not chronic opioid tolerance in dorsal root ganglia neurons and to antinociception in vivo. This suggests that opioid-induced antinociceptive tolerance may develop even in the absence of β-arrestin-2 activation, and thus significantly affect the clinical utility of biased agonists.
Keywords: morphine, antinociception, biased agonism, TRV130, mu-opioid receptor, analgesic tolerance
1. Introduction
μ-opioid receptors are known to engage scaffolding proteins called β-arrestins, which classically function to desensitize activated G-protein-coupled receptors (GPCRs) through stearic inhibition, but can also activate molecular mechanisms independent of G-protein signaling (Lefkowitz, 1998; Lohse et al., 1990). Previously, studies have shown that genetic deletion or down-regulation of β-arrestin-2, or use of G-protein-biased agonists enhances antinociception and reduces antinociceptive tolerance in rodents, thus implicating β-arrestin-2 in the mechanism of opioid-induced antinociceptive tolerance (Bohn et al., 1999; Bohn et al., 2000; DeWire et al., 2013; Grim et al., 2020; Manglik et al., 2016; Wang et al., 2016).
Primary afferent neurons of the dorsal root ganglia (DRG), specifically the thinly myelinated Aδ-and unmyelinated C-fiber neurons, are first-order components of the ascending pain pathway. These neurons generate and transduce chemical, mechanical and thermal noxious stimuli as action potentials from the periphery to second-order neurons in the spinal cord, which synapse with neurons in the pain center in the brain and with motor neurons of the spinal reflex arc (Stein and Machelska, 2011). Thus, nociceptive DRG neurons generate signals that eventually get processed in the CNS as “pain”.
While the prevailing dogma is that opioid-induced analgesia and tolerance are centrally mediated, recent studies have highlighted the critical role of nociceptive DRG neurons in the expression of antinociception and induction of opioid tolerance. Selective deletion of μ-opioid receptors from Nav1.8-containing neurons diminished the ability of morphine to mitigate inflammatory pain (Weibel et al., 2013), whereas universal elimination of μ-opioid receptors from DRG neurons abolished the acute spinal and supraspinal antinociceptive effects of opioids (Sun et al., 2019; Sun et al., 2020). Ablation of Transient Receptor Potential Vanilloid-1 (TRPV1) channel-expressing DRG neurons or conditional knockout of μ-opioid receptors on TRPV1-expressing DRG neurons reversed antinociceptive tolerance (Chen et al., 2007; Corder et al., 2017), suggesting that μ-opioid receptors on primary afferent DRG neurons could be important targets for the prevention of antinociceptive tolerance. Therefore, it is important to delineate the molecular mechanisms underlying cellular tolerance in DRG nociceptive neurons, specifically the role of β-arrestin-2.
In the present study, we investigated the role of β-arrestin-2 in the mechanism of opioid tolerance in small-diameter DRG nociceptive neurons using β-arrestin-2 knockout mice and the G-protein-biased μ-opioid receptor agonist, TRV130 (DeWire et al., 2013). We demonstrate that acute or “short-term” tolerance—manifested as a result of several hours (overnight) of opioid exposure (Williams et al., 2013)—in nociceptive DRG neurons is mediated by β-arrestin-2, whereas chronic or “long-term” tolerance—developing due to seven days of opioid exposure (Williams et al., 2013)— is independent of β-arrestin-2. Acute antinociceptive tolerance to in vivo exposure to TRV130 or morphine in mice for one day is also attenuated in the absence of β-arrestin-2 activation. However, chronic antinociceptive tolerance to morphine in either male or female mice develops independent of the β-arrestin-2 pathway. In conclusion, the findings presented in this study indicate that β-arrestin-2 is a critical mediator of acute tolerance but does not underlie chronic tolerance in DRG nociceptive neurons.
2. Materials and Methods
2.1. Drugs and Chemicals
Morphine sulfate pentahydrate and implantable morphine pellets (25 mg) were obtained from the National Institutes of Health National Institute on Drug Abuse (Bethesda, MD). (+) TRV130 was obtained from Dr. Bruce Blough (Research Triangle Institute, NC). Pyrogen-free isotonic saline was purchased from Hospira (Lake Forest, IL). Dulbecco’s modified Eagle’s medium (DMEM)/F12, Neurobasal-A medium, Ca2+ and Mg2+ -free Hank’s balanced salt solution (HBSS), 50x B-27 supplement and L-glutamine were purchased from Gibco, Thermo Fisher Scientific (Waltham, MA). Penicillin/streptomycin/amphotericin B antibiotic-antimycotic solution and laminin were purchased from Corning (Corning, NY). Papain, glial cell line-derived neurotrophic factor (GDNF) and fetal bovine serum (FBS) were purchased from Worthington Biochemical Corporation (Lakewood, NJ), Neuromics (Edina, MN) and Quality Biological, (Gaithersburg, MD), respectively. Glass cover slips were purchased from ThermoFisher Scientific (Waltham, MA). Twenty-four-well culture dishes and 35 x 10 mm petri dishes were purchased from CELLTREAT (Pepperell, MA). Collagenase from Clostridium histolyticum, poly-D-lysine, CaCl2, MgCl2, NaCl, KCl, HEPES, EGTA, NaH2PO4, glucose, Na2ATP, NaGTP, L-aspartic acid (K salt), KOH, NaOH, MgSO4 and NaHCO3 were purchased from MilliporeSigma (Burlington, MA).
2.2. Animals
All animal care and experimental procedures were conducted in accordance with procedures reviewed and approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University (Protocol # AM10322) in compliance with the US National Research Council’s Guide for the Care and Use of Laboratory Animals, the US Public Health Service’s Policy on Humane Care and Use of Laboratory Animals, and Guide for the Care and Use of Laboratory Animals.
Male and female β-arrestin-2 knockout (KO) or their wild-type (WT) littermates were separately group-housed by genotype with up to five animals per IVC cage in animal-care quarters maintained at 22°C ± 2°C on a 12-h light/dark cycle. Mice used in experimental procedures were at least 7 weeks of age. Male β-arrestin-2 mice weighed 20-25 g, and female β-arrestin-2 mice weighed 19-23 g. Mice were acclimated in the vivarium for at least one week prior to experimentation. Breeding pairs for the β-arrestin-2 mice were initially obtained from Dr. Lefkowitz (Duke University, Durham, NC) and housed within the transgenic facility at Virginia Commonwealth University. The genetic background of the β-arrestin-2 WT and KO mice used in our experimental procedures was 81% C57B6J:19% C57B6N. Mice had access to food and water ad libitum unless specified otherwise.
2.3. In vivo morphine treatment
Subcutaneously implanted continuous-release morphine pellets were used to model chronic in vivo exposure as pellets maintain a high plasma level of morphine over a longer period of time compared to intermittent injections or osmotic pumps and the tolerance induced is more robust (Dighe et al., 2009; McLane et al., 2017). Where indicated, one or two 25 mg morphine pellets were implanted subcutaneously in the dorsum of female or male β-arrestin-2 mice for 7 days, respectively. This dose was based on a previous study, where we demonstrated that a 7-day exposure to one 25 mg and two 25 mg morphine pellets in female and male β-arrestin-2 mice, respectively, substantially right-shifted the morphine dose response curve compared to wild-type mice in the warm-water tail-withdrawal assay, indicating antinociceptive tolerance (Muchhala et al., 2020). The dose of two 25 mg morphine pellets is henceforth denoted as “50 mg” in the present study. In all experiments, control mice were implanted with one or two placebo pellets. In order to implant the pellet, mice were first anesthetized with 2.5% isoflurane before shaving the hair from the base of the neck. Skin was disinfected with 10% povidone iodine (General Medical Corp, Walnut, CA) and alcohol. A 1 cm horizontal incision was made at the base of the neck and one or two pellets were inserted in the subcutaneous space. The surgical site was closed with Clay Adams Brand, MikRon AutoClip 9-mm wound clips (Becton Dickinson, Franklin Lakes, NJ) and cleansed with 10% povidone iodine. Use of aseptic surgical techniques minimized any potential contamination of the pellet, incision and subcutaneous space. Mice were allowed to recover in their home cages where they remained throughout the experiment.
A repeated injection schedule as described previously by Bohn et al. (2002) was also used to induce acute antinociceptive tolerance. Here, mice were injected subcutaneously with a high dose of morphine (100 mg/kg s.c.) or TRV130 (30 mg/kg s.c.) on Day 1. Control mice received saline. On the next day, antinociceptive tolerance was assessed using the warm-water tail-withdrawal assay (as described below) by injecting mice with either 10 mg/kg morphine s.c. or 3 mg/kg TRV130 s.c. The doses of TRV130 used in this experiment were adjusted to their morphine equivalents.
2.4. Evaluating thermal nociception
Thermal nociception was examined using the warm-water tail-withdrawal test, which represents the sensory aspects of spinally-mediated acute pain and has been classically used to test the efficacy of opioid analgesics (Mogil, 2009). In the warm-water tail-withdrawal assay, mice were gently secured in a cloth and the distal 1/3rd of the tail was immersed in a water bath warmed to 56°C ± 0.1°C. The latency to withdraw the tail from the water was recorded. A maximum cut-off of 10 s was set to prevent damage to the tail. Only naïve mice with control latency between 2 and 4 s were used in experiments. Tolerance was assessed by challenging mice with an acute subcutaneous injection of morphine or TRV130. Tail-withdrawal latency was determined 30 min after morphine or TRV130 injection and compared against baseline latency. Where indicated, antinociception was quantified as %MPE (Percent maximum possible effect), which was calculated as follows: %MPE= [(Challenge latency-baseline latency)/ (10-baseline latency)] x 100, adapted from Harris and Pierson (1964).
For the morphine cumulative dose response curves, baseline tail-withdrawal latencies were recorded as described above in 7-day placebo or morphine-pelleted mice. After testing baselines placebo-pelleted mice were injected subcutaneously every 30 min with 0.5, 0.5, 1.0, 2.0, 4.0 and 8.0 mg/kg morphine to achieve final cumulative doses of 0.5, 1.0, 2.0, 4.0, 8.0 and 16.0 mg/kg morphine. Morphine-pelleted mice were similarly tested in a cumulative fashion with doses ranging from 2.0 mg/kg to 128.0 mg/kg. Tail-withdrawal latencies were recorded every 30 min, followed immediately with an injection of the subsequent highest dose. Antinociception was quantified as %MPE.
2.5. Behavioral testing
All testing was conducted in a temperature and light-controlled room in the light phase of the 12-h light/dark cycle. Mice were acclimated to the testing room for at least 15-18 h before commencing experiments to mitigate stress to the animals and eliminate confound from potential stress-induced effects on antinociception (Sorge et al., 2014). All animals were randomly divided into control and treatment groups. Mice were excluded from experiments if they exhibited wounds from aggressive interactions with cage mates, since injury-induced activation of the endogenous opioid system could confound nociceptive assays (Corder et al., 2013).
2.6. Isolation and primary culture of dorsal root ganglia neurons
Dorsal root ganglia (DRG) cells were prepared from adult mice as previously described (Ross et al., 2012). Mice were killed via CO2 inhalation and L5 – S1 DRGs were immediately harvested using a dissecting microscope. DRGs were placed in a 35 mm dish containing Hank’s balanced salt solution (HBSS) and papain (15 U/ml) was added prior to incubation at 37°C for 18 min. After the initial incubation step, ganglia were transferred to a new 35 mm dish containing HBSS and 1.5 mg/ml collagenase from Clostridium histolyticum and incubated for 1 h at 37°C. DRGs were then transferred to a sterile 15 ml conical tube containing ice-cold (4°C) DMEM/F12 supplemented with 10% FBS and dissociated by trituration before being centrifuged at 350 x g for 5 min. The supernatant was decanted and the pellet was resuspended in neurobasal A media that contained 1% FBS, 1x B-27 supplement, 10 ng/ml GDNF, 2 mM L-glutamine, and 100 U/ml penicillin/streptomycin/amphotericin B (complete neuron media). Cells were then plated on glass cover slips (1/well) coated with laminin and poly-D-lysine. Twenty-four-well plates containing isolated DRGs were then incubated overnight at 37°C in a humidified 5% CO2/air stabilized incubator. Whole-cell patch clamp electrophysiology experiments were conducted 15-18 h later on the following day. Where indicated, neurons were exposed to 10 μM morphine sulfate pentahydrate or 200 nM TRV130 in complete neuron media for 15-18 h (overnight) prior to whole-cell patch clamp experiments to mimic prolonged opioid exposure.
Cells were also isolated from male β-arrestin-2 WT or KO mice subcutaneously implanted with 50 mg morphine pellet for 7 using the procedure described above. Isolated cells were incubated as described above in Neurobasal-A media supplemented with growth factors and antibiotic-antimycotic solution for 15-18 h.
2.7. Whole-cell patch clamp electrophysiology
Micropipettes for patch clamp experiments (2-4 MΩ) were made from pulled (Model P-97 Flaming-Brown Micropipette Puller, Sutter Instruments, Novato, CA) and fire-polished 1.5/0.84 o.d./i.d. (mm) borosilicate glass capillaries (World Precision Instruments, Sarasota, FL). The internal physiological solution was composed of 100 mM L-aspartic acid (K salt), 30 mM KCl, 4.5 mM Na2ATP, 1 mM MgCl2, 10 mM HEPES, 0.1 mM EGTA and 0.5 mM NaGTP and pH adjusted to 7.2 with 3 M KOH. Coverslips with adherent cells were transferred to a microscope stage plate continuously superfused with external physiological salt solution composed of 135 mM NaCl, 5.4 mM KCl, 0.33 mM NaH2PO4, 5 mM HEPES, 5 mM glucose, 2 mM CaCl2 and 1 mM MgCl2, and adjusted to a pH of 7.4 with 1 M NaOH. Whole-cell current-clamp recordings were made in a room temperature environment using HEKA EPC 10 (HEKA, Bellmore, NY) or Axopatch 200B (Molecular Devices, Sunnyvale, CA) amplifiers at a 10 kHz sampling frequency and 2.9 kHz low-pass Bessel filtering. Pulse generation and data acquisition were achieved with PatchMaster v2x60 (HEKA) or Clampex and Clampfit 10.2 software (Molecular Devices). The current-clamp protocol consisted of 10 pA steps, beginning at −30 pA to assess both active and passive cell properties. All current clamp recordings were performed five min after achieving whole-cell mode to allow dialysis of internal solution. Pre- “baseline” recordings were taken to ensure cell stability before beginning drug perfusion. Once neurons were deemed stable, the external solution source was exchanged for the external solution containing 3 μM morphine or 50 nM TRV130. A recording was taken immediately following this exchange, and was labeled “T0” or time zero, which served as the “baseline” recording in all studies. A recording was then taken for up to 16 min to capture neuronal responses to morphine or TRV130. Electrical properties such as threshold potential, rheobase, resting membrane potential and input resistance were extrapolated from each recording, and the difference between baseline and following drug exposure was calculated for each cell. Action potential (AP) derivatives were determined using the differential function in the PatchMaster or Clampfit software, where the derivative of the voltage with respect to time (dV/dt) was calculated in order to estimate threshold potential. Threshold potential was defined as the voltage at which dV/dt significantly deviated from zero during the course of the action potential uprise. It was used as the primary measure of neuronal excitability in our experiments. Each coverslip was discarded following drug exposure and the same process was repeated on a freshly-mounted coverslip. Cellular tolerance was assessed identically in cells either incubated with 200 nM TRV130 or 10 μM morphine overnight, or isolated from male mice implanted with two 25 mg morphine pellets for 7 days. Values reported were not corrected for junction potentials (~12 mV).
Experiments were performed only on cells with healthy morphology and stable patch. In DRG primary isolations, small-diameter neurons (< 30 pF capacitance) correspond to nociceptive Aδ fiber and C-type neurons (Abraira and Ginty, 2013; Barabas et al., 2014), therefore we preferentially selected these cells for use in our experiments. Due to the presence of multiple subtypes of small-diameter DRG neurons in our primary cultures, measures from individual neurons were considered as independent values and not replicates for data analysis. In all electrophysiology experiments ‘n’ represents the number of neurons per group and ‘N’ denotes number of animals per group.
2.8. Data and statistical analysis
Data analysis was performed in GraphPad Prism 9.0 (GraphPad Software, Inc., La Jolla, CA). Data are expressed as mean ± S.E.M. Depending on the experimental design data were analyzed using 2-tailed unpaired Student’s t-test, 2-way ANOVA or 2-way repeated measures ANOVA. In Figure 2, 2-way repeated measures ANOVA failed to detect a statistically significant interaction of the two independent variables, yet there were clear differences in the data as a result of drug administration. Therefore, we used multiple 2-tailed paired t-tests with two-stage step-up method of Benjamini, Krieger and Yekutieli, and a false discovery rate of 5%. Where specified in electrophysiology experiments, 3-way repeated-measures ANOVA was used to evaluate the main effect of three independent variables on action potential threshold, rheobase, resting membrane potential or input resistance. Bonferroni’s multiple comparisons post-hoc test was conducted only if the F value in the ANOVA table was significant (Curtis et al., 2018). An alpha level of 0.05 was pre-determined. Thus, data were statistically significant when P<0.05. The specific statistical tests used for data analysis are indicated in the text or figure legends. Sample sizes were based on our previous studies with similar experimental protocols.
Figure 2. Overnight exposure to TRV130 does not produce tolerance in β-arrestin-2 WT DRG neurons.

Representative long-pulse (100 ms) current-clamp traces of (A) naïve and (B) overnight TRV130-treated β-arrestin-2 WT DRG neurons. Action potential is generated at baseline (black) and after acute 50 nM TRV130 challenge (red). Threshold potential is extrapolated from the point on the action potential derivative trace, where the action potential differential (dV/dt) > 0. (C) Threshold potentials of naïve and overnight 200 nM TRV130-treated DRG neurons isolated from male β-arrestin-2 WT mice at baseline and after acute challenge with 50 nM TRV130. Data were analyzed by multiple 2-tailed paired t-tests with two-stage step-up method of Benjamini, Krieger and Yekutieli. The False Discovery Rate was set to 5%. *Adjusted P<0.05 and **adjusted P<0.01 vs. baseline. (D) Threshold potential change from baseline after acute 50 nM TRV130 challenge. ‘ns’ is not significant (P>0.05) by 2-tailed unpaired t-test. Naïve: N = 5, n = 9; and Overnight-treated: N = 2, n = 9. Data are mean ± S.E.M. Scatter represents individual cells.
3. Results
3.1. β-arrestin-2 knockout prevents the development of short-term tolerance in DRG neurons
We investigated whether opioid tolerance in DRG nociceptors following overnight (short-term) exposure is mediated by β-arrestin-2. For this purpose, we harvested DRG neurons from male β-arrestin-2 WT or KO mice and incubated them in media containing 10 μM morphine for 15-18 h (overnight). Cellular tolerance was assessed in only small-diameter DRG neurons (membrane capacitance < 30 pF) by challenging them with 3 μM morphine (Fig. 1). We used threshold potential, which is the membrane potential at which action potential is elicited, as the primary metric of neuronal excitability. An increase in threshold potential from baseline represented reduced neuronal excitability, whereas a cell was described as “tolerant” if the acute challenge had no effect on threshold potential. In untreated DRG neurons from both β-arrestin-2 WT and KO mice, acute 3 μM morphine significantly increased action potential threshold from baseline (Figs. 1A, 1B and 1C; Table S1). In contrast, no significant shift in action potential threshold from baseline was observed when WT neurons previously exposed to 10 μM morphine overnight were challenged with 3 μM morphine (Figs. 1A and 1C; Table S1). The shift in threshold potential from baseline was significantly decreased compared to that in naïve β-arrestin-2 WT neurons (Fig. 1D). These data together signify the development of cellular tolerance in overnight morphine-treated WT neurons. However, unlike WT neurons, β-arrestin-2 KO DRG neurons exhibited reduced threshold potentials in response to the acute 3 μM morphine challenge despite being exposed to 10 μM morphine overnight (Figs. 1B and 1C; Table S1). The shift in threshold potential in overnight morphine-treated β-arrestin-2 KO neurons was not significantly different from that in naïve β-arrestin-2 KO neurons (Fig. 1D) indicating that individual neurons devoid of β-arrestin-2 did not become tolerant to morphine. Analysis of the entire dataset in Figure 1C by 3-way repeated-measures ANOVA revealed a significant main effect of genotype versus overnight morphine treatment versus acute morphine challenge on action potential threshold [F (1, 22) = 5.73; P = 0.03; Table S1], but not on rheobase [F (1,22) = 0.000136; P = 0.90; Table S1], resting membrane potential [F (1,22) = 0.149; P = 0.70; Table S1] or input resistance [F (1,22) = 1.015; P = 0.32; Table S1].
Figure 1. β-arrestin-2 knockout abolishes tolerance to overnight morphine exposure in DRG neurons.

Representative short-pulse (10 ms) whole-cell current-clamp traces of naïve and overnight morphine-treated (A) β-arrestin-2 WT or (B) β-arrestin-2 KO DRG neurons. Action potential is generated at baseline (black) and after 16 min of acute 3 μM morphine challenge (red). Threshold potential, which is a measure of neuronal excitability, is extrapolated from the point on the action potential derivative trace, where the action potential differential (dV/dt) > 0. (C) Threshold potentials of β-arrestin-2 WT and KO DRG neurons at baseline and after bath exposure to 3 μM morphine for up to 16 min. Data analyzed by 3-way repeated-measures ANOVA with Bonferroni’s post-test. ***P<0.001 vs. baseline and ‘ns’ is not significant (P>0.05). Scatter represents individual cells (D) Threshold potential change from baseline following acute 3 μM morphine challenge for up to 16 min. Data analyzed by 2-way ANOVA with Bonferroni’s post-test. Data are mean ± S.E.M. Scatter represents individual cells. **P<0.01 and ‘ns’ is not significant (P>0.05). Naïve WT: N = 4 mice n = 7 cells; Overnight-treated WT: N = 3, n= 5; Naïve KO: N = 5, n = 7; and Overnight-treated KO: N = 3, n = 7.
Consequently, these data indicate that short-term morphine tolerance in DRG neurons is mediated by β-arrestin-2.
3.2. TRV130 does not produce short-term tolerance in DRG neurons
We next studied the effects of TRV130, a G-protein biased agonist that does not promote the recruitment of β-arrestin-2 to the μ-opioid receptor (DeWire et al., 2013; Pedersen et al., 2020). Neurons isolated from male β-arrestin-2 WT mice were incubated overnight with 200 nM TRV130 and cellular tolerance was evaluated by challenging DRG neurons (membrane capacitance < 30 pF) with 50 nM TRV130 (Fig. 2). In β-arrestin-2 WT neurons incubated in untreated media, acute exposure to 50 nM TRV130 led to statistically significant shifts in threshold potentials to more positive values compared to baseline recordings (Figs 2A and 2C; Table S2). Following overnight incubation in 200 nM TRV130, neurons challenged with 50 nM TRV130 on Day 2 continued to show statistically significant positive shifts in threshold potential compared to baseline (Figs. 2B and 2C, Table S2), indicating that overnight TRV130 exposure did not produce tolerance in individual DRG neurons. Analysis of the entire TRV130 dataset in Figure 2C by 2-way repeated-measures ANOVA did not reveal a significant main effect of overnight TRV130 treatment versus acute TRV130 challenge on action potential threshold [F (1,16) = 0.27; P = 0.61; Table S2]. Furthermore, in both naïve and overnight TRV130-treated neurons the mean threshold potential change following acute TRV130 challenge is not significantly different (Fig. 2D).
These studies show that β-arrestin-2-induced desensitization of the μ-opioid receptor mediates short-term opioid tolerance in DRG nociceptors.
3.3. Acute antinociceptive tolerance in-vivo is dependent on β-arrestin-2.
In order to test if the development of acute antinociceptive tolerance in-vivo is dependent on β-arrestin-2, we utilized a short-term tolerance injection schedule previously published by Bohn et al. (2002) and tested antinociception using the warm-water tail withdrawal assay. Morphine was assessed in both β-arrestin-2 WT and KO male mice, while the effects of TRV130 were only assessed in male WT mice as TRV130 prevents β-arrestin-2 activation. Mice were considered drug-responsive if the acute challenge dose significantly increased tail-withdrawal latency. Alternately, mice were deemed tolerant if the acute challenge dose did not significantly increase tail-withdrawal latency. Mice that received saline on Day 1 exhibited antinociception to their respective challenge dose of 10 mg/kg morphine s.c. or 3 mg/kg TRV130 s.c. on Day 2 (Fig. 3). As predicted, WT mice that received a high dose of morphine (100 mg/kg, s.c.) on Day 1 showed tolerance development when challenged on Day 2 (38.5 ± 12.6 %MPE), as compared to saline pre-treated WT controls acutely challenged with 10 mg/kg morphine on Day 2 (85.2 ± 10.1 %MPE; Fig. 3A). In β-arrestin-2 KO mice however, high dose morphine exposure on Day 1 did not lead to tolerance development when they were challenged on Day 2 (91.2 ± 8.8 %MPE vs. saline controls: 89 ± 11.0; Fig. 3A). Furthermore, 3 mg/kg TRV130 was equally effective as morphine in producing an acute antinociceptive effect (100.0 ± 0.0 %MPE; Fig 3B). A high dose of TRV130 (30 mg/kg, s.c.) given on Day 1 did not lead to tolerance development following a challenge injection on Day 2 (84.72 ± 15.3 %MPE) as compared to acute TRV130 wild type controls (100.0 ± 0.0 %MPE; Fig. 3B).
Figure 3. Acute antinociceptive tolerance in mice is mediated by β-arrestin-2.

(A) Warm-water tail-withdrawal antinociception in β-arrestin-2 WT or KO mice pre-treated with either saline or 100 mg/kg morphine s.c. on Day 1 and acutely challenged with 10 mg/kg morphine s.c. on Day 2. Data are %MPE ± S.E.M and represent Day 2 results. Scatter represents individual mice. β-arrestin-2 WT mice: N = 10/group; β-arrestin-2 KO mice: N = 9/group. Data analyzed by 2-way ANOVA with Bonferroni’s post-test. ***P<0.01 and ‘ns’ is not significant (P>0.05). (B) Warm-water tail-withdrawal antinociception in β-arrestin-2 WT mice pre-treated with either saline or 30 mg/kg TRV130 s.c. on Day 1 and acutely challenged with 3 mg/kg TRV130 s.c. on Day 2. Data are %MPE ± S.E.M and represent Day 2 results. Scatter are individual mice. β-arrestin-2 WT mice: N = 5/group. Data analyzed by 2-tailed unpaired t-test. ‘ns’ is not significant (P>0.05). WT mice injected with 100 mg/kg morphine on Day 1 respond significantly less to acute morphine challenge on Day 2 vs. saline controls, indicating tolerance. Mice devoid of β-arrestin-2 or pre-treated with TRV130 on Day 1 continue to respond to the acute morphine or TRV130 challenge on Day 2, respectively, indicating no acute antinociceptive tolerance development in absence of β-arrestin-2 activation.
Altogether, these results support the previously published finding that β-arrestin-2 mediates acute antinociceptive tolerance at the whole animal level.
3.4. Long-term exposure to morphine induces tolerance in DRG neurons independent of β-arrestin-2
We investigated whether tolerance after long-term exposure to morphine is mediated by β-arrestin-2. In order to test this, DRG neurons were collected from male β-arrestin-2 WT or KO mice subcutaneously implanted with a 50 mg morphine pellet for 7 days. As in previous studies, cellular tolerance was assessed by challenging small-diameter DRG neurons (membrane capacitance < 30 pF) with 3 μM morphine in the bath and action potential threshold was used as the indicator of cellular excitability (Fig. 4). The 3 μM morphine challenge did not appear to produce a shift in action potential threshold from baseline values of DRG neurons obtained from β-arrestin-2 WT mice exposed to 50 mg morphine pellet for 7 days (Figs. 4A and 4C; Table S3). Interestingly, 3 μM morphine also did not alter the threshold potential from baseline of DRG neurons isolated from 7-day 50 mg morphine-pelleted β-arrestin-2 KO animals (Figs. 4B and 4C; Table S3). Analysis of the entire dataset by 2-way repeated-measures ANOVA did not detect an effect of acute morphine challenge on threshold potential [F (1, 15) =1.20; P = 0.29]. The ANOVA analysis also did not reveal a significant main effect of genotype versus acute morphine challenge on action potential threshold [F (1, 15) =1.565; P = 0.23]. Furthermore, mean threshold potential change after acute morphine challenge was not significantly different between the WT and KO DRG neurons (Fig. 4D). No significant effect of genotype and acute morphine challenge on rheobase, resting membrane potential or input resistance was observed (Table S3). Altogether, these data indicated the development of morphine tolerance in DRG neurons from morphine-pelleted β-arrestin-2 WT and KO male mice.
Figure 4. β-arrestin-2 knockout does not prevent tolerance in DRG neurons isolated from chronic morphine-treated mice.

Representative current-clamp traces of DRG neurons isolated from (A) β-arrestin-2 WT or (B) β-arrestin-2 KO male mice treated with 50 mg morphine pellet for 7 days. Action potential is generated at baseline (black) and after 16 min of acute 3 μM morphine challenge (red). Threshold potential is extrapolated from the point on the action potential derivative trace, where the action potential differential (dV/dt) > 0. (C) Threshold potential values of individual neurons from morphine-pelleted WT or morphine-pelleted β-arrestin-2 KO mice at baseline and after 3 μM morphine challenge for up to 16 min. (D) Threshold potential change from baseline after acute 3 μM morphine challenge. ‘ns’ is not significant (P>0.05) by 2-tailed unpaired t-test. WT: N = 4 mice, n = 7 cells; and KO: N = 5, n = 10. Data are mean ± S.E.M. Scatter represents individual cells.
Consequently, these findings suggest that unlike short-term tolerance, β-arrestin-2 is not required for long-term tolerance to morphine in DRG neurons.
3.5. Long-term morphine tolerance to antinociception is not altered by β-arrestin-2 deletion in either male or female mice
We investigated whether β-arrestin-2 modulates the development of chronic or “long-term” antinociceptive tolerance in mice. We determined the development of tolerance over 7 days by an acute challenge of morphine (10 mg/kg morphine s.c.). Response to the challenge dose was tested in different groups of mice on Days 1, 3, 4 and 7 post pellet implantation. Note that the same cohort was not tested repeatedly. Pre-injection baseline latency was compared with post-injection latency on these days. Mice sensitive to morphine-induced antinociception exhibited increased tail-withdrawal latencies that were at or close to the 10 s maximum cutoff. Mice were regarded as tolerant if the acute morphine challenge failed to induce antinociception i.e., no statistically significant change (P>0.05) in tail-withdrawal latency from baseline. As predicted, 10 mg/kg morphine increased tail-withdrawal latency of most placebo-pelleted male mice to the maximum cutoff of 10 s (Fig. 5). In 1-day morphine-pelleted male β-arrestin-2 WT and KO mice, the baseline tail-withdrawal latency was at the 10 s maximum cutoff, indicating morphine-induced antinociception (Fig. 5A). 3 out of 7 β-arrestin-2 WT mice demonstrated less than maximum baseline responses yet continued to respond to morphine as indicated by the increased tail latency to the morphine challenge on day 3. All β-arrestin-2 KO male mice continued to exhibit maximal baseline tail-withdrawal latencies three days after morphine pellet exposure (Fig. 5B). Interestingly, after four days of morphine pellet exposure the antinociceptive effect of the morphine challenge extinguished in both male β-arrestin-2 WT and KO animals, indicating the development of morphine tolerance despite β-arrestin-2 deletion (Fig. 5C). Tolerance to 10 mg/kg morphine was observed in all male mice (7/7 WT mice and 8/8 KO mice) after 7 days of morphine exposure (Fig. 5D). Thus, β-arrestin-2 knockout neither shifted the time-course of morphine tolerance nor prevented the manifestation of morphine tolerance in male mice.
Figure 5. Long-term morphine tolerance develops to tail-withdrawal antinociception in male mice in the absence of β-arrestin-2.
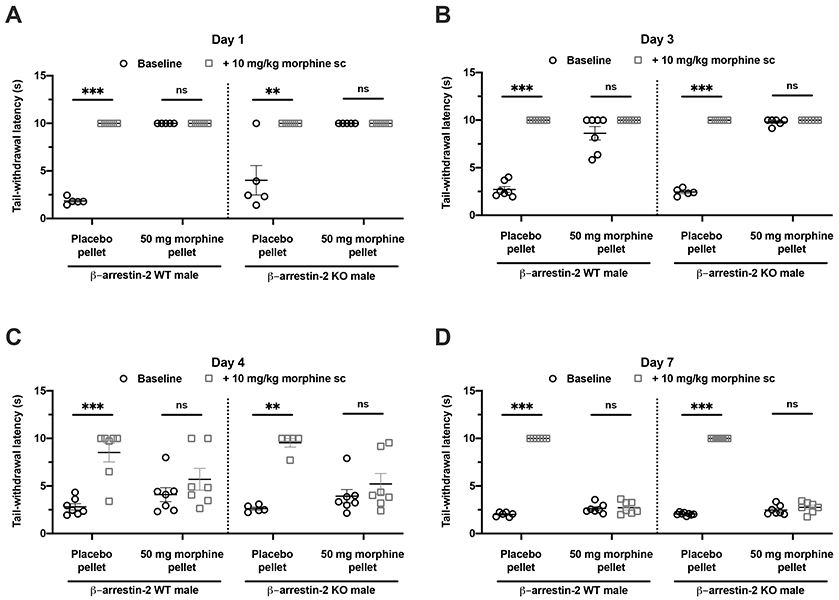
Long-term morphine tolerance was evaluated by acutely challenging male β-arrestin-2 WT (left) or KO (right) mice implanted with placebo or 50 mg morphine pellet with 10 mg/kg morphine s.c. on days (A) 1, (B) 3, (C) 4 or (D) 7, and tail-withdrawal latencies before and after challenge were compared. Data are mean ± S.E.M. Scatter represents individual animals. **P<0.01, ***P<0.001 and ‘ns’ (not significant, P>0.05) vs. baseline by 2-way repeated-measures ANOVA with Bonferroni’s post-test. Day 1: N=5/group; Day 3: N = 7 (Placebo-pelleted WT), 5 (Placebo-pelleted KO), 7 (Morphine-pelleted WT) and 6 (Morphine-pelleted KO); Day 4: N = 7 (Placebo-pelleted WT), 5 (Placebo-pelleted KO), 7 (Morphine-pelleted WT) and 7 (Morphine-pelleted KO); Day 7: N = 6 (Placebo-pelleted WT), 8 (Placebo-pelleted KO), 7 (Morphine-pelleted WT) and 7 (Morphine-pelleted KO). Both, β-arrestin-2 WT and KO mice develop antinociceptive tolerance over 7 days of morphine exposure, indicating that long-term antinociceptive tolerance in male mice is independent of β-arrestin-2.
Finally, we evaluated whether there were sex differences in antinociceptive tolerance in β-arrestin-2 KO mice. A cumulative dose-response to morphine was conducted in male and female mice (Fig. 6). Morphine-induced antinociception was quantitated as % MPE, where 100% MPE represented maximal antinociception. ED50 values of 7-day placebo-pelleted male and female KO mice were 2.90 (1.89-4.33) mg/kg and 1.56 (1.25-1.98) mg/kg, respectively. Morphine pre-treatment for 7 days produced a significant rightward shift in the dose response curve of both male and female mice (Fig. 6). The ED50 values for 7-day morphine-pelleted male and female mice were 63.9 (42.4-113.7) mg/kg and 41.82 (31.66-55.61) mg/kg, respectively. Decreased potency suggested that morphine tolerance was induced in β-arrestin-2 KO mice, irrespective of the sex.
Figure 6. Cumulative morphine dose response curve of male and female β-arrestin-2 KO mice.

Cumulative morphine dose response curves of 7-day placebo-pelleted and morphine-pelleted male and female mice were compared to evaluate the development of antinociceptive tolerance. Each point is %MPE ± S.E.M. Males: N = 6/group. Females: 7/group. The dose response curve of 50 mg morphine-pelleted male mice is significantly right-shifted compared to placebo-pelleted male mice. Similarly, the dose response curve of 25 mg morphine-pelleted female mice is significantly right-shifted compared to that of placebo-pelleted female mice. Thus, antinociceptive tolerance develops in both male and female mice despite the absence of β-arrestin-2
Consequently, these data suggest that β-arrestin-2 does not regulate long-term antinociceptive tolerance in either male or female mice.
4. Discussion
The CNS is traditionally considered as the primary site of opioid-induced antinociceptive tolerance and, therefore, research has primarily focused on delineating mechanisms underlying opioid tolerance in centrally-localized μ-opioid receptors. However, systemically-administered opioids can induce analgesia by concomitantly activating μ-opioid receptors at multiple sites along the pain pathway, including the primary afferent neurons of the DRG. μ-opioid receptors expressed on DRG primary afferent neurons are critical for regulating the influx of nociceptive stimuli to the CNS. Activation of peripheral μ-opioid receptors can prevent the sensitization of primary afferent neurons to noxious stimuli and attenuate subsequent CNS events underlying the perception of pain. Alternately, desensitization of μ-opioid receptors on DRG neurons as a result of chronic opioid exposure can render this process inactive. Recent studies have in fact demonstrated that μ-opioid receptors expressed by nociceptive neurons of the DRG profoundly contribute to the induction of antinociceptive tolerance in mice (Chen et al., 2007; Corder et al., 2017). It is therefore critical to investigate mechanisms underlying opioid tolerance in DRG neurons.
In the present study, we observed that acute tolerance in single nociceptive DRG neurons and to tail-withdrawal antinociception is prevented by genetic deletion of β-arrestin-2 or by using TRV130, a G-protein-biased agonist at the μ-opioid receptor. In contrast, long-term morphine tolerance in individual nociceptive DRG neurons and to tail-withdrawal antinociception in either male or female mice develops independently of the β-arrestin-2 pathway. Taken together the findings presented here indicate that the different phases of antinociceptive tolerance are regulated via distinct mechanisms— a rapid β-arrestin-2-dependent mechanism that mediates acute tolerance, and a slow β-arrestin-2-independent mechanism that underlies long-term tolerance—in both mice and individual DRG neurons critical in the initiation of nociceptive stimuli.
The development of G-protein-biased μ-opioid receptor agonists that preferentially reduce β-arrestin-2 activation have generated enthusiasm within the field and opened up new avenues for the treatment of pain with reduced risks such as tolerance (Madariaga-Mazón et al., 2017; Siuda et al., 2017). In fact, the G-protein biased μ-opioid receptor agonist, Olinvyk (TRV130), was recently approved by the FDA for short-term intravenous use in hospitals (U.S. Food and Drug Administration, 2020). The findings reported in the present study implicate that antinociceptive tolerance can develop in the absence of β-arrestin-2 activation. Consequently, these findings raise questions about the usefulness of G-protein-biased agonists to mitigate opioid-induced analgesic tolerance.
β-arrestins are universally expressed multi-functional adaptor proteins that prevent the coupling of GPCRs to cognate G-proteins through steric hindrance and consequently, signal transduction through membrane-delimited mechanisms is disrupted (Lohse et al., 1990). Multiple studies have implicated that this β-arrestin-2 pathway is the basis of opioid-induced analgesic tolerance (Bohn et al., 2000; DeWire et al., 2013; Grim et al., 2020; Manglik et al., 2016; Wang et al., 2016; Yang et al., 2011). Consistent with these findings, in the present study we too observed that antinociceptive tolerance in mice and cellular tolerance in nociceptive DRG neurons, specifically acute tolerance (Williams et al., 2013), is mediated by β-arrestin-2. Interestingly, in the current study tolerance induced after long-term morphine exposure (7 days) in both mice and DRG neurons was not contingent on β-arrestin-2. Altogether, the data suggests that β-arrestin-2 mediates only the acute but not the long-term phase of tolerance at the μ-opioid receptor. Indeed, Bohn et al. (2002) have previously reported that the onset of antinociceptive tolerance in the warm-water tail-withdrawal assay in mice is delayed but not prevented in the absence of β-arrestin-2. In contrast, functional deletion of β-arrestin-2 in rats prevented the development of chronic morphine tolerance to warm-water tail-withdrawal antinociception (Wang et al., 2016; Yang et al., 2011). This discrepancy could be due to species differences or disparate tolerance development models, for example daily injections vs. continuous-release subcutaneous pellets. It is well-known that the extent of tolerance produced by subcutaneous pellets is significantly greater than other techniques (Dighe et al., 2009) and therefore, tolerance produced by intermittent morphine injections might be more readily reversed compared to tolerance produced by morphine pellets.
While in the present study antinociceptive tolerance developed in the warm-water tail-withdrawal assay independently of β-arrestin-2, it has been previously reported that antinociceptive tolerance, albeit in the hot-plate assay, is attenuated in β-arrestin-2 knockout mice (Bohn et al., 2000; Raehal and Bohn, 2011). Indeed, such a discrepancy has also been previously noted by Bohn and colleagues (Bohn et al., 2002). Both tests are used to measure responsiveness to acute noxious thermal stimulus (Bannon and Malmberg, 2007). The endpoints of a hot-plate test are complex nocifensive behaviors such as hind-paw licks and/or jumps, and are thought to involve supra-spinal inputs since spinal transection prevented these behaviors in the hot-plate test (Giglio et al., 2006). Alternately, the tail-withdrawal assay measures rapid reflex action in the form of tail flexion from warm water and is predominantly spinally-mediated (Bannon and Malmberg, 2007; Irwin et al., 1951). Consequently, the differential effect of β-arrestin-2 deletion on the development of antinociceptive tolerance in the tail-withdrawal test versus the hot-plate assay could be due to discrete mechanisms of opioid tolerance in the neuronal circuitries engaged by the two tests. Indeed, studies have demonstrated previously that mechanisms of opioid-induced desensitization and tolerance vary in different neurons (Levitt and Williams, 2018).
Chronic exposure to morphine is known to induce bacterial translocation and inflammation of the gut wall and these processes have been previously implicated in the development of opioid tolerance (Kang et al., 2017; Komla et al., 2019; Meng et al., 2013; Mischel et al., 2018). Antinociceptive tolerance in mice was attenuated by modulating the gut microbiome with antibiotics or probiotics (Kang et al., 2017; Mischel et al., 2018; Zhang et al., 2019). Altering the gut microbiome also prevented morphine tolerance at the single-cell level in DRG neurons (Kang et al., 2017; Mischel et al., 2018). Conversely, colonic inflammation enhanced the rate of morphine tolerance to antinociception (Komla et al., 2019). It is therefore possible that long-term tolerance to antinociception in mice and in DRG neurons might be mediated by changes induced within the gut microbiome.
Previous studies have reported sex differences in opioid analgesia and tolerance in both humans and rodents (Bodnar and Kest, 2010; Craft et al., 1999; Kalinichev et al., 2001; Kasson and George, 1984; Lee and Ho, 2013; Mousavi et al., 2007). However, it was not known whether sex is an important variable influencing the role of β-arrestin-2 in the mechanism of antinociceptive tolerance. In the present study, we find that antinociceptive tolerance in the warm-water tail-withdrawal assay develops in both male and female mice even in the absence of β-arrestin-2, implicating that sex hormones might not interact with the β-arrestin-2 pathway to mediate antinociceptive tolerance in mice.
It is noteworthy that in the present study relatively high doses of morphine were used to investigate tolerance development in vivo in mice (25 mg and 50 mg morphine pellet for 7 days in female and male mice, respectively) and in vitro in DRG neurons (3 μM morphine for acute challenge and 10 μM morphine for overnight treatment). Previously, it has been reported in 8-week-old male C57Bl/6NCr mice that a 25 mg morphine pellet produces a peak plasma morphine concentration of 2695.3 ± 785.1 ng/mL (~3.5 μM morphine) 24 h after pellet implantation, which progressively decreased to 229.5 ± 92.5 ng/mL after 7 days (~0.4 μM morphine) (McLane et al., 2017). In humans, therapeutic doses of morphine have been reported to produce serum concentrations of 14.7-70.4 ng/mL (Netriova et al., 2006), whereas in overdose cases, wide-ranging plasma concentrations from of 113 ng/mL to 4660 ng/mL have been detected (Meissner et al., 2002; Ozaita et al., 2002). Thus, while the amount of morphine delivered to mice and isolated neurons in our experiments is on the higher end of what patients might receive in the clinic, it is comparable to what might be observed in opioid abusers.
In conclusion, the findings presented in this study implicate that antinociceptive tolerance involving peripheral μ-opioid receptors in the DRG is mediated by two disparate mechanisms—β-arrestin-2-dependent and-independent pathways—that are engaged during different phases of opioid exposure. This suggests that the contribution of β-arrestin-2-induced desensitization of μ-opioid receptors in the molecular mechanism of tolerance is more intricate and that tolerance might be mediated by other means such as microbial dysbiosis in the gut. Importantly, these findings help inform the clinical utility of chronic exposure to G-protein-biased agonists over conventional opioids for pain management and highlights considerations for the likelihood of developing analgesic tolerance.
Supplementary Material
Acknowledgments
The authors wish to thank David Stevens and Dr. Krista Scoggins for their technical assistance. This work was supported by the National Institute of Health grants: P30 DA033934, R01 DA036975, R01 DA024009.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
References
- Abraira VE, Ginty DD, 2013. The sensory neurons of touch. Neuron 79, 618–639. 10.1016/j.neuron.2013.07.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannon AW, Malmberg AB, 2007. Models of Nociception: Hot-Plate, Tail-Flick, and Formalin Tests in Rodents. Curr. Protoc. Neurosci 41, Unit 8.9. 10.1002/0471142301.ns0809s41 [DOI] [PubMed] [Google Scholar]
- Barabas ME, Mattson EC, Aboualizadeh E, Hirschmugl CJ, Stucky CL, 2014. Chemical structure and morphology of dorsal root Ganglion neurons from naive and inflamed mice. J. Biol. Chem 289, 34241–34249. 10.1074/jbc.M114.570101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar RJ, Kest B, 2010. Sex differences in opioid analgesia, hyperalgesia, tolerance and withdrawal: Central mechanisms of action and roles of gonadal hormones. Horm. Behav 58, 72–81. 10.1016/j.yhbeh.2009.09.012 [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG, 2000. μ-opioid receptor desensitization by β-arrestin-2 determines morphine tolerance but not dependence. Nature 408, 720–723. 10.1038/35047086 [DOI] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Caron MG, 2002. Differential mechanisms of morphine antinociceptive tolerance revealed in βarrestin-2 knock-out mice. J. Neurosci 22, 10494–10500. 10.1523/jneurosci.22-23-10494.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT, 1999. Enhanced morphine analgesia in mice lacking β-arrestin 2. Science (80-. ). 286, 2495–2498. 10.1126/science.286.5449.2495 [DOI] [PubMed] [Google Scholar]
- Chen S-R, Prunean A, Pan H-M, Welker KL, Pan H-L, 2007. Resistance to morphine analgesic tolerance in rats with deleted transient receptor potential vanilloid type 1-expressing sensory neurons. Neuroscience 145, 676–85. 10.1016/j.neuroscience.2006.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK, 2013. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science (80-. ). 341, 1394–1399. 10.1126/science.1239403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder G, Tawfik VL, Wang D, Sypek EI, Low SA, Dickinson JR, Sotoudeh C, Clark JD, Barres BA, Bohlen CJ, Scherrer G, 2017. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med 23, 164–173. 10.1038/nm.4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft RM, Stratmann JA, Bartok RE, Walpole TI, King SJ, 1999. Sex differences in development of morphine tolerance and dependence in the rat. Psychopharmacology (Berl). 143, 1–7. 10.1007/s002130050911 [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Alexander S, Cirino G, Docherty JR, George CH, Giembycz MA, Hoyer D, Insel PA, Izzo AA, Ji Y, MacEwan DJ, Sobey CG, Stanford SC, Teixeira MM, Wonnacott S, Ahluwalia A, 2018. Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br. J. Pharmacol 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeWire SM, Yamashita DS, Rominger DH, Liu G, Cowan CL, Graczyk TM, Chen XT, Pitis PM, Gotchev D, Yuan C, Koblish M, Lark MW, Violin JD, 2013. A G protein-biased ligand at the μ-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphines. J. Pharmacol. Exp. Ther 344, 708–717. 10.1124/jpet.112.201616 [DOI] [PubMed] [Google Scholar]
- Dighe SV, Madia PA, Sirohi S, Yoburn BC, 2009. Continuous morphine produces more tolerance than intermittent or acute treatment. Pharmacol. Biochem. Behav 92, 537–542. 10.1016/j.pbb.2009.02.004 [DOI] [PubMed] [Google Scholar]
- Giglio CA, Defino HLA, Da-Silva CA, De-Souza AS, Del Bel EA, 2006. Behavioral and physiological methods for early quantitative assessment of spinal cord injury and prognosis in rats. Brazilian J. Med. Biol. Res 39, 1613–1623. 10.1590/S0100-879X2006001200013 [DOI] [PubMed] [Google Scholar]
- Grim TW, Schmid CL, Stahl EL, Pantouli F, Ho J-H, Acevedo-Canabal A, Kennedy NM, Cameron MD, Bannister TD, Bohn LM, 2020. A G protein signaling-biased agonist at the μ-opioid receptor reverses morphine tolerance while preventing morphine withdrawal. Neuropsychopharmacology 45, 416–425. 10.1038/s41386-019-0491-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LS, Pierson AK, 1964. Some Narcotic Antagonists In The Benzomorphan Series. J. Pharmacol. Exp. Ther 143, 141–148. https://doi.org/1 February , 1964 [PubMed] [Google Scholar]
- Irwin S, Houde RW, Bennett DR, Hendershot LC, Seevers MH, 1951. The effects of morphine methadone and meperidine on some reflex responses of spinal animals to nociceptive stimulation. J. Pharmacol. Exp. Ther 101, 132–43. [PubMed] [Google Scholar]
- Kalinichev M, Easterling KW, Holtzman SG, 2001. Early neonatal experience of Long-Evans rats results in long-lasting changes in morphine tolerance and dependence. Psychopharmacology (Berl). 157, 305–312. 10.1007/s002130100806 [DOI] [PubMed] [Google Scholar]
- Kang M, Mischel RA, Bhave S, Komla E, Cho A, Huang C, Dewey WL, Akbarali HI, 2017. The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci. Rep 7, 42658. 10.1038/srep42658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasson BG, George R, 1984. Endocrine influences on the actions of morphine: IV. effects of sex and strain. Life Sci. 34, 1627–1634. 10.1016/0024-3205(84)90633-7 [DOI] [PubMed] [Google Scholar]
- Komla E, Stevens DL, Zheng Y, Zhang Y, Dewey WL, Akbarali HI, 2019. Experimental Colitis Enhances the Rate of Antinociceptive Tolerance to Morphine via Peripheral Opioid Receptors. J. Pharmacol. Exp. Ther 370, 504–513. 10.1124/jpet.119.256941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CWS, Ho IK, 2013. Sex differences in opioid analgesia and addiction: Interactions among opioid receptors and estrogen receptors. Mol. Pain 9, 1–10. 10.1186/1744-8069-9-45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefkowitz RJ, 1998. G protein-coupled receptors: III. New roles for receptor kinases and β- arrestins in receptor signaling and desensitization. J. Biol. Chem 10.1074/jbc.273.30.18677 [DOI] [PubMed] [Google Scholar]
- Levitt ES, Williams JT, 2018. Desensitization and tolerance of Mu opioid receptors on pontine kölliker-fuse neurons. Mol. Pharmacol 93, 8–13. 10.1124/mol.117.109603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ, 1990. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science (80-. ). 248, 1547–1550. [DOI] [PubMed] [Google Scholar]
- Madariaga-Mazón A, Marmolejo-Valencia AF, Li Y, Toll L, Houghten RA, Martinez-Mayorga K, 2017. Mu-Opioid receptor biased ligands: A safer and painless discovery of analgesics? Drug Discov. Today 22, 1719–1729. 10.1016/j.drudis.2017.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A, Lin H, Aryal DK, McCorvy JD, Dengler D, Corder G, Levit A, Kling RC, Bernat V, Hübner H, Huang XP, Sassano MF, Giguère PM, Löber S, Duan D, Scherrer G, Kobilka BK, Gmeiner P, Roth BL, Shoichet BK, 2016. Structure-based discovery of opioid analgesics with reduced side effects. Nature 537, 185–190. 10.1038/nature19112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLane VD, Bergquist I, Cormier J, Barlow DJ, Houseknecht KL, Bilsky EJ, Cao L, 2017. Long-term morphine delivery via slow release morphine pellets or osmotic pumps: Plasma concentration, analgesia, and naloxone-precipitated withdrawal. Life Sci. 185, 1–7. 10.1016/j.lfs.2017.07.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meissner C, Recker S, Reiter A, Friedrich HJ, Oehmichen M, 2002. Fatal versus non-fatal heroin “overdose”: blood morphine concentrations with fatal outcome in comparison to those of intoxicated drivers. Forensic Sci. Int 130, 49–54. 10.1016/s0379-0738(02)00343-2 [DOI] [PubMed] [Google Scholar]
- Meng J, Yu H, Ma J, Wang J, Banerjee S, Charboneau R, Barke RA, Roy S, 2013. Morphine Induces Bacterial Translocation in Mice by Compromising Intestinal Barrier Function in a TLR-Dependent Manner. PLoS One 8. 10.1371/journal.pone.0054040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mischel RA, Dewey WL, Akbarali HI, 2018. Tolerance to Morphine-Induced Inhibition of TTX-R Sodium Channels in Dorsal Root Ganglia Neurons Is Modulated by Gut-Derived Mediators. iScience 2, 193–209. 10.1016/j.isci.2018.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil JS, 2009. Animal models of pain: progress and challenges. Nat. Rev. Neurosci 10, 283–294. 10.1038/nrn2606 [DOI] [PubMed] [Google Scholar]
- Mousavi Z, Shafaghi B, Kobarfard F, Jorjani M, 2007. Sex differences and role of gonadal hormones on glutamate level in the nucleus accumbens in morphine tolerant rats: a microdialysis study. Eur. J. Pharmacol 554, 145–9. 10.1016/j.ejphar.2006.10.010 [DOI] [PubMed] [Google Scholar]
- Muchhala KH, Jacob JC, Alam I, Hasan S, Khan A, Kang M, Dewey WL, Akbarali HI, 2020. Rapid tolerance to morphine in the myenteric neurons of the small intestine is independent of β-arrestin-2 and mediated by PKC. bioRxiv 2020.07.17.209437 10.1101/2020.07.17.209437 [DOI] [Google Scholar]
- Netriova J, Blahova E, Johanesova Z, Brandsteterova E, Lehotay J, Serdt K, Mocak J, 2006. HPLC determination of morphine, morphine-3-glucuronide and morphine-6-glucuronide in human serum of oncological patients after administration of morphine drugs. Pharmazie 61, 528–34. [PubMed] [Google Scholar]
- Ozaita A, Escribá PV, Ventayol P, Murga C, Mayor F, García-Sevilla JA, 2002. Regulation of G Protein-Coupled Receptor Kinase 2 in Brains of Opiate-Treated Rats and Human Opiate Addicts. J. Neurochem 70, 1249–1257. 10.1046/j.1471-4159.1998.70031249.x [DOI] [PubMed] [Google Scholar]
- Pedersen MF, Wróbel TM, Märcher-Rørsted E, Pedersen DS, Møller TC, Gabriele F, Pedersen H, Matosiuk D, Foster SR, Bouvier M, Bräuner-Osborne H, 2020. Biased agonism of clinically approved μ-opioid receptor agonists and TRV130 is not controlled by binding and signaling kinetics. Neuropharmacology 166, 107718. 10.1016/j.neuropharm.2019.107718 [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM, 2011. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 10.1016/j.neuropharm.2010.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GR, Gade AR, Dewey WL, Akbarali HI, 2012. Opioid-induced hypernociception is associated with hyperexcitability and altered tetrodotoxin-resistant Na + channel function of dorsal root ganglia. Am. J. Physiol. - Cell Physiol 302, 1152–1161. 10.1152/ajpcell.00171.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuda ER, Carr R, Rominger DH, Violin JD, 2017. Biased mu-opioid receptor ligands: a promising new generation of pain therapeutics. Curr. Opin. Pharmacol 32, 77–84. 10.1016/j.coph.2016.11.007 [DOI] [PubMed] [Google Scholar]
- Sorge RE, Martin LJ, Isbester KA, Sotocinal SG, Rosen S, Tuttle AH, Wieskopf JS, Acland EL, Dokova A, Kadoura B, Leger P, Mapplebeck JCS, McPhail M, Delaney A, Wigerblad G, Schumann AP, Quinn T, Frasnelli J, Svensson CI, Sternberg WF, Mogil JS, 2014. Olfactory exposure to males, including men, causes stress and related analgesia in rodents. Nat. Methods 11, 629–632. 10.1038/nmeth.2935 [DOI] [PubMed] [Google Scholar]
- Stein C, Machelska H, 2011. Modulation of Peripheral Sensory Neurons by the Immune System: Implications for Pain Therapy. Pharmacol. Rev 63, 860–881. 10.1124/pr.110.003145 [DOI] [PubMed] [Google Scholar]
- Sun J, Chen SR, Chen H, Pan HL, 2019. μ-Opioid receptors in primary sensory neurons are essential for opioid analgesic effect on acute and inflammatory pain and opioid-induced hyperalgesia. J. Physiol 597, 1661–1675. 10.1113/JP277428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Chen SR, Pan HL, 2020. μ-Opioid receptors in primary sensory neurons are involved in supraspinal opioid analgesia. Brain Res. 1729, 146623. 10.1016/j.brainres.2019.146623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Food and Drug Administration, 2020. FDA Approves New Opioid for Intravenous Use in Hospitals, Other Controlled Clinical Settings. https://www.fda.gov/news-events/press-announcements/fda-approves-new-opioid-intravenous-use-hospitals-other-controlled-clinical-settings (accessed 1 November 2020). https://doi.org/8-7-2020
- Wang J, Xu W, Zhong T, Song Z, Zou Y, Ding Z, Guo Q, Dong X, Zou W, 2016. MiR-365 targets β-arrestin 2 to reverse morphine tolerance in rats. Sci. Rep 6, 1–11. 10.1038/srep38285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel R, Reiss D, Karchewski L, Gardon O, Matifas A, Filliol D, Becker JAJ, Wood JN, Kieffer BL, Gaveriaux-Ruff C, 2013. Mu Opioid Receptors on Primary Afferent Nav1.8 Neurons Contribute to Opiate-Induced Analgesia: Insight from Conditional Knockout Mice. PLoS One 8, e74706. 10.1371/journal.pone.0074706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Ingram SL, Henderson G, Chavkin C, von Zastrow M, Schulz S, Koch T, Evans CJ, Christie MJ, 2013. Regulation of μ-opioid receptors: Desensitization, phosphorylation, internalization, and tolerance. Pharmacol. Rev 65, 223–254. 10.1124/pr.112.005942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CH, Huang HW, Chen KH, Chen YS, Sheen-Chen SM, Lin CR, 2011. Antinociceptive potentiation and attenuation of tolerance by intrathecal β-arrestin 2 small interfering RNA in rats. Br. J. Anaesth 107, 774–781. 10.1093/bja/aer291 [DOI] [PubMed] [Google Scholar]
- Zhang L, Meng J, Ban Y, Jalodia R, Chupikova I, Fernandez I, Brito N, Sharma U, Abreu MT, Ramakrishnan S, Roy S, 2019. Morphine tolerance is attenuated in germfree mice and reversed by probiotics, implicating the role of gut microbiome. Proc. Natl. Acad. Sci 116, 13523–13532. 10.1073/pnas.1901182116 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.