

Abstract
Aberrant expression of CA125/MUC16 is associated with pancreatic ductal adenocarcinoma (PDAC) progression and metastasis. However, knowledge of the contribution of MUC16 to pancreatic tumorigenesis is limited. Here, we show that MUC16 expression is associated with disease progression, basal-like and squamous tumor subtypes, increased tumor metastasis, and short-term survival of PDAC patients. MUC16 enhanced tumor malignancy through the activation of AKT and GSK3β oncogenic signaling pathways. Activation of these oncogenic signaling pathways resulted in part from increased interactions between MUC16 and epidermal growth factor (EGF)-type receptors, which were enhanced for aberrant glycoforms of MUC16. Treatment of PDAC cells with monoclonal antibody (mAb) AR9.6 significantly reduced MUC16-induced oncogenic signaling. mAb AR9.6 binds to a unique conformational epitope on MUC16, which is influenced by O-glycosylation. Additionally, treatment of PDAC tumor-bearing mice with either mAb AR9.6 alone or in combination with gemcitabine significantly reduced tumor growth and metastasis. We conclude that the aberrant expression of MUC16 enhances PDAC progression to an aggressive phenotype by modulating oncogenic signaling through ErbB receptors. Anti-MUC16 mAb AR9.6 blocks oncogenic activities and tumor growth and could be a novel immunotherapeutic agent against MUC16-mediated PDAC tumor malignancy.
Keywords: MUC16, pancreatic ductal adenocarcinoma, Tn, Sialyl-Tn, COSMC, mAb AR9.6
Graphical abstract
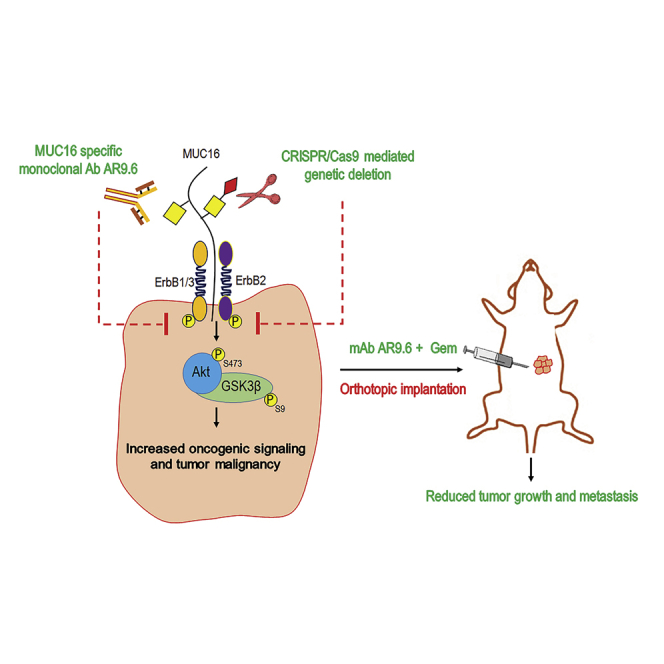
Increased expression of CA125/MUC16 enhances pancreatic ductal adenocarcinoma (PDAC) disease progression with an unknown mechanism. Here, Radhakrishnan and colleagues demonstrate that aberrant expression of MUC16 is associated with highly aggressive tumor subtypes, increased tumor progression, and metastasis, and reduced PDAC tumor-bearing animals’ survival through activation of ErbB receptors signaling axis.
Introduction
The biomarker CA125 comprises epitopes expressed on MUC16, a massive (>22,000 amino acid) membrane-bound, heavily N- and O-glycosylated cell-surface glycoprotein.1 Approximately 50%–70% of pancreatic ductal adenocarcinoma (PDAC) patients express CA125 and is associated with disease progression and metastasis.2 In the clinical setting, high and/or increasing levels of CA125/MUC16 in circulation are widely associated with poor prognosis, whereas declining values are often associated with disease stabilization or regression.3 Antibody assays that detect CA125 also use the same capture and detection antibody, which derives from the fact that such assays detect multivalent structures that contain multiple copies of the same epitope. Unique physicochemical features of mucins include tandem repeat domains that are heavily O-glycosylated with extended structures,4 which explains the fact that these are the principal multivalent components that are detected by single antibody biomarker assays. Despite the prognostic importance of CA125, there is a paucity of studies into possible biological roles of MUC16 in tumor progression.
During cancer progression, some branched O-glycan structures are truncated to produce tumor-specific, short mucin-type O-glycans Tn (GalNAc alpha1-O-Ser/Thr) and sialyl Tn (Sialic acid alpha 2-6 GalNAc alpha1-O-Ser/Thr).5,6 Overexpression of STn antigen occurs at the highest frequency in pancreatic cancer, among other solid tumors.7 Also, STn expression is observed in pancreatic intraepithelial neoplasia stage III (PanIN-3), a premalignant lesion thought to precede the development of PDAC.8 Recent studies from our laboratory demonstrated that enforced constitutive expression of truncated O-glycans (Tn and STn antigens) on mucins and other glycoproteins through knockout (KO) of COSMC enhance the malignant potential of PDAC cells,9 in part through altering the steady-state signaling pathways in cells to an oncogenic phenotype. Also, we recently demonstrated that MUC16 is one of the aberrantly O-glycosylated glycoproteins detected in tumors that are expressing truncated O-glycans.10 However, the molecular and biological mechanisms by which aberrant glycoforms of MUC16 influence the pancreatic tumorigenesis are not well understood.
Here we show that MUC16 expression is clinically associated with PDAC progression, metastasis, aggressive subtypes (squamous and basal-like), and reduced patients’ survival. We further investigated the biological functions of MUC16 in PDAC tumor growth by using CRISPR-Cas9 to genetically eliminate the expression of MUC16 in isogenic PDAC cells. The results show that MUC16 activated AKT and GSK3β oncogenic signaling pathways in tumor cells through interactions with ErbB receptors. Furthermore, antibodies specific to tandem repeats of MUC16 inhibited the activation of these pathways and malignant properties of tumor cells in vitro, and anti-MUC16 antibody alone or in combination with gemcitabine significantly inhibited tumor growth of PDAC xenografts. The results of this study show that in addition to serving as a biomarker, MUC16 has biological activities that support the progression of pancreatic cancer.
Results
MUC16 aggravates pancreatic cancer progression
Previous studies have suggested a close association between increased expression of MUC16 and PDAC progression.2,11 A Kaplan-Meyer plot evaluated the survival probability of patients expressing low MUC16 (n = 54) and high MUC16 (n = 122) from the Human Protein Atlas revealed a significantly decreased survival probability (p = 0.0041, log-rank) in patients with higher MUC16 expression (Figure S1A). Analysis of MUC16 mRNA expression from a cohort of patients at Columbia University revealed that MUC16 was significantly elevated in PDAC epithelium and PDAC stromal cells as compared to precursor lesions PanIN-1 (FDR 1.30E-02) and IPMN (FDR 2.89E-02) epithelia. Moreover, epithelial PDAC tumors show enhanced MUC16 expression compared to stromal PDAC tumors (FDR 2.24E-47; Figure 1A; Table 1).12 Immunohistochemical staining of a commercial pancreatic tissue microarray (BIC14011a; BIOMAX.US) containing tissues of normal pancreas and different stages of pancreatic tumors with anti-MUC16 monoclonal antibody (mAb) AR9.6 revealed significantly higher expression of MUC16 protein in ductal adenocarcinoma (DAC; p = 0.0402). Also, higher expression of MUC16 was found in adenosquamous carcinoma (ASC), and islet cell tumors compared to early PanINs (Figure 1B, representative immunohistochemistry [IHC] images; Figure 1C histoscores). Evaluation of matched sets of rapid autopsies (RAP) PDAC samples (Table S1) by using four independent anti-MUC16 monoclonal antibodies (OC125, AR9.6, 5B9, and 5E11)1,13 showed high levels of MUC16 protein in primary tumors as compared to normal pancreas tissues (samples 1–7; Figure 1D, heatmap of intensity score; Figure 1E, representative IHC images). A comparison of the intensity score of MUC16 epitope detection by various MUC16 antibodies against OC125 is depicted in Figure 1F. mAb AR9.6 showed significantly high epitope detection as compared to OC125 (p < 0.0001; Table S2). About 50%–70% of PDAC patients showed MUC16 expression and detected by using different antibodies (Figure 1G). Additionally, MUC16 epitopes detected by mAb AR9.6 was increased in liver metastasis; interestingly, 5E11 was higher in primary tumors as compared to liver mets, suggesting that MUC16 expressed by primary tumors shows differential post-translational processing as compared to metastases (Figures 2A and 2B, heatmap of intensity score; Figure 2C, representative images). We also found increased expression of MUC16 in metastatic sites in addition to the liver, especially in the omentum, aorta, and appendix lesions, etc. compared to primary tumors (Figure S1B, intensity score; Figure S1C, representative IHC images). Higher expression of MUC16 is associated with primary and liver mets (Figure 2D). The percent survival of RAP patients indicated that patients with low MUC16 expression (n = 31) had survived longer than patients with high MUC16 expression (n = 30; p = 0.0257; Figure 2E). Notably, protein expression of MUC16 was significantly high (p = 0.0089) in short-term survivors (n = 34) as compared to long-term survivors (n = 32) with a median survival of 274 days (Figure 2F). Analysis of the expression of MUC16 in PDAC tumor subtype classifications from TCGA PAAD dataset14 revealed significantly increased expression of MUC16 in squamous subtype (Bailey’s classifications), basal-like subtype (Moffitt classification), and quasimesenchymal subtype (Collisson classification) as compared to other subtypes (Figure 2G; Table S3). Among these, expression of MUC16 was profound in squamous subtype (n = 31) as compared to immunogenic subtype (n = 28, p < 0.001), progenitor subtype (n = 53, p < 0.001) and ADEX subtype (n = 38, p < 0.001), and compared to all the three subtypes (p = 0.0001). Also, MUC16 expression was more in the Basal-like subtype (n = 65) as compared to the classical subtype (n = 85, p < 0.001; Figure 2G).
Figure 1.

MUC16 in pancreatic cancer progression
(A) LCM-RNA-seq analysis of MUC16 in frozen human pancreas tissue sections (PDAC epithelium with IPMN [n = 19], PanIN [n = 26], and primary [n = 197]; and stroma with IPMN [n = 12], PanIN [n = 23], and primary [n = 124]). MUC16 mRNA expression for different samples were quantified by Log2 TPM (transcripts per million). (B) Immunohistochemical analysis of MUC16 in pancreatic tissue microarrays containing normal pancreas (NP, n = 10), early PanIN1 (n = 6), ductal adenocarcinoma (DAC, n = 46), adenosquamous carcinoma (ASC, n = 3), and islet cell tumors (n = 11) using anti-MUC16 antibody (AR9.6). Scale bar, 40 μm. (C) Histoscore analysis of MUC16 expression by IHC. Expression of MUC16 was compared between the early PanIN and other diseased conditions. Data were presented as the median (Dunnett’s multiple comparisons test). (D) Heatmap of IHC analysis of MUC16 expression (OC125, AR9.6, 5B9, and 5E11) in normal pancreatic tissues (n = 7) and RAP primary tumors (n = 61). (E) Representative IHC images of different anti-MUC16 antibodies (OC125, AR9.6, 5B9, and 5E11) stained normal pancreatic tissues and RAP primary tumors. Scale bar, 40 μm. (F) Graphical representation of overall expression of MUC16 (OC125, AR9.6, 5B9, and 5E11) in RAP primary tumors. Data were presented as the median (n = 61) (Dunnett’s multiple comparisons test). (G) Percent expression of MUC16 using different anti-MUC16 antibodies (OC125, AR9.6, 5B9, and 5E11) in RAP tumor samples.
Table 1.
Differential gene-expression analysis
| Compare | Where | logFC | MeanA | MeanB | t | FDR |
|---|---|---|---|---|---|---|
| PanIN versus PDA | Epithelium | 8.36 | 0.30 | 8.65 | 2.97 | 1.30e-02 |
| IPMN versus PDA | Epithelium | 7.55 | 1.10 | 8.65 | 2.55 | 2.89e-02 |
| PanIN versus PDA | Stroma | 3.72 | 0.37 | 4.08 | 3.17 | 1.10e-02 |
| IPMN versus PDA | Stroma | 2.96 | 1.12 | 4.08 | 1.98 | 1.17e-01 |
| Epi versus stroma | PanIN | −0.89 | 0.58 | −0.31 | −1.52 | 2.04e-01 |
| Epi versus stroma | IPMN | −5.08 | 3.15 | −1.93 | −3.02 | 2.52e-02 |
| Epi versus stroma | PDA | −4.45 | 8.80 | 4.35 | −23.90 | 2.24e-47 |
| Epi versus stroma | All | −4.41 | 7.73 | 3.32 | −21.46 | 2.04e-49 |
Statistical comparison of the expression of MUC16 between different groups of samples. The statistical approach used the Voom method, which is based on linear modeling. The output includes mean TPM values for each group, the log2 fold change (logFC) between the groups, t-statistic comparing the groups, and the False Discovery Rate (FDR) that corrects for multiple hypothesis testing.12 Positive log-fold changes indicate higher levels in the second group.
Figure 2.

MUC16 in pancreatic tumor metastasis
(A) Heatmap of IHC analysis of MUC16 expression (AR9.6, 5B9, and 5E11) in liver metastasis (n = 46). (B) Heatmap of comparison of MUC16 expression in matched sets of RAP primary tumors and liver mets. Higher intensity of color corresponds to high expression of MUC16 based on immunohistochemical score. (C) Representative images of IHC analysis of MUC16 expression in normal liver tissue and RAP liver mets using MUC16-specific antibodies AR 9.6, 5B9, and 5E11. Scale bar, 40 μm. (D) Percent expression of MUC16 (AR9.6) in RAP tumor tissues of primary tumor alone, liver mets alone, and primary tumor and liver mets together. (E) Survival of RAP cohort patients with high and low MUC16 (AR9.6) expression (Mantel-Cox test). (F) Histoscores of MUC16 (AR9.6) compared between short-term (n = 34) and long-term (n = 32) PDAC survivors. Data were presented as median (unpaired t test). (G) Boxplot of MUC16 expression level stratified by RNA expression dataset from Bailey class, Moffitt class, and Collisson class of PDAC. Boxplots were generated by comparing the expression of MUC16 gene among the subtypes using Mann-Whitney rank-sum test. A p value of less than 0.01 was considered statistically significant.
KO of MUC16 decreases tumorigenic features of PDAC
Our previous report showed that MUC16 has an increased number of altered O-glycans in PDAC cells.10 Hence, we eliminated MUC16 in normal branched O-glycan (wild-type [WT]) and altered truncated O-glycan (SimpleCells, SC) expressing isogenic PDAC cells by using CRISPR-Cas9 deletion constructs (Figure 3A). The loss of MUC16 expression in isogenic PDAC cells (T3M4 WT-MUC16KO, T3M4 SC-MUC16KO, Capan-2 WT-MUC16KO, and Capan-2 SC-MUC16KO) was confirmed by PCR (Figures S1D–S1F), western blotting (Figures 3B and 3C), and immunofluorescence (Figure 3D). Genetic deletion of MUC16 in isogenic PDAC cells (T3M4 WT-MUC16KO [2E4], T3M4 SC-MUC16KO [1E10], Capan-2 WT-MUC16KO [1C10], and Capan-2 SC-MUC16KO [2F9]) significantly decreased cell proliferation as compared to their isogenic parental (T3M4 and Capan-2, WT and SC) cells at each time points (p < 0.0001, two-way ANOVA with Tukey’s multiple comparisons; Figures 3E–3H). Cell-cycle analysis showed the accumulation of cells in the G0/G1 phase, as evidenced by reduced expression of G0/G1 phase checkpoint mediators, cyclin D1 (T3M4 and Capan-2), and cyclin E1 (T3M4) in WT- and SC-MUC16KO cells (Figures 3I and 3J).
Figure 3.

Genetic deletion of MUC16 in PDAC cells
(A) Schematic representation of the targeted deletion of MUC16 via CRISPR-Cas9 constructs in T3M4 and Capan-2 (WT and SC) cells with three gRNA target loci are shown as red dotted lines. (B and C) Western blotting of MUC16 in T3M4 and Capan-2 (WT and SC) parental cells and MUC16KO clones respectively. Detection of α-tubulin served as a loading control. (D). Immunofluorescence analysis of MUC16 in T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, SC-MUC16KO (1E10), Capan-2 WT, WT-MUC16KO (1C10), Capan-2 SC, and SC-MUC16KO (2F9) cells. Scale bar, 10 μm. (E–H) Cell proliferation assays in T3M4 WT, WT-MUC16KO (2E4) (E), T3M4 SC, SC-MUC16KO (1E10) (F), Capan-2 WT, WT-MUC16KO (1C10) (G), and Capan-2 SC, SC-MUC16KO (2F9) (H) cells. Data were presented as mean ± SD (n = 6; Tukey’s multiple comparisons test). (I) Western blotting of Cyclin D1 and Cyclin E1 in T3M4 WT, WT-MUC16KO clones, SC, and SC-MUC16KO clones. (J) Western blotting of Cyclin D1 in Capan-2 WT, WT-MUC16KO clones, SC, and SC-MUC16KO clones. β-actin was used as a loading control.
Further, we analyzed the in vitro and in vivo tumorigenic potential of isogenic PDAC cells with or without MUC16 (Figure 4A). MUC16KO cells showed significantly reduced migration (T3M4 WT-MUC16KO, p = 0.0104; T3M4 SC-MUC16KO, p < 0.0001; Figure 4B) and invasion (T3M4 WT-MUC16KO, p = 0.0071; T3M4 SC-MUC16KO, p < 0.0001; Figure 4C) as compared to their parental T3M4 (WT and SC) cells. We also detected a significantly reduced cell migration in T3M4 SC-MUC16KO cells compared to T3M4 WT-MUC16KO cells (p = 0.0018, Figure 4B). Additionally, we found a significantly reduced expression of matrix metalloprotease 9 (MMP9) in both T3M4 WT-MUC16KO and SC-MUC16KO cells as compared to parental cells (Figure S1G), whereas no changes in MMP2 expression (data not shown), suggesting that MUC16 regulation of MMP9 influences migration and invasion. Consistent with previous findings,9 orthotopic implantation of T3M4 SC cells resulted in significantly enhanced tumor growth (n = 14) by weight (p = 0.0019, Figure 4D) and volume (p < 0.0001, Figure 4E) and incidence of metastases to the diaphragm (50%) and lymph node (57%), as compared to T3M4 WT (Figure 4F; Tables S4). In contrast, T3M4 SC-MUC16KO cells showed significantly reduced tumor weight (p < 0.0001), volume (p < 0.0001), and metastases to peritoneum (14%, p = 0.02), diaphragm (7%, p = 0.099), lymph nodes (14%, p = 0.05), spleen (28%, p = 0.0007), and no metastasis to liver and lung as compared to T3M4 SC cells implanted tumors, (Figures 4D–4F). However, T3M4 WT-MUC16KO cells did not show any significant changes as compared to T3M4 WT cells.
Figure 4.

Genetic deletion of MUC16 reduces PDAC tumorigenicity
(A) Schema of in vitro and in vivo experiments using WT, WT-MUC16KO, SC, and SC-MUC16KO cells. (B) Cell migration assay in T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells. Data were presented as mean ± SD (n = 3; Dunnett’s multiple comparisons test). (C) Matrigel invasion assay inT3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells. Data were presented as mean ± SD (n = 3; Dunnett’s multiple comparisons test). (D and E) Tumor weight (D) and tumor volume (E) of T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells implanted orthotopic tumors. Data were presented as mean ± SEM (n = 14; Dunnett’s multiple comparisons test). (F) Analysis of tumor metastasis in T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells implanted tumor-bearing animals (Fisher’s exact test).
Aberrant glycosylation enhances the interactions between MUC16 and epidermal growth factor receptors
Ingenuity pathway analysis of truncated O-glycans expressing PDAC cells phosphoproteome dataset from our previous studies9 revealed that epidermal growth factor receptor family members have been activated in truncated O-glycan expressing PDAC cells (Table S5). Hence, we investigated the phosphorylation status of epidermal growth factor receptors in T3M4 WT and SC cells and found phosphorylation of ErbB1 (Y1173), ErbB3 (Y1289), and its downstream targets AKT (S473) and GSK3β (S9) were enhanced in SC cells as compared to WT cells (Figures S2A and S2B). Evaluation of protein-protein interactions between EGF receptors (ErbB1, ErbB2, ErbB3, and ErbB4) and MUC16 using proximity ligation assay (PLA) on T3M4 WT and SC cells revealed significant interactions between MUC16 and ErbB2 (counts/cell) in WT cells that were enhanced in SC cells (p = 0.0017; Figure 5A). Together these data support the hypothesis that interactions between MUC16 and EGF receptors activate oncogenic signaling cascades that are exacerbated by aberrant glycosylation. Therefore, we investigated the activation of EGF type receptors and consequent downstream targets in MUC16KO WT and SC PDAC cells (T3M4 and Capan-2). Consistently, there was constitutive phosphorylation of ErbB1 (Y1173), ErbB2 (Y1248), and ErbB3 (Y1289) in T3M4 and Capan-2 WT cells that was enhanced in the SC derivatives but significantly decreased or eliminated in MUC16KO WT and SC clones (T3M4 and Capan-2; Figures 5B and 5C). Similarly, phosphorylation of AKT (S473) and GSK3β (S9) was high in T3M4 and Capan-2 SC cells as compared to parental WT cells; however, there was complete inhibition of steady-state phosphorylation of AKT and GSK3β in T3M4 SC-MUC16KO cells as compared to T3M4 WT-MUC16KO cells (Figure 5D). A similar trend was observed in MUC16KO Capan-2 WT and SC cells (Figure 5E). These results were further confirmed in orthotopic tumors of T3M4 WT, T3M4 WT-MUC16KO (2E4), T3M4 SC, and T3M4 SC-MUC16KO (1E10), which showed significantly reduced expression of p-ErbB2 (p < 0.0001 and p < 0.0001; Figures 5F and 5H) and p-AKT (p = 0.0092 and p = 0.0045; Figures 5G and 5I) in T3M4 WT-MUC16KO and T3M4 SC-MUC16KO tumors as compared to control tumors, respectively. Interestingly, expression of p-ErbB2 was significantly less in T3M4 SC-MUC16KO tumors (p = 0.0068) compared to T3M4 WT-MUC16KO tumors. To confirm the clinical relevance and the posited regulatory role of MUC16 on EGFR signaling, we purified MUC16/CA125 from PDAC patients (n = 5) and applied this to T3M4 WT-MUC16KO cells (5 μg/mL for 24 h). We observed increased phosphorylation of ErbB3, ErbB2, AKT, and GSK3β in clinically isolated MUC16 treated MUC16KO cells compared to untreated cells (Figure S2C).
Figure 5.

Interaction of MUC16 with epidermal growth factor receptors
(A) Proximity ligation assay in T3M4 WT and SC cells using MUC16 and ErbB2 specific antibodies. Scale bar, 10 μm. Quantification of the number of interactions (dots/cell) quantified by blob finder 2 software. Cells incubated with a single antigen-specific antibody served as a negative control. Data were presented as mean ± SD (n = 3; unpaired t test). (B) Western blotting of T3M4 WT, WT-MUC16KO, T3M4 SC, and SC-MUC16KO cell lysates with p-ErbB3 (Y1289), ErbB3, p-ErbB2 (Y1248), ErbB2, p-ErbB1 (Y1173), and ErbB1. (C) Western blotting of Capan-2 WT, WT-MUC16KO, Capan-2 SC, and SC-MUC16KO cell lysates with p-ErbB3 (Y1289), ErbB3, p-ErbB2 (Y1248), ErbB2, p-ErbB1 (Y1173), and ErbB1. (D) Western blotting of p-AKT (S473), AKT, p-GSK3β (S9), and GSK3β in T3M4 WT, WT-MUC16KO, T3M4 SC, and SC-MUC16KO cells. (E) Western blotting of p-AKT (S473), AKT, p-GSK3β (S9), and GSK3β in Capan-2 WT, WT-MUC16KO, Capan-2 SC, and SC-MUC16KO cells. Detection of β-actin served as a loading control. (F and G) Immunohistochemical analysis of p-ErbB2 (F) and p-AKT (G) in T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells implanted mouse tumors (n = 5). Scale bar, 40 μm. (H and I) Mean histoscore of the IHC analysis of p-ErbB2 (H) and p-AKT (I) in T3M4 WT, WT-MUC16KO (2E4), T3M4 SC, and SC-MUC16KO (1E10) cells implanted mouse tumors. Data were presented as mean ± SEM (n = 5; Dunnett’s multiple comparisons test).
Anti-MUC16 antibodies treatment inhibits EGF receptors and their downstream signaling
We further evaluated the contribution of MUC16 to the constitutive activation of EGF receptors and downstream oncogenic signaling by using different anti-MUC16 mAbs to block MUC16 binding to receptors on tumor cells. We have previously reported the binding specificity of mAbs 5E1113 and B43.13 to MUC16.15 Treatment of T3M4 WT cells with anti-MUC16 antibodies (5E11, B43.13, and AR9.6; 2.5 and 5 μg/mL) for 24 h inhibited the basal level phosphorylation of AKT in a concentration-dependent manner (Figure 6A). The greatest inhibition of AKT phosphorylation was observed in cells treated with mAb AR9.6 (2.5 and 5 μg/mL). Among these antibodies, only mAb AR9.6 decreased constitutively hyperphosphorylated AKT in T3M4 SC cells compared to untreated and immunoglobulin G (IgG) control (Figure 6B). Next, we analyzed the phosphorylation of AKT upstream activator EGF receptors in mAb AR9.6-treated PDAC cells and found reduced phosphorylation of ErbB1 and ErbB2 (Figure S3A). These results prompted us to characterize the MUC16 epitope recognized by mAb AR9.6. An established epitope-mapping strategy was used that utilizes nine MUC16 constructs with overlapping sequences within the tandem repeat (TR) region of MUC16 (TR1.7), includes part of TR4, the entire Sea urchin, enterokinase and agrin (SEA), and linker domain of TR5 along with half of the SEA domain of TR6. The TR1.2, 1/4, 5/2, 5/7, 5/8, 5/9, K292, and F/R constructs represent truncated forms of this construct (Figure 6C).1 ELISA assay of MUC16 constructs that were expressed in E. coli and purified by nickel chromatography with mAb AR9.6 showed strong reactivity of mAb AR9.6 with tandem repeats TR1.7 and TR1.2. In contrast, tandem repeats 5/2, 1/4, 5/7, 5/8, 5/9, K292, F/R were unreactive (Figure 6D). This distinguishes mAb AR9.6 from the classical OC125 and M11 epitopes that were strongly reactive with the 5/2 construct. Also, incubation of proteins with anti-MUC16 mAb 5E11 served as a positive control for these reactions, which showed reactivity with proteins encoded by the TR1.7, TR1.2, 5/2, 1/4, and 5/7 constructs (Figure 6E). The incubation of proteins with a negative control showed no reactivity toward the above-mentioned TR constructs (data not shown). We found that mAb AR9.6 binds to multiple isoforms of MUC16 that are distinct from those bound by mAbs M11 and OC125. Additionally, we found that mAb 5E11 binds to a distinct isoform of MUC16 as compared to other anti-MUC16 mAbs (Figure S3B).
Figure 6.

AR9.6 mAb inhibits MUC16 induced oncogenic signaling
(A and B) Western blotting of p-AKT and AKT in T3M4 WT (A) and T3M4 SC (B) cells treated with IgG and MUC16 specific antibodies 5E11, B43.13, and AR9.6. Detection of β-actin served as a loading control. (C) Graphic representation of MUC16 tandem repeats constructs expressed in E-coli. TR1.7 includes part of TR4, the entire SEA, and the linker domain of TR5 along with half of the SEA domain of TR6 (12,660–12,923 aa). The TR1.2 (12,665–12,858 aa), 1/4 (12,665–12,785 aa), 5/2 (12,757–12,923 aa), 5/7 (12,757–12,886 aa), 5/8 (12,757–12,863 aa), 5/9 (12,757–12,839 aa), K292 (12,757–12,816 aa), and F/R (12,817–12,859 aa) constructs represent truncated forms of this construct. (D) mAb AR 9.6 epitope characterization by ELISA assay of MUC16 constructs. (E) ELISA assay of MUC16 constructs with positive control mAb 5E11. (F) Western blotting of p-ErbB3 (Y1289), ErbB3, p-ErbB1 (Y1173), ErbB1, p-GSK3β (S9), and GSK3β in MUC16KO T3M4 cells treated with TR1.2 (1 and 2 μg/mL, 14 h). (G) Western blotting of p-ErbB3 (Y1289), ErbB3, p-ErbB2 (Y1248), ErbB2, p-AKT (S473), AKT, p-GSK3β (S9), and GSK3β in MUC16KO T3M4 cells treated with TR1.2 (1 μg/mL) with or without mAb AR9.6 (5 μg/mL). Detection of β-actin and GAPDH served as the loading control. (H) Tumor weight of vehicle control (n = 11) and mAb AR9.6 (n = 13) treated T3M4 WT orthotopic tumors. Data were reported as mean ± SEM (unpaired t test). (I and J) Analysis of tumor metastasis in vehicle and mAb AR9.6 treated T3M4 WT (I) and SC (J) orthotopic tumor-bearing animals (Fisher’s exact test). (K) Representative images of live/dead cytotoxicity assay in vehicle, IgG, AR9.6, GEM, and AR9.6 + GEM treated PDAC (T3M4 and Capan-2) cells. Scale bar, 20 μm. (L and M) The percentage of cell death in vehicle, IgG, AR9.6, GEM, AR9.6 + GEM-treated T3M4 (L), and Capan-2 (M) cells. Data presented as mean ± SD (n = 3; Dunnett’s multiple comparisons test).
Next, we investigated the possible biological activity of the AR9.6 binding region on MUC16 (TR1.2) by expressing and purifying MUC16 TR1.2 from CHO cells (Figure S3C) and treating T3M4-MUC16KO cells. There were dose-dependent increases in the phosphorylation of ErbB1, ErbB3, and GSK3β upon the addition of TR1.2 to MUC16 deficient PDAC cells (Figure 6F). Pre-incubation of MUC16KO PDAC cells with mAb AR9.6 significantly inhibited TR1.2 induced phosphorylation of p-ErbB3, p-ErbB2, p-AKT, and p-GSK3β (Figure 6G). We investigated the role of glycosylation in TR1.2 binding affinity with mAb AR9.6. Treatment of TR1.2 with N-glycanase resulted in reduced mobility and binding to mAb AR9.6; however, treatment of TR1.2 with either Sialidase or O-glycanase or both significantly enhanced the binding of mAb AR9.6 with TR1.2 (Figure S3D). A similar binding trend was observed when glycosidases were applied to MUC16-positive PDAC cells (Figure S3E).
mAb AR9.6 treatment reduces in vivo tumor growth and metastasis
Next, we evaluated the preclinical in vivo therapeutic efficacy of mAb AR9.6 in an orthotopic pancreas tumor model system. mAb AR9.6-treated T3M4 WT tumor-bearing animals showed a significantly reduced tumor growth (by tumor weight, g; p = 0.01; Figure 6H), metastases to the spleen (15.34%, p = 0.04) and lymph nodes (7.69%, p = 0.003), and no metastasis to lung and diaphragm as compared to vehicle control-treated T3M4 WT tumor-bearing animals (Figure 6I). We did not observe detectable changes in tumor weight (g) between the different treatment conditions on T3M4 SC tumor-bearing animals (vehicle control and mAb AR9.6; Figure S3F). However, a significantly reduced tumor metastasis to the peritoneum (p < 0.0001), no metastasis to lung, spleen, and diaphragm, and a lower percent to the liver and lymph node were found in mAb AR9.6-treated T3M4 SC tumor-bearing animals as compared to vehicle control (Figure 6J). This finding was of great interest that mAb AR9.6 significantly reduces metastasis in tumor-bearing animals. To scrutinize the mechanism behind this, we have analyzed the expression of MMP9 in AR9.6 treated T3M4 cells. We detected a reduced expression of MMP9 in mAb AR9.6-treated PDAC cells (Figure S3G).
mAb AR9.6 and gemcitabine combination therapy reduces PDAC tumorigenesis
Since we found a significant reduction in the tumor growth and metastasis upon treatment with mAb AR9.6, we sought to analyze the potential of combination therapy of AR9.6 with gemcitabine (GEM) as it is a standard chemotherapeutic agent for PDAC. Prior to the in vivo experiments, we assessed the potential of AR9.6 + GEM in inducing in vitro cell death in PDAC cells (T3M4 and Capan-2) using live/dead cytotoxicity assay (Figure 6K). The combination of AR9.6 + GEM significantly induced cell death as compared to vehicle control (p < 0.0001 and p < 0.0001), GEM alone (p < 0.0001 and p < 0.0001), and AR9.6 alone (p < 0.0001 and p = 0.0001) in both T3M4 and Capan-2 cells, respectively (Figures 6L and 6M). To further evaluate its in vivo therapeutic potential, we treated T3M4 tumor-bearing athymic nude mice with AR9.6 + GEM (Figure 7A). Since we found that mAb AR9.6 significantly reduced tumor growth in T3M4 WT tumor (Figure 6H), we sought to analyze its effect in the later stage of tumor growth. Therefore, we treated tumor-bearing mice with mAb AR9.6, GEM or AR9.6 + GEM after 18 days of PDAC cells implantation. mAb AR9.6 + GEM combination therapy produced a maximum reduction in the tumor weight (p = 0.0032) and volume (p = 0.0115) when compared to the vehicle control group (Figures 7B and 7C). Also, animals treated with mAb AR9.6 alone showed a significant reduction in tumor weight (p = 0.0469) when compared to vehicle control (Figure 7B) even at the late time point of treatment. While there was a reduction in tumor growth, animals treated with GEM alone did not exhibit a significant reduction in tumor weight and volume probably due to the treatment at later stage of tumor growth. Treatment of tumor-bearing animals with mAb AR9.6 and GEM significantly reduced Ki-67 (p < 0.0001; Figure 7D, representative positive nuclei percentage of vehicle control; Figure S4A, representative IHC images) and CD31 (p < 0.0001) expression (Figure 7E, positive representative cells; Figure S4B, representative IHC images). Notably, the phosphorylation of ErbB2 and AKT was significantly inhibited in tumors from animals treated with mAb AR9.6 + GEM (pErbB2, p = 0.0001; p-AKT, p < 0.0001; Figures 7F and 7G representative IHC images; and Figures 7H and 7I, representative mean histoscore). Also, a significant reduction in p-ErbB2 and p-AKT expression was observed in AR9.6 alone or gemcitabine alone treated tumors as compared to vehicle control tumors (Figures 7F–7I).
Figure 7.

mAb AR9.6 plus gemcitabine treatment reduces in vivo tumor growth
(A) Schematic representation of orthotopic tumor implantation and treatment schedules of tumor-bearing mice (n = 10) with PBS, IgG, GEM, mAb AR9.6, and mAb AR9.6 plus GEM. (B and C) Tumor weight (g) (B) and tumor volume (mm3) (C) of vehicle, IgG, GEM, mAb AR9.6, and GEM + mAb AR9.6-treated T3M4 orthotopic tumors. Data presented as mean ± SEM (n = 10; Dunnett’s multiple comparisons test). (D and E) IHC analysis of Ki-67 (D) and CD31 (E) in above mentioned experimental tumors. Data presented as mean ± SEM (n = 9; Dunnett’s multiple comparisons test). (F and G) IHC analysis of p-ErbB2 (F) and p-AKT (G) in above mentioned experimental tumors. (H and I) Mean histoscore of the IHC analysis of p-ErbB2 (H) and p-AKT (I) in above mentioned experimental tumors. All treatment groups were compared against vehicle control (PBS) treated groups. Scale bar, 40 μm. Data presented as mean ± SEM (n = 9; Dunnett’s multiple comparisons test).
Discussion
MUC16 is not expressed in the normal pancreas, and aberrant expression of MUC16 is associated with pancreatic cancer progression, metastasis, and poor prognosis. Here, we show that aberrant expression of MUC16 is related to epithelial tumors, squamous, basal-like, and quasimesenchymal subtypes of PDAC, which are generally associated with an aggressive phenotype. Aberrant expression of MUC16 was also found in pancreatic adenosquamous and islet cell tumors. We found that short-term survivors of PDAC possess a higher expression of MUC16, which is in accordance with the previous reports.16 However, the pathological mechanism of MUC16 in PDAC progression and metastasis is poorly understood. Here, we observed that interactions between MUC16 and ErbB receptors facilitate PDAC progression through the activation of AKT/GSK3β oncogenic signaling that was further enhanced for aberrant glycoforms of MUC16. Genetic deletion of MUC16 in WT and to a greater extent in truncated O-glycan expressing PDAC cells significantly inhibited the activation of ErbB receptors and downstream oncogenic signaling pathways, further supporting the hypothesis that EGF-like domains on MUC16 bind and activate ErbB type receptors in a glycosylation modulated manner. As well, the genetic deletion of MUC16 significantly reduced tumor growth and metastasis (especially to the peritoneum and lymph node). These findings extend our previous report that MUC16 is upregulated in metastatic PDAC2 to show that MUC16 has pro-oncogenic activities and supports findings from other groups showing that aberrant expression of MUC16 facilitates peritoneal metastasis.17
Activation of ErbB receptors and their downstream effectors PI3K/AKT/GSK3β affect cellular processes, including cell proliferation, growth, motility, and cell survival.18 More interestingly, the squamous subtype of PDAC was shown to be associated with activation of EGF receptor and PI3K/AKT signaling cascades.19 More than 83% of the pancreatic tumors show constitutively activated AKT (S473), which portends poor prognosis.20, 21, 22 Increased expression of MUC16 in the squamous subtype of PDAC tumors along with the genetic deletion of MUC16 decreased steady-state activation of ErbB, AKT, and GSK3β oncogenic signaling strongly support our hypothesis that MUC16 promote PDAC tumorigenicity through the interaction with ErbB receptors. However, the functional consequences of normal and altered O-glycosylation of MUC16 during PDAC progression remain elusive. Generally, glycosylation can block potential sites of other post-translational modifications of proteins,23,24 and glycosylated moieties may bind directly to lectin-like motifs to facilitate or block the formation of protein complexes. We found that the association between MUC16 and ErbBs in pancreatic cancer is highly influenced by glycosylation. The results of this study support the hypothesis that normal and aberrant glycosylation influences the accessibility of domains on mucins (MUC16) that bind to and/or otherwise activate cell-surface ErbB-type receptors and consequent signaling programs, which in turn affects the malignant properties of PDAC cells. It is a well-known fact that aberrant glycoforms of MUC16 is a biomarker that is elevated in the circulation in several adenocarcinomas, and its high levels are associated with poor prognosis. Treatment of MUC16 null cells with purified MUC16 from PDAC patients’ ascites fluid induces an increased expression of phosphorylated EGF receptors and its downstream target signaling effectors, which supports our hypothesis that differential glycosylation of this biomarker creates active compounds that can bind different receptor tyrosine kinases and that these glycoforms could enhance disease progression by binding and activating signaling cascades in the local tumor microenvironment and at distant organ sites following circulatory transit.
The observed oncogenic activity of MUC16 in PDAC biology suggests a need for the development of therapeutic agents that target this tumor biomarker. In this context, we have characterized mAb AR9.6 that binds specifically to SEA domain 5 of MUC16. Binding of mAb AR9.6 to MUC16 inhibited phosphorylation of ErbB-type receptors and downstream AKT/GSK3β signaling. mAb AR9.6 was found to inhibit T3M4 WT cells implanted PDAC tumor growth and metastasis in athymic nude mice alone or in combination with gemcitabine. However, mAb AR9.6 did not affect tumor growth in T3M4 SC cells implanted athymic nude. This can be explained by the abundance of MUC16 epitope (opening of hidden epitopes) on SC cells, which may need more concentration of antibody to block the epitope in order to reduce tumor growth that needs further evaluation. AR9.6 mAb staining on clinical specimens of PDAC tumors revealed that MUC16 is not only expressed in ductal adenocarcinoma but also highly expressed in adenosquamous carcinoma and islet cell tumor. Additionally, mAb AR9.6 binding epitopes were identified in a greater number of patient samples than OC125 mAb epitopes. Further investigation of AR9.6 and other MUC16 reactive antibodies as potential therapeutic agents is warranted.
Materials and methods
Human rapid autopsy samples
De-identified sets of samples were procured from patients who underwent a Rapid Autopsy Program at the University of Nebraska Medical Center (UNMC) with proper consent and approved by Institutional Review Board (IRB 091-01-EP). A primary pancreatic tumor (n = 72), metastatic tumors (n = 46), and normal pancreas tissues (n = 7) were used for the study (Table S2).
Cell lines and culture
Human PDAC cells T3M4 and Capan-2 (WT [do not express truncated O-glycans]; COSMC KO, SC do express truncated O-glycans)9,10,25 were maintained in Dulbecco’s modified Eagle’s medium (DMEM) and RPMI medium, respectively (Hyclone, Logan, UT, USA) supplemented with 10% fetal bovine serum (FBS; Valley Biomedical, Winchester, VA) and 100 units/mL penicillin and 100 μg/mL streptomycin (Mediatech, Manassas, VA, USA). CHO cells were cultured in 80% EX CELL CHO Cloning Medium and 20% EX CELL CD CHO Fusion medium (Sigma, St. Louis, MO, USA) with 2% glutamine and 0.32 mg/mL G418 (Invitrogen, Carlsbad, CA, USA). All the cell lines were maintained at 37°C with 5% CO2 in a humidified incubator.
Genetic deletion of MUC16 in PDAC cells
The N-terminal region of the MUC16 gene was genetically deleted in T3M4 and Capan-2 (WT and SC) cells by using a pre-made CRISPR-Cas9 KO plasmid kits (Santa Cruz Biotechnology, CA, USA) as per the manufacturer’s instruction. Briefly, cells were transfected with a pool of three MUC16 guide-RNA-containing vectors (1, 5′-AACACACTCGATGGCGACTC-3′; 2, 5′-GGTGAACTCGTTACGGGCTC-3′; 3, 5′-TGTCCCAGAAACCGTTGTGC-3′), which targets MUC16 genome. MUC16 KO cells (T3M4 WT-MUC16KO, T3M4 SC-MUC16KO, Capan-2 WT-MUC16KO, and Capan-2 SC-MUC16KO) were utilized for further studies.
Cell proliferation assay
Cell proliferation assay was performed as described.26 Briefly, 1 × 103 PDAC cells (T3M4 and Capan-2) WT, WT-MUC16KO (2E4 and 1C10), SC, and SC-MUC16KO (1E10 and 2F9), respectively, were seeded into 96-well plates in quadruplicate and incubated for 24, 48, 72, and 96 h, respectively. At the end of each time, 20 μL of cell suspension was removed from each well, and 20 μL Alamar blue reagent (Invitrogen, Carlsbad, CA, USA) was added, such that Alamar blue made up 10% of the total volume of each well and incubated for further 2–4 h at 37°C. The fluorescence intensity was measured using Spectramax M5e (Molecular Devices, Sunnyvale, CA, USA) spectrofluorimeter at excitation 540 nm and emission 590 nm.
Cell migration assay
In vitro cancer cell migration assay was performed as described previously.26 Briefly, T3M4 WT, WT-MUC16KO (2E4), SC, and SC-MUC16KO (1E10) cells (5 × 104 in serum-free medium) were seeded on the top of the polyethylene terephthalate membrane (PET; 24-well insert, 8 μm; BD Biosciences, Bedford, MA, USA). After 18 h of incubation, cells that didn’t migrate through the membrane were removed with cotton swabs, and cells that migrated through the membrane were fixed and stained with Diff-Quick cell staining kit (Hema3 stat pack, Thermo Fisher Scientific, Waltham, MA, USA), and counted under a light microscope in 4 different random fields at 20× magnification.
Cell invasion assay
Tumor cell invasion assay was performed as described previously.26 In brief, T3M4 WT, WT-MUC16KO (2E4), SC, and SC-MUC16KO (1E10) cells (5 × 104 in serum-free medium) were seeded on Matrigel-coated membranes (BD Biosciences, Bedford, MA, USA) and incubated for 30 h at 37°C. After incubation, non-invading cells on the upper surface of the filter were removed with cotton swabs, and cells that invaded through the Matrigel were fixed and stained with Diff-Quick cell stain kit (Hema3 stat pack, Thermo Fisher Scientific, USA). The invaded cells were counted and analyzed.
Immunofluorescence analysis
Immunofluorescence staining of mucins and mucin glycans on cancer cells was carried out as described previously.9 Briefly, after fixing with 4% paraformaldehyde and permeabilized with 0.15% Triton X-100 in 1X PBS containing 1% BSA, PDAC cells (T3M4 and Capan-2) WT, WT-MUC16KO (2E4 and 1C10), SC, and SC-MUC16KO (1E10 and 2F9), respectively, were incubated with MUC16-specific mAb AR9.6, and control mouse IgG for 2 h at room temperature. Cells were washed and incubated with Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibodies (Jackson ImmunoResearch, West Grove, PA, USA). Cells were washed and mounted with Vectashield mounting medium with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). Immunofluorescence images were captured by using the Zeiss LSM 710 confocal laser scanning microscope (Carl Zeiss, Thornwood, NY, USA; Confocal Laser Scanning Fluorescence Microscopy Core Facility, UNMC).
Orthotopic pancreas tumor model
All the animals were housed under standard housing conditions at the University of Nebraska Medical Center animal core facilities, and all animal procedures were reviewed and approved by the UNMC institutional animal care and use committees (IACUC). MUC16-mediated tumor growth: T3M4 WT, WT-MUC16KO(2E4), T3M4 SC, and SC-MUC16KO(1E10) cells (3 × 105/30 μL PBS) were orthotopically implanted into the pancreas of athymic nu/nu mice (Crl:NU-Foxn1nu; n = 14 each group) and the tumor-bearing animals were sacrificed (day 28) and analyzed as described previously.27 mAb AR9.6 treatment: to determine the early preclinical response of AR9.6 in orthotopic tumor model, T3M4 WT and SC cells (2.5–3 × 105/30 μL PBS) implanted tumor-bearing athymic nu/nu mice were randomized (after 14 days) and treated with four doses of vehicle control (PBS, n = 11) and AR 9.6 mAb (0.5 mg, n = 13) via intraperitoneal injection (i.p.) with 72 h interval for four cycles. mAb AR9.6 plus GEM combination treatment: to determine the effect of combination therapy of mAb AR9.6 with GEM at later stages of tumor growth, T3M4 WT cells (3 × 105/30 μL PBS) implanted tumor-bearing athymic nu/nu mice were randomized (after 18 days) and treated as follows: group I, vehicle control (PBS, n = 9); group II, IgG (0.5 mg, n = 9); group III, GEM (60 mg/kg, n = 10); group IV, mAb AR9.6 (0.5 mg, n = 10), and group V, mAb AR9.6 (0.5 mg) + GEM (60 mg/kg, n = 10) four doses via i.p. with 72 h interval for four cycles. At the end of each study, the animals were sacrificed, and the tumor weight, volume, and incidence of metastases were determined.
Western blotting
T3M4 (WT, WT-MUC16KO, SC, SC-MUC16KO) and Capan-2 (WT, WT-MUC16KO, SC, SC-MUC16KO) cell lysates (radioimmunoprecipitation assay [RIPA] lysis buffer [Thermo Scientific, Rockford, IL USA] with protease and proteinase inhibitor) were resolved on 4%–20% SDS-PAGE (30–50 μg/lane) and transferred to polyvinylidene fluoride (PVDF) membrane (Thermo Scientific, Rockford, IL, USA). For analyzing the effect of various anti-MUC16 antibodies, T3M4 WT and SC cells were treated with IgG, 5E11 (provided by Dr. Ulla Mandel, University of Copenhagen, Denmark), B43.13 and AR9.6 (2.5 and 5 μg/mL; Quest PharmaTech, Canada) for 24 h. For analyzing the effect of TR1.2, T3M4 SC-MUC16KO cells (polyclonal) were treated with TR1.2 (1 and 2 μg/mL) for 14 h. For analyzing the effect of TR1.2 and AR9.6, T3M4 WT-MUC16KO cells were treated with TR1.2 (1 μg/mL) with or without mAb AR9.6 (5 μg/mL) for 14 h. At the end of each experimental time point, the cell lysates were prepared, and proteins were separated as mentioned above. The membranes were probed with the following primary and secondary antibodies as per the manufacturer’s recommendations: anti-p-AKT (S473), anti-AKT, anti-p-GSK3β (S9), anti-GSK3β, anti-p-ErbB2 (Y1248), anti-ErbB2, anti-p-ErbB3 (Y1289), anti-ErbB3, anti-cyclin D1, anti-cyclin E1, anti-GAPDH, anti-β-actin (Cell Signaling Technology, Danvers, MA, USA), anti-p-ErbB1 (Y1173; Santa Cruz Biotechnology, CA, USA), and horseradish peroxidase (HRP) conjugated goat anti-mouse IgG and goat anti-rabbit IgG (Cell Signaling Technology, Danvers, MA, USA). The signal was detected by using the enhanced chemiluminescent kit (Bio-Rad, Hercules, CA, USA).
IHC
The standard IHC method was used for the analysis of protein expression in tissue samples. Briefly, the paraffin-embedded slides were de-paraffinized with xylene, re-hydrated with series of alcohol (100% to 70%), quenched with hydrogen peroxide, and immersed in 95°C citrate buffer (pH = 6.0) for 15 min for antigen retrieval. After blocking with a universal blocker (Thermo Fisher Scientific, Waltham, MA, USA), the slides were incubated with antibodies OC125 (1:100; Abcam, Cambridge, MA, USA) AR9.6 (1:500), 5E11 (1 μg/mL), and 5B9 (1 μg/mL; provided by Dr. Ulla Mandel, University of Copenhagen, Denmark). The slides were washed and incubated with HRP conjugated secondary antibody. After 1 h, the slides were washed with Tris-buffered saline (TBS), and the antigen-antibody reaction products were developed using 3,3′-diaminobenzidine tetrahydrochloride (DAB, Vector Laboratories, Burlingame, CA, USA) substrate and then counterstained with hematoxylin. All the slides were dehydrated with alcohol series, and after xylene washes, the slides were mounted with the coverslip. Also, analysis of MUC16 expression in a pancreatic cancer tissue microarray (BIC14011a; BIOMAX.US) was performed as described above. For the analysis of p-ErbB2 (1:50; Abcam, Cambridge, MA, USA) and p-AKT (1:50; Abcam, Cambridge, MA, USA) in mouse tissue sections, the slides were processed as mentioned above. The protein expression was scored by a pathologist. The histological scoring was performed based on stain proportion (0%–100%) and intensity (0, negligible; 1, low; 2, moderate; 3, high). The histoscore was generated by multiplying the stain proportion score (1, <5%; 2, 5%–25%; 3, 26%–50%; 4, 51%–75%; 5, >75%) with the intensity score (0–3) to obtain values between 0 and 15.
Proliferation index
Tissue sections of T3M4 cells implanted tumors treated with vehicle control (PBS), IgG, GEM, mAb AR9.6, and mAb AR9.6 + GEM (n = 9) were stained with rabbit anti-Ki-67 antibody (Abcam, Cambridge, MA, USA) as described previously.28 A total of four random fields were examined in nine different tumor samples from each group. The results were expressed as a mean percentage of Ki-67-positive cells ± SEM per field (100× magnification).
Microvessel density
Tissue sections of T3M4 cells implanted tumors treated with vehicle control (PBS), IgG, GEM, mAb AR9.6, and mAb AR9.6 + GEM (n = 9) were stained with rabbit anti-CD31 antibody (Abcam, Cambridge, MA, USA), as described previously.28 The number of the CD31-positive vessel was examined under a microscope and counted. A total of four random fields were examined in nine different tumor samples from each group. The results were expressed as a mean percentage of vessels ± SEM per field (100× magnification).
Proximity ligation assay
The protein-protein interaction experiments (growth factor receptors interactions with mucins and integrin complexes) were performed by using Duo Link II (Olink Bioscience, Watertown, MA, USA) Proximity Ligation Assay kit according to manufacturer’s recommendation as described previously.29 Briefly, T3M4 WT and SC cells were grown on coverslips and then incubated with the primary antibody specific to MUC16 (AR9.6, Quest PharmaTech, Canada) and ErbB2 (Santa Cruz Biotechnology, CA, USA) and then the primary antibodies were incubated with PLA probes anti-Rabbit PLUS, anti-mouse MINUS. Experiments with omitting the primary antibody used for single recognition PLA or either one of the primary antibodies used in double recognition PLA served as a negative control. The fluorescence images were captured under Zeiss LSM 510 laser scanning confocal microscope (Carl Zeiss, Thornwood, NY, USA) at Confocal Laser Scanning Fluorescence Microscopy Core Facility, UNMC, and the signal/dots per cells were quantified by using blob finder software.
Epitope mapping
The recombinant MUC16 constructs (TR1.7, TR1.2, 1/4, 5/2, 5/7, 5/8, 5/9, K292, and F/R) were synthesized and expressed, and ELISA assays were performed as described previously.1 Briefly, MUC16 recombinant proteins were serially diluted from an initial concentration of 500 ng/mL and coated overnight at 4°C in carbonate-bicarbonate buffer (pH 9.6) in a Nunc MaxiSorp 96-well plates (Thermo Scientific, Rockford, IL USA). Then the plate was blocked with PLI-P buffer pH 7.4 (PO4, Na/K, 1% Triton, 1% BSA) for 1 h at room temperature, and incubated with mAb AR9.6 (Quest PharmaTech, Canada) at 1 μg/mL for 1 h at room temperature. Bound antibodies were detected with HRP-conjugated IgG goat anti-mouse antibody (Southern Biotech, Birmingham, AL, USA). Plates were developed with 3,3′,5,5′-Tetramethylbenzidine (TMB+) substrate (Dako, Carpinteria, CA, USA), reactions stopped with 0.5 M H2SO4, and absorbance read at 450 nm. Incubation of plates with anti-MUC16 mAb 5E11 served as a positive control, and plates incubated with anti-GalNAc-T3 mAb 2D10 (IgG) were used as a negative control.
Live/dead cytotoxicity assay
T3M4 and Capan-2 cells were treated with vehicle (PBS), IgG (10 μg/mL), AR9.6 (10 μg/mL), GEM (15 nM), and AR9.6, + GEM for 24 h and have performed live/dead cytotoxicity assay as per the manufacturer’s instruction (Invitrogen, CA, USA). Briefly, after washing with PBS, the cells were incubated with assay reagent containing 20 μL ethidium homodimer-1 (EthD-1, 2 mM) and 5 μL of calcein acetoxymethyl ester (Calcein-AM, 4 mM) in 10 mL PBS for 30 min. Images of live (green) and dead (red) cells were captured with Nikon Eclipse Ts2R fluorescent microscope and counted with ImageJ software (n = 3).
Statistical analysis
For unmatched samples, the continuous characteristics and outcomes were compared using t tests or nonparametric Wilcoxon rank-sum tests between two groups, using two-way ANOVA with Tukey’s multiple comparisons or nonparametric Kruskal-Wallis tests with Bonferroni’s multiple comparisons between more than two groups; the categorical characteristics and outcomes were compared using chi-square tests or Fisher’s exact tests between groups, and Bonferroni’s correction was used for multiple comparisons if needed. For matched samples, the continuous characteristics and outcomes were compared using paired t tests or nonparametric Wilcoxon signed-rank tests between two groups. The cell proliferation was compared between the groups by two-way ANOVA with Tukey’s multiple comparisons at each time points. Tumor cell migration, invasion, in vivo tumor weight, tumor volume, Ki-67, CD31, and live/dead cell assay was analyzed by one-way ANOVA with Dunnett’s multiple comparison test. Patient survival was plotted using the method of Kaplan-Meier and compared between groups using log-rank (Mantel-Cox) tests. Mann-Whitney rank-sum test was used between the PDAC subtype classification system. A value of p < 0.05 was considered to be statistically significant.
Acknowledgments
This work was supported by the National Cancer Institute at the National Institutes of Health R01 CA208108, NIH/SPORE Carrier Development Award (P50CA127297), and NE-DHHS/LB506 to P.R.; NIH/SPORE P50CA127297, Program Project Grant P01CA217798, Early Detection Research Network 5U01CA111294, Pancreatic Cancer Detection Consortium 5U01CA210240, and Tumor Microenvironment Network U54 CA163120 to M.A.H. and R50CA211462 to P.M.G.; The National Cancer Institute at the National Institutes of Health R01CA199064 to J.J.Y.; and The Danish National Research Foundation DNRF107 to H.C. Also, we thank Jessica Odvody, Premila Leiphrakpam, Krishti Sabloak, and Philamon Leon for their technical assistance.
Author contributions
P.R. conceived and supervised the study. D.T., S.S., and X.L. performed most of the in vitro and in vivo experiments. H.-R.L., J.A.G., P.M.G., and T.C. performed orthotopic tumor studies. B.S. scored the immunohistochemical staining. L.M.-S., H.C., C.S., and H.H.W. provided COSMC KO PDAC cells and performed anti-MUC16 mAb epitope mapping studies. H.C.M. and K.P.O. performed RNA sequencing analysis. X.L.P. and J.J.Y. performed tumor sub-type analysis with MUC16 expression. F.Q. and F.Y. performed biostatistical analysis. R.M. provided AR9.6 mAb expressing clones. U.M. provided anti-MUC16 antibodies (5E11 and 5B9) for the study. M.A.H. analyzed the data. D.T., M.A.H., and P.R wrote the manuscript.
Declaration of interests
M.A.H. and P.R. have an equity interest in OncoCare Therapeutics. R.M. is employed by Quest PharmaTech and has an equity interest in this company. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.029.
Supplemental information
References
- 1.Marcos-Silva L., Narimatsu Y., Halim A., Campos D., Yang Z., Tarp M.A., Pereira P.J.B., Mandel U., Bennett E.P., Vakhrushev S.Y. Characterization of binding epitopes of CA125 monoclonal antibodies. J. Proteome Res. 2014;13:3349–3359. doi: 10.1021/pr500215g. [DOI] [PubMed] [Google Scholar]
- 2.Remmers N., Anderson J.M., Linde E.M., DiMaio D.J., Lazenby A.J., Wandall H.H., Mandel U., Clausen H., Yu F., Hollingsworth M.A. Aberrant expression of mucin core proteins and o-linked glycans associated with progression of pancreatic cancer. Clin. Cancer Res. 2013;19:1981–1993. doi: 10.1158/1078-0432.CCR-12-2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liang C., Qin Y., Zhang B., Ji S., Shi S., Xu W., Liu J., Xiang J., Liang D., Hu Q. Oncogenic KRAS Targets MUC16/CA125 in Pancreatic Ductal Adenocarcinoma. Mol. Cancer Res. 2017;15:201–212. doi: 10.1158/1541-7786.MCR-16-0296. [DOI] [PubMed] [Google Scholar]
- 4.O’Brien T.J., Beard J.B., Underwood L.J., Dennis R.A., Santin A.D., York L. The CA 125 gene: an extracellular superstructure dominated by repeat sequences. Tumour Biol. 2001;22:348–366. doi: 10.1159/000050638. [DOI] [PubMed] [Google Scholar]
- 5.Hakomori S. Glycosylation defining cancer malignancy: new wine in an old bottle. Proc. Natl. Acad. Sci. USA. 2002;99:10231–10233. doi: 10.1073/pnas.172380699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pinho S.S., Reis C.A. Glycosylation in cancer: mechanisms and clinical implications. Nat. Rev. Cancer. 2015;15:540–555. doi: 10.1038/nrc3982. [DOI] [PubMed] [Google Scholar]
- 7.Julien S., Videira P.A., Delannoy P. Sialyl-tn in cancer: (how) did we miss the target? Biomolecules. 2012;2:435–466. doi: 10.3390/biom2040435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim G.E., Bae H.I., Park H.U., Kuan S.F., Crawley S.C., Ho J.J.L., Kim Y.S. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. 2002;123:1052–1060. doi: 10.1053/gast.2002.36018. [DOI] [PubMed] [Google Scholar]
- 9.Radhakrishnan P., Dabelsteen S., Madsen F.B., Francavilla C., Kopp K.L., Steentoft C., Vakhrushev S.Y., Olsen J.V., Hansen L., Bennett E.P. Immature truncated O-glycophenotype of cancer directly induces oncogenic features. Proc. Natl. Acad. Sci. USA. 2014;111:E4066–E4075. doi: 10.1073/pnas.1406619111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Steentoft C., Vakhrushev S.Y., Joshi H.J., Kong Y., Vester-Christensen M.B., Schjoldager K.T.-B.G., Lavrsen K., Dabelsteen S., Pedersen N.B., Marcos-Silva L. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 2013;32:1478–1488. doi: 10.1038/emboj.2013.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haridas D., Chakraborty S., Ponnusamy M.P., Lakshmanan I., Rachagani S., Cruz E., Kumar S., Das S., Lele S.M., Anderson J.M. Pathobiological implications of MUC16 expression in pancreatic cancer. PLoS ONE. 2011;6:e26839. doi: 10.1371/journal.pone.0026839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Law C.W., Chen Y., Shi W., Smyth G.K. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014;15:R29. doi: 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcos-Silva L., Ricardo S., Chen K., Blixt O., Arigi E., Pereira D., Høgdall E., Mandel U., Bennett E.P., Vakhrushev S.Y. A novel monoclonal antibody to a defined peptide epitope in MUC16. Glycobiology. 2015;25:1172–1182. doi: 10.1093/glycob/cwv056. [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas Research Network. Electronic address: andrew_aguirre@dfci.harvard.edu. Cancer Genome Atlas Research Network Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017;32:185–203.e13. doi: 10.1016/j.ccell.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Noujaim A.A., Schultes B.C., Baum R.P., Madiyalakan R. Induction of CA125-specific B and T cell responses in patients injected with MAb-B43.13--evidence for antibody-mediated antigen-processing and presentation of CA125 in vivo. Cancer Biother. Radiopharm. 2001;16:187–203. doi: 10.1089/10849780152389384. [DOI] [PubMed] [Google Scholar]
- 16.Balachandran V.P., Łuksza M., Zhao J.N., Makarov V., Moral J.A., Remark R., Herbst B., Askan G., Bhanot U., Senbabaoglu Y., Australian Pancreatic Cancer Genome Initiative. Garvan Institute of Medical Research. Prince of Wales Hospital. Royal North Shore Hospital. University of Glasgow. St Vincent’s Hospital. QIMR Berghofer Medical Research Institute. University of Melbourne, Centre for Cancer Research. University of Queensland, Institute for Molecular Bioscience. Bankstown Hospital. Liverpool Hospital. Royal Prince Alfred Hospital, Chris O’Brien Lifehouse. Westmead Hospital. Fremantle Hospital. St John of God Healthcare. Royal Adelaide Hospital. Flinders Medical Centre. Envoi Pathology. Princess Alexandria Hospital. Austin Hospital. Johns Hopkins Medical Institutes. ARC-Net Centre for Applied Research on Cancer Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature. 2017;551:512–516. doi: 10.1038/nature24462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen S.H., Hung W.C., Wang P., Paul C., Konstantopoulos K. Mesothelin binding to CA125/MUC16 promotes pancreatic cancer cell motility and invasion via MMP-7 activation. Sci. Rep. 2013;3:1870. doi: 10.1038/srep01870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Normanno N., De Luca A., Bianco C., Strizzi L., Mancino M., Maiello M.R., Carotenuto A., De Feo G., Caponigro F., Salomon D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene. 2006;366:2–16. doi: 10.1016/j.gene.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 19.Bailey P., Chang D.K., Nones K., Johns A.L., Patch A.M., Gingras M.C., Miller D.K., Christ A.N., Bruxner T.J.C., Quinn M.C., Australian Pancreatic Cancer Genome Initiative Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47–52. doi: 10.1038/nature16965. [DOI] [PubMed] [Google Scholar]
- 20.Eser S., Reiff N., Messer M., Seidler B., Gottschalk K., Dobler M., Hieber M., Arbeiter A., Klein S., Kong B. Selective requirement of PI3K/PDK1 signaling for Kras oncogene-driven pancreatic cell plasticity and cancer. Cancer Cell. 2013;23:406–420. doi: 10.1016/j.ccr.2013.01.023. [DOI] [PubMed] [Google Scholar]
- 21.Schlieman M.G., Fahy B.N., Ramsamooj R., Beckett L., Bold R.J. Incidence, mechanism and prognostic value of activated AKT in pancreas cancer. Br. J. Cancer. 2003;89:2110–2115. doi: 10.1038/sj.bjc.6601396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamamoto S., Tomita Y., Hoshida Y., Morooka T., Nagano H., Dono K., Umeshita K., Sakon M., Ishikawa O., Ohigashi H. Prognostic significance of activated Akt expression in pancreatic ductal adenocarcinoma. Clin. Cancer Res. 2004;10:2846–2850. doi: 10.1158/1078-0432.ccr-02-1441. [DOI] [PubMed] [Google Scholar]
- 23.Hollingsworth M.A., Swanson B.J. Mucins in cancer: protection and control of the cell surface. Nat. Rev. Cancer. 2004;4:45–60. doi: 10.1038/nrc1251. [DOI] [PubMed] [Google Scholar]
- 24.Schjoldager K.T., Clausen H. Site-specific protein O-glycosylation modulates proprotein processing - deciphering specific functions of the large polypeptide GalNAc-transferase gene family. Biochim. Biophys. Acta. 2012;1820:2079–2094. doi: 10.1016/j.bbagen.2012.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Steentoft C., Vakhrushev S.Y., Vester-Christensen M.B., Schjoldager K.T.-B.G., Kong Y., Bennett E.P., Mandel U., Wandall H., Levery S.B., Clausen H. Mining the O-glycoproteome using zinc-finger nuclease-glycoengineered SimpleCell lines. Nat. Methods. 2011;8:977–982. doi: 10.1038/nmeth.1731. [DOI] [PubMed] [Google Scholar]
- 26.Radhakrishnan P., Grandgenett P.M., Mohr A.M., Bunt S.K., Yu F., Chowdhury S., Hollingsworth M.A. Expression of core 3 synthase in human pancreatic cancer cells suppresses tumor growth and metastasis. Int. J. Cancer. 2013;133:2824–2833. doi: 10.1002/ijc.28322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tsutsumida H., Swanson B.J., Singh P.K., Caffrey T.C., Kitajima S., Goto M., Yonezawa S., Hollingsworth M.A. RNA interference suppression of MUC1 reduces the growth rate and metastatic phenotype of human pancreatic cancer cells. Clin. Cancer Res. 2006;12:2976–2987. doi: 10.1158/1078-0432.CCR-05-1197. [DOI] [PubMed] [Google Scholar]
- 28.Radhakrishnan P., Bryant V.C., Blowers E.C., Rajule R.N., Gautam N., Anwar M.M., Mohr A.M., Grandgenett P.M., Bunt S.K., Arnst J.L. Targeting the NF-κB and mTOR pathways with a quinoxaline urea analog that inhibits IKKβ for pancreas cancer therapy. Clin. Cancer Res. 2013;19:2025–2035. doi: 10.1158/1078-0432.CCR-12-2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Söderberg O., Gullberg M., Jarvius M., Ridderstråle K., Leuchowius K.J., Jarvius J., Wester K., Hydbring P., Bahram F., Larsson L.G., Landegren U. Direct observation of individual endogenous protein complexes in situ by proximity ligation. Nat. Methods. 2006;3:995–1000. doi: 10.1038/nmeth947. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.