

Abstract
Contrasting myelin damage through the generation of new myelinating oligodendrocytes represents a promising approach to promote functional recovery after stroke. Here, we asked whether activation of microglia and monocyte-derived macrophages affects the regenerative process sustained by G protein-coupled receptor 17 (GPR17)-expressing oligodendrocyte precursor cells (OPCs), a subpopulation of OPCs specifically reacting to ischemic injury. GPR17-iCreERT2:CAG-eGFP reporter mice were employed to trace the fate of GPR17-expressing OPCs, labeled by the green fluorescent protein (GFP), after permanent middle cerebral artery occlusion. By microglia/macrophages pharmacological depletion studies, we show that innate immune cells favor GFP+ OPC reaction and limit myelin damage early after injury, whereas they lose their pro-resolving capacity and acquire a dystrophic “senescent-like” phenotype at later stages. Intracerebral infusion of regenerative microglia-derived extracellular vesicles (EVs) restores protective microglia/macrophages functions, limiting their senescence during the post-stroke phase, and enhances the maturation of GFP+ OPCs at lesion borders, resulting in ameliorated neurological functionality. In vitro experiments show that EV-carried transmembrane tumor necrosis factor (tmTNF) mediates the pro-differentiating effects on OPCs, with future implications for regenerative therapies.
Keywords: cerebral ischemia, extracellular vesicles, GPR17 receptor, microglia, oligodendrocyte precursor cells, neuroinflammation, remyelination, tumor necrosis factor, TNF, functional recovery
Graphical abstract
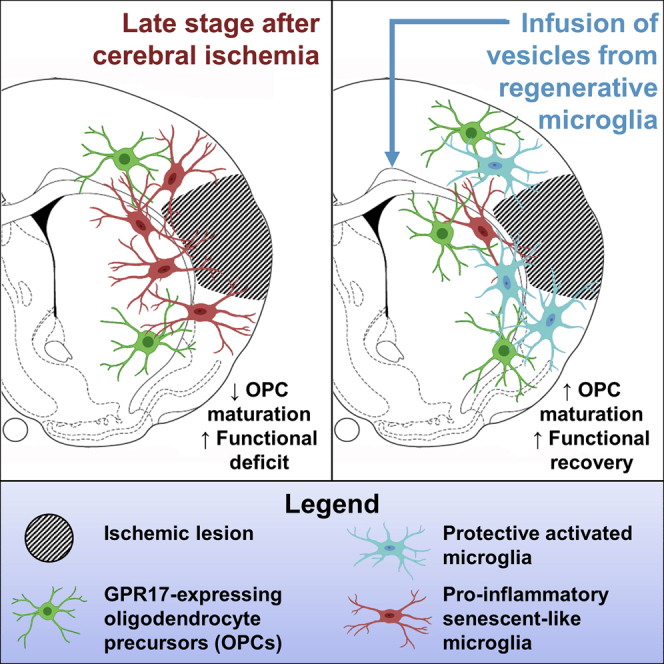
Myelin integrity is crucial for brain repair after injury. Fumagalli and colleagues showed that, early after stroke induction in mice, microglia/macrophages promote myelin reconstruction, but later, they acquire senescent-like traits and lose their pro-resolving function. Infusion of extracellular vesicles from regenerative microglia rescues this dystrophic phenotype and enhances myelin repair, favoring functional recovery.
Introduction
Ischemic stroke is a neurological disease of cardiovascular origin that still remains a leading cause of death and permanent disability worldwide.1 Early ischemic damage is triggered by the interruption of blood supply to a specific area of the brain and affects not only neurons but also oligodendrocytes, the myelin-forming glial cells enwrapping neuronal axons and ensuring impulse transmission. The resulting loss of oligodendrocyte integrity contributes to the axonal degeneration and long-term functional and cognitive deficits observed after stroke.2,3 Current therapy is limited to thrombolysis, which has, however, a narrow treatment window and high risk of cerebral hemorrhage.4 Thus, new strategies aimed at enhancing endogenous myelin repair, through the repopulation of myelin-forming oligodendrocytes, represent promising therapeutic perspectives to improve functional recovery after stroke.5, 6, 7, 8 On top of this, agents or approaches improving recovery in stroke models, like permanent middle cerebral artery occlusion (MCAo), are believed to prove efficacious also in other neurodegenerative conditions characterized by neuronal dysfunction and myelin deterioration associated to neurological and cognitive defects.9
Upon a demyelinating injury, the oligodendrocyte precursor cells (OPCs) present in the adult brain parenchyma migrate to the lesion site, proliferate, and differentiate to myelinating oligodendrocytes.10,11 Specifically, fate-mapping studies using the conditional reporter mouse line GPR17-iCreERT2:CAG-enhanced green fluorescent protein (eGFP) revealed that the subpopulation of OPCs expressing the P2Y-like G protein-coupled receptor 17 (GPR17), which is permanently labeled by GFP upon tamoxifen-induced recombination, constitutes a reserve pool persisting in the adult rodent brain for repair purposes.12 Due to the transient expression of GPR17, that is highest at the pre-oligodendrocyte stage and then disappears in fully differentiated oligodendrocytes,13 these transgenic reporter mice represent the only means to study in vivo the role of GPR17 in OPC differentiation. Tamoxifen-induced recombination in this mouse line indeed results in the permanent labeling of the OPCs that express GPR17 at the very moment of tamoxifen administration; fluorescent labeling persists even after GPR17 downregulation and is also transmitted to cell progeny, thus allowing to follow cells’ terminal maturation.12 By exploiting GPR17-iCreERT2:CAG-eGFP mice, it has been demonstrated that this pool of cells (hereafter referred to as GPR17-expressing or GFP+ OPCs) actively reacts to ischemic damage by increasing their proliferation rate and migratory ability in order to accumulate at injury borders.14 However, the spontaneous maturation capabilities of GFP+ OPCs recruited at the site of damage appear very limited.14 Data coming from experimental models of multiple sclerosis highlight the importance of a permissive local environment for the maturation of this pool of progenitors.15,16 Thus, the remyelination failure observed after the initial reaction of these precursors to ischemic damage may be due to the unfavorable local inflammatory milieu, which is mainly sustained by brain resident microglia and blood-borne macrophages.17 However, the role of microglia/macrophages after stroke is very complex, since these cells are able not only to limit but also to aid repair mechanisms.18 At early stages after ischemic injury, microglia exert protective functions by containing detrimental astrocyte activation,19 by limiting neutrophil infiltration within the lesion,20 and by reducing excitotoxic injury to neurons.21 On the contrary, at later time points, microglia/macrophages acquire a detrimental pro-inflammatory phenotype hindering brain repair processes.18,22 Pharmacological modulation of microglial polarization could influence oligodendrogenesis and OPC differentiation, improving fiber connectivity and long-term functional recovery after stroke.7,8 Furthermore, pro-regenerative microglia have been shown to support remyelination in other experimental models of neurodegenerative disorders, such as multiple sclerosis23 and Alzheimer’s disease.24
Growing evidence indicates novel mechanisms through which immune cells can influence the response of neighboring cells in the brain. Recent studies have attributed to extracellular vesicles (EVs) the capability to mediate intercellular exchanges among brain cells.25 Specifically, microglia utilize EVs to regulate the level of synaptic proteins in recipient neurons,26 to control migration and maturation of OPCs,27 or to propagate inflammatory and degenerative signals in response to tissue damage and disease.28, 29, 30 Of note, the molecular composition and function of microglial EVs reflect the activation state of donor cells.31 Whereas EVs derived from inflammatory microglia have been shown to block remyelination, EVs produced by pro-regenerative cells efficiently promoted myelin repair,27 thus emerging as promising tools to foster regenerative processes following brain injuries. However, how microglia/macrophages orchestrate the response of the pool of GFP+ OPCs following ischemic stroke, as well as the specific impact of microglia-derived EVs in this process, has never been explored.
In this study, we investigated the contribution of microglia/macrophages to the response of GFP+ OPCs after ischemic stroke, using GPR17-iCreERT2:CAG-eGFP reporter mice subjected to MCAo. We demonstrate that microglia/macrophages exert a beneficial action on GFP+ OPCs in the early injury phase after MCAo, whereas their activation becomes detrimental at a later stage. Importantly, we show that infusion of EVs derived from pro-regenerative microglia favors a pro-resolving phenotype and rescues dystrophic, senescent-like traits of resident immune cells, leading to GFP+ OPC differentiation and increased functional recovery. Finally, we provide initial clues of the involvement of transmembrane tumor necrosis factor (tmTNF) in the mechanism underlying the beneficial effect of microglial EVs on OPC maturation.
Results
Activation of microglia/macrophages increases over time after cerebral ischemia
To characterize microglia/macrophage activation in GPR17-iCreERT2:CAG-eGFP reporter mice after cerebral ischemia, we performed immunohistochemistry (IHC) analysis for the microglial/macrophagic marker ionized calcium-binding adapter molecule 1 (Iba1) at ischemic lesion boundaries (0−500 μm from the lesion border) and in the corresponding region of the intact contralateral (contra) hemisphere at days 1, 3, 7, and 14 post-MCAo (Figure 1A). Starting from day 3 post-MCAo, a statistically significant increase in Iba1-expressing (Iba1+) cell density at the boundary of the ischemic lesion was found compared to the corresponding region of the intact contralateral hemisphere (Figures 1B and 1E). Moreover, Iba1+ cells continued to accumulate at injury borders up to day 14 post-MCAo (Figures 1B and 1E). Notably, at both day 3 and day 14 post-MCAo, the majority of Iba1+ cells co-expressed the scavenger receptor CD68 (Figure S1), indicating ongoing phagocytic activity. In particular, at day 3 post-MCAo, CD68 expression was diffused in the whole cell body, whereas at day 14 post-MCAo, CD68 was mainly detectable in defined intracellular structures reminiscent of phagolysosomes, suggesting different phases of the phagocytic process taking place during the early or late stage after ischemia (Figure S1).
Figure 1.

Microglia/macrophage activation at the boundary of the ischemic lesion increases over time after MCAo
(A) Schematic representation of the experimental protocol exploited to study microglial activation following MCAo. (B) Representative images of Iba1+ cells at the boundary of the ischemic lesion (0−500 μm) at days 1, 3, 7, and 14 after MCAo and in the corresponding region of the contralateral hemisphere at day 1 post-MCAo. Scale bars, 50 μm. Magnifications show ischemia-induced modifications of Iba1+ cell morphology; scale bars, 25 μm. (C) Representative images of CD16/32+ cells at the boundary of the ischemic lesion (0−500 μm) at days 1, 3, 7, and 14 after MCAo and in the corresponding region of the contralateral hemisphere at day 1 post-MCAo. Scale bars, 50 μm. (D) Representative images of Ym1+ cells at the boundary of the ischemic lesion (0−500 μm) at days 1, 3, 7, and 14 after MCAo and in the corresponding region of the contralateral hemisphere at day 1 post-MCAo. Scale bars, 50 μm. (E) Quantification of the density of Iba1+ cells at the boundary of the ischemic lesion (0−500 μm) and in the corresponding region of the contralateral hemisphere at days 1, 3, 7, and 14 after MCAo (n = 3). Data are expressed as mean ± SE. Two-way ANOVA (interaction p < 0.0001, time p < 0.0001, MCAo p < 0.0001) followed by Tukey’s post hoc analysis (p values relative to multiple comparisons are reported in the tables). (F) Quantification of the density of Iba1+ cells co-expressing the pro-inflammatory marker CD16/32 at the boundary of the ischemic lesion (0−500 μm) and in the corresponding region of the contralateral hemisphere at day 1, 3, 7, and 14 after MCAo (n = 3). Data are expressed as mean ± SE. Two-way ANOVA (interaction p < 0.0001, time p < 0.0001, MCAo p < 0.0001) followed by Tukey’s post hoc analysis (p values relative to multiple comparisons are reported in the tables). (G) Quantification of the density of Iba1+ cells co-expressing the pro-regenerative marker Ym1 at the boundary of the ischemic lesion (0−500 μm) and in the corresponding region of the contralateral hemisphere at days 1, 3, 7, and 14 after MCAo (n = 3). Data are expressed as mean ± SE. Two-way ANOVA (interaction p = 0.0967, time p = 0.2763, MCAo p < 0.0001) followed by Tukey’s post hoc analysis (p values relative to multiple comparisons are reported in the tables).
Microglia/macrophage activation was also characterized by progressive morphological modifications (Figures 1B and S1), exhibiting a highly ramified, resting structure in the healthy contralateral hemisphere and hypertrophic and ameboid shape in the ipsilateral (ipsi) hemisphere starting from day 3 post-MCAo, reflecting intense inflammatory activity.32 Of note, at day 14 post-MCAo, Iba1+ cells acquired a dystrophic morphology with fragmented cell processes and reduced cell volume (Figures 1B and S1), reminiscent of senescent-like cells.32 Microglia/macrophage activation was paralleled by the increase of GPR17-expressing OPCs, labeled by GFP expression (GFP+ cells), at injury borders (Figure S2), consistent with stroke-induced GFP+ cell proliferation and migration.14 A statistically significant positive correlation between Iba1+ and GFP+ cell densities was found, suggesting a possible functional inter-relationship between these two cell subsets (Figure S2).
Although it is known that microglia/macrophages are able to acquire a broad spectrum of activation states, resulting in a variety of immune phenotypes,33,34 specific cell markers are useful to get information on detrimental phenotype, e.g., Fc gamma receptors CD16/32, and pro-regenerative status, e.g., chitinase 3-like 3, Ym1.35,36 On this basis, we performed Iba1 co-staining with CD16/32 or Ym1 to quantify pro-inflammatory and pro-regenerative cells, respectively. At the border of the ischemic lesion, pro-inflammatory cells co-expressing Iba1 and CD16/32 sharply increased over time, up to day 14 post-MCAo (Figures 1C, 1F, and S3), whereas the number of pro-regenerative cells, double positive for Iba1 and Ym1, after an initial increase at day 3 post-MCAo, remained constant (Figures 1D, 1G, and S3). Interestingly, at day 3 post-MCAo, a considerable fraction of Iba1+ cells co-expressed both CD16/32 and Ym1 (Figure S3).
Taken together, these results delineated two distinct temporal windows in the cellular response following MCAo, namely an early phase (day 3 post-MCAo), when hypertrophic pro-inflammatory and pro-regenerative Iba1+ cells co-exist at the border of the ischemic lesion, and a late stage (day 14 post-MCAo), when dystrophic, senescent-like, pro-inflammatory cells dominate the peri-infarct area.
At early stages after ischemia, microglia/macrophages exert beneficial effects on the response of GFP+ OPCs
Our initial characterization data suggest that microglia/macrophages exert differential effects depending on the phenotype that they acquire at different times of ischemic progression. To evaluate the impact of microglia/macrophages on GFP+ OPC response after MCAo, we exploited a previously described approach based on the administration of gadolinium chloride (GdCl3), a cytotoxic agent capable of specifically inducing apoptosis of phagocytic myeloid cells.23,37 GPR17-iCreERT2:CAG-EeGFP reporter mice were treated with four daily intranasal administrations of GdCl3 starting from 2 h before MCAo (Figure 2A). At day 3 post-MCAo, a partial but statistically significant reduction of Iba1+ cell density was observed at the border of the ischemic lesion in GdCl3-treated animals compared to those receiving vehicle (Figures 2B and 2C), suggesting that this cytotoxin was able to induce a partial microglia/macrophages depletion during the early phase after ischemia. This treatment was equally effective in depleting both Iba1+ and CD16/32+ pro-inflammatory and Iba1+ and Ym1+ pro-regenerative cells (Figures 2B, 2C, and S4). Interestingly, the early partial depletion of microglia/macrophages was paralleled by a significant reduction of GFP+ OPC density, mainly affecting early precursors co-expressing the neural/glial antigen 2 (NG2) at the border of the ischemic lesion (Figures 2B, 2D, and S5). The percentage of GFP+/Olig2+ on the total Olig2+ oligodendroglial population is, instead, the same in mice treated with vehicle or GdCl3 (Figure S5), excluding variations in GFP expression. Of note, the decrease of early GFP+ OPC density may be due to impaired microglia-dependent proliferation of these cells, as also suggested by the smaller percentage of GFP+ OPCs incorporating 5-bromo-2′-deoxyuridine (BrdU), observed in GdCl3-treated animals as compared to the vehicle group (Figures 2B and 2D). Accordingly, a positive linear correlation between the density of Iba1+ and GFP+ cells was found at this time point (Figure 2E). Importantly, no changes in propidium iodide (PI)-stained cells were detected after exposure of primary OPC cultures to GdCl3 (Figure S5), thus excluding direct oligo-toxicity of this compound. In addition, damage to myelin structure was exacerbated upon the partial depletion of microglia/macrophages during the early phase after MCAo, as indicated by an increased g-ratio and reduction of both myelin thickness and myelinated axon density in the corpus callosum of GdCl3-treated animals as compared to vehicle-treated littermates (Figures 2F and 2G). Conversely, no significant changes in axon diameter were detected between vehicle and GdCl3 groups (Figures 2F and 2G), excluding a direct effect of early microglia/macrophages reduction also on axonal integrity. Taken together, these results suggest that, during the early phase after cerebral ischemia, microglia/macrophages are beneficial for the response of GFP+ OPCs and contribute to limiting myelin damage.
Figure 2.

Partial depletion of microglia/macrophages in the early phase after MCAo impairs GPR17-expressing (GFP+) OPC response and exacerbates myelin damage
(A) Schematic representation of the experimental protocol exploited for early depletion of microglia/macrophages after MCAo. (B) Representative images of cells stained for Iba1, CD16/32, Ym1, GFP, and GFP/BrdU at the boundary of the ischemic lesion (0−500 μm) at day 3 post-MCAo following intranasal administration of GdCl3 or vehicle. Arrowheads indicate cells double positive for GFP and BrdU. Scale bars, 50 μm. (C) Quantification of the density of total Iba1+, Iba1+ and CD16/32+, and Iba1+ and Ym1+ cells at the boundary of the ischemic lesion (0−500 μm) at day 3 post-MCAo following intranasal administration of GdCl3 or vehicle (n = 4). Data are expressed as mean ± SE. Student’s t test. (D) Quantification of the density of GFP+ OPCs and of the percentage of GFP+ cells incorporating BrdU at the boundary of the ischemic lesion (0−500 μm) at day 3 post-MCAo following intranasal administration of GdCl3 or vehicle (n = 4). Data are expressed as mean ± SE. Student’s t test. (E) Scatterplot representation of the linear correlation between the densities of Iba1+ cells (x axis) and GFP+ OPCs (y axis) at the boundary of the ischemic lesion (0−500 μm) at day 3 post-MCAo. Green dots correspond to GdCl3-treated animals, whereas blue dots represent vehicle-treated animals. For correlation analysis, two-tailed Pearson test was used. (F) Representative electron micrographs showing myelinated axons in the ipsilateral corpus callosum (CC) of ischemic mice at day 3 post-MCAo following intranasal administration of GdCl3 or vehicle and in the corresponding region of the contralateral hemisphere of vehicle-treated animals. Scale bars, 1 μm. (G) Quantification of g-ratio, myelin thickness, axon diameter, and myelinated axon density in the ipsilateral corpus callosum of ischemic mice at day 3 post-MCAo following intranasal administration of GdCl3 or vehicle and in the corresponding region of the contralateral hemisphere of vehicle-treated animals (n = 3; 300 fibers/experimental condition have been analyzed). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis.
At later stages after ischemia, the partial depletion of microglia/macrophages increases the response of GFP+ OPCs but has no impact on their maturation
To achieve microglia/macrophage depletion at later stages after cerebral ischemia, when pro-inflammatory cells dramatically increase at injury borders, GPR17-iCreERT2:CAG-EGFP reporter mice received four daily doses of GdCl3, starting from day 14 post-MCAo (Figure 3A). At day 17 post-MCAo, GdCl3 treatment was able to partially but significantly reduce Iba1+ cell density in the ischemic penumbra (Figures 3B and 3C). At this time point, GdCl3 treatment depleted only Iba1+ and CD16/32+ pro-inflammatory microglia/macrophages, whereas Iba1+ and Ym1+ pro-regenerative cells were not affected (Figures 3B, 3C, and S4). Under these conditions, we found a statistically significant increase in the density of GFP+ OPCs at the border of the ischemic lesion, compared to vehicle-treated animals (Figures 3B and 3D), and a negative correlation between the densities of Iba1+ and GFP+ cells (Figure 3E). It is worth noting that, despite that strong, reactive astrogliosis is commonly reported after microglia depletion approaches,19,38 no changes in glial fibrillary acidic protein (GFAP) immunoreactivity were detected following GdCl3 administration at both early and late stages after MCAo (Figure S5). To analyze the impact of the late partial depletion of microglia/macrophages on long-term GFP+ OPC differentiation, an additional experimental group was sacrificed at day 42 post-MCAo, 4 weeks after treatment with GdCl3 or vehicle (Figure 3A). Results show that the increase in the density of GFP+ OPCs was maintained in GdCl3-treated animals also at this time point but that the differentiation capability of these cells did not improve, as no differences in the percentage of GFP+ cells co-expressing the mature oligodendroglial marker glutathione S-transferase (GST)π were detected (Figures 3F and 3G). Accordingly, myelin density in the ipsilateral corpus callosum was not affected by late microglia/macrophages depletion (Figures 3H and 3I). These results indicate that, at late stages after MCAo, the pharmacological depletion of pro-inflammatory Iba1+ cells could improve the capacity of GFP+ OPCs to stack at injury borders but was not able to ameliorate their terminal maturation.
Figure 3.

Partial depletion of microglia/macrophages at the late stage after MCAo promotes a GFP+ OPC response but has no impact on their maturation
(A) Schematic representation of the experimental protocol exploited for late depletion of microglia/macrophages after MCAo. (B) Representative images of cells stained for Iba1, CD16/32, Ym1, and GFP at the boundary of the ischemic lesion (0−500 μm) at day 17 post-MCAo following intranasal administration of GdCl3 or vehicle. Scale bars, 50 μm. (C) Quantification of the density of total Iba1+, Iba1+ and CD16/32+, and Iba1+ and Ym1+ cells at the boundary of the ischemic lesion (0−500 μm) at day 17 post-MCAo following intranasal administration of GdCl3 or vehicle (n = 5). Data are expressed as mean ± SE. Student’s t test. (D) Quantification of the density of GFP+ OPCs at the boundary of the ischemic lesion (0−500 μm) at day 17 post-MCAo following intranasal administration of GdCl3 or vehicle (n = 6). Data are expressed as mean ± SE. Student’s t test. (E) Scatterplot representation of the linear correlation between the densities of Iba1+ cells (x axis) and GFP+ OPCs (y axis) at the boundary of the ischemic lesion (0−500 μm) at day 17 post-MCAo. Green dots correspond to GdCl3-treated animals, whereas blue dots represent vehicle-treated animals. For correlation analysis, two-tailed Pearson test was used. (F) Representative images of cells stained for GFP and GSTπ at the boundary of the ischemic lesion (0−500 μm) at day 42 post-MCAo following intranasal administration of GdCl3 or vehicle. Scale bars, 50 μm. Magnifications show cells co-expressing GFP and GSTπ: scale bars, 25 μm. (G) Quantification of the density of total GFP+ OPCs and GFP+ and GSTπ+ cells at the boundary of the ischemic lesion (0−500 μm) at day 42 post-MCAo following intranasal administration of GdCl3 or vehicle (n = 5−6). Data are expressed as mean ± SE. Student’s t test. (H) Representative images of myelin visualized using FluoroMyelin Red stain in the corpus callosum (delimited by white dashed lines) at the boundary of the ischemic lesion (0−500 μm) at day 42 post-MCAo, following intranasal administration of GdCl3 or vehicle. Scale bars, 50 μm. (I) Quantification of the percentage of FluoroMyelin+ area in the corpus callosum at the boundary of the ischemic lesion (0−500 μm) at day 42 post-MCAo, following intranasal administration of GdCl3 or vehicle (n = 5−6). Data are expressed as mean ± SE and normalized to vehicle set to 100.
Infusion of microglia-derived EVs modifies the phenotype of microglia/macrophages at lesion borders
Based on the data described above, we reasoned that reprogramming dystrophic and pro-inflammatory microglia/macrophages could be useful to enhance tissue remodeling and functional recovery after cerebral ischemia. Very relevant, microglial EVs have been shown to modify the phenotype and morphology of recipient microglia/macrophages in vivo,28 with different final effects depending on the activation state of the donor cells.39,40 On this basis, EVs collected from in vitro-manipulated pro-regenerative (interleukin [IL]-4 EVs) or pro-inflammatory (i-EVs) microglia were infused into the ipsilateral corpus callosum of GPR17-iCreERT2:CAG-EGFP reporter mice through osmotic minipumps, starting from day 14 post-MCAo and allowing chronic delivery for 1 week (Figures 4A and S6). The tunable-resistive pulse sensing (TRPS) technique showed no statistically significant differences in EV concentration and size distribution between i-EVs and IL-4 EVs (Figure S6). IHC analysis was performed at day 28 post-MCAo to evaluate the impact of the infusion of microglial EVs on resident microglia/macrophages in the peri-infarct region. Results show no variations in global Iba1+ cell density after infusion of IL-4 EVs, i-EVs, or vehicle alone (Figures 4B−4D). A slight but significative increase was detected in the density of pro-inflammatory cells co-expressing Iba1 and CD16/32 after infusion of i-EVs as compared to vehicle administration (Figures 4B and 4D). The density of Iba1+ and Ym1+ pro-regenerative microglia/macrophages was instead found to be higher after infusion of IL-4 EVs compared to i-EVs and vehicle treatment (Figures 4C and 4D). Moreover, morphological analysis revealed that both IL-4 EVs and i-EVs induced prominent modifications of Iba1+ cell branching when compared to vehicle-treated animals. Namely, an increase in the number of branchpoints and ramification index (Figure 4E), indicative of a recovery from a dystrophic shape to a functional one,32 was observed. In addition, exposure to IL-4 EVs resulted in markedly increased Iba1+ cell volume and surveilled cell territory compared to vehicle and i-EVs (Figure 4E), which is coherent with a switch toward pro-resolving functions. Globally, these data suggest that IL-4 EV infusion had a significant impact on microglia/macrophages at injury boundaries, promoting their pro-regenerative activation.
Figure 4.

Infusion of pro-regenerative microglia-derived EVs at late stages after ischemia promotes a beneficial polarization of microglia/macrophages
(A) Schematic representation of the experimental protocol exploited for the infusion of microglia-derived EVs after MCAo. (B) Representative images of cells stained for Iba1 and CD16/32 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. (C) Representative images of cells stained for Iba1 and Ym1 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bar, 50 μm. (D) Quantification of the density of total Iba1+, Iba1+ and CD16/32+, and Iba1+ and Ym1+ cells at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (n = 4). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis. (E) Quantification of Iba1+ microglia/macrophage number of branchpoints, ramification index, cell volume, and cell territory at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (130−150 cells from 3−4 animals/experimental condition have been analyzed). Data are expressed as mean ± SE. Kruskal-Wallis test followed by Dunn’s post hoc analysis.
Infusion of pro-regenerative microglia-derived EVs promotes GFP+ OPC maturation after cerebral ischemia
In addition to the aforementioned effects of microglial EVs on Iba1+ cells, our recent evidence indicates that EVs produced by microglia are also able to directly influence OPCs surrounding myelin lesions.27 Thus, we analyzed the impact of microglia-derived EV infusion on the differentiation of GFP+ OPCs in the peri-infarct area.
At day 28 post-MCAo, IL-4 EVs were able to increase the density of GFP+ cells in the ischemic penumbra when compared to animals receiving either i-EVs or vehicle alone (Figures 5A and 5B). The amount of GFP+ OPCs co-expressing the early differentiation marker NG2 did not differ among all of the experimental conditions (Figures 5C and 5D), whereas the percentages of GFP+ cells co-expressing GPR17 (indicative of a pre-oligodendrocyte stage) or the mature myelinating oligodendrocyte marker GSTπ were significantly increased after IL-4 EV infusion compared to vehicle or i-EV conditions (Figures 5E−5H). Furthermore, the administration of IL-4 EVs resulted in a significant increase of FluoroMyelin stain in the ipsilateral corpus callosum of ischemic mice (Figures 5I and 5J), indicating that efficient myelin repair occurred. These results suggest that the infusion of microglial IL-4 EVs at late stages after MCAo was able to promote a selective response to ischemic damage of the pool of OPCs expressing GPR17 and to foster their terminal maturation, contributing to enhanced remyelination.
Figure 5.

Infusion of pro-regenerative microglia-derived EVs at late stages after ischemia enhances GFP+ OPC differentiation
(A) Representative images of GFP+ OPCs at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. (B) Quantification of the density of GFP+ OPCs at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (n = 4−9). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis. (C) Representative images of cells stained for GFP and NG2 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. Magnifications show cells co-expressing GFP and NG2: scale bars, 25 μm. (D) Quantification of the percentage of GFP+ OPCs co-expressing NG2 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (n = 4−9). Data are expressed as mean ± SE. (E) Representative images of cells stained for GFP and GPR17 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. Magnifications show cells co-expressing GFP and GPR17: scale bars, 25 μm. (F) Quantification of the percentage of GFP+ OPCs co-expressing GPR17 at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (n = 3−4). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis. (G) Representative images of cells stained for GFP and GSTπ at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. Magnifications show cells co-expressing GFP and GSTπ: scale bars, 25 μm. (H) Quantification of the percentage of GFP+ OPCs co-expressing GSTπ at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs or vehicle (n = 4−9). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis. (I) Representative images of myelin visualized using FluoroMyelin Red stain in the corpus callosum (delimited by white dashed lines) at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle. Scale bars, 50 μm. (J) Quantification of the percentage of FluoroMyelin+ area in the corpus callosum at the boundary of the ischemic lesion (0−500 μm) at day 28 post-MCAo, following infusion of i-EVs, IL-4 EVs, or vehicle (n = 4). Data are expressed as mean ± SE and normalized to vehicle set to 100. One-way ANOVA followed by Tukey’s post hoc analysis.
Infusion of pro-regenerative microglia-derived EVs fosters functional recovery of ischemic mice
The most invalidating consequences of cerebral ischemia are the long-lasting persistence of cognitive and motor disability in patients.41,42 However, a reliable analysis of these deficits is difficult to be implemented in ischemic mice, due to the extremely high rate of spontaneous recovery and to compensatory mechanisms involving the intact contralateral hemisphere, which occur within the first week after ischemia, thus making long-term evaluations very challenging.43 In this respect, one important feature of stroke-induced deficit is commonly referred to as “neglect” and consists in the inability to perceive stimuli coming from the region contralateral to the ischemic hemisphere, with important consequences for spatial cognition and explorative behaviors.44 Of note, such a defect is maintained in rodent models of cerebral ischemia, manifesting as a turning preference toward the ipsilateral side, with a consequent reduction in the percentage of contralateral turns.43
To evaluate the impact of IL-4 EVs on the turning preference of ischemic mice, a Y-maze test45 was performed at day 14 post-MCAo, before minipump implantation, and at day 28 post-MCAo, after IL-4 EVs or vehicle infusion (Figures 6A and 6B). As expected, at day 14 post-MCAo, ischemic animals displayed a significative reduction in the percentage of contralateral turns as compared to sham-operated controls (Figure 6C). No significant differences in locomotor activity and spontaneous alternation were detected at day 14 post-MCAo between ischemic mice and sham-operated controls (Figures 6D and 6E). Interestingly, at day 28 post-MCAo, vehicle-treated animals still maintained a significant impairment of contralateral turns, whereas in mice receiving IL-4 EVs, a reversion of turning preference was observed, which restored functionality up to that observed in sham-operated controls (Figure 6F). Also at this time point, no changes in locomotor activity and spontaneous alternation among the three experimental groups were recorded (Figures 6G and 6H).
Figure 6.

Infusion of pro-regenerative microglia-derived EVs at late stages after ischemia promotes functional recovery of ischemic mice
(A) Schematic representation of the experimental protocol exploited for behavioral analysis before and after the infusion of microglia-derived EVs after MCAo. (B) Schematic representation of the Y-maze test to evaluate the turning preference of ischemic mice. (C) Quantification of the percentage of contralateral turns made in the Y-maze test at day 14 post-MCAo by MCAo mice (n = 14) and sham-operated controls (n = 5). Data are expressed as mean ± SE. Student’s t test. (D) Quantification of the locomotor activity of MCAo mice (n = 14) and sham-operated controls (n = 5) during the Y-maze test at day 14 post-MCAo. Data are expressed as mean ± SE. (E) Quantification of the percentage of spontaneous alternations made in the Y-maze test at day 14 post-MCAo by MCAo mice (n = 14) and sham-operated controls (n = 5). Data are expressed as mean ± SE. (F) Quantification of the percentage of contralateral turns made in the Y-maze test at day 28 post-MCAo by sham-operated controls (n = 5) and MCAo mice after infusion of IL-4 EVs (n = 7) or vehicle (n = 7). Data are expressed as mean ± SE. One-way ANOVA followed by Tukey’s post hoc analysis. (G) Quantification of the locomotor activity of sham-operated controls (n = 5) and MCAo mice after infusion of IL-4 EVs (n = 7) or vehicle (n = 7) during the Y-maze test at day 28 post-MCAo. Data are expressed as mean ± SE. (H) Quantification of the percentage of spontaneous alternations made in the Y-maze test at day 28 post-MCAo by sham-operated controls (n = 5) and MCAo mice after infusion of IL-4 EVs (n = 7) or vehicle (n = 7). Data are expressed as mean ± SE. (I) Representative images of NeuN+ tissue at day 28 post-MCAo, following infusion of IL-4 EVs or vehicle. Green dashed lines delineate the area of NeuN staining in the ipsilateral hemisphere. White dashed lines correspond to the projection of the NeuN+ area in the intact contralateral hemisphere. Scale bars, 1 mm. (J) Representative images of the ischemic region in hematoxylin-eosin (H&E) stained sections at day 28 post-MCAo, following infusion of IL-4 EVs or vehicle. Black dashed lines delineate the ischemic core. Scale bars, 400 μm. (K) Quantification of the percentage of NeuN+ tissue loss at day 28 post-MCAo after infusion of IL-4 EVs or vehicle (n = 5). Data are expressed as mean ± SE. Student’s t test. (L) Quantification of the percentage of H&E-labeled tissue loss at day 28 post-MCAo after infusion of IL-4 EVs or vehicle (n = 5). Data are expressed as mean ± SE. Student’s t test.
In addition, at day 28 post-MCAo, infusion of IL-4 EVs resulted in a significant reduction of the percentage of ischemia-induced neuronal tissue loss, mainly affecting the motor cortex, as compared to vehicle-treated mice (Figures 6I−6L), possibly contributing to the functional amelioration observed.
tmTNF is involved in the beneficial effects exerted by microglial EVs on OPC maturation
To explore the direct effects of IL-4 EVs on the pool of GFP+ cells, OPCs were isolated from GPR17-iCreERT2:CAG-EeGFP mice and exposed in vitro to either IL-4 EVs or medium alone (CTRL) for 72 h.
Notably, after in vitro exposure to 4-hydroxytamoxifen (OH-TAM), no significant differences were detected in the percentage of GFP+ cells between CTRL and IL-4 EV-treated cultures (Figures 7A and 7B). Immunofluorescence analysis showed that IL-4 EVs significantly increased the percentage of mature myelin basic protein-expressing (MBP+) cells compared to CTRL (Figures 7A−7C), demonstrating a direct pro-differentiating effect of IL-4 EVs. Moreover, the fraction of GFP+ cells co-expressing MBP was significantly higher in cultures exposed to IL-4 EVs with respect to CTRL, whereas no significant changes were observed in the GFP negative pool of progenitors (Figure 7D). This suggests that the subpopulation of GFP+ OPCs, namely the GPR17-expressing pool of cells,12 is indeed more prone to differentiate and more responsive to the pro-differentiating action of microglial EVs. Accordingly, in both CTRL and IL-4 EV conditions, the percentage of MBP+ cells was significantly higher in the GFP+ OPC subset as compared to the GFP negative one (Figure 7D).
Figure 7.

Direct effects of microglial IL-4 EVs on GFP+ OPC maturation in vitro
(A) Representative images showing cells expressing GFP and MBP in primary OPC cultures from GPR17-iCreERT2:CAG-EGFP mice exposed to IL-4 EVs or medium alone (CTRL). Scale bars, 50 μm. (B) Quantification of the percentage of GFP+ cells in primary OPC cultures from GPR17-iCreERT2:CAG-EGFP mice exposed to IL-4 EVs or CTRL. Data are expressed as mean ± SE (n = 9 coverslips from 3 independent experiments). (C) Quantification of the total percentage of MBP+ cells in primary OPC cultures from GPR17-iCreERT2:CAG-EGFP mice exposed to IL-4 EVs or CTRL. Data are expressed as mean ± SE (n = 9 coverslips from 3 independent experiments). Student’s t test. (D) Quantification of the percentage of GFP+ and GFPneg cells co-expressing MBP in primary OPC cultures from GPR17-iCreERT2:CAG-EGFP mice exposed to IL-4 EVs or CTRL. Data are expressed as mean ± SE (n = 9 coverslips from 3 independent experiments). One-way ANOVA followed by Tukey’s post hoc analysis. (E) Representative images showing MBP+ cells in primary OPC cultures exposed to IL-4 EVs or CTRL in the presence or absence of the selective solTNF inhibitor XPro1595 (XPro) and of the nonselective TNF inhibitor etanercept (ETN). Scale bars, 50 μm. (F) Quantification of the total percentage of MBP+ cells in primary OPC cultures exposed to IL-4 EVs or CTRL in the presence or absence of XPro or ETN. Data are expressed as mean ± SE (n = 9 coverslips from 3 independent experiments). One-way ANOVA followed by Tukey’s post hoc analysis.
Recent evidence indicates that TNF is increased in EVs derived from activated microglia46 and macrophages.47 In particular, EVs derived from these myeloid cells are enriched in the transmembrane form (tmTNF) of the cytokine,47 which promotes oligodendrocyte differentiation and remyelination both in vitro and in vivo via activation of TNFR2 receptor,48 suggesting that TNF may play a role in the pro-differentiating effects of IL-4 EVs. To address this issue, primary OPCs were exposed to IL-4 EVs in the presence of the selective soluble TNF (solTNF) inhibitor XPro1595 (XPro) or the nonselective solTNF and tmTNF blocker etanercept (ETN). Interestingly, whereas XPro did not inhibit the increase in the percentage of MBP+ cells induced by IL-4 EVs, ETN counteracted the pro-differentiating action of IL-4 EVs (Figures 7E and 7F). These data provide concrete clues of a possible involvement of the tmTNF/TNFR2 axis in the mechanism underlying the beneficial effect of microglial EVs on OPC maturation.
Discussion
Despite representing only a subset of all Olig2+ oligodendrocyte lineage cells,12 the subpopulation of OPCs expressing the GPR17 receptor (indicated as GFP+ cells in this study) has attracted great interest for its capacity to rapidly respond to injury and trigger reparative mechanisms after cerebral ischemia14 or upon other neurodegenerative conditions.15,16 However, the spontaneous differentiation capability of this pool of progenitors is often counteracted by local inflammation, which makes their pro-myelinating attempts ineffective.14, 15, 16 Our previous data have suggested the importance of locally released factors in addressing the reparative function of the GFP+ cells, but the type of cells and mechanisms at the basis of this in situ regulation were unclear. Here, we uncover the pivotal role of the crosstalk between microglia/macrophages and GFP+ OPCs in shaping brain repair and functional recovery after ischemic stroke.
We demonstrate that Iba1+ immune cells exert a beneficial action on the pool of GFP+ OPCs in the early injury phase, whereas their pro-resolving properties are lost at late disease stages. The change in Iba1+ effector cell function is clearly reflected by morphological alterations, with microglia/macrophages acquiring a hypertrophic and ameboid shape in the early post-ischemic phase and then becoming dystrophic at late stages. Furthermore, we show that EVs collected from microglia cultured with a pro-regenerative stimulus are able to both counteract the late ineffective phenotype of resident immune cells and foster the differentiation of GFP+ OPCs to mature functional cells, leading to increased neurological recovery. By in vitro experiments, we provide evidence for a direct beneficial effect exerted by microglial EVs on the pool of GFP+ OPCs and highlight EV-carried tmTNF as a putative pivotal player in mediating microglia-to-OPC communication.
Early after ischemia, microglia/macrophages enhance OPC density and limit myelin damage
Although it is known that neuroinflammation mediated by peripherally derived macrophages and their CNS resident counterpart microglia greatly influences neuronal network activity in the ischemic-injured brain,21,49 its exact impact on oligodendrocytes during the acute and chronic phases after ischemic stroke still remains unclear. This is a key point to be addressed, considering the importance of myelin repair in neurological recovery11 and the potential of immune cells (particularly microglia) in supporting myelin regeneration in different neurological disorders, such as multiple sclerosis.50,51
By IHC for phenotypic markers and pharmacological immune cell depletion, we show that Iba1+ cells are rapidly activated after stroke, with a persistent response evolving over time. At early stages after stroke, pro-inflammatory and pro-regenerative Iba1+ cells co-exist and are committed to a protective function, with a large fraction of cells co-expressing both inflammatory and pro-resolving markers. The presence of this mixed phenotype is in agreement with recent evidence displacing the canonical dichotomous classification of microglia activation in favor of a higher diversity of functional states52 and may help to explain the lack of selectivity of GdCl3 treatment (which was previously shown to specifically target pro-inflammatory cells23,37) observed at the early stage after ischemia. On the contrary, at late phases of the disease, the pro-inflammatory component gradually dominates the site of injury. This is in line with previous observations by other groups.22,53,54 Specifically, the activation of local microglia was suggested to precede and dominate over the infiltration of peripheral macrophages during the first days post-stroke, when they assume a protective phenotype,18 having them a limited capacity to acquire a pro-inflammatory state.55 Conversely, blood-derived macrophages were reported to contribute to delayed post-ischemic inflammation and injury,56,57 as their depletion reduces brain atrophy volume and neurological deficits 14 days after transient ischemia.58 However, infiltrating macrophages were also shown to be essential for maintaining a pro-regenerative anti-inflammatory polarization of Iba1+ immune cells early after ischemic brain injury, therefore contributing to the endogenous protective events at this early time point.22,35 Although our study did not discriminate between microglia and blood-borne macrophages, results from our depletion experiments highlight a novel protective action of both immune cells early after ischemia, namely, to sustain the rapid reaction of GFP+ OPCs at the lesion site14 and to limit myelin damage. Indeed, early after GdCl3-mediated immune cell depletion, the number of GFP+ OPCs was significantly reduced, in line with a role for microglia/macrophages in promoting OPC proliferation,23 as also supported by our data showing a reduced percentage of GFP+ OPCs incorporating BrdU in GdCl3-treated mice. However, since previous results also demonstrated a positive impact of immune cells on OPC migration,27 it cannot be completely excluded that a lack of chemoattractive molecules released by microglia/macrophages might contribute to the impaired recruitment of GFP+ cells observed. The reduction of both myelin thickness and myelinated axon density observed in GdCl3-treated animals further indicated the critical contribution of these immune cells in preserving myelin integrity at early times after ischemia.
At late stages after ischemic injury, Iba1+ immune cells lose their pro-regenerative capacity
In addition to the data above, we show that, at later stages after MCAo, depletion of pro-inflammatory Iba1+ immune cells was paralleled by an increase in GFP+ OPC density, suggesting that chronicization of the pro-inflammatory response of Iba1+ cells potentiates damage progression at delayed times after ischemic injury.18,59 Despite this effect, no improvement of oligodendroglial differentiation was detected in these conditions, indicating that the remaining microglial/macrophagic cells did not retain their ability to efficiently sustain OPC maturation. In line with this consideration, at later stages after ischemia, Iba1+ cell morphology appeared to be dystrophic, resembling that of senescent cells during physiological aging or chronic neurodegenerative conditions.32 Dystrophic morphology of microglia/macrophages has supposed to have been induced by their overstimulation and has been frequently associated with impaired capacity of these cells to exert pro-regenerative and neuroprotective functions.32 Hence, to promote structural and functional recovery at late stages after stroke, therapeutic approaches should be aimed at selectively restoring microglia/macrophage regenerative effector functions rather than simply depleting these myeloid cells, which inevitably leads to the loss of their beneficial effects.
An EV-based strategy to prevent immune cell detrimental function and foster differentiation of GFP+ OPCs
Among the strategies able to redirect microglia/macrophages toward beneficial functions,60 microglial EVs recently emerged as a promising approach able to modulate the phenotype of recipient myeloid cells in experimental models of neurological disorder.28,39,40 Consistently, here, we showed that microglial EVs are able to induce significant modifications in recipient Iba1+ cells in the ischemic penumbra, with different responses depending on the activation state of donor cells as previously demonstrated.30,40,61 Importantly, EVs derived from pro-regenerative microglia were able to restore the pro-resolving activities of recipient microglia/macrophages at late stages after ischemia, thus contributing to create the permissive local environment required for efficient repair processes, including oligodendroglial maturation and remyelination.16,51 This is in line with our recent evidence that EVs collected from pro-regenerative microglia have the capacity, when infused in vivo, to enhance the endogenous reparative response of OPCs at focal myelin lesions.27 The present study significantly extends these findings, by demonstrating that IL-4 EVs specifically stimulate the response of GFP+ OPCs at ischemic injury borders and markedly rescued neurological deficits in ischemic mice.
Several issues remain to be explored, including how EVs are taken up by recipient cells62 and what the molecular mechanisms are by which they exert their pro-differentiating effects on OPCs. Intriguingly, our data suggest that TNF, previously shown to be enriched in EVs derived from activated microglia,27,46 may, at least in part, contribute to the pro-differentiating effects of IL-4 EVs by activating oligodendroglial TNFR2.48,63 This is in line with very recent results showing that release of TNF by microglia in response to demyelinating injury is essential to start myelin repair processes.64 Nevertheless, the lipid cargo of microglial EVs was also demonstrated to enhance OPC differentiation,27 suggesting that a combination of factors, rather than a single molecule, might be responsible for EV-mediated beneficial effects.
Taken together, the data presented in this study advance our understanding of the complex interaction between microglia/macrophages and OPCs after stroke and pave the way for developing EV-based strategies65 aimed at implementing myelin repair and tissue recovery after stroke.
Materials and methods
Animals and experimental procedures
The procedures concerning animal care, surgery, and sacrifice were performed in accordance with national (D.L. n.26, 2014) and international laws and policies (EU Directive 2010/63/EU) and approved and authorized by the National Ministry of Health-University of Milan Committee (approval number 868/2016-PR). Moreover, all protocols used were in accordance with ARRIVE guidelines. GPR17-iCreERT2:CAG-EGFP reporter mice14 have been used, bred, and housed at the animal facility of the Department of Pharmacological and Biomolecular Sciences in Milan. After genotyping, performed on DNA extracts from tail clippings as described,12 11-week-old, double-transgenic GPR17-iCreERT2:CAG-EGFP mice received 10 mg tamoxifen (Sigma-Aldrich, Taufkirchen, Germany), dissolved in 10% ethanol and 90% corn oil, three times by gavage, once every second day to induce recombination. After 3 weeks of washout, mice were anesthetized with ketamine (80 mg/kg) and xylazine (16 mg/kg) and underwent permanent MCAo as previously described.14,66 Briefly, a vertical midline skin incision was made between the right orbit and ear. The temporal muscle was excised, and the oblique edge of a drill bit was used to thin an approximate 1-mm circle of bone overlying the MCA. The thinned bone and the dura mater were incised with a 29-gauge needle tip, and the right MCA was exposed and permanently occluded by means of microbipolar coagulation (SAMED MB122), just proximal to the origin of the olfactory branch. Sham-operated mice underwent the same surgical procedures with the exception of artery occlusion. The retracted temporalis muscle was allowed to fall back into place and sutured. The animals were then allowed to recover from anesthesia. Male and female mice have been equally employed in all of the experiments. The mortality rate we observed in the MCAo procedure was 10%. For microglia characterization, mice were sacrificed at days 1, 3, 7, and 14 post-MCAo (n = 3 mice for each time point), whereas for microglia/macrophage depletion and EV infusion experiments, mice were sacrificed at days 17, 28, and 42 post-MCAo (n = 4−9 mice for each time point; see details below).
Depletion of microglia/macrophage cell populations
To achieve microglia/macrophages depletion, we chose a strategy based on the administration of GdCl3.23,37 Briefly, GPR17-iCreERT2:CAG-EGFP mice received daily intranasal doses of GdCl3 (10 mg/kg/day; Sigma-Aldrich, Taufkirchen, Germany), dissolved in water, added with 40% PEG300 or vehicle. A first group of mice have been treated starting from 2 h before MCAo until day 3 after the surgery (early depletion). A second group of mice received GdCl3 from day 14 to day 17 post-MCAo (late depletion). Both groups were sacrificed immediately at the end of the treatment. Finally, a third group of animals have been exposed to the late depletion protocol and sacrificed at day 42 (n = 5 mice per experimental condition). To evaluate GFP+ OPC proliferation, animals included in the early depletion experiments also received daily intraperitoneal injections of the DNA synthesis marker BrdU (Sigma-Aldrich, Taufkirchen, Germany), dissolved in saline (50 mg/kg/day), starting from 4 h after MCAo until sacrifice.
Primary microglia cultures
Mixed glial cell cultures, containing both astrocytes and microglia, were established from C57BL/6 wild-type mice (P2) (Charles River, Lecco, Italy) and maintained for 10 days in the presence of South American fetal bovine serum (Life Technologies, Monza, Italy) that optimizes microglia expansion. Microglia were harvested by orbital shaking for 40 min at 1,300 rpm and re-plated on poly-l-ornithine-coated tissue-culture dishes (50 μg/mL; Sigma-Aldrich, Taufkirchen, Germany). To minimize the activation, pure microglia (>98%)26 were kept for 24 h in low-serum (1%) medium. Cells were then stimulated with a cocktail of T helper cell type 1 (Th1) cytokines, i.e., 20 ng/mL IL-1β (PeproTech, Milan, Italy), 20 ng/mL TNF (PeproTech, Milan, Italy), and 25 ng/mL interferon (IFN)-γ (Sigma-Aldrich, Taufkirchen, Germany) or with 20 ng/ml IL-4 (R&D, Milan, Italy) for 48 h. At the end of treatment, microglia were washed and stimulated with ATP to increase EV production, as described.26 Primary murine microglia (MG) were maintained in the absence of stimuli (NS), under inflammatory (i-MG) or pro-regenerative (IL-4 MG) conditions for 48 h. Real-time PCR have been performed, as follows, to check the expression of pro-inflammatory and pro-regenerative markers (Figure S6).
RNA isolation and quantification
After ATP stimulation, cell supernatant was collected for EV isolation (see below), and donor cells were lysed with TRIzol reagent (Life Technologies, Monza, Italy). Total RNA was extracted using Direct-zol RNA Micro-Prep (Zymo Research, Irvine, CA, USA), according to the manufacturer’s instructions. RNA was then pretreated with RQ1 DNase (Promega, Milan, Italy) for eliminating genomic DNA contamination. Retrotranscription of 400 ng RNA was performed with the SensiFAST cDNA synthesis kit (Bioline, London, UK). For real-time PCR, several mixes were prepared according to the number of interested genes. Each mix included Master Mix 2× (Life Technologies, Monza, Italy), 250 nM probe (Arg1 Mm00475988_m1; Ym1 Mm00657889_mH; IL-1β Mm00434228_m1; TNF Mm00443258_m1; inducible nitric oxide synthase [iNOS] Mm00440502_m1; Rpl13a Mm05910660_g1; Life Technologies, Monza, Italy), and 20 ng of cDNA. Gene expression was analyzed with the TaqMan Gene Expression Assay and normalized to housekeeping gene Rpl13a expression using the CFX96 real-time PCR system (Bio-Rad, Segrate, Italy) following the manufacturer’s protocol. Data are presented as mean of log2 (fold change) ± SE.
Isolation and quantification of microglia-derived EVs
To isolate EVs, polarized microglia were stimulated with 1 mM ATP for 30 min in KRH (125 mM NaCl, 5 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO, 2 mM CaCl2, 6 mM d-glucose, and 25 mM HEPES/NaOH, pH 7.4). The culture supernatant was collected, and EVs were pelleted at 100,000 × g after preclearing from cells and debris at 300 × g, as previously described.67 EV pellets were resuspended and used immediately after isolation. EV size and concentration were measured by TRPS technique, using an Izon qNano instrument (Izon, Christchurch, New Zealand). EVs produced by 1 × 106 microglia in 1 h were resuspended in a volume of 100 μL. A reagent kit from Izon (Izon EV Reagent Kit) was used for both pretreating the pore and suspending EVs in order to prevent EV binding to the pore or spontaneous EV aggregation. NP300 nanopore (Izon, Christchurch, New Zealand) was employed, and CPC200 (carboxylated polystyrene particles, supplied by Izon and diluted following the manufacturer’s instructions) was used as standards. In each experiment, the same applied voltage, pressure, and pore stretch values were set for all sample and calibration particle recordings. Data acquisition and analysis were performed using Izon Control Suite software (version [v.]3.3).
EV intracerebral infusion
At day 14 post-MCAo, mice received infusion of IL-4 EVs (n = 9), i-EVs (n = 4), or vehicle (n = 9). Briefly, approximately 2 × 108 EVs, produced by 1.5 × 106 microglia and dissolved in 150 μL of sterile saline, were infused in the ipsilateral corpus callosum of ischemic mice (coordinates from bregma: 1.0 mm rostral, 1.0 mm lateral, 2 mm deep) using osmotic mini-pumps (1007D equipped with Brain Infusion Kit 3; Alzet, Cupertino, CA, USA) over 7 days at 0.5 μL/h delivery rate. To limit EV degradation, minipumps were filled with freshly isolated EVs and implanted within a few hours. With the use of this method, stability of microglial EVs into the minipumps has been reported for more than 4 days, and the presence of injected GFP-labeled EVs within the tissue, as well as their interaction with both microglia and OPCs at the injury site, has been previously demonstrated.27 Vehicle-treated animals were obtained with delivery of saline solution alone. Mice were sacrificed at day 28 post-MCAo.
Behavioral assessment
To evaluate persistent asymmetries in mice turning preference before and after infusion of IL-4 EVs, the Y-maze test was performed.45 Briefly, animals have been positioned in the middle of a Y-shaped maze, consisting of three plastic arms at a 120° angle from each other, and left free to explore for 8 min, during which, the number and sequence of entries in each arm have been manually recorded by an operator blinded to the experimental groups. It has been considered an entry when all four limbs were within the new arm, and one spontaneous alternation has been counted when the animal entered all three different arms consecutively. Locomotor activity (total number of entries in a new arm), percent alternation (number of spontaneous alternations/number of total triads × 100), and percent of contralateral turns (number of contralateral turns when entering a new arm/number of total turns × 100) have been calculated. First, stroke-induced alterations have been assessed on ischemic mice (MCAo; n = 14) at day 14 post-MCAo, before EV administration. As controls, sham-operated mice (n = 5), undergoing the same surgical procedures of MCAo mice with exception of artery occlusion, have been utilized. Immediately after this first behavioral evaluation, MCAo animals have been randomly assigned using the RANDBETWEEN function of Excel (Microsoft, Redmond, WA, USA) to infusion of IL-4 EVs (n = 7) or vehicle (n = 7), as described above. Analysis of variance (ANOVA), followed by Helmert contrast analysis, excluded any bias in the assignment of MCAo mice to vehicle or IL-4 EV group (p > 0.05). At day 28 post-MCAo, the Y-maze test has been repeated to evaluate the impact of IL-4 EV infusion on mice-turning preference with respect to both vehicle-treated animals and sham-operated controls.
IHC analysis
At the day of the sacrifice, after anesthesia with ketamine (80 mg/kg) and xylazine (16 mg/kg), mice were perfused with 0.01 M phosphate-buffered saline (PBS) and then 4% paraformaldehyde in 0.01 M PBS for at least 25 min. Brains were removed, post-fixed 1 h in the same solution, and then cryoprotected in 30% sucrose solution until precipitation at 4°C. Coronal sections of 20 μm thickness have been collected using a microtome and incubated with the following primary antibodies: chicken anti-GFP (1:1400; Aves Labs, Tigard, OR, USA), rat anti-BrdU (1:200; Abcam, Cambridge, UK), rat anti-CD68 (1:400; Bio-Rad, Segrate, Italy), rat anti-CD16/CD32 (1:200; BD Biosciences, Milan, Italy), rabbit anti-GSTπ (1:500; MBL International, Woburn, MA, USA), rabbit anti-Iba1 (1:30,000; Wako, Japan), rabbit anti-NeuN (1:100; Cell Signaling Technologies, Danvers, MA, USA), rabbit anti-NG2 (1:2,000; Millipore, Milan, Italy), rabbit anti-GFAP (1:500; Dako, Glostrup, Denmark), rabbit anti-GPR17 (1:2,500; custom antibody produced by Primm, Milan, Italy),68 and rabbit anti-Ym1 (1:100; STEMCELL Technologies, Cambridge, UK). Incubation with primary antibodies was made overnight at 4°C in 0.01 M PBS with 1% normal goat serum (Dako, Glostrup, Denmark) and 0.3% Triton X-100. The sections were exposed for 2 h at room temperature to the corresponding secondary antibodies (all goat) conjugated with Alexa Fluor 488, 555, or 633 (1:600; Life Technologies, Monza, Italy). For rabbit anti-NG2, GPR17, and Iba1, the signal intensity was enhanced using the High Sensitivity Tyramide Signal Amplification kit (PerkinElmer, Monza, Italy) following the manufacturer’s instructions. For double-GFP/BrdU labeling, staining of BrdU was performed last, after fixing sections with 4% paraformaldehyde for 10 min and incubating them with HCl 2 M for 45 min at 37°C. Nuclei were labeled with Hoechst 33258 (0.06 μg/mL; Life Technologies, Monza, Italy). For myelin staining, FluoroMyelin Red stain (1:300; Life Technologies, Monza, Italy) has been used following the manufacturer’s instructions. For the quantitative analysis, the peri-infarct region (0−500 μm from the boundary of the ischemic lesion) and its corresponding area in the contralateral hemisphere have been considered. Images were acquired at ×20 magnification (647.04 × 647.04 μm) using a confocal microscope (merge of 6 μm z stack at 2 μm intervals; A1R; Nikon, Tokyo, Japan). Iba1 staining was performed using 633 fluorophore, and the false color green has been applied in representative images to better display co-localization with other markers in red. To visualize brain damage, hematoxylin-eosin (H&E; Sigma-Aldrich, Taufkirchen, Germany) staining has been performed and images acquired by means of a slide scanner (NanoZoomer S60; Hamamatsu Photonics, Hamamatsu City, Japan). The number of cells that were GFP-positive; double-positive GFP and NG2, GFP and GPR17, GFP and GSTπ, GFP and BrdU, Iba1 and CD16/32, Iba1 and Ym1, and Iba1 and CD68; and triple-positive Iba1 and CD16/32 and Ym1; the densitometric analysis of GFAP and FluoroMyelin staining; and the area of NeuN+- and H&E-stained tissue were manually quantified by a blinded investigator, using the software Fiji/ImageJ, on three slices per mouse taken from −1.00 to 0.00 mm from bregma. For analysis of tissue loss, the area of NeuN or H&E staining has been manually measured for each slice, both in the ipsilateral hemisphere and in the intact contralateral one, and the percentage of tissue loss has been calculated as % tissue loss = (area contralateral – area ipsilateral)/area contralateral × 100.
Morphological analysis of microglia/macrophages
Analysis of microglia/macrophage cell morphology was carried out on 20 μm-thick brain sections stained for Iba1.69 For characterization, images were acquired at ischemic injury borders at day 3 and day 14 post-MCAo and in the corresponding region of the contralateral hemisphere at day 3 post-MCAo. To evaluate the impact of EV infusion on Iba1+ cell morphology, images were acquired at day 28 post-MCAo at lesion boundaries in animals treated with IL-4 EVs, i-EVs, or vehicle. Analysis was carried out by exploiting the semi-automatic MATLAB-based tool 3DMorph.70 130−150 cells from 3−4 animals for each experimental condition have been analyzed. Among morphological parameters considered were number of branchpoints and ramification index, calculated as cell surface area/4π × ((3 × cell volume)/(4π))2/3 and gaining score 1 for round-shaped cells and higher values as cell ramification increases, both reflecting the complexity of cell branching. Moreover, we analyzed cell volume, which gives indications about the intensity of cell activation, and cell territory, which is proportional to cell volume and to the number and the length of cell processes and is indicative of the brain area surveilled by each individual cell.
Transmission electron microscopy (TEM)
To perform TEM analysis, mice receiving early intranasal administration of GdCl3 or vehicle (n = 3) were transcardially perfused with 2% paraformaldehyde and 2.5% glutaraldehyde in 0.15 M sodium cacodylate buffer (pH 7.4; Sigma-Aldrich, Taufkirchen, Germany). Then, tissues were post-fixed for an additional 24 h at 4°C, and processed as described.71 Briefly, coronal sections of 100 μm thickness were collected using a vibratome, and selected areas of the ipsilateral corpus callosum adjacent to the ischemic damage, together with the corresponding regions of the contralateral hemisphere, were manually dissected using a stereoscope. Samples were post-fixed in 2% osmium tetroxide, stained using 1% uranyl acetate for 45 min, dehydrated, and embedded in Spurr’s epoxy resin baked at 60°C for 48 h. Finally, thin sections (70 nm) were cut using a Leica EM UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany), stained with a saturated solution of uranyl acetate in 20% ethanol and 1% lead citrate, and grids were observed using a Philips CM10 TEM (FEI, Eindhoven, Netherland). For quantitative analysis, images at ×13,500 magnification were taken and g-ratio, myelin thickness, axon diameter, and myelinated axon density were calculated on 300 axons for each experimental condition using the software Fiji/ImageJ as described.27
Primary OPC cultures
Primary OPCs were isolated from total brain of GPR17-iCreERT2:CAG-EGFP mice at postnatal day 2 (P2). Briefly, brains were dissected from pups and maintained at 4°C in Tissue Storage Solution (Miltenyi Biotec, Bologna, Italy) for 3 h during genotyping (performed on tail biopsies as described above). Only brains deriving from double-transgenic GPR17-iCreERT2:CAG-EGFP mice were pulled together and dissociated into single-cell suspensions using the Papain-based Neural Tissue Dissociation Kit (NTDK; Miltenyi Biotec, Bologna, Italy). Then, platelet-derived growth factor receptor (PDGFR)α+ OPCs were sorted by magnetic-activated cell sorting (MACS) separation after incubation with anti-PDGFRα magnetic microbeads (Miltenyi Biotec, Bologna, Italy) following the manufacturer’s instructions. PDGFRα+ cells (approximately 250,000 cells obtained from each pup) have been cultured on poly-DL-ornithine (Sigma-Aldrich, Taufkirchen, Germany)-coated 24-well plates (30,000 cells/well) in OPC proliferation medium containing Neurobasal (Life Technologies, Monza, Italy), 2% B27 (Life Technologies, Monza, Italy), 1% L-glutamine (Euroclone, Pero, Italy), 1% penicillin/streptomycin (Euroclone, Pero, Italy), 10 ng/mL PDGF-AA (Sigma-Aldrich, Taufkirchen, Germany), 10 ng/mL fibroblast growth factor (FGF)2 (Space Import Export, Milan, Italy), and 1 μM 4-hydroxitamoxifen (to induce GFP expression; Sigma-Aldrich, Taufkirchen, Germany). After 2 days, cells were switched to differentiation medium containing DMEM (Euroclone, Pero, Italy), 1% N-2 supplement (Life Technologies, Monza, Italy), 2% B27, 0.01% BSA (Sigma-Aldrich, Taufkirchen, Germany), 1% L-glutamine, 1% penicillin/streptomycin, and 10 ng/mL T3 (Sigma-Aldrich, Taufkirchen, Germany), and 4 h later, CTRL or IL-4 EVs were added to OPC culture (1:2 OPC-microglia ratio) in the presence or absence of the nonselective TNF inhibitor ETN (200 ng/mL; Wyeth, Madison, NJ, USA) or XPro (200 ng/mL; Xencor, Monrovia, CA, USA), a selective solTNF inhibitor.72 Cells have been differentiated for 3 days and then fixed at room temperature with 4% paraformaldehyde (Sigma-Aldrich, Taufkirchen, Germany) in 0.01 M PBS containing 0.12 M sucrose (Sigma-Aldrich, Taufkirchen, Germany) for immunocytochemistry. Under these culture conditions, contaminating astrocytes and microglia were routinely less than 1% each. To evaluate GdCl3 toxicity in vitro, primary OPCs have been cultured in proliferating conditions in the presence of GdCl3 concentrations demonstrated to be toxic for microglial cultures, i.e., 270 μM and 1 mM,73 or vehicle alone (CTRL) for 72 h. Cells were then fixed as described above and processed for immunocytochemistry.
Immunocytochemistry
Labeling was performed incubating cells overnight at 4°C with the following primary antibodies diluted in goat serum dilution buffer (GSDB; 450 mM NaCl [Sigma-Aldrich, Taufkirchen, Germany], 20 mM sodium phosphate buffer, pH 7.4, 15% goat serum [Life Technologies, Monza, Italy], and 0.3% Triton X-100 [Sigma-Aldrich, Taufkirchen, Germany]): rat anti-MBP (1:200; Millipore, Milan, Italy) and chicken anti-GFP (1:1,400; Aves Labs, Tigard, OR, USA). Cells were then incubated for 1 h at room temperature with the following secondary antibodies: goat anti-rat conjugated to Alexa Fluor 555 and goat anti-chicken conjugated to Alexa Fluor 488 (1:600 in GSDB; Life Technologies, Monza, Italy). In GdCl3-treated cultures, OPC viability has been assessed by incubating cells for 15 min at room temperature with PI (1 μg/mL; Sigma-Aldrich, Taufkirchen, Germany). Nuclei were labeled with Hoechst 33258 (0.3 μg/mL; Life Technologies, Monza, Italy). Coverslips were finally mounted with a fluorescent mounting medium (Dako, Glostrup, Denmark) and analyzed using an inverted fluorescence microscope (200 M; Zeiss, Jena, Germany) connected to a PC equipped with AxioVision software (Zeiss, Jena, Germany). For cell counts, 30 fields were acquired at ×20 magnification (0.07 mm2/field; three coverslips for each experimental condition deriving from three independent experiments). Analysis was performed using Fiji/ImageJ software.
Statistical analysis
For each experimental procedure, the proper sample size has been calculated by means of G∗Power 3.1 software,74 estimating effect size and standard deviation based on previously published data14,23,27,45 and fixing the alpha value (type 1 error) at the level of 5% (p = 0.05) and the power at 80%.
Data are expressed as mean ± standard error (SE). Statistical analysis was performed using Prism 7 software (GraphPad, San Diego, CA, USA). Gaussian distribution of the values in each experimental group has been assessed using Shapiro-Wilk (for n < 8) or D’Agostino-Pearson (for n > 8) normality tests. For all comparisons between two groups with a normal distribution, unpaired Student’s t test was performed, whereas for groups without normal distribution, nonparametric Mann-Whitney test was used. For multiple comparison testing, depending on the experimental design, one-way or two-way ANOVA, accompanied by Tukey’s post hoc test, was used for groups with normal distribution, whereas nonparametric Kruskal-Wallis test, followed by Dunn’s post hoc analysis, was performed when normal distribution of the values could not be assumed. For correlation analysis, two-tailed Pearson test was used. Differences were considered significant for p value <0.05.
Data availability
Further information and data that support the findings of this study are available from the corresponding author upon reasonable request.
Acknowledgments
We are grateful to Prof. Roberta Brambilla (University of Miami, Miami, FL, USA) for fruitful discussion and Dr. David E. Szymkowski (Xencor, Monrovia, CA, USA) for providing XPro1595. We thank Prof. Flavia Antonucci (University of Milan, Milan, Italy) for help with the Y-maze apparatus, Dr. Norma Lattuada (University of Milan, Milan, Italy) for technical support with electron microscopy analysis, and Dr. Martina Gabrielli (CNR, Institute of Neuroscience, Milan, Italy) for assistance with characterization of EVs. Part of this work was carried out at NOLIMITS, an advanced imaging facility established by Università degli Studi di Milano. This study was supported by Fondazione Cariplo, Italy (grant number 2015-0910 to M.F.), Università degli Studi di Milano (Piano di Sostegno alla Ricerca, 2015-2017 to M.F.), and by Italian Ministry of University and Research-MIUR project “Dipartimenti di Eccellenza 2018-2022” to Department of Pharmacological and Biomolecular Sciences.
Author contributions
M.F. conceived and supervised the project. S.R., P.G., M.P.A., C.V., and M.F. designed experiments. S.R., P.G., E.B., L.C., and M.L. carried out experiments, data acquisition, and analysis. M.C. performed brain ischemic surgery. S.R., P.G., L.S., M.P.A., C.V., and M.F. contributed to data interpretation and discussion. S.R. and M.F. wrote the original draft. All coauthors critically revised and edited the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.009.
Supplemental information
References
- 1.Benjamin E.J., Muntner P., Alonso A., Bittencourt M.S., Callaway C.W., Carson A.P., Chamberlain A.M., Chang A.R., Cheng S., Das S.R. Heart Disease and Stroke Statistics−2019 Update: A Report From the American Heart Association. Circulation. 2019;139:e56–e528. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Zhao L.-R., Willing A. Enhancing endogenous capacity to repair a stroke-damaged brain: An evolving field for stroke research. Prog. Neurobiol. 2018;163-164:5–26. doi: 10.1016/j.pneurobio.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rost N.S., Cougo P., Lorenzano S., Li H., Cloonan L., Bouts M.J.R.J., Lauer A., Etherton M.R., Karadeli H.H., Musolino P.L. Diffuse microvascular dysfunction and loss of white matter integrity predict poor outcomes in patients with acute ischemic stroke. J. Cereb. Blood Flow Metab. 2018;38:75–86. doi: 10.1177/0271678X17706449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marshall R.S. Progress in intravenous thrombolytic therapy for acute stroke. JAMA Neurol. 2015;72:928–934. doi: 10.1001/jamaneurol.2015.0835. [DOI] [PubMed] [Google Scholar]
- 5.Duncan I.D., Brower A., Kondo Y., Curlee J.F., Jr., Schultz R.D. Extensive remyelination of the CNS leads to functional recovery. Proc. Natl. Acad. Sci. USA. 2009;106:6832–6836. doi: 10.1073/pnas.0812500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang R., Chopp M., Zhang Z.G. Oligodendrogenesis after cerebral ischemia. Front. Cell. Neurosci. 2013;7:201. doi: 10.3389/fncel.2013.00201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han L., Cai W., Mao L., Liu J., Li P., Leak R.K., Xu Y., Hu X., Chen J. Rosiglitazone Promotes White Matter Integrity and Long-Term Functional Recovery After Focal Cerebral Ischemia. Stroke. 2015;46:2628–2636. doi: 10.1161/STROKEAHA.115.010091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gelosa P., Bonfanti E., Castiglioni L., Delgado-Garcia J.M., Gruart A., Fontana L., Gotti M., Tremoli E., Lecca D., Fumagalli M. Improvement of fiber connectivity and functional recovery after stroke by montelukast, an available and safe anti-asthmatic drug. Pharmacol. Res. 2019;142:223–236. doi: 10.1016/j.phrs.2019.02.025. [DOI] [PubMed] [Google Scholar]
- 9.Kremer D., Göttle P., Hartung H.-P., Küry P. Pushing Forward: Remyelination as the New Frontier in CNS Diseases. Trends Neurosci. 2016;39:246–263. doi: 10.1016/j.tins.2016.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Crawford A.H., Tripathi R.B., Richardson W.D., Franklin R.J.M. Developmental Origin of Oligodendrocyte Lineage Cells Determines Response to Demyelination and Susceptibility to Age-Associated Functional Decline. Cell Rep. 2016;15:761–773. doi: 10.1016/j.celrep.2016.03.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Franklin R.J.M., Ffrench-Constant C. Regenerating CNS myelin - from mechanisms to experimental medicines. Nat. Rev. Neurosci. 2017;18:753–769. doi: 10.1038/nrn.2017.136. [DOI] [PubMed] [Google Scholar]
- 12.Viganò F., Schneider S., Cimino M., Bonfanti E., Gelosa P., Sironi L., Abbracchio M.P., Dimou L. GPR17 expressing NG2-Glia: Oligodendrocyte progenitors serving as a reserve pool after injury. Glia. 2016;64:287–299. doi: 10.1002/glia.22929. [DOI] [PubMed] [Google Scholar]
- 13.Fumagalli M., Daniele S., Lecca D., Lee P.R., Parravicini C., Fields R.D., Rosa P., Antonucci F., Verderio C., Trincavelli M.L. Phenotypic changes, signaling pathway, and functional correlates of GPR17-expressing neural precursor cells during oligodendrocyte differentiation. J. Biol. Chem. 2011;286:10593–10604. doi: 10.1074/jbc.M110.162867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonfanti E., Gelosa P., Fumagalli M., Dimou L., Viganò F., Tremoli E., Cimino M., Sironi L., Abbracchio M.P. The role of oligodendrocyte precursor cells expressing the GPR17 receptor in brain remodeling after stroke. Cell Death Dis. 2017;8:e2871. doi: 10.1038/cddis.2017.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppolino G.T., Marangon D., Negri C., Menichetti G., Fumagalli M., Gelosa P., Dimou L., Furlan R., Lecca D., Abbracchio M.P. Differential local tissue permissiveness influences the final fate of GPR17-expressing oligodendrocyte precursors in two distinct models of demyelination. Glia. 2018;66:1118–1130. doi: 10.1002/glia.23305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lecca D., Raffaele S., Abbracchio M.P., Fumagalli M. Regulation and signaling of the GPR17 receptor in oligodendroglial cells. Glia. 2020;68:1957–1967. doi: 10.1002/glia.23807. [DOI] [PubMed] [Google Scholar]
- 17.Iadecola C., Anrather J. The immunology of stroke: from mechanisms to translation. Nat. Med. 2011;17:796–808. doi: 10.1038/nm.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hu X., Li P., Guo Y., Wang H., Leak R.K., Chen S., Gao Y., Chen J. Microglia/macrophage polarization dynamics reveal novel mechanism of injury expansion after focal cerebral ischemia. Stroke. 2012;43:3063–3070. doi: 10.1161/STROKEAHA.112.659656. [DOI] [PubMed] [Google Scholar]
- 19.Jin W.N., Shi S.X.Y., Li Z., Li M., Wood K., Gonzales R.J., Liu Q. Depletion of microglia exacerbates postischemic inflammation and brain injury. J. Cereb. Blood Flow Metab. 2017;37:2224–2236. doi: 10.1177/0271678X17694185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otxoa-de-Amezaga A., Miró-Mur F., Pedragosa J., Gallizioli M., Justicia C., Gaja-Capdevila N., Ruíz-Jaen F., Salas-Perdomo A., Bosch A., Calvo M. Microglial cell loss after ischemic stroke favors brain neutrophil accumulation. Acta Neuropathol. 2019;137:321–341. doi: 10.1007/s00401-018-1954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Szalay G., Martinecz B., Lénárt N., Környei Z., Orsolits B., Judák L., Császár E., Fekete R., West B.L., Katona G. Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat. Commun. 2016;7:11499. doi: 10.1038/ncomms11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajan W.D., Wojtas B., Gielniewski B., Gieryng A., Zawadzka M., Kaminska B. Dissecting functional phenotypes of microglia and macrophages in the rat brain after transient cerebral ischemia. Glia. 2019;67:232–245. doi: 10.1002/glia.23536. [DOI] [PubMed] [Google Scholar]
- 23.Miron V.E., Boyd A., Zhao J.W., Yuen T.J., Ruckh J.M., Shadrach J.L., van Wijngaarden P., Wagers A.J., Williams A., Franklin R.J.M., Ffrench-Constant C. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat. Neurosci. 2013;16:1211–1218. doi: 10.1038/nn.3469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behrendt G., Baer K., Buffo A., Curtis M.A., Faull R.L., Rees M.I., Götz M., Dimou L. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia. 2013;61:273–286. doi: 10.1002/glia.22432. [DOI] [PubMed] [Google Scholar]
- 25.Budnik V., Ruiz-Cañada C., Wendler F. Extracellular vesicles round off communication in the nervous system. Nat. Rev. Neurosci. 2016;17:160–172. doi: 10.1038/nrn.2015.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Prada I., Gabrielli M., Turola E., Iorio A., D’Arrigo G., Parolisi R., De Luca M., Pacifici M., Bastoni M., Lombardi M. Glia-to-neuron transfer of miRNAs via extracellular vesicles: a new mechanism underlying inflammation-induced synaptic alterations. Acta Neuropathol. 2018;135:529–550. doi: 10.1007/s00401-017-1803-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lombardi M., Parolisi R., Scaroni F., Bonfanti E., Gualerzi A., Gabrielli M., Kerlero de Rosbo N., Uccelli A., Giussani P., Viani P. Detrimental and protective action of microglial extracellular vesicles on myelin lesions: astrocyte involvement in remyelination failure. Acta Neuropathol. 2019;138:987–1012. doi: 10.1007/s00401-019-02049-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verderio C., Muzio L., Turola E., Bergami A., Novellino L., Ruffini F., Riganti L., Corradini I., Francolini M., Garzetti L. Myeloid microvesicles are a marker and therapeutic target for neuroinflammation. Ann. Neurol. 2012;72:610–624. doi: 10.1002/ana.23627. [DOI] [PubMed] [Google Scholar]
- 29.Joshi P., Turola E., Ruiz A., Bergami A., Libera D.D., Benussi L., Giussani P., Magnani G., Comi G., Legname G. Microglia convert aggregated amyloid-β into neurotoxic forms through the shedding of microvesicles. Cell Death Differ. 2014;21:582–593. doi: 10.1038/cdd.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nigro A., Colombo F., Casella G., Finardi A., Verderio C., Furlan R. Myeloid Extracellular Vesicles: Messengers from the Demented Brain. Front. Immunol. 2016;7:17. doi: 10.3389/fimmu.2016.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drago F., Lombardi M., Prada I., Gabrielli M., Joshi P., Cojoc D., Franck J., Fournier I., Vizioli J., Verderio C. ATP Modifies the Proteome of Extracellular Vesicles Released by Microglia and Influences Their Action on Astrocytes. Front. Pharmacol. 2017;8:910. doi: 10.3389/fphar.2017.00910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savage J.C., Carrier M., Tremblay M.È. Morphology of Microglia Across Contexts of Health and Disease. Methods Mol. Biol. 2019;2034:13–26. doi: 10.1007/978-1-4939-9658-2_2. [DOI] [PubMed] [Google Scholar]
- 33.Ransohoff R.M., Perry V.H. Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 2009;27:119–145. doi: 10.1146/annurev.immunol.021908.132528. [DOI] [PubMed] [Google Scholar]
- 34.Colonna M., Butovsky O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017;35:441–468. doi: 10.1146/annurev-immunol-051116-052358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perego C., Fumagalli S., Zanier E.R., Carlino E., Panini N., Erba E., De Simoni M.G. Macrophages are essential for maintaining a M2 protective response early after ischemic brain injury. Neurobiol. Dis. 2016;96:284–293. doi: 10.1016/j.nbd.2016.09.017. [DOI] [PubMed] [Google Scholar]
- 36.Villa A., Gelosa P., Castiglioni L., Cimino M., Rizzi N., Pepe G., Lolli F., Marcello E., Sironi L., Vegeto E., Maggi A. Sex-Specific Features of Microglia from Adult Mice. Cell Rep. 2018;23:3501–3511. doi: 10.1016/j.celrep.2018.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Van Steenwinckel J., Schang A.-L., Krishnan M.L., Degos V., Delahaye-Duriez A., Bokobza C., Csaba Z., Verdonk F., Montané A., Sigaut S. Decreased microglial Wnt/β-catenin signalling drives microglial pro-inflammatory activation in the developing brain. Brain. 2019;142:3806–3833. doi: 10.1093/brain/awz319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wylot B., Mieczkowski J., Niedziolka S., Kaminska B., Zawadzka M. Csf1 Deficiency Dysregulates Glial Responses to Demyelination and Disturbs CNS White Matter Remyelination. Cells. 2019;9:99. doi: 10.3390/cells9010099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Casella G., Colombo F., Finardi A., Descamps H., Ill-Raga G., Spinelli A., Podini P., Bastoni M., Martino G., Muzio L., Furlan R. Extracellular Vesicles Containing IL-4 Modulate Neuroinflammation in a Mouse Model of Multiple Sclerosis. Mol. Ther. 2018;26:2107–2118. doi: 10.1016/j.ymthe.2018.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grimaldi A., Serpe C., Chece G., Nigro V., Sarra A., Ruzicka B., Relucenti M., Familiari G., Ruocco G., Pascucci G.R. Microglia-Derived Microvesicles Affect Microglia Phenotype in Glioma. Front. Cell. Neurosci. 2019;13:41. doi: 10.3389/fncel.2019.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stinear C. Prediction of recovery of motor function after stroke. Lancet Neurol. 2010;9:1228–1232. doi: 10.1016/S1474-4422(10)70247-7. [DOI] [PubMed] [Google Scholar]
- 42.Stinear C.M. Prediction of motor recovery after stroke: advances in biomarkers. Lancet Neurol. 2017;16:826–836. doi: 10.1016/S1474-4422(17)30283-1. [DOI] [PubMed] [Google Scholar]
- 43.Balkaya M.G., Trueman R.C., Boltze J., Corbett D., Jolkkonen J. Behavioral outcome measures to improve experimental stroke research. Behav. Brain Res. 2018;352:161–171. doi: 10.1016/j.bbr.2017.07.039. [DOI] [PubMed] [Google Scholar]
- 44.Vallar G., Calzolari E. Unilateral spatial neglect after posterior parietal damage. Handb. Clin. Neurol. 2018;151:287–312. doi: 10.1016/B978-0-444-63622-5.00014-0. [DOI] [PubMed] [Google Scholar]
- 45.Saadoun S., Waters P., Bell B.A., Vincent A., Verkman A.S., Papadopoulos M.C. Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice. Brain. 2010;133:349–361. doi: 10.1093/brain/awp309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang Y., Boza-Serrano A., Dunning C.J.R., Clausen B.H., Lambertsen K.L., Deierborg T. Inflammation leads to distinct populations of extracellular vesicles from microglia. J. Neuroinflammation. 2018;15:168. doi: 10.1186/s12974-018-1204-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Soni S., O’Dea K.P., Tan Y.Y., Cho K., Abe E., Romano R., Cui J., Ma D., Sarathchandra P., Wilson M.R., Takata M. ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J. 2019;33:6442–6455. doi: 10.1096/fj.201802386R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madsen P.M., Motti D., Karmally S., Szymkowski D.E., Lambertsen K.L., Bethea J.R., Brambilla R. Oligodendroglial TNFR2 mediates membrane TNF-dependent repair in experimental autoimmune encephalomyelitis by promoting oligodendrocyte differentiation and remyelination. J. Neurosci. 2016;36:5128–5143. doi: 10.1523/JNEUROSCI.0211-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kronenberg G., Uhlemann R., Richter N., Klempin F., Wegner S., Staerck L., Wolf S., Uckert W., Kettenmann H., Endres M., Gertz K. Distinguishing features of microglia- and monocyte-derived macrophages after stroke. Acta Neuropathol. 2018;135:551–568. doi: 10.1007/s00401-017-1795-6. [DOI] [PubMed] [Google Scholar]
- 50.Davies C.L., Miron V.E. Distinct origins, gene expression and function of microglia and monocyte-derived macrophages in CNS myelin injury and regeneration. Clin. Immunol. 2018;189:57–62. doi: 10.1016/j.clim.2016.06.016. [DOI] [PubMed] [Google Scholar]
- 51.Lloyd A.F., Miron V.E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 2019;15:447–458. doi: 10.1038/s41582-019-0184-2. [DOI] [PubMed] [Google Scholar]
- 52.Stratoulias V., Venero J.L., Tremblay M.È., Joseph B. Microglial subtypes: diversity within the microglial community. EMBO J. 2019;38:e101997. doi: 10.15252/embj.2019101997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Perego C., Fumagalli S., De Simoni M.G. Temporal pattern of expression and colocalization of microglia/macrophage phenotype markers following brain ischemic injury in mice. J. Neuroinflammation. 2011;8:174. doi: 10.1186/1742-2094-8-174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Peruzzotti-Jametti L., Donegá M., Giusto E., Mallucci G., Marchetti B., Pluchino S. The role of the immune system in central nervous system plasticity after acute injury. Neuroscience. 2014;283:210–221. doi: 10.1016/j.neuroscience.2014.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Durafourt B.A., Moore C.S., Zammit D.A., Johnson T.A., Zaguia F., Guiot M.C., Bar-Or A., Antel J.P. Comparison of polarization properties of human adult microglia and blood-derived macrophages. Glia. 2012;60:717–727. doi: 10.1002/glia.22298. [DOI] [PubMed] [Google Scholar]
- 56.Dimitrijevic O.B., Stamatovic S.M., Keep R.F., Andjelkovic A.V. Absence of the chemokine receptor CCR2 protects against cerebral ischemia/reperfusion injury in mice. Stroke. 2007;38:1345–1353. doi: 10.1161/01.STR.0000259709.16654.8f. [DOI] [PubMed] [Google Scholar]
- 57.Breckwoldt M.O., Chen J.W., Stangenberg L., Aikawa E., Rodriguez E., Qiu S., Moskowitz M.A., Weissleder R. Tracking the inflammatory response in stroke in vivo by sensing the enzyme myeloperoxidase. Proc. Natl. Acad. Sci. USA. 2008;105:18584–18589. doi: 10.1073/pnas.0803945105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ma Y., Li Y., Jiang L., Wang L., Jiang Z., Wang Y., Zhang Z., Yang G.Y. Macrophage depletion reduced brain injury following middle cerebral artery occlusion in mice. J. Neuroinflammation. 2016;13:38. doi: 10.1186/s12974-016-0504-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.An C., Shi Y., Li P., Hu X., Gan Y., Stetler R.A., Leak R.K., Gao Y., Sun B.L., Zheng P., Chen J. Molecular dialogs between the ischemic brain and the peripheral immune system: dualistic roles in injury and repair. Prog. Neurobiol. 2014;115:6–24. doi: 10.1016/j.pneurobio.2013.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fumagalli M., Lombardi M., Gressens P., Verderio C. How to reprogram microglia toward beneficial functions. Glia. 2018;66:2531–2549. doi: 10.1002/glia.23484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Garzetti L., Menon R., Finardi A., Bergami A., Sica A., Martino G., Comi G., Verderio C., Farina C., Furlan R. Activated macrophages release microvesicles containing polarized M1 or M2 mRNAs. J. Leukoc. Biol. 2014;95:817–825. doi: 10.1189/jlb.0913485. [DOI] [PubMed] [Google Scholar]
- 62.van Niel G., D’Angelo G., Raposo G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018;19:213–228. doi: 10.1038/nrm.2017.125. [DOI] [PubMed] [Google Scholar]
- 63.Raffaele S., Lombardi M., Verderio C., Fumagalli M. TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells. 2020;9:2145. doi: 10.3390/cells9102145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cunha M.I., Su M., Cantuti-Castelvetri L., Müller S.A., Schifferer M., Djannatian M., Alexopoulos I., van der Meer F., Winkler A., van Ham T.J. Pro-inflammatory activation following demyelination is required for myelin clearance and oligodendrogenesis. J. Exp. Med. 2020;217:e20191390. doi: 10.1084/jem.20191390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.EL Andaloussi S., Mäger I., Breakefield X.O., Wood M.J.A. Extracellular vesicles: biology and emerging therapeutic opportunities. Nat. Rev. Drug Discov. 2013;12:347–357. doi: 10.1038/nrd3978. [DOI] [PubMed] [Google Scholar]
- 66.Storini C., Bergamaschini L., Gesuete R., Rossi E., Maiocchi D., De Simoni M.G. Selective inhibition of plasma kallikrein protects brain from reperfusion injury. J. Pharmacol. Exp. Ther. 2006;318:849–854. doi: 10.1124/jpet.106.105064. [DOI] [PubMed] [Google Scholar]
- 67.Gabrielli M., Battista N., Riganti L., Prada I., Antonucci F., Cantone L., Matteoli M., Maccarrone M., Verderio C. Active endocannabinoids are secreted on extracellular membrane vesicles. EMBO Rep. 2015;16:213–220. doi: 10.15252/embr.201439668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bonfanti E., Bonifacino T., Raffaele S., Milanese M., Morgante E., Bonanno G., Abbracchio M.P., Fumagalli M. Abnormal Upregulation of GPR17 Receptor Contributes to Oligodendrocyte Dysfunction in SOD1 G93A Mice. Int. J. Mol. Sci. 2020;21:2395. doi: 10.3390/ijms21072395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Magni G., Boccazzi M., Bodini A., Abbracchio M.P., van den Maagdenberg A.M., Ceruti S. Basal astrocyte and microglia activation in the central nervous system of Familial Hemiplegic Migraine Type I mice. Cephalalgia. 2019;39:1809–1817. doi: 10.1177/0333102419861710. [DOI] [PubMed] [Google Scholar]
- 70.York E.M., LeDue J.M., Bernier L.P., MacVicar B.A. 3DMorph automatic analysis of microglial morphology in three dimensions from ex vivo and in vivo imaging. eNeuro. 2018;5 doi: 10.1523/ENEURO.0266-18.2018. ENEURO.0266-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murru L., Vezzoli E., Longatti A., Ponzoni L., Falqui A., Folci A., Moretto E., Bianchi V., Braida D., Sala M. Pharmacological Modulation of AMPAR Rescues Intellectual Disability-Like Phenotype in Tm4sf2-/y Mice. Cereb. Cortex. 2017;27:5369–5384. doi: 10.1093/cercor/bhx221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Karamita M., Barnum C., Möbius W., Tansey M.G., Szymkowski D.E., Lassmann H., Probert L. Therapeutic inhibition of soluble brain TNF promotes remyelination by increasing myelin phagocytosis by microglia. JCI Insight. 2017;2:e87455. doi: 10.1172/jci.insight.87455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mizgerd J.P., Molina R.M., Stearns R.C., Brain J.D., Warner A.E. Gadolinium induces macrophage apoptosis. J. Leukoc. Biol. 1996;59:189–195. [PubMed] [Google Scholar]
- 74.Charan J., Kantharia N.D. How to calculate sample size in animal studies? J. Pharmacol. Pharmacother. 2013;4:303–306. doi: 10.4103/0976-500X.119726. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Further information and data that support the findings of this study are available from the corresponding author upon reasonable request.