

Abstract
Suicide gene therapies provide a unique ability to target cancer cells selectively, often based on modification of viral tropism or transcriptional regulation of therapeutic gene expression. We designed a novel suicide gene therapy approach wherein the gene product (herpes simplex virus thymidine kinase or yeast cytosine deaminase) is phosphorylated and stabilized in expression by the extracellular signal-regulated kinase (ERK), which is overactive in numerous cancers with elevated expression or mutation of receptor tyrosine kinases or the GTPase RAS. In contrast to transcriptional strategies for selectivity, regulation of protein stability by ERK allows for high copy expression via constitutive viral promoters, while maintaining tumor selectivity in contexts of elevated ERK activity. Thus, our approach turns a signaling pathway often coopted by cancer cells for survival into a lethal disadvantage in the presence of a chimeric protein and prodrug, as highlighted by a series of in vitro and in vivo examples explored here.
Keywords: FRA1, glioblastoma, herpes simplex virus thymidine kinase, yeast cytosine deaminase, ganciclovir, 5-fluorocytosine, extracellular signal-regulated kinase, ERK
Graphical abstract

Suicide gene products convert prodrugs to cytotoxic substances, and their selective expression is typically based on viral tropism or specific promoters. Day and colleagues designed a suicide gene with selectivity based on the ERK signaling pathway, which is frequently overactive in cancers and involved in acquired resistance to targeted therapy.
Introduction
Gene-directed enzyme prodrug therapy, often referred to as suicide gene therapy, is a two-step approach wherein cells are first virally transduced with an exogenous gene encoding an enzyme, followed by administration of an innocuous prodrug that is converted by the enzyme into a cytotoxic product.1 This approach has entered clinical trials in some tumor types, such as glioblastoma multiforme (GBM), as an alternative or adjuvant to conventional chemotherapy, often via local administration of viral particles carrying payload encoding the suicide gene.2,3 Suicide gene therapy offers design advantages for specific targeting of cancer cells through vector modification at multiple levels (e.g., viral tropism, viral replication mechanisms, and transcriptional regulation of the gene to engineer specificity of expression).4 One particular approach is the use of transcriptional promoters for the suicide gene that leverage tissue specificity or tumor-specific oncogenic factors.5,6 As an example, a phase I clinical trial used a human ERBB2 promoter-driven suicide gene to target HER2-overexpressing breast cancer.7 One limitation of transcription-regulated targeting is that reliance on endogenous promoters often produces insufficient copies of the transcript needed to induce high levels of toxicity.8
For many years, specificity has been engineered into other classes of cancer therapies (small molecule drugs and antibodies) by designing them to bind to specific signaling proteins that are overexpressed or mutated in cancer. Many such drugs target the canonical receptor tyrosine kinase (RTK)/RAS pathway due to the high frequency with which it is pathologically altered in cancer. For example, RTK/RAS signaling is dysregulated in ∼75% of GBM, pancreatic ductal adenocarcinoma (PDAC), and non-small cell lung cancer (NSCLC) tumors.9, 10, 11 In a subset of cancers, inhibitors of this pathway are clinically effective (at least for a time), with one well-known example being inhibitors of the epidermal growth factor receptor (EGFR) in NSCLC.12 Unfortunately, in many instances targeting alterations in the RTK/RAS pathway is ineffective, either due to “undruggable” properties of the intended target (e.g., KRAS, recent progress in this area notwithstanding) or resistance via bypass signaling or secondary mutations.13,14 These challenges motivate development of alternative approaches to target cancers displaying activation of the RTK/RAS pathway.
Here, we describe a novel cancer therapy approach that combines elements of conventional suicide gene therapy and signaling pathway-specific targeting. Our design involves a suicide gene product that is post-translationally stabilized by active extracellular signal-regulated kinase 1/2 (ERK1/2, referred to throughout as ERK), a key downstream effector in the RTK/RAS pathway.15,16 In this approach, viral vectors encode a fusion of the suicide gene herpes simplex virus-1 thymidine kinase (HSV1tk, referred to throughout as HSVtk) with a domain from the transcription factor fos-related antigen 1 (FRA1) that is doubly phosphorylated by ERK to slow degradative turnover. The basic design represents an adaptation of a previously described live-cell fluorescent reporter of ERK activity,17 but that design was also modified here to generate a number of variants useful for specific control experiments. Also generated was a variant in which HSVtk was substituted with yeast cytosine deaminase (yCD) as an alternative enzyme. In the studies presented here, we validated that our suicide gene therapy produces ERK-dependent cell killing in cancer cells in a number of in vitro and in vivo experiments and that it demonstrates selectivity between transformed (high ERK) and normal non-cancer cells (low ERK) when both are transduced by the same virus preparation. Moreover, the advantageous ERK selectivity of our approach was accompanied by cancer cell toxicity comparable to that observed with conventional constitutive expression of an unmodified suicide gene. In addition, we highlight scenarios for which the ERK-dependent suicide gene might be an improvement to unmodified or tissue-specific targeting therapies, particularly in the context of ERK-driven therapeutic resistance.
Results
HSVtk expression vector design exploits ERK-mediated stabilization of the FRA1 transcription factor
The central design element of our suicide gene approach relies on the same native interaction between ERK and the transcription factor FRA1 that was leveraged in the design of a live-cell fluorescent ERK activity reporter by Albeck et al.17 The FRA1 PEST domain (amino acids 163–271, rich in proline [P], glutamic acid [E], serine [S], and threonine [T]) promotes FRA1 degradation, but ERK-mediated phosphorylation of serines 252 and 265 slows degradation and stabilizes FRA1.18 We exploited this natural interaction by fusing the FRA1 PEST domain to the C-terminus of HSVtk, which converts the prodrug ganciclovir (GCV) into a cytotoxic, acyclic nucleotide precursor that causes double-stranded breaks when integrated in DNA.19 This was accomplished by excising the gene encoding mVenus from one of the plasmids constituting the FIRE (FRA1-based Integrative Reporter of ERK) system described by Albeck et al.17 and replacing it with a gene encoding HSVtk (Figure 1). The sequence encoding the N-terminal nuclear localization signal (NLS) from SV40 Large T-antigen included in the original design of the FIRE reporter was retained to promote sequestration of the suicide gene product in the nucleus, mimicking the normal subcellular localization observed for FRA1. Hereafter, the labels “NLS-” and “-FIRE” are appended before and after protein names to indicate N-terminal NLS fusion and C-terminal FRA1 PEST fusion, respectively. The resulting novel suicide gene product is designed to leverage differences in ERK activity between transformed (tumor) and surrounding normal tissue cells, as have been reported for example in GBM and PDAC tumors.20,21 Several alternative retroviral expression vector inserts were also generated for use as controls or to provide flexibility in cell selection (Figure S1; Table S1). One version of note replaces the FRA1 PEST domain with an alternative PEST (d2) that is not regulated (stabilized) by ERK.
Figure 1.
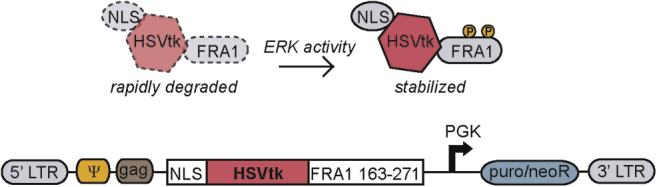
ERK-dependent suicide gene therapy is designed to selectively target cancer cells
Schematic demonstrating stabilization of herpes simplex virus thymidine kinase (HSVtk) protein expression via ERK-mediated phosphorylation of the FRA1 PEST domain, based on the reporter developed by Albeck et al.17 The retroviral vector insert design is also shown. The insert includes an in-frame nuclear localization signal (NLS), HSVtk, and the FRA1 domain, along with viral packaging signals and an antibiotic resistance cassette.
ERK-regulated HSVtk fusion selectively kills cells with elevated ERK activity
To evaluate ERK-dependent selectivity of the suicide gene therapy, we transduced U87MG GBM cells stably expressing the variant III mutant of the EGFR (EGFRvIII), referred to hereafter as U87MG for simplicity, with virus encoding the NLS-HSVtk-FIRE insert described in Figure 1. For control experiments, cells were alternatively transduced with a vector incorporating the d2 PEST domain (NLS-HSVtk-d2) or an empty vector (EV) (Figure 2A). Note that EGFRvIII expression was chosen as a cell background of interest for this study because EGFRvIII is found in ∼25% of GBM tumors and can promote ERK activity in some contexts22 (Figure S2A). It should be noted, though, that multiple kinds of somatic gene alterations found in GBM cells are capable of promoting ERK activity and that EGFRvIII expression is therefore not singularly responsible for the tendency of GBM tumor cells to display ERK activity that is elevated compared to non-transformed cells.23 U87MG cells expressing NLS-HSVtk-FIRE were sensitive to GCV, confirming catalytic activity of the HSVtk fusion protein (Figure S2B). Next, U87MG cells transduced with the different vectors were treated with or without the mitogen-activated kinase kinase (MEK) inhibitor CI-1040 to modulate ERK activity (Figure 2B). Western blotting using an antibody against phosphorylated FRA1 (specific to the ERK-regulated serine 265 within the PEST domain) confirmed that MEK inhibition antagonized phosphorylation of NLS-HSVtk-FIRE, endogenous FRA1, and ERK. Importantly, MEK inhibition also led to decreased cell death in response to GCV in cells expressing NLS-HSVtk-FIRE but not in cells expressing NLS-HSVtk-d2 (Figure 2C). GCV treatment had no influence on cell death in EV-transduced cells (Figure S2C).
Figure 2.

Increased ERK activity in cells expressing NLS-HSVtk-FIRE leads to greater ganciclovir (GCV)-mediated cell death and DNA damage
(A) Lysates from U87MG cells transduced with NLS-HSVtk-FIRE, NLS-HSVtk-d2, or an empty vector (EV) were analyzed by western blotting using antibodies against the indicated proteins. RNA prepared in parallel was analyzed via RT-PCR using primers for the indicated transcripts. (B) Cells from (A) were treated with 4 μM CI-1040 or DMSO for 48 h prior to lysis and analysis by western blotting using antibodies against the indicated proteins. (C) Cells pre-treated as described in (B) were treated for an additional 4 days with 50 μM GCV or DMSO, and cell death was measured by flow cytometry. (D) Cells treated as described in (C) were lysed 2 days after starting treatment with GCV and analyzed by western blotting using antibodies against the indicated proteins, with pH2A.X signal quantified by densitometry and normalized to GCV-treated values in cells expressing NLS-HSVtk-FIRE. (E) U87MG cells expressing NLS-HSVtk-FIRE were pre-treated for 48 h with 4 μM CI-1040, or DMSO, followed by treatment for 48 h with 50 μM GCV or DMSO. Fixed cells were stained for pH2A.X, vimentin, and DNA. Scale bar, 50 μm. pH2A.X signal intensities were calculated for n > 100 cells across three biological replicates and normalized to mean intensity of the GCV-treated condition. For (C)–(E), the indicated statistical comparisons between GCV-treated cells with or without CI-1040 were based on the Student’s t test. (F) Human pancreas ductal epithelial (HPDE) cells were transduced with a vector encoding KRASG12V or an EV. Cells were treated with 50 μM GCV or DMSO and analyzed by western blotting using antibodies against the indicated proteins. Indicated signals were quantified by densitometry and normalized to DMSO-treated values in EV cells. “pFIRE” denotes pFRA1 signal detected at the molecular weight corresponding to FRA1 domain-fused HSVtk. Statistical comparisons based on Tukey’s post hoc testing following two-way ANOVA are shown. (G) RNA from untreated HPDE cells described in (F) were analyzed via qRT-PCR using primers for KRAS. Statistical comparisons from Student’s t test are shown. Throughout the figure panels, representative blots and images are shown, and error bars indicate mean ± SEM for three replicates, unless otherwise noted; ∗p < 0.05 for the indicated comparisons using statistical methods described for each panel.
To further confirm trends expected based on insert design, cells treated as in Figure 2C were analyzed for levels of phosphorylated H2A histone family member X (H2A.X), a marker of DNA double-stranded breaks (Figures 2D, 2E, and S2D). In cells expressing NLS-HSVtk-FIRE, but not in those expressing NLS-HSVtk-d2, MEK inhibition reduced H2A.X phosphorylation in response to GCV. Similar trends were observed in the glioma-initiating cell (GIC) line G88, which expresses EGFRvIII, when it was transduced with NLS-HSVtk-FIRE (Figure S2E). Further reinforcing the ERK dependence of the HSVtk fusion protein containing the FRA1 PEST domain, expression of constitutively active MEK (MEK1DD, a phosphomimetic mutant of MEK1) in U87MG cells led to increased phosphorylation of NLS-HSVtk-FIRE, endogenous FRA1, and ERK (Figure S2F).
To further demonstrate the ERK selectivity for NLS-HSVtk-FIRE, we engineered normal human pancreatic ductal epithelial cells to express mutant KRAS (G12V), which promoted phosphorylation of ERK and NLS-HSVtk-FIRE (Figures 2F and 2G). The ERK-dependent increase in NLS-HSVtk-FIRE phosphorylation corresponded with increased DNA damage in response to GCV. These data provide an example of a therapeutic window created by a common oncogene that can be leveraged by our system.
The ERK dependence we engineered for HSVtk expression can also be applied to other suicide genes. To demonstrate the modularity of the system, we generated retroviral constructs with yCD taking the place of HSVtk (Figure S2G). yCD converts the prodrug 5-fluorocytosine (5-FC) into the commonly used chemotherapeutic 5-fluorouracil. U87MG transduced with vectors encoding yCD or yCD-FIRE were treated with or without EGF and fibroblast growth factor 2 (FGF2) to generate populations with different levels of ERK activity. As expected, elevated ERK activity promoted yCD-FIRE expression, but not yCD expression, and preferentially increased cell sensitivity to 5-FC for cells expressing yCD-FIRE (Figures S2H and S2I).
Comparisons of the ERK-dependent HSVtk expression vector to unmodified HSVtk reveal their comparable toxicity
While results from Figure 2 suggest that FRA1 PEST domain fusion provided ERK-based selectivity for HSVtk-mediated cell killing, we suspected that fusion to the PEST domain could also antagonize HSVtk expression compared to unmodified HSVtk. NLS fusion may also alter HSVtk expression in unanticipated or undesirable ways. To make the appropriate comparisons, we transduced U87MG cells with versions of expression vectors encoding HSVtk, NLS-HSVtk, and NLS-HSVtk-FIRE that were engineered for co-expression of GFP via an internal ribosomal entry site (Figure 3A). With this system, differences in HSVtk protein expression across cells with equal GFP expression can be attributed to translational or post-translational mechanisms. As expected, cells flow-sorted for equal GFP expression displayed approximately equal HSVTK transcript expression (Figure 3A). Flow-sorted cells expressing NLS-HSVtk were surprisingly less sensitive to GCV than cells expressing HSVtk (Figure 3B). However, cells expressing NLS-HSVtk-FIRE were slightly more sensitive to GCV than cells expressing unmodified HSVtk. Increased cell death and DNA damage in response to GCV were also observed in cells expressing NLS-HSVtk-FIRE versus NLS-HSVtk, further confirming the ability of the FRA1 PEST domain to enhance suicide gene-mediated toxicity in the context of elevated ERK activity (Figures 3C and 3D).
Figure 3.

NLS-HSVtk-FIRE expression leads to comparable levels of GCV-mediated cell death as unmodified HSVtk in glioblastoma cells with constitutive ERK activity
(A) Schematic of insert for retroviral vector encoding NLS-HSVtk-FIRE with in-frame IRES-driven GFP expression, used for fluorescence-activated cell sorting. Pre- and post-sort GFP intensity histograms for U87MG cells transduced with HSVtk, NLS-HSVtk, NLS-HSVtk-FIRE, or an empty vector encoding only GFP (EV). The parental cell line was used as a GFP-negative control. RNA extracts from post-sorted cells were analyzed via qRT-PCR using primers for HSVTK. Statistical comparisons from Tukey’s post hoc testing following one-way ANOVA are shown (n.s. = not significant). (B) Sorted cells expressing HSVtk, NLS-HSVtk, or NLS-HSVtk-FIRE were treated for 4 days with the indicated concentrations of GCV or DMSO, and cell number was evaluated by counting nuclei. (C) Sorted cells expressing NLS-HSVtk, with or without the FRA1 domain, were treated for 3 days with the indicated concentrations of GCV or DMSO, and cell death was measured by flow cytometry. (D) Cells described in (C) were treated for 24 h with 50 μM GCV or DMSO. Lysates were analyzed by western blotting using antibodies against the indicated proteins, with pH2A.X signal quantified by densitometry. For (C) and (D), statistical comparisons between GCV-treated cells expressing NLS-HSVtk or NLS-HSVtk-FIRE were based on Student’s t test. (E) GFP-sorted versions of the indicated cells were treated for 3 days with 1 μM GCV or DMSO, and cell death was measured by flow cytometry. Statistical comparisons between GCV-treated cells from Tukey’s post hoc testing following one-way ANOVA are shown. (F) GFP-sorted versions of the indicated cells were treated for 24 h with 50 μM GCV or DMSO, and lysates were analyzed by Luminex assay for active caspase-3. (G) Sorted versions of the indicated cells were treated for 2 days with the indicated concentrations of GCV or DMSO, and cell death was measured by flow cytometry. For (F) and (G), statistical comparisons between GCV-treated cells expressing HSVtk or NLS-HSVtk-FIRE were based on Student’s t test. For figure panels with phenotypic measurements of GCV response, cells were treated for 3–4 days. For lysate collection, cells were treated for 24 h to avoid significant loss of adherent cells. Throughout the figure panels, representative blots are shown, and error bars indicate mean ± SEM for three replicates; ∗p < 0.05 for the indicated comparisons using statistical methods described for each panel.
Next, we compared GCV-mediated cell death with unmodified HSVtk to nucleus-localized HSVtk, with or without the FRA1 domain (Figure 3E). Cells expressing NLS-HSVtk-FIRE were more sensitive to GCV than cells expressing NLS-HSVtk or HSVtk alone (Figures 3E and 3F). However, increased sensitivity with NLS-HSVtk-FIRE expression was not maintained at higher concentrations of GCV, suggesting that the advantage of the FIRE fusion can be overwhelmed or saturated at sufficiently high GCV concentrations (Figure 3G). In G88 cells transduced with the GFP-encoding vectors, differences in GCV response between NLS-HSVtk and NLS-HSVtk-FIRE expression were maintained (Figures S3A–S3D). However, unmodified HSVtk was more effective than NLS-HSVtk-FIRE with respect to cell death induction, potentially due to the lower phosphorylated ERK levels in these cells compared to U87MG (Figures S3E and S3F). Taken together, these data suggest that incorporation of the ERK-selective FRA1 PEST domain in the suicide gene product induces toxicity that is comparable to, if not greater than, that observed for unmodified HSVtk in contexts of high ERK activity.
Nuclear localization of ERK in response to DNA damage may augment the efficacy of NLS-HSVtk-FIRE
While the sorting based on GFP expression in Figure 3 controlled for transgene integration across different HSVtk-encoding vectors, it did not allow for direct assessment of HSVtk expression. To determine the effect of the NLS and FRA1 PEST domain fusion on HSVtk protein stability and expression, we generated versions of the previously described vectors including an N-terminal 3×FLAG tag (Figure 4A). In U87MG cells engineered to express FLAG-tagged HSVtk, with or without NLS or FRA1 PEST domains, fluorescence microscopy revealed that proteins displayed anticipated subcellular localization patterns and that fusion with the FRA1 PEST domain reduced HSVtk expression, with or without the NLS fusion (Figures 4B and S4A). Expression differences were also apparent by western blotting (Figure 4C). Given that FRA1 PEST domain fusion likely promotes increased proteasomal degradation of HSVtk, we probed the effect of the proteasomal inhibitor MG132 on HSVtk-FIRE expression. While MG132 treatment promoted expression of HSVtk-FIRE, but not unmodified HSVtk, overall FLAG-tagged protein expression was still significantly lower in cells expressing HSVtk-FIRE than unmodified HSVtk (Figure S4B). This could potentially be explained by incomplete proteasomal inhibition. Additionally, we measured FLAG-tagged HSVtk abundance in response to increased ERK activation by exogenous growth factors (Figure 4D). As expected, FLAG-tagged HSVtk expression increased in response to ERK activation only for the NLS-HSVtk-FIRE fusion. However, even this augmented expression was still substantially lower than observed for HSVtk alone.
Figure 4.

Effectiveness of the ERK-dependent suicide gene is not explained by HSVtk abundance alone
(A) Schematic of insert for retroviral vector encoding FLAG-tagged NLS-HSVtk-FIRE with in-frame IRES-driven GFP expression, used for detection of HSVtk protein abundance. (B) U87MG cells expressing FLAG-tagged HSVtk, HSVtk-FIRE, NLS-HSVtk, or NLS-HSVtk-FIRE were stained for pFRA1, FLAG, and DNA. Quantification of total and nuclear immunofluorescence intensities, in addition to GFP, were calculated for n > 100 cells across three biological replicates. Scale bar, 50 μm. (C) Cells described in (B) were lysed and analyzed by western blotting using antibodies against the indicated proteins, with FLAG and GFP signals quantified by densitometry. For (B) and (C), statistical comparisons from Tukey’s post hoc testing following one-way ANOVA are shown. (D) U87MG cells expressing FLAG-tagged HSVtk or NLS-HSVtk-FIRE were treated with or without EGF and FGF2 for 4 days. Lysates were analyzed using antibodies against the indicated proteins, with FLAG and GFP signals quantified by densitometry. Statistical comparisons between ligand-treated and -untreated conditions were based on Student’s t test. (E) Schematic of mutated serine residues (S252, S265) in the S2D version of the FRA1 domain. U87MG cells expressing FLAG-tagged NLS-HSVtk fused to either the wild-type (WT) or S2D mutant FRA1 domain were treated with or without EGF and FGF2 for 4 days. Lysates were analyzed using antibodies against the indicated proteins, with FLAG and GFP signals quantified by densitometry. Statistical comparisons from Tukey’s post hoc testing following one-way ANOVA are shown. Identical cells, without ligand treatment, were treated with the indicated concentrations of GCV or DMSO, and cell viability was measured by MTS assay. (F) U87MG cells expressing FLAG-tagged NLS-HSVtk-FIRE were treated for 24 h with 50 μM GCV or DMSO. Cells were stained for pERK, FLAG, and DNA. Quantification of total and nuclear immunofluorescence intensities, in addition to GFP, were calculated for n > 100 cells across three biological replicates. Scale bar, 20 μm. Statistical comparisons from Student’s t test are shown. Throughout the figure panels, representative blots and images are shown, and error bars indicate mean ± SEM for three replicates, unless otherwise noted. For boxplots, median, first/third quartiles, minimum/maximum, and outlier values are displayed for the indicated number of cells across replicates; ∗p < 0.05 for the indicated comparisons using statistical methods described for each panel.
HSVtk contains an endogenous, N-terminal NLS,24 but the results in Figure 4B suggest that this sequence is insufficient to confine FLAG-tagged HSVtk to the nucleus. We generated versions of the HSVtk fusion proteins lacking the first 33 amino acids (referred to as HSVtkΔ1–33) to directly probe the effects of the endogenous NLS on subcellular localization, protein expression, or GCV-induced toxicity (Figure S4C). U87MG cells engineered to express endogenous NLS-null versions of the HSVtk-FIRE fusions did not display substantially altered HSVtk subcellular localization (Figure S4D). Interestingly, there was an increase in overall HSVtk expression upon deletion of the endogenous NLS, but relative levels of FRA1 PEST domain phosphorylation were decreased (Figure S4E). Ultimately, deletion of the endogenous NLS had only a modest impact on GCV-induced toxicity, tending to make cells marginally less sensitive to GCV (Figure S4F). Together with the results in Figure 3, these data suggest that protein expression levels are not the sole determinant of GCV-mediated cell killing.
In certain cellular contexts (e.g., strong drivers of ERK activity, minimal phosphatase activity against the PEST domain), it is conceivable that expression of NLS-HSVtk-FIRE could be comparable to HSVtk. To determine if that it is indeed possible, we generated cells expressing a mutant of NLS-HSVtk-FIRE in which the operative ERK-phosphorylated serines in the FRA1 PEST domain were mutated to aspartic acid, which serves as a phosphomimetic (“S2D FIRE”; Figure 4E). Fusion of HSVtk with the S2D mutant FRA1 domain significantly increased HSVtk expression when compared to the wild-type FRA1 domain (Figure 4E). However, the mutated FRA1 domain only marginally increased response to GCV in U87MG cells (Figure 4E), and the ∼6-fold increase in expression compared to wild type was still much lower than the previously measured fold difference between unmodified HSVtk and NLS-HSVtk-FIRE (Figure 4C; ∼45-fold difference in GFP-normalized FLAG signal). Thus, even when the FRA1 PEST domain is effectively maximally phosphorylated, the NLS-HSVtk-FIRE fusion still results in lower HSVtk expression than unmodified HSVtk.
To explain the comparable GCV toxicities observed for HSVtk and NLS-HSVtk-FIRE, we hypothesized that previously described mechanisms of ERK-driven resistance to DNA damage25, 26, 27 might be at play. Interestingly, we noticed that GCV treatment caused an increase in nuclear localization of phosphorylated ERK (Figures 4F and S4G). Increased nuclear localization of phosphorylated ERK was coincident with an increase in FLAG-tagged HSVtk abundance in cells expressing NLS-HSVtk-FIRE. This potential feed-forward effect offers one possible explanation as to why inclusion of the FRA1 domain for nucleus-localized HSVtk promotes response to GCV. Increased GCV-mediated toxicities with the FRA1 PEST fusion (compared to unmodified HSVtk) were lost without the NLS (Figure S4H). Overall, these data suggest that the relatively low expression of NLS-HSVtk-FIRE is potentially compensated for by DNA damage-induced shuttling of active ERK to the nucleus, which stabilizes fusion protein expression and produces cell toxicities comparable to that observed with more abundant (but more diffusely distributed) HSVtk.
Co-culture experiments provide in vitro proof-of-concept for ERK-dependent suicide gene selectivity
In real tumors, transformed cells displaying elevated ERK activity will exist in the vicinity of normal cells, and both populations of cells may be transduced by therapeutic viral particles carrying a suicide gene payload. To mimic this scenario in vitro, we set out to identify a pair of tissue type-matched normal and cancer cells displaying differential ERK activities. Neural stem cells (NSCs) are responsible for generation of multiple cell types in the nervous system and have been proposed as the cell of origin for gliomas.28,29 In fact, NSCs transform into GICs with expression of GBM-associated oncogenes.30 Therefore, we hypothesized that mixed populations of NSCs and GICs, such as the G88 cells used previously, would be a reasonable in vitro model for evaluating the ERK selectivity of NLS-HSVtk-FIRE. NSCs derived from human embryonic stem cells31 and G88 cells had significantly different levels of ERK activation, with G88 cells exhibiting increased whole-cell and nuclear levels of phosphorylated ERK (Figures 5A and S5A). The two types were transduced separately using the vector encoding FLAG-tagged NLS-HSVtk-FIRE and GFP and then evaluated for cell death response to GCV (Figures 5B and S5B). G88 cells were more responsive to GCV, but this difference could have been due to the lower apparent transduction efficiency of NSCs (which presumably divide more slowly than G88s and are therefore less susceptible to retroviral transduction and GCV-induced, cell division-dependent death). Even when G88 and NSC transduction efficiencies were brought into closer alignment through the use of concentrated virus preparations, NSCs remained minimally responsive to GCV (Figure 5B). We also noticed that NSCs and G88 cells with similar percentages of cells transduced displayed different GFP distributions, with GFP-positive NSCs having a GFP intensity that was lower than G88s on average (Figure S5B). Therefore, to exclude the possibility that viral integration differences explain differential GCV response, we sorted cells with equal GFP intensities and again evaluated response to GCV (Figures 5C and S5C). Even after sorting, NSCs exhibited less DNA damage and cell death in response to GCV than G88 cells. Next, dye-labeled and flow-sorted G88 cells were mixed with flow-sorted NSCs, and the combined population was treated with GCV. Consistent with previous trends, GCV treatment led to enrichment of NSCs within the mixed, live-cell population (Figure S5D).
Figure 5.

NLS-HSVtk-FIRE suicide gene therapy demonstrates selectivity in vitro using mixed cultures of cells with differential ERK activity
(A) Lysates generated from neural stem cells (NSCs) and G88 cells were analyzed by western blotting using antibodies against the indicated proteins, with pERK and pFRA1 signals quantified by densitometry. (B) Virus packaged with 3×FLAG-NLS-HSVtk-FIRE-IRES-GFP insert was added to NSC and G88 cells for 24 h. 2 days later, cells were treated for 24 h with 10 μM GCV or DMSO. Cell death and GFP intensity were analyzed by flow cytometry. Identical experiments were performed using 12× concentrated virus. (C) NSC and G88 cells were flow sorted as outlined in Figure S5B and treated for 24 h with 10 μM GCV or DMSO. Lysates were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry. Statistical comparisons from Tukey’s post hoc testing following two-way ANOVA are shown. (D) NSC and G88 cells expressing Cerulean or mRFP1, respectively, were plated alone or mixed and imaged 24 h later. Scale bar, 100 μm. Virus described in (B) was added for 24 h, followed by treatment for 48 h with 10 μM GCV or DMSO. Percentage of Cerulean- or mRFP1-positive live cells in each condition was determined by flow cytometry. Gating strategies are shown in Figure S5E. Levels of cell death and GFP intensities are shown in Figure S5F. (E) PD3077 murine pancreas cancer cells with heterogeneous expression of FLAG-tagged HSVtk-FIRE and doxycycline (DOX)-inducible ERK2 shRNA were treated for 2 days with 5 μg/mL DOX, and cells were stained for FLAG. Cell clusters with approximately equal GFP expression, with (yellow, low ERK2) or without (white, high ERK2) detectable dsRed expression, are highlighted. Scale bar, 500 μm. (F) U87MG cells expressing FLAG-tagged NLS-HSVtk-FIRE and DOX-inducible ERK2 shRNA were treated for 2 days with or without 5 μg/mL DOX. Lysates were analyzed by western blotting using antibodies against the indicated proteins, with signals quantified by densitometry and normalized to condition without DOX. (G) U87MG cells expressing FLAG-tagged NLS-HSVtk-FIRE and DOX-inducible control or ERK2 shRNA were pre-treated for 2 days with or without 5 μg/mL DOX. Pre-treated cells were treated for 3 days with 10 μM GCV or DMSO, and cell death was measured by flow cytometry. (H) U87MG cells expressing FLAG-tagged NLS-HSVtk-FIRE and inducible ERK2 shRNA were pre-treated for 2 days with or without 5 μg/mL DOX, followed by treatment for 24 h with 10 μM GCV or DMSO. Lysates were analyzed by western blotting using antibodies against the indicated proteins, with pH2A.X signals quantified by densitometry. Statistical comparisons from Tukey’s post hoc testing following two-way ANOVA are shown. (I) Cells described in (G) were mixed at a 1:1 ratio and pre-treated for 4 days with or without 5 μg/mL DOX, followed by treatment for 24 h with 10 μM GCV or DMSO. Cells were stained for pH2A.X and DNA, and nuclear pH2A.X intensities in cells with or without detectable dsRed expression were quantified for n > 100 cells across three biological replicates. White arrow highlights cell with especially high dsRed expression. Note that dsRed was only expressed from the ERK2 shRNA vector. Scale bar, 50 μm. Throughout the figure panels, representative blots and images are shown, and error bars indicate mean ± SEM for three replicates, unless otherwise noted. For boxplots, median, first/third quartiles, minimum/maximum, and outlier values are displayed for the indicated number of cells across replicates; ∗p < 0.05 for the indicated comparisons using Student’s t test except when otherwise indicated.
In order to mimic the simultaneous retroviral transduction of normal and tumor cells that would occur in vivo, NSC and G88 cells were engineered with stable expression of the fluorescent proteins Cerulean and mRFP1, respectively (Figure 5D). Fluorescent protein-expressing NSC and G88 cells were plated, transduced with virus encoding NLS-HSVtk-FIRE, and treated with GCV or vehicle. Although they were transduced less efficiently (as expected), Cerulean-expressing NSCs were substantially enriched in the live-cell fraction following GCV treatment (Figures 5D and S5E). Overall levels of GCV-mediated cell death were consistent with the trends of enrichment, where conditions with higher percentages of G88 cells exhibited greater cell death (Figure S5F). These data reflect the net effects of cell-intrinsic viral transduction efficiency and ERK-dependent selectivity of the suicide gene therapy and demonstrate the concept of preferential tumor cell killing after retroviral transduction of a mixed population.
As an alternative co-culture approach, cancer cells were engineered with a doxycycline-inducible short hairpin RNA (shRNA) targeting ERK2, which was chosen over ERK1 because phosphorylated ERK2 is typically more abundant in cancer cells than phosphorylated ERK1. In this model system, cells with normal (no doxycycline) or reduced ERK2 expression (doxycycline-treated) are isogenic, creating a more tightly controlled comparison between the two cell populations. The LT3REPIR shRNA vector we used also encodes doxycycline-inducible expression of dsRed in tandem with the hairpin, stratifying populations of cells with high and low ERK2 (Figure 5E). To validate this approach, murine PDAC cells expressing both the inducible ERK2 shRNA and FLAG-tagged HSVtk-FIRE were treated with doxycycline and analyzed via immunofluorescence microscopy (Figures 5E and S5G). Cells expressing dsRed (ERK2 knockdown) appeared to display lower FLAG signal than those lacking dsRed, when comparing across cells with similar NLS-HSVtk-FIRE transduction (GFP signal). U87MG and G88 cells engineered in this way displayed decreased ERK2 and FLAG-tagged NLS-HSVtk-FIRE expression in response to doxycycline (Figures 5F and S5H). The observed NLS-HSVtk-FIRE expression differences in U87MG cells corresponded to differences in GCV response, with decreased cell death or increased cell viability due to doxycycline treatment in cells expressing ERK2 shRNA, but not control shRNA (Figures 5G, S5I, and S5J). Similar effects with respect to cell death were observed in G88 cells (Figure S5K). Differences in phenotypic response to GCV were consistent with population-level measurements of DNA damage (Figures 5H and S5H). Moreover, in a mixed population of cells expressing either inducible dsRed/ERK2 shRNA or control shRNA (lacking dsRed expression), dsRed-positive cells exhibited lower H2A.X phosphorylation than dsRed-negative cells in response to co-treatment of doxycycline and GCV (Figures 5I and S5L). Note that cells engineered with inducible dsRed/ERK2 shRNA expression were nearly 100% dsRed-positive by flow cytometry in response to doxycycline.
Using ERK-dependent suicide gene therapy to overcome resistance to kinase inhibitors and chemotherapy
Across oncology, there are numerous examples where tumor cells adapt to and resist various therapies by increasing signaling through the ERK pathway. The NLS-HSVtk-FIRE suicide gene product may represent an alternative approach to leverage the tendency of cancer cells to utilize this resistance mechanism. As one example, GBM cells have been shown to use ERK-dependent bypass signaling to resist RTK inhibition32 (Figure 6A). In cells transduced to express NLS-HSVtk-FIRE, ERK bypass signaling would presumably promote suicide gene expression and make cells more susceptible to GCV-mediated death. Consistent with this conceptual model, treatment of U87MG cells expressing NLS-HSVtk-FIRE with inhibitors of EGFR and MET (which must be combined in this cell background to fully antagonize ERK activity and produce some degree of cell death32, 33, 34) and GCV was more effective than EGFR and MET inhibitors alone or GCV alone (Figure 6A).
Figure 6.

Potential applications of the NLS-HSVtk-FIRE therapy to overcome common therapeutic resistance mechanisms in cancer
(A) U87MG cells were treated for t ≤ 48 h with 10 μM gefitinib (G) + 3 μM PHA665752 (P) and lysates were analyzed by western blotting with antibodies against the indicated proteins. U87MG cells transduced with NLS-HSVtk-FIRE or EV were treated for 48 h with 10 μM GCV, 10 G + 3 μM P, 10 μM GCV + 10 μM G + 3 μM P, or DMSO. Cell death was measured by flow cytometry. Statistical comparisons from Tukey’s post hoc testing following two-way ANOVA are shown. (B) HPAF-II pancreas cancer cells expressing NLS-HSVtk-FIRE were treated with 10 ng/mL TGF-β and 50 ng/mL HGF for 3 days. Lysates were analyzed by western blotting with antibodies against the indicated proteins. Signals quantified by densitometry are provided in Figure S6A. (C) PD3077 and PD7242 murine pancreas cancer cells expressing NLS-HSVtk-FIRE were treated for 48 h with 100 μM GCV or DMSO. Lysates were analyzed by western blotting with antibodies against the indicated proteins. Signals quantified by densitometry are provided in Figure S6C. (D) PD3077 and PD7242 cells described in (C) were treated for 48 h with the indicated concentrations of Abraxane, 100 μM GCV, or DMSO, and cell death was measured by flow cytometry. The indicated statistical comparison between the two GCV-treated cell lines was based on the Student’s t test. Throughout the figure panels, representative blots and images are shown, and error bars indicate mean ± SEM for three replicates, unless otherwise noted; ∗p < 0.05 for the indicated comparisons using statistical methods described for each panel.
Another example centers on epithelial-mesenchymal transition (EMT), which promotes chemoresistance in a number of carcinomas.35,36 Given that EMT depends upon ERK activity in multiple cancer cell settings,37,38 an ERK-dependent suicide gene therapy could potentially represent an approach for combating EMT-driven chemoresistance. To test whether conditions that drive EMT could mediate differences in suicide gene product expression, we transduced HPAF-II human PDAC cells to express NLS-HSVtk-FIRE and treated them with the growth factors transforming growth factor β (TGF-β) and hepatocyte growth factor (HGF), which cooperate to drive robust EMT in many cell lines. Decreased E-cadherin expression and increased vimentin expression were observed, as expected, along with elevated phosphorylation of ERK and NLS-HSVtk-FIRE (Figures 6B and S6A). In addition, we identified two tumor cell lines from an autochthonous KPC mouse model of PDAC (KRAS G12D expression and loss of one p53 allele via Pdx1-cre) exhibiting either baseline mesenchymal characteristics or baseline epithelial characteristics (Figure S6B).39 PD3077 (mesenchymal) and PD7242 (epithelial) cells were transduced to express NLS-HSVtk-FIRE using an identical viral preparation, and response to GCV was compared (Figures 6C and S6C). PD3077 cells displayed greater levels of ERK activity, NLS-HSVtk-FIRE phosphorylation, and DNA damage in response to GCV than PD7242 cells. Consistent with the reported effects of EMT on chemoresistance, the transduced PD3077 cells were substantially more resistant to Abraxane than PD7242 counterparts (Figures 6D and S6D). However, PD3077 cells were more sensitive to GCV than were PD7242 cells, demonstrating that (in this one example) NLS-HSVtk-FIRE could be useful for the selective targeting of carcinoma cells that have undergone EMT.
In vivo testing of the ERK-dependent suicide gene-mediated selectivity and efficacy
As an initial in vivo test of the ERK-responsive suicide gene system, we injected NU/NU mice subcutaneously with U87MG cells engineered and flow sorted as described in Figure 3A and allowed tumors to form for 6 days. Mice were then treated with GCV or vehicle for an additional 12 days (Figures 7A and S7A). Tumors expressing NLS-HSVtk-FIRE were as responsive to GCV as tumors with unmodified HSVtk. Tumors expressing NLS-HSVtk were less responsive to GCV than the other versions tested, consistent with in vitro trends.
Figure 7.

In vivo feasibility studies to test the selectivity and efficacy of the ERK-dependent suicide gene therapy
(A) Subcutaneous xenograft tumors were formed in mice using GFP-sorted U87MG cells described in Figure 3A. After 6 days, mice were treated for the indicated times with 50 mg/kg GCV or vehicle. Representative excised tumors from each group are shown. Scale bar, 10 mm. Tumor volumes were measured every 2–3 days, with average size of HSVtk-expressing GCV-treated groups shown (n = 8 tumors per group). Arrow indicates treatment start. Tumor growth curves for EV- and vehicle-treated groups are shown in Figure S7A. Statistical comparisons from one-way ANOVA are shown. (B) Subcutaneous xenograft tumors were formed in mice using U87MG cells expressing FLAG-tagged NLS-HSVtk-FIRE and DOX-inducible ERK2 shRNA described in Figure 5F. At the indicated days (arrows), DOX and GCV treatments began, with tumor volumes measured every 2–3 days. dsRed flux (photons per second, p/s) was measured by an IVIS imager 3 days after starting DOX administration and plotted for each group (n = 6 or 8 tumors per group). (C) Lysates generated from excised subcutaneous tumors described in (B) were analyzed by western blotting using antibodies against the indicated proteins. ERK2 and pH2A.X signals were quantified by densitometry. (D) Representative images of stained sections from formalin-fixed paraffin-embedded subcutaneous tumors, using antibodies against active caspase-3. Scale bar, 1 cm. Active caspase-3 signal quantification performed for > 3 fields of view per tumor (n = 3 per group). For (B)–(D), statistical comparisons from Tukey’s post hoc testing following two-way ANOVA are shown. (E) H&E-stained and GFP antibody-stained brain sections from tumor-bearing mice (G34 cell line, orthotopic implantation) injected with concentrated retrovirus encoding the indicated transgene with in-frame IRES-GFP expression. White dashed line indicates tumor location. Scale bar, 1 mm. Fractions of animals with GFP-positive tumors are plotted. (F) Representative images are shown of orthotopic tumors injected with NLS-HSVtk-FIRE concentrated retrovirus, as described in (E), with GFP-positive tumors treated with GCV or PBS (vehicle). White dashed line indicates tumor location on H&E-stained images. Scale bar, 1 mm. Higher magnification images of GFP-positive regions with pH2A.X staining are also included (scale bar, 200 μm), and percentage pH2A.X-positive staining of GFP-positive tumors in response to GCV is plotted. Representative images for EV virus injections are shown in Figure S7E. A staining summary of all mice for in situ delivery experiments is provided in Table S3. Throughout the figure panels, representative blots and images are shown, and error bars indicate mean ± SEM for three replicates, unless otherwise noted; ∗p < 0.05 for the indicated comparisons using statistical methods described for each panel.
We next utilized the inducible ERK2 shRNA system, introduced in Figures 5E–5I, to test whether differences in ERK activation in subcutaneous xenografts would mediate differential GCV response. U87MG cells expressing doxycycline-inducible ERK2 shRNA and FLAG-tagged NLS-HSVtk-FIRE were injected subcutaneously in NU/NU mice, and tumors were allowed to form. After 13 days, a subset of mice was switched to drinking water containing doxycycline to induce ERK2 shRNA expression (Figures 7B and S7B). After 3 days of doxycycline, all mice (drinking water with or without doxycycline) began treatment with GCV or vehicle, and tumor sizes were monitored. In both groups (+/− doxycycline), treatment with GCV significantly reduced tumor volume compared to vehicle. However, tumors from GCV- and doxycycline-treated mice were significantly larger than tumors from mice treated with GCV but not doxycycline (Figures 7B, S7C, and S7D). Furthermore, analysis of excised tumors suggested that ERK2 shRNA expression antagonized H2A.X phosphorylation and caspase-3 activation in response to GCV (Figures 7C and 7D). Thus, decreased ERK2 expression in tumors expressing NLS-HSVtk-FIRE impaired GCV-mediated tumor reduction and cell killing in vivo.
As a preliminary test of in situ delivery of therapeutic retroviral particles, we injected concentrated preparations of viruses packaged with NLS-HSVtk-FIRE or EV in mouse brains containing human GIC-derived GBM tumors. The GFP-encoding versions of these vectors were used in order to monitor viral delivery. 9 out of 15 total tumors were positive for GFP signal (83.3% of EV, 44.4% of NLS-HSVtk-FIRE vector; Figure 7E). Only tumors treated with the NLS-HSVtk-FIRE virus displayed detectable levels of phosphorylated H2A.X in response to GCV treatment (Figures 7F and S7E). While administration will need to be optimized, these data do suggest that in situ delivery of viruses armed with the ERK-regulated HSVtk construct can promote DNA damage in tumors treated with GCV.
Discussion
The ERK-dependent, post-translationally regulated suicide gene therapy developed here offers an alternative approach for selectively targeting cancers with elevated RTK/RAS pathway activity. While there are at least a few previously described suicide gene strategies based upon post-translational mechanisms,40,41 these strategies have almost exclusively focused on low oxygen tension (hypoxia response) found in tumors versus normal tissues, which are arguably less cancer specific than aberrant ERK activity. We demonstrated the selectivity offered by the ERK-regulated FRA1 PEST domain using two different gene-directed enzyme prodrug therapy systems (HSVtk/GCV and yCD/5-fluorocytosine; outlined in Figure 8). The modular design should allow for adaptation to other suicide gene strategies. Characterization will be necessary for other suicide gene adaptations, as NLS or FRA1 PEST domain fusions may impact intrinsic enzymatic activity or disrupt optimal nuclear/cytoplasmic distributions for specific suicide gene/prodrug combinations. Indeed, the different fusions of HSVtk created here displayed non-intuitive trends with respect to GCV-mediated cell death. In particular, it was perhaps surprising that cell death in response to GCV was comparable between NLS-HSVtk-FIRE and unmodified HSVtk, even with substantially lowered HSVtk expression when fused to the FRA1 PEST domain. While we suggested these differences are partially related to DNA damage-induced nuclear translocation of active ERK, there are potentially other contributing factors. For example, HSVtk can exist in dimeric and monomeric states, and dimers may be less active.42 It is conceivable that the FRA1 PEST domain fusion impairs HSVtk dimerization formation in a way that promotes HSVtk activity. Of course, further characterization would be necessary to address this possibility.
Figure 8.

Modular design of suicide gene therapy can be adapted to multiple gene-directed enzyme prodrug therapy (suicide gene) pairs, selectively targeting cancer cells with elevated ERK activity
The suicide gene therapy approach outlined in the current study exploits increased ERK activity found in tumor cells, with most of the cell line models studied harboring activating mutations in EGFR or KRAS. Analogous FRA1 domain fusion proteins across two suicide gene products, HSVtk and yeast cytosine deaminase (yCD), were demonstrated to have similar differential cytotoxic effects between cells with high or low ERK activation.
In the current study, our gene therapy was exclusively delivered to cells via replication-incompetent retrovirus. While there are several advantages to retroviral delivery, including selective infection of dividing cells, the suicide gene could be packaged into virtually any viral vector. Perhaps the most actively investigated commercial vectors are recombinant adeno-associated viruses (rAAVs), due to their limited immunogenicity and ease of manufacturing at scale.43 The primary limitation of rAAVs is the upper limit on size of packaged insert (<5 kb), but this would not be a concern for the NLS-HSVtk-FIRE gene (∼1.5 kb). The size of NLS-HSVtk-FIRE is also compatible with self-complementary AAVs, which have lower packing limits (∼3.3 kb) but yield increased transgene expression. Another characteristic of rAAVs is the relatively low frequency of transgene integration in host cell genomes. While the resultant limited duration of transgene expression can be desirable for some rAAV therapies, it may limit the window to treat patients with an HSVtk/GCV-based approach to one shorter than is ideal. Adenoviral vectors similarly have been used to deliver suicide gene therapies, with a number of ongoing clinical trials using HSVtk (e.g., NCT03603405, NCT03004183). NLS-HSVtk-FIRE is also compatible with packaging by adenovirus (limit of ∼8.5 kb). However, unlike AAV, adenoviruses often elicit an immune response, potentially generating undesirable inflammatory responses in patients.44 Despite the safety issues related to host cell genome integration, retroviral-based suicide gene therapies are in commercial development. In particular, Tocagen has developed a retroviral replicating vector that delivers an engineered version of yCD, which has been tested in GBM clinical trials.45,46 Ultimately, further testing and optimization, including use of preclinical orthotopic models with an intact immune response, will be necessary to determine the most translationally relevant vector for the novel transgene design outlined here.
The efficacy of ERK-dependent suicide gene therapy will also be impacted by limitations of viral delivery systems as a whole. Experiments testing the intracranial delivery of our vector suggested that only a fraction of the tumor was successfully transduced, potentially due to limited transport of virus particles into the tissue. A high degree of locally diffuse spreading of tumors such as GBM (which may not even be fully recapitulated in the orthotopic model system used in this study) may exacerbate potential transport issues, especially for non-replicative viruses. Adapting this therapy for use in replication-competent viruses or using it in combination with methods such as convection-enhanced delivery47 could improve efficacy for intracranial applications. The trade-off between increased viral propagation and safety could be rebalanced by combining the NLS-HSVtk-FIRE gene insert design with existing approaches for transcriptionally based or viral tropism-based selectivity. Of course, the bystander effect reported for suicide gene therapy approaches (e.g., diffusion of phosphorylated GCV to neighboring cells not expressing NLS-HSVtk-FIRE) could also help to overcome limitations of tumor cells transduction by viruses but would at the same time limit selectivity.48,49
Therapies targeting RTK/RAS signaling (including effectors such as RAF) often fail due to reactivation of ERK signaling.50,51 Even strategies to directly inhibit ERK (e.g., the small molecule SCH772984) may fail to durably suppress ERK signaling due to the possibility of ERK mutations that impair drug binding.52,53 The NLS-HSVtk-FIRE suicide gene may provide an alternative adjuvant approach to combat this potential resistance mechanism because our design does not perturb activation of ERK directly. Furthermore, tumor-infiltrating immune cells that rely on ERK activation for proper cell function54 would be spared by combining the ERK-dependent suicide gene with cancer cell-specific viral tropism, which is not possible with freely diffusible inhibitors of MEK/ERK.
Materials and methods
Cell culture and reagents
U87MG cells expressing EGFRvIII (Dr. Frank Furnari) were maintained as adherent cultures in DMEM supplemented with 10% fetal bovine serum (VWR), 1 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). G88 and G34 GICs (Dr. Jakub Godlewski), both of which express endogenous EGFRvIII,55 were maintained in either suspension cultures in Neurobasal Medium with B27 and N-2 supplements (Gibco) with 50 ng/mL human recombinant EGF (PeproTech) and human recombinant basic FGF (PeproTech) or as adherent cultures in conditions identical to NSCs when used for co-culture comparisons. NSCs (Dr. Dimitris Placantonakis) were derived from human embryonic stem cells as described previously31 and maintained as adherent cultures on laminin (Gibco)-coated tissue culture plates in DMEM-F12 containing 2.5 mM L-glutamine and HEPES buffer, supplemented with B27 and N-2 supplements (Gibco), 1.6 g/mL glucose, 20 μg/mL bovine insulin, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco), with 50 ng/mL human recombinant EGF (PeproTech) and human recombinant basic FGF (PeproTech). Normal human pancreatic ductal epithelial cells (Dr. Kimberly Kelly) and HPAF-II (Dr. Carl June) cells were maintained as adherent cultures in RPMI supplemented with 10% fetal bovine serum (VWR), 1 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco). PD3077 and PD7242 (Dr. Ben Stanger) cells were maintained in DMEM supplemented with 10% fetal bovine serum, 1 mM L-glutamine, 2 mM GlutaMAX, 0.086 mg/mL gentamicin, 100 units/mL penicillin, and 100 μg/mL streptomycin (Gibco).
Plasmids and suicide gene vector cloning
HSV1TK (Dr. Maria Castro, Addgene #22053) and FCY1 (Dr. John McCusker, Addgene #35104) genes were isolated by PCR of plasmid DNA and inserted into the retroviral pMSCV vector using standard bacterial cloning techniques. A comprehensive list of variants of these vectors is listed in Table S1, with source of genetic material for N-terminal tags (e.g., NLS and 3×FLAG) and PEST domains referenced. The S2D mutant FRA1 domain was generated by Q5 site-directed mutagenesis (New England Biolabs) using the manufacturer’s recommendations. Final versions of cloned plasmids were verified by Sanger sequencing (Eurofins). pMSCV-NLS-mVenus-FIRE-puro and pMSCV-NLS-mCerulean-d2-puro vectors,17 the vectors primarily used as a starting point for cloning the suicide gene vectors in this study, were generously provided by Dr. John Albeck. Retroviral plasmids pBABE-Cerulean-hygro and pBABE-mRFP1-hygro (Dr. Kevin Janes) were used to generate NSCs and G88 cells expressing fluorescent proteins. Retroviral plasmid pBABE-KRAS-G12V-puro (Dr. Channing Der, Addgene #12544) was used to generate human pancreatic epithelial cells expressing mutant KRAS. The retroviral plasmid pBABE-MEK1DD-hygro was created by standard subcloning procedures from pBABE-MEK1DD-puro56 (Dr. William Hahn) and used to generate U87MG cells expressing constitutively active MEK. Oligonucleotides for ERK2-targeting shRNA (targeting sequence: 5′-TTTAAGATCTGTATCCTGGCTG-3′) were cloned into the lentiviral plasmid LT3REPIR (Dr. Scott Lowe) and used to generate cells with doxycycline-inducible shRNA expression with simultaneous induction of dsRed expression. Non-targeting control shRNA was cloned into lentiviral plasmid TET-pLKO-control shRNA (Dr. Dmitri Wiederschain, Addgene #21915), which lacks inducible RFP expression, and used to generate U87MG cells for admixed experiments. EGFRvIII shRNA (targeting sequence: 5′-GAAAGGTAATTATGTGGTG-3′) and matched control shRNA-expressing retroviral plasmids were provided by Dr. Frank Furnari and described previously.57
Viral production, concentration, and cellular selection
Lentivirus was produced by calcium phosphate-mediated co-transfection of LentiX 293T (Takara Bio) cells with lentiviral plasmids (LT3REPIR, TET-pLKO-control shRNA) and packaging vectors pCMV-VSVg, pMDL-gp-RRE, and pRSV-Rev. Virus was harvested 48 h post-transfection and used to infect target cells. Retrovirus was similarly produced by calcium phosphate-mediated transfection of LentiX 293T with retroviral plasmids (pBABE, pMSCV) and packaging vectors pCMV-VSVg and pUMVC. Virus was harvested 48 h post-transfection and used to infect target cells. For transduction of GICs, LentiX 293T cells were cultured in serum-free medium. For intracranial injections of concentrated retrovirus, supernatant was concentrated by ultracentrifugation at 10,000 rpm and 4 h at 4°C, using 10% sucrose-cushioned tubes in a Beckman ultracentrifuge equipped with a SW-28 rotor. Cells transduced with virus encoding for antibiotic resistance were selected in 0.5-2 μg/mL puromycin or 350-500 μg/mL G418 (Gemini Biosciences). G88 cells transduced with virus were selected by fluorescence-activated cell sorting (FACS) with a FACSAria Fusion cell sorter (BD Biosciences).
Antibodies
Antibodies against pERK (Thr 202/Tyr 204, #4377), ERK (#4695), pFRA1 (Ser 265, #5841), pH2A.X (Ser 139, #9718), FLAG (#8146), pEGFR (Tyr 1068, #3777), EGFR (#2232), and PARP (#9542) were purchased from Cell Signaling Technology. Antibodies against GAPDH (sc-32233) and vimentin (sc-37373) were purchased from Santa Cruz Biotechnology. The antibody against GFP (ab6673) was purchased from Abcam. The antibody against E-cadherin (13-1900) was purchased from Invitrogen. The antibody against yCD (2485-4906) was purchased from Bio Rad. The antibody against active caspase-3 (559565) was purchased from BD Biosciences. The antibody against dsRed (632496, Takara Bio) was a gift from Dr. Patrice Guyenet. Infrared dye-conjugated secondary antibodies for western blotting were purchased from Rockland Immunochemicals (anti-mouse IgG IRDye700 conjugated, 610-130-121; anti-rabbit IgG DyLight800 conjugated, 611-145-002). Anti-rabbit, -mouse, and -goat secondary antibodies conjugated to Alexa Fluor 488, 546, or 647 for immunofluorescence microscopy or immunohistochemistry were purchased from Life Technologies. All antibodies were used according to manufacturers’ recommendations.
Inhibitors and growth factors
CI-1040 (LC Laboratories), trametinib (ApexBio), GCV (ApexBio), 5-fluorocytosine (Alfa Aesar), gefitinib (LC Laboratories), PHA665752 (Santa Cruz Biotechnology), and MG132 (EMD Millipore) were reconstituted in DMSO for use in vitro. Abraxane (Dr. Steven Albelda) and doxycycline (Sigma) were reconstituted in sterile, ultra-pure water for use in vitro. Recombinant human EGF and FGF2 (PeproTech) for treating GBM cells were reconstituted according to manufacturer’s recommendation and used at a final concentration of 50 ng/mL, unless otherwise indicated. Recombinant human TGF-β and HGF (PeproTech) for treating PDAC cells were reconstituted according to manufacturer’s recommendation and used at final concentrations of 10 ng/mL and 50 ng/mL, respectively.
Western blotting
Whole-cell lysates were prepared, and western blotting was performed as described previously.37 Nitrocellulose membranes were imaged on a LI-COR Odyssey CLx system. Densitometry measurements were calculated using Image Studio software version 5.2.5.
Reverse transcription PCR (RT-PCR)
RNA was extracted from cells using the RNeasy kit (QIAGEN). Equal amounts of RNA were reverse transcribed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems). For qualitative RT-PCR to detect presence or absence of HSVTK, PCR products were run on a 1% agarose gel stained with ethidium bromide and imaged with an Alpha Innotech gel documentation station. Quantitative RT-PCR (qRT-PCR) was performed using SYBR Green PCR Master Mix (Applied Biosystems) on a QuantStudio 3 or StepOnePlus Real-Time PCR System. Relative amounts of mRNA were determined using the comparative CT method using GAPDH as an endogenous reference. Primer sequences are listed in Table S2.
Immunofluorescence microscopy
Cells were plated on glass coverslips and maintained in 6-well dishes. After indicated treatments, cells were fixed in 4% paraformaldehyde in PBS for 20 min, followed by permeabilization with 0.25% Triton X-100 in PBS for 5 min, and stained overnight at 4°C with antibodies at the manufacturer’s recommended dilution. The next day, samples were washed and stained with secondary antibodies and the DNA dye Hoechst (Life Technologies). Samples were imaged using a Zeiss AxioObserver Z1 widefield microscope with a 10×, 20×, 40×, or 63× objective and Axiocam 506 digital camera. To quantify subcellular localization of immunofluorescence images, nuclei were selected using the Hoechst signal as a mask in the ImageJ software. Similarly, total cell area was selected using the GFP signal as a mask. The integrated density of each signal within these regions, reported per cell, was used to calculate nuclear intensities or ratios of nuclear to total intensities. Unless otherwise indicated, quantified intensities or ratio of intensities were reported for n > 100 cells, across three biological replicates, for each condition.
Flow cytometry and FACS
For cell death assays, cells were plated in 6- or 24-well dishes and treated with inhibitors. At the end of an experiment, floating and adherent cells were collected, resuspended in PBS containing TO-PRO-3 (Life Technologies), and analyzed within 1 h of collection on a FACSCalibur cytometer (BD Biosciences). Flow sorting of GFP-positive cells was performed by staff at the University of Virginia (UVA) Flow Cytometry core on a FACSAria Fusion cell sorter (BD Biosciences). Experiments sorting populations of cells with approximately equal GFP expression used a half-log gate with overlap across all GFP+ populations, similar to those indicated in the corresponding figures.
Active caspase-3 Luminex assay
Whole-cell lysates were prepared in a standard cell extraction buffer (Life Technologies) supplemented with protease and phosphatase inhibitors (Millipore Sigma). Lysates were cleared by centrifugation at 20,000 × g and 4°C for 10 min, and total protein concentrations were determined by micro-bicinchoninic assay (Thermo Fisher Scientific). The MILLIPLEX Caspase 3 (Active) MAPmate Cell Signaling Assay kit (46-604MAG, Millipore Sigma) was used according to the manufacturer’s recommendations. Multiplexed beads in 96-well plates were measured with a MAGPIX instrument, and median fluorescence intensities were recorded.
Co-culture experiments with NSCs and GICs
NCSs and G88 GICs were engineered to express Cerulean and mRFP1, respectively, using retroviral pBABE vectors. NSC only, G88 only, and a mixed population (4:1, NSC to G88) of cells were seeded in 24-well plates, with each condition plated in triplicate. One day after plating, virus concentrated using Lenti-X Concentrator reagent (Takara Bio) was added to the 24-well plate and incubated with cells for 24 h. After confirming GFP expression by microscopy, cells were treated with GCV or DMSO for 2 days. At the completion of the experiment, the entire well contents were collected and analyzed by flow cytometry using TO-PRO-3 (Thermo Fisher Scientific) to detect dead cells. The autofluorescence of dead cells in the same channel as mRFP1 detection impaired our ability to determine which fluorescent proteins were expressed in the dead cell fraction. For experiments involving cells stained with lipophilic DiI, NSC and G88 cells transduced with FLAG-tagged NLS-HSVtk-FIRE and sorted for equal GFP were incubated for 20 min at 37°C with either 5 μM DiI (Invitrogen) or DMSO as a control. Cells were then washed twice with fresh medium to remove excess dye and plated in 24-well plates. One day after plating, cells were treated with the indicated concentrations of GCV or DMSO for 2 days. At the completion of the experiment, dead cells were washed and aspirated from the 24-well plates. Remaining adherent, live cells were stained with Hoechst diluted in PBS and imaged within 30 min.
Mouse subcutaneous tumor xenografts
2.0 × 106 engineered U87MG cells were injected subcutaneously into each flank of 4- to 8-week-old female, NU/NU mice (Charles River). For experiments comparing addition of NLS or FRA1 domain to HSVtk, animals were randomly assigned to groups of GCV or vehicle treatment, once tumors reached an average volume of 50 mm3. GCV was resuspended in sterile PBS and administered daily at 50 mg/kg by intraperitoneal injection. For experiments with doxycycline-inducible ERK2 shRNA expression, animals were randomly assigned to groups receiving 1 mg/mL doxycycline in their drinking water or normal drinking water once tumors reached an average volume of 100 mm3. After 3 days of doxycycline administration, an IVIS Spectrum imager was used to measure dsRed expression to confirm appropriate induction of ERK2 shRNA. GCV was then administered daily at 50 mg/kg by intraperitoneal injection. For all subcutaneous xenografts, tumors were measured with calipers before and during treatment, and tumor volumes were calculated as π/6 × A × B2, where A and B are the larger and smaller tumor diameters, respectively. Excised tumors were homogenized in lysis buffer before western blotting, as described previously, or fixed in 10% formalin prior to paraffin embedding. All experiments were approved by the UVA Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
Intracranial tumor xenografts and in situ retrovirus delivery
750 G34 cells were stereotactically injected into the right striatum of 4- to 8-week-old, female BALB/c SCID mice. One week after tumor cell injections, animals were randomly assigned to groups receiving stereotactic injections of concentrated EV or NLS-HSVtk-FIRE virus, each using versions of transgene inserts encoding for IRES-GFP (N = 11 EV; N = 10 NLS-HSVtk-FIRE). After recovery from the second surgical procedure, animals were randomly assigned to treatment groups of GCV (N = 8 or 9) or vehicle (N= 2). Drugs were prepared at concentrations listed above for subcutaneous experiments. Animals were treated daily for at least 3 days prior to euthanasia. Harvested mouse brains were fixed in 10% formalin and paraffin-embedded. Sections from all animals were stained with hematoxylin and eosin (H&E), and those with detectable tumors (N= 7/11 EV, N = 9/10 NLS-HSVtk-FIRE) were analyzed by immunohistochemistry using antibodies against GFP (Abcam, ab6673) and pH2A.X (Cell Signaling Technology, #9718), plus Hoechst DNA stain. All experiments were approved by the UVA Institutional Animal Care and Use Committee and performed in accordance with NIH guidelines.
Immunohistochemistry
Charged slides with sections from formalin-fixed, paraffin-embedded tissues were generated by the Research Histology Core at UVA. Antigen-retrieved tissues, prepared by the Biorepository and Tissue Research Facility at UVA, were permeabilized with 0.10% Triton X-100 in PBS for 15 min, blocked for 1 h at room temperature, and stained overnight at 4°C with antibodies at the manufacturer’s recommended dilution. The next day, samples were washed and stained with secondary antibodies conjugated to Alexa Fluor dyes and the DNA dye Hoechst (Life Technologies). Antibody-stained sections were imaged using a Zeiss AxioObserver Z1 widefield microscope with a 5×, 10×, or 20× objective and Axiocam 506 digital camera. H&E-stained slides were imaged using a BioTek Cytation 5 Cell Imaging Multi-Mode Reader with a 5× objective.
GCV and 5-fluorocytosine titration MTS and cell number assays
Cells expressing HSVtk, yCD, or indicated fusion protein versions of these enzymes, were seeded in 96-well plates. For MTS assays, cells treated with GCV or 5-fluorocytosine were incubated with medium including the MTS reagent (Promega) at the manufacturer-recommended concentration. Conversion of the MTS reagent into a soluble formazan product was allowed to proceed for 1–4 h, after which time absorbance at 490 nm was measured. To ensure signal linearity with respect to time, the absorbance was measured at three different times. Values from the median time point were reported. For cell number assays, cells treated with GCV were washed and incubated with TO-PRO-3 diluted in 0.25% Triton X-100 in PBS. 96-well plates were scanned using a LI-COR Odyssey CLx system. Densitometry measurements (for TO-PRO-3 signal) were calculated using Image Studio software version 5.2.5 and normalized to DMSO-treated conditions.
Statistical analysis
Statistical comparisons between two groups/conditions were made using a two-tailed Student’s t test calculated in Microsoft Excel, where significance was determined by a p value < 0.05. Statistical analyses for higher-order comparisons were made using one- or two-way ANOVA performed using GraphPad Prism, with reported p values calculated by Tukey’s post hoc comparisons. Significance was determined by a p value < 0.05. Details of all statistical comparisons, including values for n, are found in the figure legends. Unless stated otherwise in the figure legend, all data are representative of results obtained from three independent replicates. Figure legends also list definitions for dispersion and precision measurements, stating error bar definitions throughout.
Acknowledgments
We thank the following core facilities at the University of Virginia (UVA) for assistance: Flow Cytometry Core Facility, Molecular Imaging Core, Research Histology Core, and Biorepository and Tissue Research Facility. We also thank Robert Norgard for cloning the LT3REPIR vector encoding inducible ERK2 shRNA and Alice Walsh for cloning the hygromycin-selectable version of pBABE-MEK1DD. This work was supported by UVA Coulter Foundation Translational Research Fund, National Science Foundation CBET grant no. 1511853 (M.J.L.); American Cancer Society Research Scholar grant no. RSG-15-010-01-CDD (M.J.L.); National Cancer Institute 1R21CA252576-01 (M.J.L. and B.P.); NSF GRFP grant no. DGE-1321851 (E.K.D.); and UVA Cancer Center Support Grant from the National Cancer Institute (P30CA044579).
Author contributions
Conceptualization, E.K.D., B.P., and M.J.L.; Investigation, E.K.D., A.C., A.P., T.R., A.X., and Q.Z.; Writing – Original Draft, E.K.D. and M.J.L.; Writing – Review & Editing, E.K.D., B.P., and M.J.L.; Funding Acquisition, E.K.D., B.P., and M.J.L.; Supervision, B.P. and M.J.L.
Declaration of interests
E.K.D., B.P., and M.J.L. have filed a non-provisional U.S. patent application (Ser. No. 16/669,191) related to the ERK-dependent suicide gene, and adaptations thereof, outlined in this work.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.019.
Supplemental information
References
- 1.Greco O., Dachs G.U. Gene directed enzyme/prodrug therapy of cancer: historical appraisal and future prospectives. J. Cell. Physiol. 2001;187:22–36. doi: 10.1002/1097-4652(2001)9999:9999<::AID-JCP1060>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 2.Klatzmann D., Valéry C.A., Bensimon G., Marro B., Boyer O., Mokhtari K., Diquet B., Salzmann J.L., Philippon J., Study Group on Gene Therapy for Glioblastoma A phase I/II study of herpes simplex virus type 1 thymidine kinase “suicide” gene therapy for recurrent glioblastoma. Hum. Gene Ther. 1998;9:2595–2604. doi: 10.1089/hum.1998.9.17-2595. [DOI] [PubMed] [Google Scholar]
- 3.Rainov N.G. A phase III clinical evaluation of herpes simplex virus type 1 thymidine kinase and ganciclovir gene therapy as an adjuvant to surgical resection and radiation in adults with previously untreated glioblastoma multiforme. Hum. Gene Ther. 2000;11:2389–2401. doi: 10.1089/104303400750038499. [DOI] [PubMed] [Google Scholar]
- 4.Thomas C.E., Ehrhardt A., Kay M.A. Progress and problems with the use of viral vectors for gene therapy. Nat. Rev. Genet. 2003;4:346–358. doi: 10.1038/nrg1066. [DOI] [PubMed] [Google Scholar]
- 5.Saukkonen K., Hemminki A. Tissue-specific promoters for cancer gene therapy. Expert Opin. Biol. Ther. 2004;4:683–696. doi: 10.1517/14712598.4.5.683. [DOI] [PubMed] [Google Scholar]
- 6.Robson T., Hirst D.G. Transcriptional Targeting in Cancer Gene Therapy. J. Biomed. Biotechnol. 2003;2003:110–137. doi: 10.1155/S1110724303209074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pandha H.S., Martin L.A., Rigg A., Hurst H.C., Stamp G.W., Sikora K., Lemoine N.R. Genetic prodrug activation therapy for breast cancer: A phase I clinical trial of erbB-2-directed suicide gene expression. J. Clin. Oncol. 1999;17:2180–2189. doi: 10.1200/JCO.1999.17.7.2180. [DOI] [PubMed] [Google Scholar]
- 8.Karjoo Z., Chen X., Hatefi A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv. Drug Deliv. Rev. 2016;99(Pt A):113–128. doi: 10.1016/j.addr.2015.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., Cancer Genome Atlas Research Network Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell. 2018;173:321–337.e10. doi: 10.1016/j.cell.2018.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gschwind A., Fischer O.M., Ullrich A. The discovery of receptor tyrosine kinases: targets for cancer therapy. Nat. Rev. Cancer. 2004;4:361–370. doi: 10.1038/nrc1360. [DOI] [PubMed] [Google Scholar]
- 11.Downward J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 2003;3:11–22. doi: 10.1038/nrc969. [DOI] [PubMed] [Google Scholar]
- 12.Linardou H., Dahabreh I.J., Bafaloukos D., Kosmidis P., Murray S. Somatic EGFR mutations and efficacy of tyrosine kinase inhibitors in NSCLC. Nat. Rev. Clin. Oncol. 2009;6:352–366. doi: 10.1038/nrclinonc.2009.62. [DOI] [PubMed] [Google Scholar]
- 13.Engelman J.A., Zejnullahu K., Mitsudomi T., Song Y., Hyland C., Park J.O., Lindeman N., Gale C.M., Zhao X., Christensen J. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science. 2007;316:1039–1043. doi: 10.1126/science.1141478. [DOI] [PubMed] [Google Scholar]
- 14.Engelman J.A., Jänne P.A. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin. Cancer Res. 2008;14:2895–2899. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 15.Liu F., Yang X., Geng M., Huang M. Targeting ERK, an Achilles’ Heel of the MAPK pathway, in cancer therapy. Acta Pharm. Sin. B. 2018;8:552–562. doi: 10.1016/j.apsb.2018.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ryan M.B., Der C.J., Wang-Gillam A., Cox A.D. Targeting RAS-mutant cancers: is ERK the key? Trends Cancer. 2015;1:183–198. doi: 10.1016/j.trecan.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albeck J.G., Mills G.B., Brugge J.S. Frequency-modulated pulses of ERK activity transmit quantitative proliferation signals. Mol. Cell. 2013;49:249–261. doi: 10.1016/j.molcel.2012.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Basbous J., Chalbos D., Hipskind R., Jariel-Encontre I., Piechaczyk M. Ubiquitin-independent proteasomal degradation of Fra-1 is antagonized by Erk1/2 pathway-mediated phosphorylation of a unique C-terminal destabilizer. Mol. Cell. Biol. 2007;27:3936–3950. doi: 10.1128/MCB.01776-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fillat C., Carrió M., Cascante A., Sangro B. Suicide gene therapy mediated by the Herpes Simplex virus thymidine kinase gene/Ganciclovir system: fifteen years of application. Curr. Gene Ther. 2003;3:13–26. doi: 10.2174/1566523033347426. [DOI] [PubMed] [Google Scholar]
- 20.Pham N.A., Schwock J., Iakovlev V., Pond G., Hedley D.W., Tsao M.S. Immunohistochemical analysis of changes in signaling pathway activation downstream of growth factor receptors in pancreatic duct cell carcinogenesis. BMC Cancer. 2008;8:43. doi: 10.1186/1471-2407-8-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hutter G., Sailer M., Azad T.D., von Bueren A.O., Nollau P., Frank S., Tostado C., Sarvepalli D., Ghosh A., Ritz M.F. Reverse phase protein arrays enable glioblastoma molecular subtyping. J. Neurooncol. 2017;131:437–448. doi: 10.1007/s11060-016-2316-5. [DOI] [PubMed] [Google Scholar]
- 22.Lorimer I.A., Lavictoire S.J. Activation of extracellular-regulated kinases by normal and mutant EGF receptors. Biochim. Biophys. Acta. 2001;1538:1–9. doi: 10.1016/s0167-4889(00)00129-4. [DOI] [PubMed] [Google Scholar]
- 23.Brennan C.W., Verhaak R.G., McKenna A., Campos B., Noushmehr H., Salama S.R., Zheng S., Chakravarty D., Sanborn J.Z., Berman S.H., TCGA Research Network The somatic genomic landscape of glioblastoma. Cell. 2013;155:462–477. doi: 10.1016/j.cell.2013.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degrève B., Johansson M., De Clercq E., Karlsson A., Balzarini J. Differential intracellular compartmentalization of herpetic thymidine kinases (TKs) in TK gene-transfected tumor cells: molecular characterization of the nuclear localization signal of herpes simplex virus type 1 TK. J. Virol. 1998;72:9535–9543. doi: 10.1128/jvi.72.12.9535-9543.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang D., Wu D., Hirao A., Lahti J.M., Liu L., Mazza B., Kidd V.J., Mak T.W., Ingram A.J. ERK activation mediates cell cycle arrest and apoptosis after DNA damage independently of p53. J. Biol. Chem. 2002;277:12710–12717. doi: 10.1074/jbc.M111598200. [DOI] [PubMed] [Google Scholar]
- 26.Wei F., Yan J., Tang D. Extracellular signal-regulated kinases modulate DNA damage response - a contributing factor to using MEK inhibitors in cancer therapy. Curr. Med. Chem. 2011;18:5476–5482. doi: 10.2174/092986711798194388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Golding S.E., Rosenberg E., Neill S., Dent P., Povirk L.F., Valerie K. Extracellular signal-related kinase positively regulates ataxia telangiectasia mutated, homologous recombination repair, and the DNA damage response. Cancer Res. 2007;67:1046–1053. doi: 10.1158/0008-5472.CAN-06-2371. [DOI] [PubMed] [Google Scholar]
- 28.Gilbertson R.J., Rich J.N. Making a tumour’s bed: glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer. 2007;7:733–736. doi: 10.1038/nrc2246. [DOI] [PubMed] [Google Scholar]
- 29.Sanai N., Alvarez-Buylla A., Berger M.S. Neural stem cells and the origin of gliomas. N. Engl. J. Med. 2005;353:811–822. doi: 10.1056/NEJMra043666. [DOI] [PubMed] [Google Scholar]
- 30.Koso H., Takeda H., Yew C.C., Ward J.M., Nariai N., Ueno K., Nagasaki M., Watanabe S., Rust A.G., Adams D.J. Transposon mutagenesis identifies genes that transform neural stem cells into glioma-initiating cells. Proc. Natl. Acad. Sci. USA. 2012;109:E2998–E3007. doi: 10.1073/pnas.1215899109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Modrek A.S., Golub D., Khan T., Bready D., Prado J., Bowman C., Deng J., Zhang G., Rocha P.P., Raviram R. Low-Grade Astrocytoma Mutations in IDH1, P53, and ATRX Cooperate to Block Differentiation of Human Neural Stem Cells via Repression of SOX2. Cell Rep. 2017;21:1267–1280. doi: 10.1016/j.celrep.2017.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Day E.K., Sosale N.G., Xiao A., Zhong Q., Purow B., Lazzara M.J. Glioblastoma Cell Resistance to EGFR and MET Inhibition Can Be Overcome via Blockade of FGFR-SPRY2 Bypass Signaling. Cell Rep. 2020;30:3383–3396.e7. doi: 10.1016/j.celrep.2020.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walsh A.M., Kapoor G.S., Buonato J.M., Mathew L.K., Bi Y., Davuluri R.V., Martinez-Lage M., Simon M.C., O’Rourke D.M., Lazzara M.J. Sprouty2 Drives Drug Resistance and Proliferation in Glioblastoma. Mol. Cancer Res. 2015;13:1227–1237. doi: 10.1158/1541-7786.MCR-14-0183-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Furcht C.M., Buonato J.M., Skuli N., Mathew L.K., Muñoz Rojas A.R., Simon M.C., Lazzara M.J. Multivariate signaling regulation by SHP2 differentially controls proliferation and therapeutic response in glioma cells. J. Cell Sci. 2014;127:3555–3567. doi: 10.1242/jcs.150862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zheng X., Carstens J.L., Kim J., Scheible M., Kaye J., Sugimoto H., Wu C.C., LeBleu V.S., Kalluri R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature. 2015;527:525–530. doi: 10.1038/nature16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fischer K.R., Durrans A., Lee S., Sheng J., Li F., Wong S.T., Choi H., El Rayes T., Ryu S., Troeger J. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature. 2015;527:472–476. doi: 10.1038/nature15748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Buonato J.M., Lazzara M.J. ERK1/2 blockade prevents epithelial-mesenchymal transition in lung cancer cells and promotes their sensitivity to EGFR inhibition. Cancer Res. 2014;74:309–319. doi: 10.1158/0008-5472.CAN-12-4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Buonato J.M., Lan I.S., Lazzara M.J. EGF augments TGFβ-induced epithelial-mesenchymal transition by promoting SHP2 binding to GAB1. J. Cell Sci. 2015;128:3898–3909. doi: 10.1242/jcs.169599. [DOI] [PubMed] [Google Scholar]
- 39.Aiello N.M., Maddipati R., Norgard R.J., Balli D., Li J., Yuan S., Yamazoe T., Black T., Sahmoud A., Furth E.E. EMT Subtype Influences Epithelial Plasticity and Mode of Cell Migration. Dev. Cell. 2018;45:681–695.e4. doi: 10.1016/j.devcel.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wright C.M., Wright R.C., Eshleman J.R., Ostermeier M. A protein therapeutic modality founded on molecular regulation. Proc. Natl. Acad. Sci. USA. 2011;108:16206–16211. doi: 10.1073/pnas.1102803108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cho S.H., Oh B., Kim H.A., Park J.H., Lee M. Post-translational regulation of gene expression using the ATF4 oxygen-dependent degradation domain for hypoxia-specific gene therapy. J. Drug Target. 2013;21:830–836. doi: 10.3109/1061186X.2013.829073. [DOI] [PubMed] [Google Scholar]
- 42.Wurth C., Thomas R.M., Folkers G., Scapozza L. Folding and self-assembly of herpes simplex virus type 1 thymidine kinase. J. Mol. Biol. 2001;313:657–670. doi: 10.1006/jmbi.2001.5060. [DOI] [PubMed] [Google Scholar]
- 43.Naso M.F., Tomkowicz B., Perry W.L., 3rd, Strohl W.R. Adeno-Associated Virus (AAV) as a Vector for Gene Therapy. BioDrugs. 2017;31:317–334. doi: 10.1007/s40259-017-0234-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dewey R.A., Morrissey G., Cowsill C.M., Stone D., Bolognani F., Dodd N.J., Southgate T.D., Klatzmann D., Lassmann H., Castro M.G., Löwenstein P.R. Chronic brain inflammation and persistent herpes simplex virus 1 thymidine kinase expression in survivors of syngeneic glioma treated by adenovirus-mediated gene therapy: implications for clinical trials. Nat. Med. 1999;5:1256–1263. doi: 10.1038/15207. [DOI] [PubMed] [Google Scholar]
- 45.Cloughesy T.F., Landolfi J., Vogelbaum M.A., Ostertag D., Elder J.B., Bloomfield S., Carter B., Chen C.C., Kalkanis S.N., Kesari S. Durable complete responses in some recurrent high-grade glioma patients treated with Toca 511 + Toca FC. Neuro-oncol. 2018;20:1383–1392. doi: 10.1093/neuonc/noy075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cloughesy T.F., Landolfi J., Hogan D.J., Bloomfield S., Carter B., Chen C.C., Elder J.B., Kalkanis S.N., Kesari S., Lai A. Phase 1 trial of vocimagene amiretrorepvec and 5-fluorocytosine for recurrent high-grade glioma. Sci. Transl. Med. 2016;8:341ra75. doi: 10.1126/scitranslmed.aad9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Allard E., Passirani C., Benoit J.P. Convection-enhanced delivery of nanocarriers for the treatment of brain tumors. Biomaterials. 2009;30:2302–2318. doi: 10.1016/j.biomaterials.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 48.van Dillen I.J., Mulder N.H., Vaalburg W., de Vries E.F., Hospers G.A. Influence of the bystander effect on HSV-tk/GCV gene therapy. A review. Curr. Gene Ther. 2002;2:307–322. doi: 10.2174/1566523023347733. [DOI] [PubMed] [Google Scholar]
- 49.Tanaka T., Duflot-Dancer A., Tiraby M., Piccoli C., Tiraby G., Yamasaki H., Mesnil M. Bystander effect from cytosine deaminase and uracil phosphoribosyl transferase genes in vitro: a partial contribution of gap junctions. Cancer Lett. 2009;282:43–47. doi: 10.1016/j.canlet.2009.02.050. [DOI] [PubMed] [Google Scholar]
- 50.Samatar A.A., Poulikakos P.I. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat. Rev. Drug Discov. 2014;13:928–942. doi: 10.1038/nrd4281. [DOI] [PubMed] [Google Scholar]
- 51.Poulikakos P.I., Zhang C., Bollag G., Shokat K.M., Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morris E.J., Jha S., Restaino C.R., Dayananth P., Zhu H., Cooper A., Carr D., Deng Y., Jin W., Black S. Discovery of a novel ERK inhibitor with activity in models of acquired resistance to BRAF and MEK inhibitors. Cancer Discov. 2013;3:742–750. doi: 10.1158/2159-8290.CD-13-0070. [DOI] [PubMed] [Google Scholar]
- 53.Jha S., Morris E.J., Hruza A., Mansueto M.S., Schroeder G.K., Arbanas J., McMasters D., Restaino C.R., Dayananth P., Black S. Dissecting Therapeutic Resistance to ERK Inhibition. Mol. Cancer Ther. 2016;15:548–559. doi: 10.1158/1535-7163.MCT-15-0172. [DOI] [PubMed] [Google Scholar]
- 54.Vella L.J., Pasam A., Dimopoulos N., Andrews M., Knights A., Puaux A.L., Louahed J., Chen W., Woods K., Cebon J.S. MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells. Cancer Immunol. Res. 2014;2:351–360. doi: 10.1158/2326-6066.CIR-13-0181. [DOI] [PubMed] [Google Scholar]
- 55.Mineo M., Ricklefs F., Rooj A.K., Lyons S.M., Ivanov P., Ansari K.I., Nakano I., Chiocca E.A., Godlewski J., Bronisz A. The Long Non-coding RNA HIF1A-AS2 Facilitates the Maintenance of Mesenchymal Glioblastoma Stem-like Cells in Hypoxic Niches. Cell Rep. 2016;15:2500–2509. doi: 10.1016/j.celrep.2016.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Boehm J.S., Zhao J.J., Yao J., Kim S.Y., Firestein R., Dunn I.F., Sjostrom S.K., Garraway L.A., Weremowicz S., Richardson A.L. Integrative genomic approaches identify IKBKE as a breast cancer oncogene. Cell. 2007;129:1065–1079. doi: 10.1016/j.cell.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 57.Mukasa A., Wykosky J., Ligon K.L., Chin L., Cavenee W.K., Furnari F. Mutant EGFR is required for maintenance of glioma growth in vivo, and its ablation leads to escape from receptor dependence. Proc. Natl. Acad. Sci. USA. 2010;107:2616–2621. doi: 10.1073/pnas.0914356107. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.