

Abstract
Ex vivo gene correction of hematopoietic stem and progenitor cells (HSPCs) has emerged as a promising therapeutic approach for treatment of inherited human blood disorders. Use of engineered nucleases to target therapeutic transgenes to their endogenous genetic loci addresses many of the limitations associated with viral vector-based gene replacement strategies, such as insertional mutagenesis, variable gene dosage, and ectopic expression. Common methods of nuclease-mediated site-specific integration utilize the homology-directed repair (HDR) pathway. However, these approaches are inefficient in HSPCs, where non-homologous end joining (NHEJ) is the primary DNA repair mechanism. Recently, a novel NHEJ-based approach to CRISPR-Cas9-mediated transgene knockin, known as homology-independent targeted integration (HITI), has demonstrated improved site-specific integration frequencies in non-dividing cells. Here we utilize a HITI-based approach to achieve robust site-specific transgene integration in human mobilized peripheral blood CD34+ HSPCs. As proof of concept, a reporter gene was targeted to a clinically relevant genetic locus using a recombinant adeno-associated virus serotype 6 vector and single guide RNA/Cas9 ribonucleoprotein complexes. We demonstrate high levels of stable HITI-mediated genome editing (∼21%) in repopulating HSPCs after transplantation into immunodeficient mice. Our study demonstrates that HITI-mediated genome editing provides an effective alternative to HDR-based transgene integration in CD34+ HSPCs.
Keywords: hematopoietic stem and progenitor cells, HSPCs, gene editing, genome editing, gene therapy, homology-independent targeted integration, HITI, non-homologous end joining, NHEJ, homology-directed repair, HDR, leukocyte adhesion deficiency type 1, LAD-1, adeno-associated virus serotype 6, AAV6, ribonucleoprotein, RNP
Graphical abstract
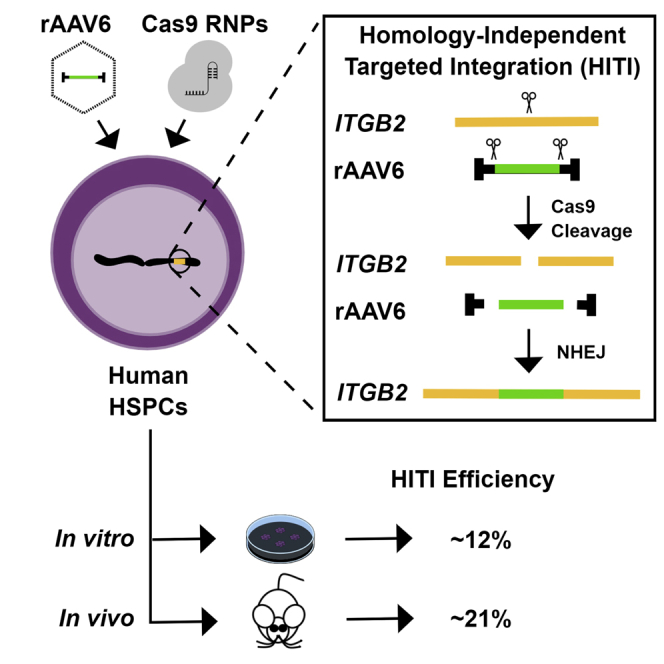
Common methods of genome editing utilize the homology-directed repair (HDR) pathway. However, these approaches are inefficient in human hematopoietic stem and progenitor cells (HSPCs), where non-homologous end joining (NHEJ) is the primary DNA repair mechanism. Bloomer et al. demonstrate stable high-level genome editing (∼21%) in human repopulating HSPCs using a novel NHEJ-based approach known as homology-independent targeted integration (HITI).
Introduction
Transplanted human hematopoietic stem and progenitor cells (HSPCs) have the remarkable ability to reconstitute and maintain a functional blood system for the lifespan of an individual. Allogeneic transplantation of HSPCs is employed in the clinic for therapeutic correction of numerous inherited hematologic, metabolic, and immunologic disorders as well as some forms of hematologic cancer. However, allogeneic stem cell transplantation suffers from significant drawbacks, including limited availability of human leukocyte antigen-matched donors, the need for toxic myeloablative conditioning regimens, and the risk of complications such as graft versus host disease or graft failure.1
Gene therapy approaches utilizing autologous stem cell transplantation, in which a patient’s endogenous HSPCs are isolated, genetically modified ex vivo, and subsequently reinfused, have emerged as an alternative to allogeneic stem cell transplantation. Current ex vivo gene therapy strategies utilize integrating viral vectors, such as retrovirus- and lentivirus-derived vectors, to splice functional copies of a therapeutic transgene into the genome of recipient HSPCs. Although clinically effective,2, 3, 4 the use of integrating viral vectors presents several pitfalls, such as variable gene dosage, the potential for insertional mutagenesis, and, in some instances, ectopic expression.5, 6, 7, 8 For these reasons, alternative approaches to ex vivo gene addition in autologous HSPCs are being developed.
The recent emergence of programmable engineered nuclease technologies is a promising alternative to integrating viral vectors by facilitating site-specific integration of a therapeutic transgene cassette into a defined sequence within the target cell genome.9,10 Such technologies include zinc-finger nucleases (ZFNs), transcription activator-like effector nucleases (TALENs), and the clustered regularly interspaced short palindromic repeats (CRISPR)-CRISPR-associated protein 9 (Cas9) system. With these approaches, a recombinant DNA endonuclease is targeted to a cognate site within the target cell genome to induce a site-specific DNA double-strand break (DSB). The targeted DSB is subsequently repaired by one of two highly conserved DNA repair pathways: non-homologous end joining (NHEJ) or homology-directed repair (HDR).11,12 During the NHEJ process, the DNA DSB is enzymatically processed and ligated, often resulting in small insertions or deletions (indels) at the site of the DSB. In contrast, repair by HDR requires a homologous donor template, such as a sister chromatid. In the presence of an exogenous homologous donor template, DSB-induced HDR can be exploited to introduce precise genetic alterations and/or insertion of homology arm-flanked transgenes.13,14
Recently, several groups have utilized an HDR-based approach to introduce transgenes into human CD34+ HSPCs derived from cord blood (CB) or mobilized peripheral blood (MPB). Although efficient site-specific transgene integration has been reported in cultured HSPCs (up to 50% positive in CB and MPB), there was a significant reduction in the observed frequency of edited cells upon transplantation into immunodeficient mice for analysis of repopulating potential (≤25% positive in CB and ∼10% positive in MPB).15, 16, 17, 18, 19, 20, 21 Although CB-derived HSPCs are efficiently edited ex vivo, MPB-derived HSPCs are a more clinically relevant source of blood stem cells for autologous transplantation. However, utilization of MPB-derived HSPCs for non-viral gene addition strategies and transplantation will require advances in the efficiency of gene addition and expression post-engraftment.
Primary CD34+ HSPCs are quiescent cells in which, because of their protracted G0/G1 phasing, NHEJ is the primary DNA repair pathway.22 We hypothesized that an NHEJ-based approach to gene addition in HSPCs would facilitate efficient site-specific transgene integration in these cells. Recently, a novel NHEJ-based method for CRISPR-Cas9-mediated gene addition, known as homology-independent targeted integration (HITI), has demonstrated improved site-specific transgene integration frequencies in dividing and non-dividing cells of non-hematopoietic tissues.23,24 We developed a HITI-based approach that utilizes a recombinant adeno-associated virus serotype 6 (rAAV6) vector in combination with electroporated single guide RNA (sgRNA)/Cas9 ribonucleoprotein (RNP) complexes to achieve robust, site-specific transgene integration in human MPB CD34+ HSPCs. As proof of principle, we demonstrate efficient HITI-mediated reporter gene integration at a clinically relevant genomic locus in primary human CD34+ HSPCs as well as high-level transgene expression 18 weeks post-transplant within immunodeficient mice.
Results
As a proof of principle, we evaluated the ability of HITI to efficiently insert a reporter gene construct at a clinically relevant genetic locus in human MPB-derived HSPCs. CRISPR-Cas9-mediated DSB formation was used to target an rAAV6-packaged copGFP expression cassette to the first non-coding exon of the ITGB2 gene (Figure 1). Loss-of-function mutations within ITGB2, which encodes the β2 integrin subunit CD18, are associated with an inherited immunodeficiency syndrome known as leukocyte adhesion deficiency type 1 (LAD-1).25 The first non-coding exon of the ITGB2 gene was chosen as a proof-of-principle target site because introduction of a promoterless ITGB2 open reading frame construct at this location would place the therapeutic transgene under transcriptional control of the endogenous ITGB2 promoter and have the potential for phenotypic correction regardless of the nature of downstream mutations in individuals suffering from LAD-1.
Figure 1.

Schematic of HITI-mediated recombination at the ITGB2 locus
Top: the human ITGB2 gene is represented schematically. A 20-nt Cas9 target site, designated ITGB2-ts, was chosen within ITGB2 exon 1 (E1). Target site orientation is indicated by a light blue arrowhead, and scissors denote the Cas9 cleavage site. The relative locations of nested PCR primers flanking the Cas9 target site are indicated by purple arrows. For HITI-mediated recombination, an rAAV6 donor template bearing a CMV promoter-driven copGFP reporter gene flanked by ITGB2-ts sites (target site orientation is indicated by gray arrowheads) was constructed and packaged within rAAV6 capsids. Center: human CD34+ HSPCs are transduced with rAAV6-copGFP and subsequently electroporated with Cas9 effector molecules. In the presence of sgRNA/Cas9 RNP complexes, the ITGB2-ts-flanked reporter cassette and the endogenous ITGB2 target gene are concomitantly cleaved, promoting NHEJ-mediated transgene insertion at the Cas9-induced genomic DNA DSB. Bottom: NHEJ-mediated transgene insertion can yield forward- or reverse-orientation inserts. Reverse-orientation insertion recreates intact ITGB2-ts sites that promote re-cleavage by Cas9. Red arrows represent forward-orientation, 5′ junction-specific PCR primers. Blue arrows represent forward-orientation, 3′ junction-specific PCR primers. Green arrows represent reverse-orientation, 5′ junction-specific PCR primers. Orange arrows represent reverse-orientation, 3′ junction-specific primers.
Efficient HITI-mediated genome editing in vitro
A 20-nt guide RNA target sequence was chosen within ITGB2 exon 1 (Figure 2A). This sequence was fused with an S. pyogenes-specific Cas9 sgRNA scaffold sequence under transcriptional control of the human U6 promoter within the rAAV6-copGFP vector construct. In addition, a 2′-O-methyl 3′-phosphorothioate-modified sgRNA was chemically synthesized. To evaluate the efficiency of DSB formation at the chosen target site, CD34+ HSPCs isolated from five healthy adult donors were electroporated with sgRNA/Cas9 RNPs, and the frequency of targeted indel formation was determined by T7 endonuclease 1 (T7E1) cleavage assay (Figures 2B and 2C). Robust indel formation was observed, with an average indel frequency of 62% among the five donor samples. Cell survival 4 days post-electroporation (EP) was slightly diminished in Cas9- and sgRNA/Cas9 recipient cells compared with mock-electroporated cells; however, average cellular viability remained in excess of 80% (Figure 2D). In addition, deep sequencing of five randomly selected homologous off-target sites (up to 3-base mismatches) showed no evidence of Cas9 promiscuity (Figures 2E and 2F).
Figure 2.

Cas9 target site validation
(A) Schematic of the sgRNA/Cas9 RNP target site in non-coding E1 of the ITGB2 gene. The TSS is represented by a black arrow. The start codon within E2 is indicated by an asterisk. (B) Representative gel image of the T7 endonuclease I (T7EI) cleavage assay for quantification of sgRNA/Cas9 RNP-mediated indels 4 days post-electroporation (EP). Expected amplicon sizes are indicated. (C) Indel frequencies as measured by T7E1 cleavage assay (n = 5 independent donors). (D) Viability of mock-treated (electroporated only), Cas9-treated, and sgRNA/Cas9 RNP-treated cells 4 days post-EP (n = 5 independent donors). (E and F) Off-target analysis of the ITGB2 sgRNA/Cas9 RNP complex. Shown are percent sequencing reads with indels (E) and summary of individual indel size and frequency (F) at the ITGB2 on-target site and five in silico-predicted off-target (OT) sites. Total indel frequencies quantified by T7EI assay (C) and deep sequencing (E) were comparable at the ITGB2 on-target site. No indels were observed at the selected OT sites. Sequences for each target site are displayed in blue font; for OT sites, bases differing from the ITGB2 target sequence are indicated by lowercase letters. Respective protospacer adjacent motif (PAM) sequences (NGG) are highlighted in red font. Dashes represent deleted bases. In (C) and (D), results are displayed as mean ± SEM; ns, not significant by one-way repeated-measures ANOVA tests.
We next evaluated the ability of electroporated Cas9 or sgRNA/Cas9 RNPs to direct integration of an rAAV6-packaged copGFP reporter gene construct at the ITGB2 locus in MPB-derived HSPCs (Figure 3). CD34+ cells were mock-transduced or transduced with rAAV6 2 days prior to EP to allow sufficient time for double-strand DNA conversion of the single-stranded rAAV6 vector genome, a prerequisite for RNP-mediated cleavage. Two days post-transduction, samples were electroporated in the absence of RNPs (designated “rAAV6”), electroporated with Cas9 in the absence of synthetic sgRNA (designated “rAAV6+Cas9”), or electroporated with sgRNA/Cas9 RNP complexes (designated “rAAV6+RNP”) (Figure 3A).
Figure 3.

Efficient HITI-mediated genome editing in human CD34+ HSPCs
(A) Schematic of the experimental design. Human MPB CD34+ HSPCs were thawed and transduced with rAAV6 for 48 h. Cells were either electroporated alone (rAAV6), with Cas9 (rAAV6+Cas9), or with sgRNA/Cas9 RNPs (rAAV6+RNP) and cultured for downstream analyses up to 28 days post-EP. Mock-treated cells were electroporated only. (B) Gel images of in-out PCR to detect forward direction integration at the 5′ and 3′ junction ends 4 days post-EP. Expected PCR product sizes are labeled. (C) Percent GFP-expressing cells up to 28 days post-EP (n = 3 independent donors). Results are displayed as mean ± SEM. (D) Representative flow cytometry plots of copGFP expression on day 4 and day 28 post-EP. For (C) and (D), see also Figure S1.
To detect site-specific integration of the copGFP reporter gene in the forward orientation, we used nested “in-out PCR” to amplify vector-genomic DNA junction sequences at the 5′ and 3′ junctions of the integrated transgene (Figure 3B; see Figure 1, bottom panel for relative primer location). Amplicons of the expected size were observed in CD34+ HSPCs treated with rAAV6+RNP but not in control samples or in HSPCs treated with vector in combination with Cas9 in the absence of synthetic sgRNA (rAAV6+Cas9) (Figure 3B). A lack of detectable site-specific integration in rAAV6+Cas9-treated cells suggests that the timing, stability, or abundance of ITGB2-targeted sgRNA expressed from the rAAV6 vector may be insufficient for promoting HITI and is consistent with previous reports showing superior levels of genome editing in HSPCs using RNPs.19,26 Accordingly, subsequent experiments evaluating HITI in MPB-derived HSPCs utilized the rAAV6+RNP protocol.
To evaluate the persistence of transgene expression in CD34+ HSPCs edited by HITI, treated cells were cultured up to 28 days post-EP and periodically sampled for flow cytometry analysis of copGFP expression (Figures 3C and 3D). Efficient rAAV6 transduction of MPB-derived HSPCs (>35% copGFP-positive cells) was observed on day 4. The levels of reporter gene expression observed in cells transduced with rAAV6 in the absence of RNP gradually decreased to less than 1% of total by day 28. In contrast, the levels of reporter gene expression observed in cells treated with rAAV6+RNP stabilized by day 14, with an average of 11% copGFP-positive cells observed during the remaining duration of the time course (Figure 3C). To determine the frequency of edited alleles within the bulk rAAV6+RNP-treated HSPC cellular population, droplet digital PCR (ddPCR) was performed on day 4 post-EP (Figure S1). In-out primer sets specific for forward or reverse orientation transgene insertion (Figure S1A) revealed that an average of ∼6% of target site alleles bore transgene insertions, with 63% of total insertions occurring in the forward orientation (Figures S1B and S1C). Considering that 11% of the bulk rAAV6+RNP recipient cells demonstrated persistent reporter gene expression upon prolonged culture (Figure 3C), the observation that 6% of target alleles were edited is consistent with predominantly mono-allelic transgene integration.
To determine the frequency of HITI-mediated gene addition on a per-cell basis and to characterize the myeloid-erythroid differentiation potential of manipulated HSPC progenitor cells, we performed colony-forming unit (CFU) assays on treated samples (Figure 4). Consistent with integration frequencies determined by flow cytometry analyses of bulk gene-edited CD34+ cells, we observed an average of 12% copGFP-positive colonies derived from HSPCs treated with rAAV6+RNP (Figure 4A). In contrast, no copGFP-expressing colonies were detected in mock- or rAAV6-treated CFU cultures. Reporter gene-positive colonies arising from erythroid- and myeloid lineage-restricted progenitors were identified (Figure 4B). Compared with mock- and rAAV6-treated samples, rAAV6+RNP-recipient cells demonstrated no significant difference in the relative frequency of myeloid or erythroid progenitors (Figure 4C). Consistent with previously published data indicating that one or few nuclease-induced DSBs after precise genome editing have a limited effect on cellular function,27 we observed no significant difference in clonogenic output between rAAV6- and rAAV6+RNP-treated cells; however, compared with mock-treated HSPCs (i.e., electroporated but not transduced with rAAV6 vectors), rAAV6- and rAAV6+RNP-treated cells showed an overall decrease in the total number of CFUs (Figure 4D). This finding is consistent with recent observations demonstrating reduced clonogenic output after genome editing with rAVV6 donor templates,28,29 attributed to rAAV6-mediated exacerbation of p53 activation, delayed cellular proliferation, increased apoptosis, and upregulated inflammatory response in transduced HSPCs relative to mock controls.27
Figure 4.

High-frequency HITI-mediated genome editing in human CD34+ progenitors
(A) Percent GFP-expressing colonies from colony-forming unit (CFU) assays of mock-, rAAV6-, and rAAV6+RNP-treated CD34+ HSPCs (n = 3 independent donors). (B) Representative images of bright-field and fluorescence microscopy showing GFP expression in burst-forming unit-erythroid (BFU-E) and CFU-granulocyte-macrophage (CFU-GM) colonies from rAAV6+RNP-treated cells. (C) Relative percent of each type of colony scored (n = 3 independent donors). CFU-granulocyte-erythrocyte-monocyte-megakaryocyte (CFU-GEMM) colonies were not detected. (D) Normalized numbers of colonies (n = 3 independent donors). In (A), (C), and (D), results are displayed as mean ± SEM; ∗p < 0.05 by one-way repeated-measures ANOVA tests.
Efficient HITI-mediated genome editing in repopulating HSPCs
To evaluate the engraftment potential and transgene integration frequency in repopulating, HITI-edited HSPCs in vivo, we transplanted immunodeficient non-obese diabetic (NOD)-severe combined immunodeficiency (SCID)-Il2Rgnull (NSG) mice with vector-transduced HSPCs immediately following EP (Figure 5A). Given the lack of transgene integration with either control group in our initial in vitro studies (i.e., mock and rAAV6 controls; Figure 4A) and the previously documented toxicity of rAAV6 in long-term repopulating hematopoietic stem cells,15,27, 28, 29 we deemed CD34+ cells transduced with rAAV6 to be a baseline control more similar to rAAV6+RNP-treated cells for assessment of editing efficiency in human repopulating cells. Murine bone marrow (BM) was collected 18 weeks post-transplantation and analyzed for the presence of CD45+ human cells via flow cytometry. Human cell engraftment (defined as more than 0.1% CD45+ cells) was detected in 5 of 6 NSG mice within the rAAV6 group and all 6 NSG mice within the rAAV6+RNP group (Figure 5B; see Figure S2 for the gating strategy). No statistically significant difference in the levels of engraftment was noted between the two conditions. Myeloid (CD13+ cell) and lymphoid (CD20+ B cell) lineages as well as a substantial percentage of CD34+ progenitors were detected within the total CD45+ cellular population, with B cells representing the majority of engrafted human cells, as typically observed in this model (Figure 5C; see Figure S2 for the gating strategy).29,30
Figure 5.

Efficient HITI-mediated genome editing in human repopulating HSPCs
(A) Schematic of human CD34+ HSPC transplants into NSG mice. CD34+ HSPCs were treated with rAAV6 (red circles) or rAAV6+RNP (blue circles) and transplanted into NSG mice after EP. (B) Human cell engraftment as measured by human CD45 expressing cells in the BM of recipient NSG mice 18 weeks post-transplantation. (C) Percent cellular composition of CD13+ myeloid cells, CD20+ B cells, and CD34+ hematopoietic progenitor cells in the total human cell populations. (D) Percent GFP-expressing cells in the total human cell populations, CD13+ myeloid cells, CD20+ B cells, and CD34+ hematopoietic progenitor cells. (E) Representative flow cytometry plots of GFP expression in the total human cell populations. (F) Gel image of in-out PCR to detect forward direction integration at the 5′ junction end in all six NSG mice in the rAAV6+RNP group. NC, negative control. Expected PCR product size is labeled. In (B)–(D), results are displayed as mean ± SEM; unpaired t tests. For (B)–(E), see also Figure S2.
The frequency of HITI-mediated reporter gene knockin in repopulating HSPCs was determined via flow cytometry analysis of copGFP expression. An average of 21% (median, 10.7%; range, 2.0%–57.3%) of engrafted CD45+ cells demonstrated copGFP expression within the rAAV6+RNP group, whereas cells transduced with rAAV6 alone showed background levels of reporter gene activity (Figures 5D and 5E; see Figure S2 for the gating strategy). Cells expressing copGFP were represented roughly equally (as a percentage of total) among the CD13+, CD20+, and CD34+ compartments of the CD45+ cellular population. The specificity of target site integration was confirmed by nested in-out PCR performed on genomic DNA isolated from murine BM harvested from rAAV6+RNP recipient mice. An amplicon of the expected size (665 bp) was observed in all mice, indicating detection of forward-oriented integration of the copGFP reporter cassette at the ITGB2 locus (Figure 5F; see Figure 1, bottom panel, for relative primer location).
Characterization of genome editing outcomes
To provide detailed characterization of genome editing outcomes (including insert orientation, insertion heterozygosity, and the nature of vector-genomic DNA junction sequences) within HITI-edited CD34+ HSPCs, copGFP-positive cells treated with rAAV6+RNP in vitro were individually sorted into separate wells of 96-well plates containing methylcellulose medium. After 14 days, copGFP-expressing colonies were collected for genomic DNA extraction and PCR analysis. Site-specific integration and the orientation of transgene insertion relative to the ITGB2 promoter were determined by in-out PCR using primers specific to the forward or reverse orientation of transgene integration (see Figure 1, bottom panel, for relative primer location). Site-specific integration was detected in all copGFP-expressing colonies tested, with several clones displaying heterogeneity of junction sequences, as evidenced by amplicon length polymorphism (Figure 6A). Consistent with the HITI design-based placement of transgene-flanking guide RNA target sites to favor re-cleavage of reverse-orientation insertions,23 the majority of alleles (71.4%) demonstrated integration in the forward orientation (Figure 6B). This observation is consistent with the estimate of 63% of forward-oriented alleles as determined by ddPCR of bulk-treated cells (Figure S1). Sanger sequencing of PCR amplicons derived from 5′ and 3′ junction sequences of representative forward-orientation and reverse-orientation transgene insertions revealed three structural classes of junction sequences: (1) junctions displaying precise joining of genomic and vector DNAs at their respective Cas9 cleavage sites (25% of total junctions), (2) junctions bearing only small indels within the genome or vector sequences indicative of limited end trimming prior to joining (46% of total junctions), and (3) complex junctions containing partial inverted terminal repeat (ITR) vector sequences immediately preceding the vector-encoded Cas9 target site sometimes associated with small indels (29% of total junctions) (Figure 6C). The detailed structure of the junction sequences is depicted in Figure S3, and the extent of end trimming at transgene junctions is summarized in Table S1.
Figure 6.

Characterization of HITI-mediated transgene insertions
(A) Gel images of in-out PCR to detect forward- or reverse-orientation insertion at the 5′ and 3′ integration junctions. Human CD34+ HSPCs treated with rAAV6+RNP were plated in methylcellulose medium in individual wells of a 96-well plate. Isolated copGFP-expressing colonies were collected, and DNA was extracted for PCR analysis. Expected PCR products are labeled. Control a, no DNA template; control b, DNA from an rAAV6-treated HSPC colony. See also Figure S3. (B) Percent forward and reverse insertion orientations. (C) Junction structural classes as determined by Sanger sequencing of junction-derived PCR amplicons. (D) PCR amplicons derived from primers flanking the ITGB2-ts target site to detect mono- or bi-allelic editing. For non-edited clones, a single band of 633 bp is expected. For mono-allelic clones, two bands, 633 bp and 2,708 bp, are expected (top bands appear faint because of competition with lower-molecular-weight amplicons). For bi-allelic clones, a single band of 2,708 bp is expected. (E) Percent mono- or bi-allelic edited clones.
The frequency of mono-allelic versus bi-allelic integration events was determined by examining the pattern of PCR amplicon products obtained with genomic DNA-specific primers flanking the integration site (Figure 6D; see Figure 1, top panel, for relative primer location). A PCR amplicon of 633 bp represents an “empty” targeted insertion site, whereas an amplicon of 2,708 bp represents an allele bearing a transgene integration event. Twelve of thirteen clones examined (i.e., 92.3%) demonstrated evidence of mono-allelic insertion (Figure 6E). Clone 5 demonstrated bi-allelic editing, with one insertion in the forward orientation and the other insertion in the reverse orientation.
Discussion
To date, HDR-based strategies were the primary means of introducing transgenes at engineered nuclease-mediated DSBs within human blood stem cells. However, efficient transgene integration has proven challenging using these approaches15, 16, 17, 18, 19, 20, 21 because of limited HDR activity in quiescent HSPCs.22,31 This is especially true in human MPB-derived CD34+ HSPCs, which are the principal source of autologous blood stem cells used in the clinic. In contrast, NHEJ is the predominant DNA repair pathway in primary HSPCs.22 Here we show that homology-independent (i.e., NHEJ-based) targeted integration23,24 is an effective alternative to HDR-based strategies to support efficient site-specific transgene knockin in human MPB-derived HSPCs, as shown in various terminally differentiated, post-mitotic cell types of the murine brain and retina.24
Several studies have noted much lower efficiencies of genome editing in repopulating HSPCs after transplantation in vivo compared with more mature hematopoietic progenitors assayed in vitro.15, 16, 17, 18, 19, 20, 21,32 Using the HITI-based approach to gene addition, we achieved comparable transgene integration efficiencies in progenitors and repopulating HSPCs transplanted into immunodeficient NSG mice, with an average 21% (median, 10.7%; range, 2.0%–57.3%) of BM-resident CD45+ cells displaying stable reporter gene activity at more than 4 months post-transplantation. However, even with purposeful design and placement of the Cas9 target sites flanking the integrating transgene to favor a desired sequence orientation, a small percentage of stably integrated sequences occurred in the opposite (undesired) orientation. In our study, in the absence of position-sensitive cis-acting effects, the insert orientation is nonconsequential because integrated transgene constructs utilized cloned heterologous promoter elements to drive transgene expression; however, when an endogenous promoter is required for transgene expression, improper transgene orientation could decrease the overall efficiency of phenotypic correction. This shortcoming is, in most cases, not anticipated to limit clinical translation of the HITI approach because the majority of alleles (71%) demonstrated integration in the forward orientation, and the overall transgene expression (average 21%) was well above levels required for therapeutic benefits in several inherited blood disorders. For instance, pre-clinical evaluation of viral vector-mediated ITGB2 gene therapy indicates that levels as low as 5% of leukocyte marking can reverse the LAD-1 phenotype in a canine model of the disease.33 In our study, 83% (5 of 6) of the immunodeficient mice transplanted with HITI-edited HSPCs demonstrated transgene expression in more than 5% of the engrafted CD45+ human cell population. These observations suggest clinical relevance of this approach in LAD-1 and other disorders requiring comparable levels of correction for therapeutic benefits (e.g., chronic granulomatous disease34) or in which expression of the therapeutic gene in corrected cells and their progeny provides a selective survival and growth advantage after transplantation (e.g., Fanconi anemia35, 36, 37 and X-linked SCID2,38, 39, 40).
Of further relevance to the potential clinical benefit of the HITI approach for targeted gene addition into human HSPCs, the vast majority (∼71%) of total sequenced junctions demonstrated faithful (i.e., precise) joining of transgene and genomic DNA sequences at the Cas9 cleavage site or minor alterations restricted to small insertions or deletions at the site of Cas9 cleavage within the donor template and/or genomic DNA target site (Figure S3). This limited DNA end trimming evident at many of the transgene junction sequences and the observation of small non-templated insertions within some junction sequences are reflective of the NHEJ DNA repair mechanism utilized in the HITI approach. Junctions with minor alterations are, in most cases, not expected to interfere with transgene expression or limit clinical translation of the HITI approach when the possibility of end trimming is carefully taken into account when selecting a Cas9 target site within the endogenous gene of interest and during design of HITI donor templates. For instance, in our study, because constraints on the number of suitable sgRNA target sequences available for Cas9-mediated DSB formation upstream of the endogenous ITGB2 open reading frame, the Cas9 cleavage site was located 15 bp downstream of the transcription start site (TSS) of the ITGB2 messenger RNA (mRNA; transcript variant 1, accession number NM_000211.5); therefore, only end trimming of more than 15 bp would impinge upon the TSS. Inspection of 28 vector-genomic DNA junction sequences revealed that all instances of end trimming at Cas9-induced, genomic DNA blunt ends displayed only restricted end trimming (range, 2–9 bp), with the majority of events limited to 6 bp or less (Table S1; Figure S3). In all cases, the endogenous ITGB2 TSS and upstream promoter elements were preserved. Trimming of vector sequences was similarly restricted to small stretches of 1–17 bp in most instances; however, end trimming in excess of 200 bp was observed at one of the junction sequences isolated from two clones (clones 3 and 11; Table S1; Figure S3). The CMV promoter-enhancer sequences encoded within the reporter transgene sequences of the donor template begin 101 bp downstream of the Cas9-cleavage site; therefore, the extended end trimming observed at the forward-orientation, 5′ junction of clone 11 (Figure S3A) and at the reverse-orientation, 3′ junction of clone 3 (Figure S3D) resulted in deletion of a portion of the CMV enhancer. However, partial loss of enhancer elements did not silence reporter gene expression. Because the CFU clones utilized for junction analysis were sorted for copGFP expression prior to analysis, it is possible that longer end-trimming events sufficient to silence reporter gene expression were selected against. Thus, based on the possibility of donor sequence end trimming, short stretches of non-essential DNA sequences flanking the HITI transgene expression cassette should be incorporated into the vector construct to ensure preservation of donor gene function after integration. The remainder (29%) of sequenced junctions contained small stretches of flanking vector DNA, some with more complex rearrangement; their effect on transgene expression and phenotypic correction will require further testing with rAAV6 donor vectors lacking a heterologous promoter sequence. Retention of rAAV6 vector sequences at some integrant junctions is consistent with previous reports describing the ability of rAAV6 vectors to integrate at nuclease-induced DSBs in vitro and in vivo.41, 42, 43 Although AAV-based vectors are utilized in more than 100 clinical trials with no reported vector-related adverse events, the genotoxicity profile of AAV in the context of nuclease-mediated genome editing in human HSPCs remains poorly characterized. Additional preclinical work, preferably in large animal models where prolonged follow-up of in vivo hematopoiesis is more practical, will further evaluate the probability of cis activation of cellular oncogenes and clonal expansion following AAV integration at Cas9-induced DNA breaks.
The HITI-based gene editing strategy also has practical advantages for CRISPR-mediated approaches to gene addition in HSPCs utilizing recombinant AAV particles to provide the required donor sequence. Current HDR-based strategies and an alternate method based on homology-mediated end joining (HMEJ) recently described for targeted integration in various cellular systems, including non-dividing cells,44 typically employ a minimum of approximately 0.8–1 kb of homologous DNA sequences within the rAAV construct to promote effective homology-directed recombination. Given the limited packaging capacity of rAAV6 vectors (∼5 kb) and the need to retain approximately 0.3 kb of wild-type AAV genomic sequences for vector rescue, replication, and packaging, HITI-based rAAV6 vectors provide a functionally significant increase in total exogenous sequence capacity compared with HDR- and HMEJ-based rAAV6 constructs. Furthermore, construction of rAAV6 donor templates for HITI-based recombination is markedly simplified by avoiding the need for multi-step cloning or inefficient multi-fragment ligation of transgene and flanking homology arm sequences. In the HMEJ approach, placement of the Cas9 target sites outside of the homology arms in the donor template could also result in cleavage and insertion of the gene of interest, along with the homology arms, into the genomic cut site and impair expression of the integrated transgene. However, the long homology arms incorporated in the design of HDR and HMEJ donor templates may result in higher percentages of inserts in the desired orientation at the chosen genomic locus compared with HITI-treated cells. Another limitation of the HITI approach relative to homology-mediated methods is its lack of suitability for correction of point mutations or other forms of gene conversion that may be the preferred form of phenotypic correction in some instances. Future work comparing head-to-head safety and efficacy parameters of each method will be required before therapeutic applications in human HSPCs.
In conclusion, we achieved efficient site-specific transgene integration in human MPB-derived CD34+ HSPCs utilizing a HITI-based approach to targeted gene addition. This method provides an effective alternative to HDR-based transgene integration for treatment of monogenic human diseases affecting the hematopoietic system and warrants further pre-clinical evaluation and development.
Materials and methods
HITI donor template and recombinant AAV production
The recombinant AAV HITI donor template construct (pSR898B) contains a cytomegalovirus (CMV) promoter/enhancer-driven copGFP reporter gene (derived from pmaxGFP, Lonza) and SV40-derived polyadenylation sequences immediately downstream of a Cas9-specific sgRNA sequence (bearing a unique 20-mer, 5′-GCTTTGCTACCAGTCTGCCC-3′, complimentary to sequences within exon 1 of the human ITGB2 gene) under transcriptional control of the human U6 promoter. The copGFP reporter cassette is flanked by 20-nt ITGB2 sgRNA target sequences (top-strand orientation) so that an approximately 2.1-kb, blunt-end DNA fragment is generated in the presence of target site-specific Cas9 RNP complexes. All sequences are within a baculovirus shuttle plasmid derivative of plasmid pFBGR45 for baculovirus-mediated production of rAAV in insect cells. The vector-encoding sequences of pSR898B were incorporated into a recombinant baculovirus using the Bac-to-Bac Baculovirus Expression System (Thermo Fisher Scientific) according to the manufacturer’s instructions and subsequently packaged within rAAV6 capsids using a baculovirus-mediated rAAV production and immunoaffinity column purification protocol as described previously.46
Isolation and culture of human CD34+ HSPCs
Human CD34+ HSPCs were obtained from healthy male and female volunteers (age, 31–60 years) after informed consent in accordance with the Declaration of Helsinki and under an institutional review board-approved clinical protocol (https://clinicaltrials.gov/ct2/show/NCT00001529; ClinicalTrials.gov: NCT00001529). Peripheral blood mobilization of HSPCs was induced via subcutaneous injection of 10 μg/kg granulocyte-colony stimulating factor (G-CSF, Filgrastim, Amgen) for 5 days, followed by leukapheresis using a Cobe Spectra Apheresis System (Terumo BCT). The mononuclear cell concentrates were enriched in CD34+ HSPCs using a CliniMACS Plus instrument (Miltenyi Biotec) and cryopreserved prior to use. All CD34+ HSPCs were cultured in StemSpan SFEM II medium (STEMCELL Technologies) supplemented with stem cell factor (SCF; 100 ng/mL), thrombopoietin (TPO; 100 ng/mL), Fms-like tyrosine kinase 3 ligand (Flt3-ligand; 100 ng/mL), UM729 (0.8 μM, STEMCELL Technologies), and 1% penicillin-streptomycin (GIBCO). Cells were cultured at 37°C, 5% CO2, and 5% O2.
Cas9 RNP and EP
A 100-nt sgRNA bearing a unique 20-mer (5′-GCUUUGCUACCAGUCUGCCC-3′) specific to exon 1 of the human ITGB2 gene was chemically synthesized (Agilent Technologies). For enhanced stability, 3 nt at each termini bore 2′-O-methyl 3′-phosphorothioate-modified bases. Recombinant S. pyogenes Cas9 protein was purchased from PNA BIO.
Two days prior to EP, CD34+ HSPCs were thawed and plated at 0.5 × 106 cells/mL. Cells were incubated at 37°C, 5% CO2, and 5% O2. After 48 h, sgRNA/Cas9 RNP complexes were prepared by incubating 3.1 μM sgRNA and 1.3 μM Cas9 (∼2.5:1 ratio) in MaxCyte EP buffer (Hyclone) on ice for 30 min. CD34+ HSPCs were collected, and 0.75 × 106–3.5 × 106 cells were resuspended in pre-incubated sgRNA/Cas9 RNP complexes. EP was performed using the MaxCyte STX transfection system (program HPSC34-3) in an OC-25x3 or OC-100 processing assembly. Following EP, CD34+ HSPCs were plated at a concentration of 0.5 × 106–1.0 × 106 cells/mL and incubated at 37°C, 5% CO2, and 5% O2.
T7 Endonuclease I cleavage assay
Cas9 RNP-mediated indel formation was quantified by T7 endonuclease I cleavage assay. Four days post-EP, genomic DNA was extracted from treated HSPCs using a NucleoSpin tissue kit (Macherey-Nagel) according to the manufacturer’s protocol. Genomic DNA was amplified by PCR using forward and reverse primers flanking the sgRNA target site (ITGB2-F and ITGB2-R; primer sequences are detailed in Table S2). The PCR reaction contained 100 ng of each primer, 1× Ranger Mix (Bioline), 250 ng genomic DNA, and water to a final volume of 25 μL. The following PCR program was used: step 1, 95°C for 3 min; step 2, 98°C for 15 s; step 3, 65°C for 30 s; step 4, 72°C for 2 min; step 5, repeat steps 2–4 for a total of 35 cycles; step 6, 72°C for 5 min; step 7, 4°C. PCR amplicons were purified using Quantum Prep PCR Kleen spin columns (Bio-Rad). To denature and re-anneal amplicons, 200 ng of purified amplicons were added to 1× NEBuffer 2 (New England Biolabs) and water to a final volume of 20 μL. The following thermocycler condition was used: step 1, 95°C for 5 min; step 2, 95°C–85°C, ramp −2°C/s; step 3, 85°C–25°C, ramp −0.1°C/s; step 4, 22°C. The heterocomplexed PCR products were incubated with 10 units (1 μL) of T7 endonuclease I (New England Biolabs) at 37°C for 15 min and subjected to electrophoresis on a 4%–20% polyacrylamide Tris-borate-EDTA (TBE) gel (Invitrogen). The gels were stained using SYBR Green I nucleic acid gel stain (Invitrogen) and visualized using a Benchtop 3UV transilluminator (UVP). Densitometric quantification of DNA bands was done using ImageJ software. Indel frequencies were calculated using the formula 100 × (1 – [1 – fraction cleaved](1/2)), where the fraction cleaved = ([density of digested products] / [density of digested products + density of undigested parental band]) as described previously.47
Analysis of off-target Cas9 activity in human HSPCs
The Cas-OFFinder on-line search tool48 (available at http://www.rgenome.net/cas-offinder/) was used to search the human genome (build GRCh38/hg38) for potential Streptococcus pyogenes Cas9 off-target sites (PAM = 5′-NGG-3′) using the unique, target-specific portion of the sgRNA used in this study (i.e., 5′-GCTTTGCTACCAGTCTGCCC-3′) as a query sequence while allowing for up to 3 mismatched base pairs between the sgRNA and candidate target sites. Twenty-one potential off-target sites with three mismatches were identified. No sites harboring 1 or 2 mismatches were found. Five potential autosomal off-target sites were chosen at random for PCR amplification and deep sequencing. The five potential off-target sites as well as the on-target site were PCR amplified from bulk genomic DNA isolated 4 days post-EP from CD34+ HSPCs that had undergone gene editing at the ITGB2 locus. Primer pairs used to amplify the on-target and potential off-target sites are shown in Table S3. Off-target PCR amplicons were generated using Ranger Mix PCR Mastermix (Meridian BioScience) and the following thermocycler parameters: 95°C for 3 min followed by 37 cycles of 98°C for 15 s, 57°C for 30 s, and 72°C for 1 min, with a final extension of 72°C for 5 min. PCR amplification of the on-target site within the ITGB2 gene utilized a similar protocol but with an annealing temperature gradient between 57°C and 65°C (optimal amplification was observed at 62.1°C). PCR amplicons were purified using Quantum Prep PCR Kleen spin columns (Bio-Rad) and subjected to blunt end repair, 3′ adenylation, and ligation of indexing adapters using a TruSeq Nano DNA library prep kit (Illumina) according to the manufacturer’s instructions. Library-prepped amplicons were normalized and pooled for multiplexed, short-read sequencing (2 × 300-bp paired-end reads) on an Illumina MiSeq system. Paired-end reads were filtered for quality and merged using FLASH49 open source software (https://ccb.jhu.edu/software/FLASH/). Merged reads were aligned to the hg38 reference genome using BWA Aligner50 (http://bio-bwa.sourceforge.net/), followed by variant calling using VarScan251 (http://dkoboldt.github.io/varscan/). The percentage of indel-containing reads within a 100-bp window spanning the putative Cas9 cleavage site of each target sequence was calculated.
HITI-mediated genome editing
Two days prior to EP, CD34+ HSPCs were thawed and allowed to recover in fresh StemSpan medium for 1 h at 37°C, 5% CO2, and 5% O2. After the recovery period, ∼3 × 106 cells were collected and transduced with rAAV6 at a multiplicity of infection (MOI) of 100,000–300,000 vector copies/cell for 1 h in a Falcon 15-mL conical centrifuge tube (Corning). The cells and inoculum were then transferred to a 6-well cell culture plate (Corning) at a density of 1 × 106 cells/mL. Cells were incubated at 37°C, 5% CO2, and 5% O2 for 48 h. CD34+ HSPCs were then collected and electroporated with sgRNA/Cas9 RNPs as described above. After EP, CD34+ HSPCs were cultured for up to 28 days at 37°C in the presence of 5% CO2 and 5% O2.
Integration analysis
To confirm site-specific transgene integration, genomic DNA was extracted from treated HSPCs 4 days post-EP using a NucleoSpin tissue kit (Macherey-Nagel) according to the manufacturer’s protocol. Genomic DNA was subjected to nested PCR using an in-out PCR approach (one transgene-specific primer and one genomic locus-specific primer) to detect forward direction integration at the 5′ and 3′ junction ends (primer sequences are detailed in Table S4).
Outer PCR reactions contained 100 ng of each primer, 1× Q5 High-Fidelity Master Mix (New England Biolabs), 250 ng genomic DNA, and water to a final volume of 25 μL. PCR amplicons were purified using Quantum Prep PCR Kleen Spin and diluted 1/100 in water. Inner PCR reactions contained 100 ng of each primer, 1× Q5 High-Fidelity Master Mix, 1 μL of diluted PCR amplicons, and water to a final volume of 25 μL. The following PCR program was used for all PCR reactions: step 1, 95°C for 3 min; step 2, 98°C for 15 s; step 3, annealing for 30 s (annealing temperatures for the respective primer sets are detailed in Table S4); step 4, 72°C for 1 min; step 5, repeat steps 2–4 for a total of 35 cycles; step 6, 72°C for 5 min; step 7, 4°C.
ddPCR
To quantify allelic integration via HITI, genomic DNA was extracted from samples of treated HSPCs collected on day 4 post-EP and subjected to ddPCR. A hexachloro-fluorescein (HEX) reference assay detecting the promoter region of the ITGB2 gene and two 6-carboxy-fluorescein (FAM) assays detecting HITI integration in the forward or reverse orientation were designed using Primer3Plus (primer and probe sequences are detailed in Table S5).
Genomic DNA was digested with EcoRV-HF (20 U/μg genomic DNA, New England Biolabs) at 37°C for 1 h. ddPCR reactions contained 1× reference primer/probe mix synthesized at a 3.6 ratio (900 nM primer and 250 nM HEX-labeled probe), 1× target primer/probe mix (forward or reverse) synthesized at a 3.6 ratio (FAM-labeled probe), 1× ddPCR Supermix, 30–50 ng of digested DNA, and water for a total of 22 μL. Droplets were generated on an automated droplet generator (Bio-Rad) according to the manufacturer’s protocol. The following ddPCR program was used: step 1, 95°C for 10 min, ramp 2°C/s; step 2, 94°C for 30 s, ramp 2°C/s; step 3, annealing temperatures of 58.5°C for forward orientation detection and 62°C for reverse orientation detection for 30 s, ramp 2°C/s; step 4, 72°C 1 min, ramp 2°C/s; step 5, repeat steps 2–4 for 50 cycles; step 6, 98°C for 10 min, ramp 2°C/s; step 7, 4°C, ramp 2°C/s. Droplets were analyzed on a QX-200 droplet reader (Bio-Rad) and analyzed using QuantaSoft (Bio-Rad).
CFU assay
Between 1 and 24 h post-EP, CD34+ HSPCs were collected and plated in MethoCult H4435 Enriched (STEMCELL Technologies) at a concentration of 250–600 cells/mL. Between days 12–14, colonies were counted and scored based on morphological features using an EVOS XL Core transmitted-light microscope (Thermo Fisher Scientific) and analyzed for copGFP fluorescence using an EVOS FL fluorescent microscope (Thermo Fisher Scientific).
Transplantation of CD34+ HSPCs into NSG mice
Twelve- to 30-week-old female NSG mice (Jackson Laboratory, stock number 005557) were sublethally irradiated (280 cGy) 24 h before tail vein injection. CD34+ HSPCs treated with rAAV6 or rAAV6+RNP were collected immediately after EP, and ∼300,000 cells were transplanted into six NSG mice per condition. Animals were housed and handled in accordance with the guidelines set by the Committee on Care and Use of Laboratory Animals of the Institute of Laboratory Animal Resources, National Research Council (DHHS publication NIH 85-23), and the protocol was approved by the Animal Care and Use Committee of the National Heart, Lung, and Blood Institute.
Assessment of human cell engraftment and HITI-mediated genome editing in NSG mice
Levels of human cell engraftment were assessed 18 weeks post-transplantation. Briefly, tibiae, femora, and pelvises were harvested from individual mice, and BM was flushed from each bone into 2 mL of Iscove's Modified Dulbecco's Medium (IMDM) using an insulin syringe with a 29G needle. Samples were then centrifuged at 1,200 rpm for 10 min. Red blood cells were lysed by 5- to 10-min incubation at room temperature with Ammonium-Chloride-Potassium (ACK) lysis buffer (Quality Biological), and the remaining cells were collected by centrifugation at 1,200 rpm for 10 min. Cells were stained for 30 min on ice with the antibody panel CD45 allophycocyanin (APC) (BD Pharmingen), CD20 phycoerythrin-cyanin 7 (PE-Cy7) (BD Biosciences), CD34 PE-Cy5 (BD Pharmingen), and CD13 PE (BD Pharmingen) and analyzed on a Fortessa flow cytometer (Becton Dickinson). Human engraftment was defined as more than 0.1% of CD45+ cells. Additionally, cells from the six mice in the rAAV6+RNP group were stained with CD45 APC (BD Pharmingen) and sorted by flow cytometry using BD FACSAria II or BD FACSAria Fusion instruments. Following sorting, cells were spun down at 3,000 rpm for 30 min, and genomic DNA was extracted as described previously. Genomic DNA was subjected to nested PCR using an in-out PCR approach to detect forward direction integration at the 5′ junction end (primer sequences are detailed in Table S4).
Characterization of genome editing outcomes
Between 1–3 days post-EP, CD34+ HSPCs were sorted for copGFP expression, and single cells were seeded into 96-well plates containing MethoCult H4435 Enriched methylcellulose-based medium (STEMCELL Technologies) as described previously.52 On day 14, genomic DNA was extracted from copGFP-expressing colonies. Briefly, wells with GFP-expressing colonies were filled with 200 μL PBS, and the contents were transferred to individual Eppendorf tubes. Cells were centrifuged at 3,000 rpm for 10 min and washed with 300 μL PBS. Cell pellets were resuspended in 25 μL DNA extraction buffer (1 mg/mL Proteinase K, 50 mmol/L Tris-HCl [pH 8.0], 10 mmol/L EDTA, 100 mmol/L NaCl, and 1% Triton X-100). Cells were incubated at 55°C for 12–18 h and at 95°C for 5 min.
To analyze the direction of transgene integration, colonies were subjected to nested PCR using an in-out PCR approach to detect integration in the forward and reverse direction at the 5′ and 3′ junction ends (primer sequences are detailed in Table S4). Outer PCR reactions contained 100 ng of each primer, 1× Q5 High-Fidelity Master Mix, 2 μL genomic DNA, and water to a final volume of 25 μL. PCR amplicons were purified using Quantum Prep PCR Kleen Spin and diluted 1/100 in water. Inner PCR reactions contained 100 ng of each primer, 1× Q5 High-Fidelity Master Mix, 1 μL of diluted PCR amplicons, and water to a final volume of 25 μL. PCR reactions were run using the program described previously (annealing temperatures for the respective primer sets are detailed in Table S4).
Additionally, to quantify the proportion of mono- and bi-allelic integrations, colonies were subjected to nested PCR using PCR primers flanking the sgRNA target site at the ITGB2 locus (primer sequences are detailed in Table S2). Outer and inner PCR reactions contained reagents as described previously. The following PCR program was used for outer and inner PCR: step 1, 95°C for 3 min; step 2, 98°C for 15 s; step 3, annealing for 30 s (annealing temperatures for the respective primer sets are detailed in Table S2); step 4, 72°C for 5 min; step 5, repeat steps 2–4 for a total of 35 cycles; step 6, 72°C for 5 min; step 7, 4°C. PCR amplicons were sequenced by a commercial vendor (Eurofins Genomics).
Statistical analysis
Results were analyzed with GraphPad Prism software (version 8.4.3) using unpaired Student’s t tests or one-way repeated-measures ANOVA tests. Results are displayed as mean ± SEM. ∗p < 0.05; ns, not significant.
Acknowledgments
The authors thank all members of the Larochelle Lab for helpful discussions; Laurakay Bruhn, PhD, and Daniel Ryan, PhD, of Agilent Technologies for providing chemically modified sgRNAs; Mara Pavel-Dinu, PhD, for assisting with ddPCR primer and probe design as well as Steven Shema and the NCI CCR Genomics Core for performing ddPCR experiments; Yusheng Li, PhD, and the NHLBI Sequencing Core Facility for conducting high-throughput sequencing of PCR amplicons at off-target sites; Mehdi Pirooznia, MD, PhD, and the NHLBI Bioinformatics and Computational Biology Core Facility for analysis of high-throughput sequencing data; Keyvan Keyvanfar and the NHLBI Flow Cytometry Core Facility for performing sorting of gene-edited HSPCs; David Stroncek, MD, and the NIH Department of Transfusion Medicine and Cell Processing Section staff for apheresis, selection, and cryopreservation of human CD34+ cells; Richard Gustafson, RN, and the outpatient clinic nursing staff for recruiting normal volunteers and providing G-CSF administration teaching to healthy subjects; and Bernadette Alisantosa, DVM, James Hawkins, DVM, and the mouse core facility staff for excellent animal care. This work was supported by the Intramural Research Program of the National Heart, Lung, and Blood Institute, National Institutes of Health (Z99 HL999999).
Author contributions
H.B., R.H.S., and A.L. designed the experiments. H.B. performed the experiments. H.B. and W.H. harvested and processed murine bone marrow for flow cytometry analysis. R.H.S. designed and constructed rAAV plasmid reagents. H.B., R.H.S., and A.L. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.010.
Supplemental information
References
- 1.Mohty B., Mohty M. Long-term complications and side effects after allogeneic hematopoietic stem cell transplantation: an update. Blood Cancer J. 2011;1:e16. doi: 10.1038/bcj.2011.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Ravin S.S., Wu X., Moir S., Anaya-O’Brien S., Kwatemaa N., Littel P., Theobald N., Choi U., Su L., Marquesen M. Lentiviral hematopoietic stem cell gene therapy for X-linked severe combined immunodeficiency. Sci. Transl. Med. 2016;8:335ra57. doi: 10.1126/scitranslmed.aad8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aiuti A., Cattaneo F., Galimberti S., Benninghoff U., Cassani B., Callegaro L., Scaramuzza S., Andolfi G., Mirolo M., Brigida I. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N. Engl. J. Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 4.Biffi A., Montini E., Lorioli L., Cesani M., Fumagalli F., Plati T., Baldoli C., Martino S., Calabria A., Canale S. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- 5.Toscano M.G., Frecha C., Benabdellah K., Cobo M., Blundell M., Thrasher A.J., García-Olivares E., Molina I.J., Martin F. Hematopoietic-specific lentiviral vectors circumvent cellular toxicity due to ectopic expression of Wiskott-Aldrich syndrome protein. Hum. Gene Ther. 2008;19:179–197. doi: 10.1089/hum.2007.098. [DOI] [PubMed] [Google Scholar]
- 6.Ranzani M., Cesana D., Bartholomae C.C., Sanvito F., Pala M., Benedicenti F., Gallina P., Sergi L.S., Merella S., Bulfone A. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat. Methods. 2013;10:155–161. doi: 10.1038/nmeth.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun C.J., Boztug K., Paruzynski A., Witzel M., Schwarzer A., Rothe M., Modlich U., Beier R., Göhring G., Steinemann D. Gene therapy for Wiskott-Aldrich syndrome--long-term efficacy and genotoxicity. Sci. Transl. Med. 2014;6:227ra33. doi: 10.1126/scitranslmed.3007280. [DOI] [PubMed] [Google Scholar]
- 8.Persons D.A., Hargrove P.W., Allay E.R., Hanawa H., Nienhuis A.W. The degree of phenotypic correction of murine beta -thalassemia intermedia following lentiviral-mediated transfer of a human gamma-globin gene is influenced by chromosomal position effects and vector copy number. Blood. 2003;101:2175–2183. doi: 10.1182/blood-2002-07-2211. [DOI] [PubMed] [Google Scholar]
- 9.Li H., Yang Y., Hong W., Huang M., Wu M., Zhao X. Applications of genome editing technology in the targeted therapy of human diseases: mechanisms, advances and prospects. Signal Transduct. Target. Ther. 2020;5:1. doi: 10.1038/s41392-019-0089-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gaj T., Gersbach C.A., Barbas C.F., 3rd ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering. Trends Biotechnol. 2013;31:397–405. doi: 10.1016/j.tibtech.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Johnson R.D., Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J. 2000;19:3398–3407. doi: 10.1093/emboj/19.13.3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mao Z., Bozzella M., Seluanov A., Gorbunova V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair (Amst.) 2008;7:1765–1771. doi: 10.1016/j.dnarep.2008.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gomez-Ospina N., Scharenberg S.G., Mostrel N., Bak R.O., Mantri S., Quadros R.M., Gurumurthy C.B., Lee C., Bao G., Suarez C.J. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019;10:4045. doi: 10.1038/s41467-019-11962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pavel-Dinu M., Wiebking V., Dejene B.T., Srifa W., Mantri S., Nicolas C.E., Lee C., Bao G., Kildebeck E.J., Punjya N. Gene correction for SCID-X1 in long-term hematopoietic stem cells. Nat. Commun. 2019;10:1634. doi: 10.1038/s41467-019-09614-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.V., Lill G.R., Kaufman M.L. Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep. 2018;23:2606–2616. doi: 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Ravin S.S., Reik A., Liu P.Q., Li L., Wu X., Su L., Raley C., Theobald N., Choi U., Song A.H. Targeted gene addition in human CD34(+) hematopoietic cells for correction of X-linked chronic granulomatous disease. Nat. Biotechnol. 2016;34:424–429. doi: 10.1038/nbt.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dever D.P., Bak R.O., Reinisch A., Camarena J., Washington G., Nicolas C.E., Pavel-Dinu M., Saxena N., Wilkens A.B., Mantri S. CRISPR/Cas9 β-globin gene targeting in human haematopoietic stem cells. Nature. 2016;539:384–389. doi: 10.1038/nature20134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang J., Exline C.M., DeClercq J.J., Llewellyn G.N., Hayward S.B., Li P.W., Shivak D.A., Surosky R.T., Gregory P.D., Holmes M.C., Cannon P.M. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat. Biotechnol. 2015;33:1256–1263. doi: 10.1038/nbt.3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Genovese P., Schiroli G., Escobar G., Tomaso T.D., Firrito C., Calabria A., Moi D., Mazzieri R., Bonini C., Holmes M.C. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510:235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mohrin M., Bourke E., Alexander D., Warr M.R., Barry-Holson K., Le Beau M.M., Morrison C.G., Passegué E. Hematopoietic stem cell quiescence promotes error-prone DNA repair and mutagenesis. Cell Stem Cell. 2010;7:174–185. doi: 10.1016/j.stem.2010.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Suzuki K., Izpisua Belmonte J.C. In vivo genome editing via the HITI method as a tool for gene therapy. J. Hum. Genet. 2018;63:157–164. doi: 10.1038/s10038-017-0352-4. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki K., Tsunekawa Y., Hernandez-Benitez R., Wu J., Zhu J., Kim E.J., Hatanaka F., Yamamoto M., Araoka T., Li Z. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature. 2016;540:144–149. doi: 10.1038/nature20565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.van de Vijver E., Maddalena A., Sanal Ö., Holland S.M., Uzel G., Madkaikar M., de Boer M., van Leeuwen K., Köker M.Y., Parvaneh N. Hematologically important mutations: leukocyte adhesion deficiency (first update) Blood Cells Mol. Dis. 2012;48:53–61. doi: 10.1016/j.bcmd.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cromer M.K., Vaidyanathan S., Ryan D.E., Curry B., Lucas A.B., Camarena J., Kaushik M., Hay S.R., Martin R.M., Steinfeld I. Global Transcriptional Response to CRISPR/Cas9-AAV6-Based Genome Editing in CD34+ Hematopoietic Stem and Progenitor Cells. Mol. Ther. 2018;26:2431–2442. doi: 10.1016/j.ymthe.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schiroli G., Conti A., Ferrari S., Della Volpe L., Jacob A., Albano L., Beretta S., Calabria A., Vavassori V., Gasparini P. Precise Gene Editing Preserves Hematopoietic Stem Cell Function following Transient p53-Mediated DNA Damage Response. Cell Stem Cell. 2019;24:551–565.e8. doi: 10.1016/j.stem.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pavani G., Laurent M., Fabiano A., Cantelli E., Sakkal A., Corre G., Lenting P.J., Concordet J.P., Toueille M., Miccio A., Amendola M. Ex vivo editing of human hematopoietic stem cells for erythroid expression of therapeutic proteins. Nat. Commun. 2020;11:3778. doi: 10.1038/s41467-020-17552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Romero Z., Lomova A., Said S., Miggelbrink A., Kuo C.Y., Campo-Fernandez B., Hoban M.D., Masiuk K.E., Clark D.N., Long J. Editing the Sickle Cell Disease Mutation in Human Hematopoietic Stem Cells: Comparison of Endonucleases and Homologous Donor Templates. Mol. Ther. 2019;27:1389–1406. doi: 10.1016/j.ymthe.2019.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huntsman H.D., Bat T., Cheng H., Cash A., Cheruku P.S., Fu J.F., Keyvanfar K., Childs R.W., Dunbar C.E., Larochelle A. Human hematopoietic stem cells from mobilized peripheral blood can be purified based on CD49f integrin expression. Blood. 2015;126:1631–1633. doi: 10.1182/blood-2015-07-660670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hao S., Chen C., Cheng T. Cell cycle regulation of hematopoietic stem or progenitor cells. Int. J. Hematol. 2016;103:487–497. doi: 10.1007/s12185-016-1984-4. [DOI] [PubMed] [Google Scholar]
- 32.Schiroli G., Ferrari S., Conway A., Jacob A., Capo V., Albano L., Plati T., Castiello M.C., Sanvito F., Gennery A.R. Preclinical modeling highlights the therapeutic potential of hematopoietic stem cell gene editing for correction of SCID-X1. Sci. Transl. Med. 2017;9:eaan0820. doi: 10.1126/scitranslmed.aan0820. [DOI] [PubMed] [Google Scholar]
- 33.Bauer T.R., Jr., Allen J.M., Hai M., Tuschong L.M., Khan I.F., Olson E.M., Adler R.L., Burkholder T.H., Gu Y.C., Russell D.W., Hickstein D.D. Successful treatment of canine leukocyte adhesion deficiency by foamy virus vectors. Nat. Med. 2008;14:93–97. doi: 10.1038/nm1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kang E.M., Choi U., Theobald N., Linton G., Long Priel D.A., Kuhns D., Malech H.L. Retrovirus gene therapy for X-linked chronic granulomatous disease can achieve stable long-term correction of oxidase activity in peripheral blood neutrophils. Blood. 2010;115:783–791. doi: 10.1182/blood-2009-05-222760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Río P., Navarro S., Guenechea G., Sánchez-Domínguez R., Lamana M.L., Yañez R., Casado J.A., Mehta P.A., Pujol M.R., Surrallés J. Engraftment and in vivo proliferation advantage of gene-corrected mobilized CD34+ cells from Fanconi anemia patients. Blood. 2017;130:1535–1542. doi: 10.1182/blood-2017-03-774174. [DOI] [PubMed] [Google Scholar]
- 36.Río P., Navarro S., Wang W., Sánchez-Domínguez R., Pujol R.M., Segovia J.C., Bogliolo M., Merino E., Wu N., Salgado R. Successful engraftment of gene-corrected hematopoietic stem cells in non-conditioned patients with Fanconi anemia. Nat. Med. 2019;25:1396–1401. doi: 10.1038/s41591-019-0550-z. [DOI] [PubMed] [Google Scholar]
- 37.Román-Rodríguez F.J., Ugalde L., Álvarez L., Díez B., Ramírez M.J., Risueño C., Cortón M., Bogliolo M., Bernal S., March F. NHEJ-Mediated Repair of CRISPR-Cas9-Induced DNA Breaks Efficiently Corrects Mutations in HSPCs from Patients with Fanconi Anemia. Cell Stem Cell. 2019;25:607–621.e7. doi: 10.1016/j.stem.2019.08.016. [DOI] [PubMed] [Google Scholar]
- 38.Cavazzana-Calvo M., Hacein-Bey-Abina S., Fischer A. Gene therapy of X-linked severe combined immunodeficiency. Curr. Opin. Allergy Clin. Immunol. 2002;2:507–509. doi: 10.1097/00130832-200212000-00005. [DOI] [PubMed] [Google Scholar]
- 39.Gaspar H.B., Parsley K.L., Howe S., King D., Gilmour K.C., Sinclair J., Brouns G., Schmidt M., Von Kalle C., Barington T. Gene therapy of X-linked severe combined immunodeficiency by use of a pseudotyped gammaretroviral vector. Lancet. 2004;364:2181–2187. doi: 10.1016/S0140-6736(04)17590-9. [DOI] [PubMed] [Google Scholar]
- 40.Hacein-Bey-Abina S., Fischer A., Cavazzana-Calvo M. Gene therapy of X-linked severe combined immunodeficiency. Int. J. Hematol. 2002;76:295–298. doi: 10.1007/BF02982686. [DOI] [PubMed] [Google Scholar]
- 41.Miller D.G., Petek L.M., Russell D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- 42.Hanlon K.S., Kleinstiver B.P., Garcia S.P., Zaborowski M.P., Volak A., Spirig S.E., Muller A., Sousa A.A., Tsai S.Q., Bengtsson N.E. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019;10:4439. doi: 10.1038/s41467-019-12449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson C.E., Wu Y., Gemberling M.P., Oliver M.L., Waller M.A., Bohning J.D., Robinson-Hamm J.N., Bulaklak K., Castellanos Rivera R.M., Collier J.H. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019;25:427–432. doi: 10.1038/s41591-019-0344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yao X., Wang X., Hu X., Liu Z., Liu J., Zhou H., Shen X., Wei Y., Huang Z., Ying W. Homology-mediated end joining-based targeted integration using CRISPR/Cas9. Cell Res. 2017;27:801–814. doi: 10.1038/cr.2017.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Urabe M., Ding C., Kotin R.M. Insect cells as a factory to produce adeno-associated virus type 2 vectors. Hum. Gene Ther. 2002;13:1935–1943. doi: 10.1089/10430340260355347. [DOI] [PubMed] [Google Scholar]
- 46.Smith R.H., Levy J.R., Kotin R.M. A simplified baculovirus-AAV expression vector system coupled with one-step affinity purification yields high-titer rAAV stocks from insect cells. Mol. Ther. 2009;17:1888–1896. doi: 10.1038/mt.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guschin D.Y., Waite A.J., Katibah G.E., Miller J.C., Holmes M.C., Rebar E.J. A rapid and general assay for monitoring endogenous gene modification. Methods Mol. Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- 48.Bae S., Park J., Kim J.S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Magoč T., Salzberg S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics. 2011;27:2957–2963. doi: 10.1093/bioinformatics/btr507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koboldt D.C., Zhang Q., Larson D.E., Shen D., McLellan M.D., Lin L., Miller C.A., Mardis E.R., Ding L., Wilson R.K. VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012;22:568–576. doi: 10.1101/gr.129684.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bak R.O., Dever D.P., Porteus M.H. CRISPR/Cas9 genome editing in human hematopoietic stem cells. Nat. Protoc. 2018;13:358–376. doi: 10.1038/nprot.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.