

Abstract
Glaucoma is the leading cause of irreversible vision loss, affecting more than 70 million individuals worldwide. Circulatory disturbances of aqueous humor (AH) have long been central pathological contributors to glaucomatous lesions. Thus, targeting the AH outflow is a promising approach to treat glaucoma. However, the epigenetic mechanisms initiating AH outflow disorders and the targeted treatments remain to be developed. Studying glaucoma patients, we identified GDF7 (growth differentiation factor 7) hypomethylation as a crucial event in the onset of AH outflow disorders. Regarding the underlying mechanism, the hypomethylated GDF7 promoter was responsible for the increased GDF7 production and secretion in primary open-angle glaucoma (POAG). Excessive GDF7 protein promoted trabecular meshwork (TM) fibrosis through bone morphogenetic protein receptor type 2 (BMPR2)/Smad signaling and upregulated pro-fibrotic genes, α-smooth muscle actin (α-SMA) and fibronectin (FN). GDF7 protein expression formed a positive feedback loop in glaucomatous TM (GTM). This positive feedback loop was dependent on the activated TET (ten-eleven translocation) enzyme, which kept the GDF7 promoter region hypomethylated. The phenotypic transition in TM fortified the AH outflow resistance, thus elevating the intraocular pressure (IOP) and attenuating the nerve fiber layer. This methylation-dependent mechanism is also confirmed by a machine-learning model in silico with a specificity of 84.38% and a sensitivity of 89.38%. In rhesus monkeys, we developed GDF7 neutralization therapy to inhibit TM fibrosis and consequent AH outflow resistance that contributes to glaucoma. The neutralization therapy achieved high-efficiency control of the IOP (from 21.3 ± 0.3 to 17.6 ± 0.2 mmHg), a three-fold improvement in the outflow facility (from 0.1 to 0.3 μL/min · mmHg), and protection of nerve fibers. This study provides new insights into the epigenetic mechanism of glaucoma and proposes an innovative GDF7 neutralization therapy as a promising intervention.
Keywords: DNA methylation, glaucoma, trabecular meshwork, neutralizing antibody, fibrosis, computational modeling
Graphical abstract
Zhou and colleagues reported that GDF7 methylation, as a novel epigenetic event, triggered trabecular meshwork fibrosis in vitro and in vivo. For treatment purpose, the GDF7 neutralizing antibody was developed and proved to be safe and effective, thus holding great promise in the targeted therapy of glaucoma.
Introduction
Glaucoma, a main cause of permanent vision impairment, affects more than 70 million individuals worldwide.1,2 Disruption of aqueous humor (AH) outflow is believed to be the major cause of primary open-angle glaucoma (POAG), which resulted in elevation of intraocular pressure (IOP).3 If left untreated, elevated IOP will result in progressive loss of retinal ganglion cells and axons in the optic nerve leading to irreversible vision loss. First-degree relatives of POAG patients have a 4 to 10 times higher disease prevalence than that of the general population.4,5 Familial aggregation studies of POAG support a substantial heritable component; however, a simple mode of inheritance is not robust for identifying POAG susceptibility genes. Therefore, genome-wide association studies were recruited and successfully identified common variants at more than 100 genetic loci that associate with POAG.6,7
However, POAG is also complex clinically. The lead genetic variants for POAG together explain less than 5% of high IOP variance, indicating the heterogeneous genetic basis of POAG. Moreover, the relationship between IOP elevation and retinal ganglion cell degeneration is not simple, as many individuals have IOP elevation without optic nerve damage, and some patients develop optic nerve degeneration without elevated IOP.8,9 With the consideration of the complexity, rather than simple inheritance, POAG susceptibility is influenced by DNA sequence variants in many genes, environmental factors, and their interactions.10 Integration of chromatin annotation maps of POAG supports the importance of altered transcriptional regulation. Thus, the investigation of epigenetic changes in POAG is of interest.
Previous studies mainly focus on the general methylation or acetylation level among the whole genome, without identifying specific genes or loci.11,12 Also, the epigenetic mechanisms and therapies in maintaining the AH outflow remain to be defined. Mapping the dysregulated DNA methylation loci in POAG patients would be pivotal in understanding the pathogenesis and suggest promising clinical applications. Since the DNA methylation landscape is predominantly tissue specific,13 trabecular meshwork (TM) samples, the crucial component of the AH outflow passway,14 should be recruited in epigenetic screening.
This study, which assesses the role of aberrant DNA methylation in POAG patients, aims to illustrate whether dysregulated DNA methylation is responsible for outflow resistance of AH and visual impairments. Growth differentiation factor 7 (GDF7) hypomethylation is a major contributor of elevated AH outflow resistance by promoting TM fibrosis and closely related with clinical outcomes. DNA hypomethylation-induced overexpression of GDF7 could be enhanced in a ten-eleven translocation (TET)-dependent positive feedback loop. By activating bone morphogenetic protein receptor type 2 (BMPR2)/Smad signaling, excessive GDF7 protein fortifies the TM fibrosis with increased α-smooth muscle actin (α-SMA), fibronectin (FN), and collagen expression. A machine-learning model is then applied to validate the methylation-dependent mechanism in silico. Furthermore, GDF7 neutralization therapy is developed in rhesus monkeys to prevent TM fibrosis and thus improve the AH outflow facility, control the IOP elevation, and protect retinal neurons.
This study provides novel epigenetic insights into the pathogenesis of glaucoma by deciphering DNA methylation. Moreover, the GDF7 neutralization therapy launches a groundbreaking paradigm shift in the management of glaucoma.
Results
GDF7 hypomethylation was identified in POAG
Dysregulated DNA methylation is responsible for various biological and pathological processes, including cell fibrosis and morphophysiological variation.15,16 The excavation of the DNA methylation disturbance underlying glaucoma might open a new view for the fibrosis of TM. To identify the DNA methylation disturbance in POAG, the TM samples were collected from eight POAG patients and eight matched normal controls (NCs). The glaucomatous TM (GTM) samples were obtained during trabeculectomy from POAG patients. The normal TM (NTM) samples were obtained from the eye bank in the Zhongshan Ophthalmic Center (see Materials and methods). The inclusion criteria of POAG patients and NC are stated in Materials and methods (patients’ information summarized in Table S1).
As reported in bioinformatic analysis, Pearson’s correlation coefficients between the same experiments were 0.9985/0.9985 (POAG/NC), indicating conformity among the samples. With the use of a genome-wide DNA methylation microarray, we identified 810 aberrantly methylated genes and 1,294 CpG sites (|β-difference| > 0.2 and p < 0.05) in POAG patients (Figure 1A). The top 20 differentially methylated genes were listed in the heatmap (Figure 1B).
Figure 1.

GDF7 hypomethylation was the crucial event in glaucoma
(A) Diagram of aberrantly methylated regions in primary open angle glaucoma (POAG). From the outside in, the first layer presents the chromosomal information; the second and third layers present the different methylation sites in POAG patients and healthy controls (CONs), respectively; and the fourth and fifth layers present aberrantly methylated promoters and untranslated regions (UTRs) in POAG patients. (B) The top 20 differentially methylated sites in glaucomatous trabecular meshwork (GTM) samples (presented in reference name). As normalized to the controls, the relative methylation levels of the target regions were represented in the pseudocolor (n = 8 per group). (C) The disrupted methylation genes in POAG patients enriched in seven biological pathways as analyzed by Kyoto Encyclopedia of Genes and Genomes (KEGG). (D) The dysregulated methylation levels of five candidates were confirmed by bisulfite sequencing PCR (BSP) in trabecular meshwork (TM) samples and cells (n = 3 per group). (E) Decrease in methylation level of the GDF7 promoter was confirmed in eight GTM samples compared to healthy controls by BSP (n = 8 per group). (F) Ninety GTM samples were obtained from trabeculectomy (clinical information in Table S2) to validate the expression of GDF7 by quantitative real-time PCR and ELISA. Note: the readouts were normalized to the eight normal TM (NTM), due to the limitation of the donations we can get. (G) The GDF7 methylation level in response to 5-aza-2′-deoxycytidine (DAC) treatment was tested by BSP, and the change in GDF7 mRNA level was measured by real-time PCR in TM cells (n = 3 per group). (H) Correlation analysis between GDF7 methylation level and clinical manifestations in POAG patients (n = 8). The data represent the mean ± SD. Compared with NTM samples: ∗p < 0.05, ∗∗p < 0.01. Compared with NTM cells: #p < 0.05, ##p < 0.01. NC, normal control; IOP, intraocular pressure; CDR, cup/disc ratio; RNFL, retinal nerve fiber layer.
Assisted by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis, the dysregulated methylation sites in POAG patients are primarily enriched in seven biological pathways. As shown in Figure 1C, disturbed DNA methylation acquires a pivotal role in tissue fibrosis from the aspects of cellular adhesion and extracellular matrix (ECM) dynamics. Based on these findings, five candidate methylation sites related to fibrotic signaling were selected for further validation: GDF7 (fold change = 3.36, adjusted p value = 0.017), Col1A1 (fold change = 3.01, adjusted p value = 0.025), transforming growth factor (TGF)-β1 (fold change = 2.69, adjusted p value = 0.039), fibroblast growth factor (FGF)12 (fold change = −4.69, adjusted p value = 0.03), and MMP-27 (fold change = −1.68, adjusted p value = 0.042).
Bisulfite sequencing PCR (BSP) assay was conducted in TM samples and primary TM cells (see Materials and methods) to confirm the DNA methylation levels of target genes. As indicated in Figure 1D, the disturbed DNA methylation levels of the five candidate sites were in conformity with the microarray results. GDF7 methylation level showed a fold change > 2 with p < 0.05. However, the other four candidates did not present as much significance in methylation change. Based on these results, GDF7 hypomethylation was nominated as a promising regulator in POAG.
Decreased methylation of the GDF7 promoter was the crucial event in POAG
As presented in the diagram, the dysregulated methylation site is located in the CpG islands of the GDF7 promoter region (Figure S5A). Therefore, the DNA methylation level of the GDF7 promoter was further tested in eight GTM samples. As analyzed by BSP, the DNA methylation level of the GDF7 promoter was decreased in GTM samples compared with NC (Figure 1E).
Since the dysregulated methylation level in the promoter would largely affect the transcription activity,17 the GDF7 expression was tested in TM samples by real-time PCR. To eliminate the individual difference, an additional 90 GTM samples (clinical information of patients in Table S2) were recruited in the validation. Results presented that the mRNA levels of GDF7 were significantly increased in GTM samples compared with NC. In addition, the increase of GDF7 protein was confirmed in homogenates of GTM samples, as measured by enzyme-linked immunosorbent assay (ELISA) (Figure 1F). To better define the role of DNA hypomethylation in increased GDF7 expression, NTM cells were treated with 0.5 μM 5-aza-2′-deoxycytidine (DAC), a general inhibitor of DNA methyltransferase, for 72 h.18 DAC-induced demethylation in the GDF7 gene was validated by a BSP assay, and the transcription of GDF7 was significantly enhanced in DAC-treated TM cells, indicating that the epigenetic upregulation of GDF7 expression in GTM was methylation dependent (Figure 1G).
We then analyzed the correlation between the methylation level of the GDF7 promoter and the clinical manifestations: IOP elevation, retinal nerve fiber layer (RNFL) attenuation, cup/disc ratio (CDR) increase, and visual field (VF) defects in patients. IOP elevation, the direct manifestation of AH outflow resistance, negatively correlated with the GDF7 methylation level in POAG patients. The mean RNFL thickness is a commonly used measure to infer the status of retinal ganglion cells in glaucoma diagnosis and progression.19 As shown in Figure 1H, the mean RNFL thickness is positively correlated with the methylation level of the GDF7 promoter in POAG patients. However, there was no significant correlation between GDF7 methylation level and CDR or VF defects (Figure 1H).
Overexpressed GDF7 protein promoted TM fibrosis
Fibrotic TM was nominated as the predominant pathological event in AH outflow resistance.20,21 Increased accumulation of collagen was observed in the GTM samples and cells with Masson staining (first lane of Figures S1A and S1B). Also, pro-fibrotic markers,22 including α-SMA, FN, and Col I, were significantly upregulated in GTM samples and cultured GTM cells. By contrast, the marker of NTM cells, N-cadherin (N-cad), was significantly decreased in patients (Figures S1A and S1B). To exclude potential individual differences, a fluorescence image gallery of TM samples from POAG patients and healthy donors was presented in Figure S3A. Moreover, real-time PCR and immunoblot results confirmed the TM fibrosis in POAG (Figures S2A−S2C). These changes suggested an important role of fibrosis in GTM.
Given that GDF7 protein was overexpressed in GTM samples (Figure 1F), we wondered whether excessive GDF7 expression may be a pivotal player in TM fibrosis. To this end, recombinant human GDF7 protein (rhGDF7) was used. NTM cells were treated with 5 ng/mL rhGDF7 in culture medium for 72 h and then washed off before testing. As determined by the immunofluorescence (IF) assay, rhGDF7 treatment dramatically upregulated the expression level of fibrotic markers α-SMA and FN and downregulated N-cad expression in TM cells. Moreover, rhGDF7 augmented the accumulation of collagen in TM cells (Figures 2A and S4A). The disturbed expression of these fibrosis-related markers was also confirmed by real-time PCR and western blot (WB) (Figures 2B−2D).
Figure 2.

Excessive GDF7 protein-promoted fibrosis in cultured TM cells
(A) In cultured TM cells, the expression level of fibrosis-related markers was presented with immunofluorescence, and the collagen accumulation in TM cells was presented by Masson stain (n = 3 per group). (B) The mRNA levels of N-cadherin (N-cad), α-smooth muscle actin (α-SMA), and fibronectin (FN) were tested in recombinant human GDF7 (rhGDF7)-treated TM cells by quantitative real-time PCR assay (n = 3 per group). (C and D) As presented in western blot results, the protein levels of N-cad, α-SMA, and FN were measured in NTM cells with or without rhGDF7 treatment (n = 3 per group). Scale bars, 20 μm. The data represent the mean ± SD. Compared with NTM cells: ∗∗p < 0.01. Compared with rhGDF7 group: #p < 0.05, ##p < 0.01. nGDF7, GDF7 neutralizing antibody; Col I, collagen I.
Then we made GDF7 neutralizing antibody (nGDF7; details in Materials and methods) to specify whether nGDF7 could inhibit rhGDF7-induced fibrotic phenotypic change in TM cells. In rhGDF7-treated TM cells, the decrease of the N-cad could be prevented by nGDF7. Meanwhile, the rhGDF7-induced increase of α-SMA and FN could be inhibited by nGDF7 compared with the NC antibody. Also, the rhGDF7-induced accumulation of collagen in TM cells was significantly suppressed by nGDF7, as shown in Masson staining (Figure 2A). At transcriptional and translational levels, nGDF7 significantly suppressed Col I, α-SMA, and FN expression and rescued N-cad expression. These results showed that neutralizing GDF7 effectively repressed rhGDF7-induced fibrosis in TM cells (Figures 2B−2D). Taken collectively, TM fibrosis was promoted by excessive GDF7 protein and could be inhibited by GDF7 neutralization.
TET enzyme maintained GDF7 hypomethylation and facilitated transcription
DNA methylation in the form of 5-methylcytosine can be actively reversed to unmodified cytosine by TET dioxygenase,23 which is the most ubiquitous demethylation mechanism in eukaryotes. We wondered whether glaucomatous GDF7 hypomethylation was dependent on dysregulated TET activity. Therefore, we checked TET activity in TM samples. As measured by chemiluminescence, TET enzyme was dramatically activated in the GTM samples and cells as compared with NC (Figure 3A).
Figure 3.

Ten-eleven translocation (TET) activation promoted glaucomatous GDF7 production
(A) The activity of the TET enzyme was measured in nucleus extracts from TM samples/cells by chemiluminescence assay (n = 3 per group). (B) BSP assay was recruited to measure the methylation level of the GDF7 promoter in response to dimethyloxallyl glycine (DMOG) in NTM and GTM cells (n = 3 per group). (C) All of the RNA fragments that bond to the ETS1 protein were pulled down by ChIP and then tested by qPCR. The amplification of different regions in the GDF7 promoter was conducted with specially designed primers (details in Table S7; n = 3 per group). (D) Luciferase activity was measured to indicate the binding affinity between the GDF7 promoter and ETS1 (n = 3 per group). (E) The binding between ETS1 and the GDF7 promoter from NTM or GTM cells was tested by luciferase reporter assay (n = 3 per group). (F) As measured by ChIP-qPCR, the binding between the ETS1 protein and the GDF7 promoter presented in GTM cells compared with NTM cells in response to DMOG (n = 3 per group). (G) The transcription activity of the GDF7 promoter region in NTM cells and GTM was tested by the luciferase reporter assay. The transcription activity disturbance in response to DMOG was also measured (n = 3 per group). (H) GDF7 secretion was tested in conditioned medium by ELISA. GDF7 secretion was dramatically increased in GTM cells, which was effectively prevented by DMOG treatment (n = 3 per group). The data were represented as mean ± SD. Compared with NTM samples: ∗∗p < 0.01. Compared with NTM cells: ##p < 0.01. Compared with vector or scramble: @p < 0.05, @@p < 0.01. Compared with GTM cells: $p < 0.05, $$p < 0.01. siETS1, siRNA of ETS1.
Dimethyloxallyl glycine (DMOG), inhibitor of the TET enzyme, was employed to define the role of TET in the demethylation of the GDF7 promoter. In DMOG-treated NTM cells, there was no significant change in the methylation level of the GDF7 promoter region as measured by BSP, but in GTM cells, the methylation level of the GDF7 promoter was increased by 4 h DMOG treatment as compared to vehicle. Results indicated that the activated TET enzyme is required for the maintenance of the GDF7 promoter hypomethylation status in glaucoma but not the initiator of DNA demethylation during the pathological transition from NTM to GTM (Figure 3B).
To illustrate how the decreased methylation level in the GDF7 promoter facilitates the transcription of the GDF7 gene, reverse-chromatin immunoprecipitation (ChIP) was used to identify the transcription factors binding to the hypomethylated GDF7 promoter (detailed binding site presented in Figure S5A). Proteins pulled down by reverse ChIP probes were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) spectrometry. Three transcription factors were identified to bind with the GDF7 promoter in GTM cells: ETS1, Foxo1, and KDM3A (Figure S5B). The increased binding between ETS1 and region 2 of the GDF7 promoter in GTM cells was confirmed by ChIP-quantitative PCR (qPCR) compared with NC (Figure 3C). However, the other two transcription factors have similar binding affinity with the glaucomatous and normal GDF7 promoter, which lacked significance in pathological scenarios (Figures S5C and S5D).
To explore the binding between the ETS1 and GDF7 promoter, human ETS1 plasmid and luciferase reporter with the GDF7 promoter were co-transfected into TM cells. Luciferase activity was increased in ETS1-overexpressed cells. In ETS1 transient knockdown cells, the luciferase activity was suppressed, indicating that ETS1 can directly bind with the GDF7 promoter to enhance the gene transcription (Figures S5E and 3D). To understand the changes in transcription activity, region 2 of the GDF7 promoter from NTM and GTM cells was cloned into the luciferase reporter, respectively. Increased luciferase activity confirmed the increased transcription activity of the glaucomatous GDF7 promoter (Figure 3E). These results indicated that ETS1 can bind to the hypomethylated GDF7 promoter to facilitate gene transcription activity in POAG patients.
But whether GDF7 overexpression in POAG was mediated by the TET enzyme is unclear. As measured by ChIP-qPCR, DMOG treatment had no effect on the binding between the ETS1 and GDF7 promoter in NTM cells. However, the increased binding between ETS1 and GDF7 in GTM cells can be inhibited by DMOG (Figure 3F). Also, the role of DMOG in regulating GDF7 transcription was measured. The luciferase reporter assay confirmed that DMOG significantly inhibited transcription activity of the glaucomatous GDF7 promoter compared with that of vehicle-treated cells (Figure 3G). We further tested the essential role of TET in GDF7 production and secretion in glaucoma. As shown in Figure 1F, the GDF7 secretion was increased in the supernatant of GTM cells, and the glaucoma-induced overproduction of GDF7 was prevented by DMOG treatment (Figures 3H and S5F), indicating that TET was involved in GDF7 overexpression. Conclusively, the active TET enzyme maintained hypomethylation status in the GDF7 promoter to facilitate the gene transcription in POAG.
Then the role of TET in TM fibrosis was validated. In GTM cells, DMOG effectively inhibited the expression of Col I, α-SMA, and FN, as determined by the IF assay, WB, and real-time PCR, and the expression of N-cad was rescued by DMOG in GTM cells (Figures S6A−S6D). DMOG exerted no effect on expression of these markers in NTM cells. These results indicated that TET is crucial for the TM fibrosis in glaucoma.
TET-dependent positive feedback loop of GDF7 expression
To further elucidate the mechanism of methylation-dependent GDF7 overexpression in POAG, we subcultured the rhGDF7-treated NTM cells twice (P3 cells) without rhGDF7 in the culture medium. After 10–12 days, P3 cells and supernatants were collected. Interestingly, GDF7 secretion remained increased in P3 cell supernatants compared with NC (806 ± 13 pg/mL versus 349 ± 21 pg/mL, p < 0.05), indicating that exogenous GDF7 protein can trigger the expression of endogenous GDF7 in TM cells. To validate the hypothesis, GDF7 was transiently knocked down with small interfering RNAs (siRNAs; siGDF7). In P3 cells, GDF7 secretion was suppressed by GDF7 knockdown (742 ± 9 pg/mL), indicating a regulatory role of GDF7 protein in gene transcription (Figure 4A). Then we tested how exogenous GDF7 protein activates endogenous gene expression. We theorized that this regulatory effect lies in disturbed methylation in the GDF7 gene. As determined by the BSP assay, the methylation level of the GDF7 promoter remained decreased in P3 cells, and siGDF7 significantly increased the methylation level of the GDF7 promoter (Figure 4B). Also, siGDF7 effectively inhibited upregulation of α-SMA and FN, and expression of N-cad was regained by GDF7 knockdown in P3 cells as presented in IF (Figure 4C). The knockdown efficiency was validated by both real-time PCR and WB. In concordance, the upregulation of Col I, α-SMA, and FN in P3 cells was inhibited by GDF7 knockdown with an observed increase in N-cad at mRNA and protein levels (Figures S4A−S4C). These results indicated that the exogenous GDF7 protein demethylates the GDF7 promoter to trigger the endogenous production of GDF7, thus forming a positive feedback loop to maintain the fibrotic phenotype in TM cells.
Figure 4.

DMOG broke the positive feedback loop of GDF7 expression
(A) The conditioned medium from P3 cells was collected and measured by ELISA. The GDF7 protein concentration was presented (n = 3 per group). (B) The methylation level of the GDF7 promoter and mRNA level of the GDF7 gene were measured by BSP and RT-PCR, respectively, in P3 cells (n = 3 per group). (C) The expression of pro-fibrotic genes, α-SMA and FN, and the level of N-cad were presented by immunofluorescence labeling in P3 cells (n = 3 per group). Scale bars, 20 μm. The data were represented as mean ± SD. Compared with NTM cells: ∗∗p < 0.01. Compared with P3 cells: #p < 0.05, ##p < 0.01. Compared with siRNA of GDF7 (siGDF7)-treated P3 cells: $$p < 0.01.
As stated above, TET was pivotal in maintaining the demethylation status of the GDF7 promoter and in promoting the transcription of GDF7. Therefore, we wondered whether TET also participates in the positive feedback loop of GDF7 expression. After treatment with DMOG for 4 h, the methylation level of the GDF7 promoter was increased in P3 cells, as measured by BSP assay. Also, DMOG inhibited the GDF7 production, as detected by real-time PCR (Figure 4B). Next, the role of DMOG in fibrosis of P3 cells was tested. With the use of IF, we observed upregulation in N-cad after DMOG treatment. DMOG also inhibited the overexpression of fibrotic markers, α-SMA and FN, in P3 cells (Figures 4C and S7A). Consistently, DMOG prevented dysregulation of the fibrosis-related markers in P3 cells, as confirmed by real-time PCR and immunoblot (Figures S7B−S7D). These results indicated that the TET enzyme has a crucial role in the positive feedback expression of endogenous GDF7 and TM fibrosis.
GDF7 promoted TM fibrosis via BMPR2/Smad1, -5, and -9 signaling
Given that nGDF7 blocked the pro-fibrotic effect of the exogenous GDF7 protein as shown above, we assumed that the secreted GDF7 protein might function via binding to receptors on the plasma membrane. In order to figure out the GDF7 receptor, we conducted bioinformatics analysis to annotate the proteins interacting with GDF7. The results suggested that GDF7 closely interacted with BMPR2. Therefore LDN-193189 (LDN), the specific inhibitor of BMPR2, was employed to further illustrate the interaction between GDF7 and BMPR2 signaling.
As measured by IF, the rhGDF7-induced upregulation of α-SMA and FN was prevented by 5 nM LDN treatment for 24 h, and LDN could prevent the N-cad decrease in rhGDF7-treated TM cells. Also, LDN inhibited collagen deposition in rhGDF7-treated cells (Figures 5A and S8A). Consistently, the rhGDF7-induced TM fibrosis was inhibited by LDN with decreased α-SMA and FN, as measured by WB and real-time PCR (Figures 5B and S8B). However, LDN treatment has no effect in NTM cells (Figures 5A, 5B, S8A, and S8B), indicating that overproduction of GDF7 promoted TM fibrosis through activating BMPR2 in glaucoma.
Figure 5.

GDF7 promoted fibrosis through the BMPR2/Smad1, -5, and -9 pathway
(A) Fibrosis was determined by immunofluorescence labeling with N-cad, α-SMA, and FN together with Masson staining in cultured TM cells. (B) The mRNA levels of GDF7 and fibrosis-related markers N-cad, α-SMA, and FN were tested by quantitative real-time PCR in four groups of TM cells (n = 3 per group). (C and D) The ratio between phosphorylated and total Smad1, -5, and -9 and Smad4 protein was measured and analyzed by immunoblot in TM cells (n = 3 per group). Scale bars, 20 μm. The data were represented as mean ± SD. Compared with rhGDF7 group: ∗p < 0.05, ∗∗p < 0.01. Compared with NTM cells: #p < 0.05.
Activated BMPR2 works through its downstream effectors. The phosphorylation of Smad protein marks the activation of the BMPR2 signal. As shown in WB, Smad proteins were hyperphosphorylated in GTM samples compared with NC (Figure S8C), presenting the activation of the BMPR2/Smad signal in GTM. Then we examined whether the BMPR2/Smad signal was activated in response to rhGDF7. As measured by immunoblots, Smad1/-5/-9 and Smad4 were phosphorylated by rhGDF7 to their active forms. The rhGDF7-induced phosphorylation of Smad proteins could be inhibited by LDN (Figures 5C and 5D). Taken collectively, GDF7 promoted TM fibrosis through BMPR2/Smad signaling.
In silico validation of the GDF7/BMPR2/Smad signal in POAG
The artificial neural network (ANN)-based model (sketched in Figure 6A) was established to test the role of GDF7/BMPR2/Smad signaling in the POAG pathological mechanism and clinical manifestation. The methylation levels of 6 markers in this pathway (GDF7, BMPR2, Smad1, Smad5, Smad9, and Smad4) were used to predict 4 major clinical manifestations of POAG. As presented, our model predicted the attenuated RNFL with area under the curve (AUC) 85.9%, a high IOP with AUC 82.9%, a high CDR with AUC 92.4%, and VF defects with AUC 87.2% (Figure 6B). The confusion matrixes of 4 clinical manifestations are presented in Figure 6C, and the sums of all false positive and false negative cases were below 20% (Figure 6D). The evaluation indices are presented in Table S3. In all, these two observations indicated that our model is well trained and robust.
Figure 6.

Validation of GDF7 methylation in POAG pathogenesis with the ANN model
(A) Study pipeline for agent training, validation, and testing. Methylation levels of GDF7, BMPR2, Smad1, Smad5, Smad9, and Smad4 were enrolled in the training library to predict four binary POAG-related outcomes (high IOP, attenuation of RNFL thickness, high CDR, and visual field [VF] defects). (B) The ANN-based model was used to predict the four binary POAG-related outcomes. The accuracy of the model was presented by area under the curve (AUC). (C and D) Summary of the confusion matrices in the four outcome networks was displayed. (E) The contribution of input indexes (methylation levels of GDF7, Smad1, Smad5, Smad9, and Smad4) in predicting clinical outcomes related to POAG was calculated. TP, true positive; TN, true negative; FP, false positive; FN, false negative.
Then we analyzed the contribution of 6 methylation markers to the clinical outcomes of POAG. GDF7 methylation level acquired the largest contribution to POAG-related outcomes (contribution factor −1.00 in RNFL, −0.80 in IOP, −0.33 in CDR, and −1.00 in VF). Such a phenomenon is consistent with our previous finding that GDF7 hypomethylation was the critical event in POAG development. BMPR2 methylation level contributed the least (contribution factor 0.00 in RNFL, 0.00 in IOP, 0.02 in CDR, and 0.18 in VF) in predicting the clinical outcomes. This result also fit the biological function of the BMPR2 receptor, which is primarily regulated through ligand binding and post-translational modifications24 rather than DNA methylation. Meanwhile, the methylation level of Smad1 (contribution factor −0.42 in RNFL, −0.54 in IOP, −0.20 in CDR, and −0.35 in VF), Smad9 (contribution factor −0.06 in RNFL, −0.42 in IOP, −0.18 in CDR, and 0.03 in VF), and Smad4 (contribution factor 0.20 in RNFL, 0.24 in IOP, 0.13 in CDR, and 0.22 in VF) substantially contributed to the clinical manifestations (Figure 6E).
GDF7 neutralization effectively inhibited TM fibrosis in rhesus monkeys
Each eye of the eight monkeys had a baseline IOP below 20 mmHg, and no ocular abnormalities in the anterior segment, fundus, or the RNFL thickness were observed before the experiment (Figures S9A−S9F). 2.5 μg rhGDF7 was delivered into the anterior chamber three times on weeks 0, 2, and 4. After 10 weeks’ treatment of rhGDF7 (sketched in Figure 7A), the AH outflow facility was dramatically decreased (c = 0.154 ± 0.003 μL/min · mmHg) compared with that of control (CON) eyes (c = 0.342 ± 0.007 μL/min · mmHg, p < 0.05). If left untreated, the outflow facility of rhGDF7 eyes remained low during our observation period (Figure 7B). Meanwhile, the IOP was gradually elevated in rhGDF7-treated monkey eyes, from 16.39 ± 1.73 to 21.12 ± 0.25 mmHg (p < 0.05). During the observation period, the IOP of the rhGDF7-treated monkeys continuously exceeded that of the CON monkeys (p < 0.05) (Figure 7C). The intraocular effect of rhGDF7 treatment in monkey eyes lasted for 7 months, which further confirmed the positive feedback loop of GDF7 expression in vivo. Concurrently, the RNFL thickness, a direct index of glaucomatous neuronal lesions, was reduced in response to rhGDF7 treatment, especially in nasal (54.56 ± 1.62 μm versus 61.33 ± 1.33 μm in CON, p < 0.05) and temporal (61.53 ± 1.35 μm versus 69.93 ± 0.98 μm in CON, p < 0.05) parts of the fundus (Figures S10A and S10B). Conclusively, the intraocular accumulation of the GDF7 protein disrupted the AH outflow, increased the IOP, and caused neuronal lesions in monkeys.
Figure 7.

GDF7 neutralization protected TM function in rhesus monkeys
(A) Simplified workflow of animal experiment. In brief, rhGDF7 was delivered into the anterior chamber three times on the first days of weeks 0, 2, and 4. The nGDF7 was injected into the anterior chamber for three times on the first days of weeks 10, 12, and 14. Ophthalmic examinations were conducted every 2 weeks throughout the entire process. The TM samples were harvested on the end of week 28. (B) The aqueous humor (AH) outflow facility was measured throughout the whole experiment. (C) Curves presented the IOP change in response to rhGDF7 delivery or/and nGDF7 treatment in monkey eyes (n = 4 per group). (D) Immunofluorescence labeling with N-cad, α-SMA, and FN was conducted in monkey TM tissues, and collagen accumulation was determined by Masson trichrome staining (n = 4 per group). Scale bars, 20 μm.
The nGDF7 was delivered into the anterior chamber of monkey eyes three times on the first days of weeks 10, 12, and 14 in the nGDF7 group. 18 weeks after nGDF7 treatment, the rhGDF7 reduced outflow facility was greatly recovered (0.31 ± 0.005 μL/min · mmHg in the rhGDF7 + nGDF7 group versus c = 0.124 ± 0.009 μL/min · mmHg in the rhGDF7 group, p < 0.05) (Figure 7B). Also, the IOP elevation was inhibited by nGDF7 (IOP = 17.30 ± 0.12 mmHg) compared with that of rhGDF7-treated eyes (IOP = 21.81 ± 0.54 mmHg, p < 0.05) (Figure 7C). Moreover, the rhGDF7-induced attenuation of the RNFL thickness was suppressed by the nGDF7 (Figures S10A and S10B). Simple delivery of nGDF7 into normal eyes presented no significant difference. Therefore, the GDF7 neutralizing treatment effectively blocked the rhGDF7-induced damage in AH outflow.
Mechanistically, intracameral delivery of exogenous GDF7 protein activated the endogenous expression of GDF7 via a positive feedback loop. The nGDF7 effectively prevented GDF7 overexpression in monkey TM samples, as measured by immunoblot (Figure S11B). These results indicated that GDF7 neutralization exerted protective effects via interrupting the positive feedback loop. Moreover, various disturbances of fibrotic markers were observed in rhGDF7-treated TM tissues, which could be markedly inhibited by the neutralizing antibody. As measured by the IF assay and WB, the rhGDF7-induced increase in α-SMA and FN was suppressed by nGDF7. Also, nGDF7 prevented the rhGDF7-induced loss of N-cad (Figures 7D and S11A). Meanwhile, the accumulation of collagen in TM tissues was inhibited by GDF7 neutralization, as measured by Masson staining compared with rhGDF7-treated eyes (Figure 7D). In addition, the nGDF7 significantly suppressed the phosphorylation of Smad1, -5, and -9 and Smad4, thus inhibiting the activation of BMPR2/Smad signaling in monkey eyes (Figure S11C).
Taken collectively, intracameral GDF7 delivery resulted in glaucoma-like TM fibrosis and clinical manifestations in monkey models. GDF7 neutralizing treatment effectively prevented the fibrotic phenotype and improved AH outflow through TM.
Discussion
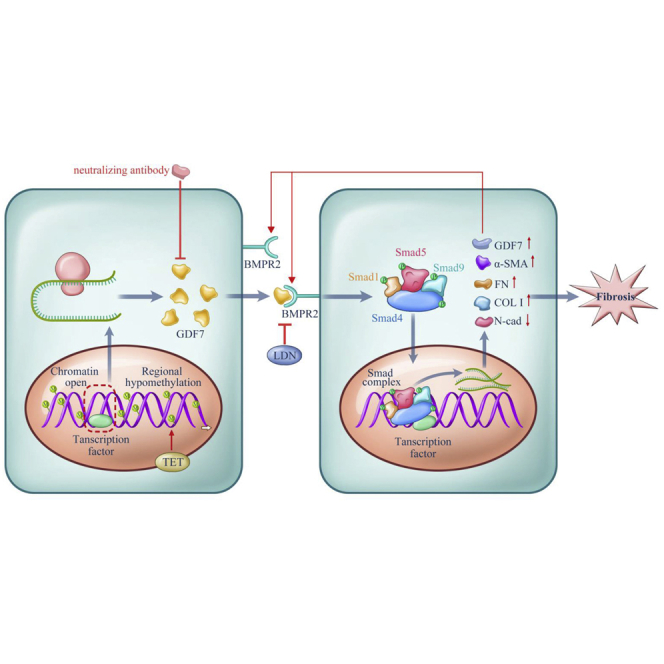
A TM fibrosis-induced disturbance in AH outflow is an important cause of retinal ganglion cell death and thus is involved in the pathological process of glaucoma.25,26 Here, for the first time, we identified GDF7 hypomethylation as the crucial contributor of AH outflow resistance in glaucoma patients. Mechanistically, the demethylation of the GDF7 promoter facilitated the transcription of the GDF7 gene and then increased production of the GDF7 protein. Locally accumulated GDF7 protein promoted TM fibrosis, thus declining the outflow facility of AH and elevating the IOP. rhGDF7-induced phenotypic change is comparable to the glaucomatous manifestations in patients. For therapeutic purposes, we developed the nGDF7 to effectively inhibit glaucomatous damage in rhesus monkey eyes (summarized in Figure 8).
Figure 8.

Proposed model for the methylation-dependent pathology in POAG
Hypomethylation in the GDF7 gene gave rise to the increased GDF7 mRNA transcription via facilitating transcription factors binding to open chromatin. The TET enzyme sustained GDF7 overexpression by keeping the promoter region hypomethylated and open. Excessive GDF7 proteins activated BMPR2 and phosphorylated the downstream effectors, Smad1, -5, and -9 and Smad4. The phosphorylated Smad complex translocated into the nucleus and further promoted the overexpression of GDF7 and fibrosis markers, including Col I, α-SMA, and FN. Meanwhile, the expression of N-cad was suppressed, which remains to be explored in further studies. The imbalance between these proteins triggered TM fibrosis, which leads to the function failure in TM and ended up in IOP elevation. The accumulated GDF7 protein also activated the TET enzyme via an unknown mechanism, thus forming a positive feedback loop. Based on the aforementioned mechanism, the nGDF7 could impede the pro-fibrotic effect of GDF7. Therefore, GDF7 neutralization halts the progression toward fibrosis and maintains a normal AH through the TM.
There is one concern about the different preliminary sampling procedures: NTM was obtained from healthy donors, and GTM was obtained during trabeculectomy. Such difference may introduce biases on the following analyses, especially the microarray results. To minimize the biases, we established an identical isolation procedure after the preliminary sampling. Specifically, the isolation was carried out by two experienced ophthalmologists to remove all of the adjacent tissues under an optical microscope and only retained the translucent and meshwork-shaped tissue. Conformity of isolation procedures in both NTM and GTM secured the purity of TM cells in our samples, thus reducing the possible bias in the following analysis.
We also identified a positive feedback loop in the epigenetic regulation of GDF7 expression. This positive feedback regulation of GDF7 perfectly matches the clinical manifestations, including the gradual IOP elevation and continual RNFL attenuation. These clinical manifestations were attributed to the consistent and irreversible circulatory disturbance of the AH.27 In regenerative tissues, such as TM, the transient environmental impact or temporary post-translational regulation cannot fit into this continuous clinical pattern. Only with the help of a positive feedback loop can the regional methylation disturbance in GDF7 lead to an increase in the magnitude of the AH circulatory perturbation. In our study, DMOG, the inhibitor of the TET enzyme, interrupted the GTM fibrosis by increasing the methylation level of the GDF7 promoter (Figure 5C). Therefore, we ascribed the positive feedback loop to activation of TET enzymes in our scenario. However, the detailed landscape of this positive feedback loop requires further studies.
In this study, DMOG was recruited to inhibit the activity of the TET enzyme and further illustrate the role of the TET enzyme in POAG. It should be noted that DMOG, as a synthetic analog of α-ketoglutarate, can also induce the hypoxia-inducible factor (HIF) signal. However, as indicated by the manufacturer (Sigma-Aldrich) and published data, the inhibit function of the DMOG is dose specific. The DMOG dose inhibiting TET is 10 times different with the dose to regulate HIF.28,29 Also, many studies used DMOG as a TET inhibitor to inhibit TET activity both in vivo and in vitro.30, 31, 32 Based on this information, we assume that DMOG functioned mainly as a TET inhibitor in this study.
The GDF7 positive feedback loop in glaucoma patients depends largely on the hyperactivation of the TET enzyme. Three regulatory mechanisms have been implicated in the modulation of TET activity. First, TET activity can be determined by the levels and type of the transcript produced, as reported by the Good et al.33 and Jiang et al.34 groups. A variety of the enhancer and promoter can regulate the expression of transcripts to encode TET proteins with different catalytic activity. Second, TET activity can also be regulated by post-translational modifications (e.g., ubiquitination, acetylation, phosphorylation, GlcNAcylation, and PARylation).23,33,34 Third, TET activity was related to the expression of multiple microRNAs.35 It would be interesting to identify the specific regulator of the TET enzyme in future studies, which can provide more basic insights into the pathogenesis of GDF7 hypomethylation-related POAG.
The rhGDF7 protein was used to illustrate the role of GDF7 in cultured TM cells and in monkey eyes. However, we are not able to distinguish the exogenous GDF7 protein from the endogenous one. The protein tags36 (His or Myc) and radioactivity labeling are both effective methods but can only be achieved in vitro. The fluorescent label is a theoretically feasible method to monitor the exogenous GDF7 protein in vivo. However, the molecular weight of the fluorescent tag (GFP or mCherry) is relatively large,37 so it will induce the TM dysfunction bias by blocking the AH outflow physically. Another limitation of the fluorescent-label method is that we cannot exclude the tag bias in causing conformation and functional domain disturbance of the protein. Hence, it would be meaningful to develop a novel method to detect rhGDF7 from endogenous protein in future studies.
Based on these mechanistic findings, we developed the nGDF7, which was effective in rhesus monkeys. In past decades, many efforts have been made in targeting DNA methylation, but general methylation regulators, such as DAC, influence the entire family of DNA methyltransferases, which can bring about severe side effects.38,39 However, the nGDF7 specifically targeted the GDF7 protein and blocked its detrimental effect regarding TM fibrosis. In addition, the neutralizing treatment was very safe in eyeballs. Intracameral injection is widely used in clinical routines to treat infection, post-operation inflammation, and cystoid macular edema with great safety and efficacy.40 Due to the relatively large molecular weight of our antibody (45 kDa), it cannot pass through the blood-ocular barrier or enter other tissues. This critical characteristic will greatly protect patients from possible circulatory side effects. Moreover, unlike small molecules, antibody elimination occurs mostly through receptor-mediated endocytosis, which is dependent on a limited number of receptors.41 With a relatively slow elimination rate, the half-life of the neutralizing antibody was significantly longer (9–14 days) compared with traditional anti-glaucoma drugs (2–4 h).42 Therefore, the effective dose of the nGDF7 can be quite low (0.05 mg/injection), which might also be safe for patients. In addition, GDF7 neutralization achieved high-efficiency control of the IOP, improved the outflow facility, and protected nerve fibers in primates, as reported in our results. Therefore, with great safety and efficacy, the nGDF7 appears to be a promising therapy in POAG patients.
We checked fibrosis in the three random NTM and GTM samples. As presented in Figure S3A, we found that the fibrosis in GTM #1 and #3 was obvious, and GTM #2 was not that obvious. This individual difference was consistent with the published literature, which suggested other pathological findings in the TM of POAG patients. Notably, cross-linked actin network (CLAN) formation in TM tissues acquired the most attention due to the following evidence. First, the formation of CLANs has been found more commonly in the cultured GTM cells compared to the NTM cells.43,44 Second, the increased CLAN formation was also witnessed in glaucomatous human eyes in situ.45 Third, the formation of CLANs was involved in AH outflow resistance.46 Given this evidence, it will be interesting to confirm the function and investigate the underlying mechanism of the CLAN formation in POAG.
This study identified GDF7 methylation as the novel epigenetic event in the pathogenesis of glaucoma. The ANN model confirmed the methylation-dependent regulatory mechanism via GDF7/BMPR2/Smad signaling in silico. Furthermore, this model provides new insights into the detection of POAG and the recognition of clinical manifestations with specific methylation markers. The nGDF7 proved to have great safety and efficacy in the management and treatment of glaucoma, and this has launched a groundbreaking paradigm shift.
Materials and methods
Patients and CONs
Nighty-eight POAG patients and eight NCs were enrolled in this study. The POAG group recruited patients with an age between 18 and 80 years and an adherence to the diagnostic criteria of POAG (simplified version is shown in Table S4).47,48 The inclusion criteria comprised patients who needed to undergo trabeculectomy for uncontrolled IOP under maximal-tolerated topical medical therapy. Subjects with a visual acuity of <20/40; a spherical refraction of <−10.0 or >+3.0 diopters (Ds); a history of ocular surgery; evidence of retinal or macular pathology; or a history of pseudoexfoliation syndrome, diabetes mellitus, uveitis, systemic collagenopathy, and objective neurologic signs, or use of topical or systemic corticosteroids, were excluded. The patients that met the inclusion criteria of POAG without trabeculectomy were excluded from our study. Eight patients were randomly picked from the POAG group for genome-wide DNA methylation screening with the microarray. The samples from the rest of the 90 patients were used for the validation of GDF7 expression.
The healthy donors were recruited from the Eye Bank of Zhongshan Ophthalmic Center (Guangzhou, Guangdong, China). These donors were required to have no known ocular diseases, no family history of glaucoma, and a normal optic nerve head appearance. Eight healthy donors were matched with POAG subjects in DNA methylation screening regarding the sex and age using a frequency-matching method.
This study was approved by the Ethics Committee for Human Experimentation at Zhongshan Ophthalmic Center, Sun Yat-sen University (Guangzhou, China; 2017KYPJ021), and it was performed in accordance with the Declaration of Helsinki. Patients or the first-degree relatives of the healthy donors were well informed about the study and potential risk. Written, informed consent was obtained from all of the subjects for collection of TM samples and clinical data.
Data collection
The demographic data (including age, sex, and medical history) of POAG patients and healthy donors were recorded. Visual acuity, IOP, VF, slit lamp, optical coherence tomography (OCT), ophthalmoscope, and gonioscopy examinations were carried out in POAG patients.
TM sampling
NTM samples were obtained from the leftover tissue after cornea transplantation. In all cases, death to preservation time was less than 12 h. NTM samples were isolated from the juxta-canalicular and corneoscleral regions of the human eye.
The GTM samples were obtained according to standard surgical procedures. The excision was conducted at least 0.5 mm away from the limbus to avoid any damage to the corneal epithelium.49 The procedures were executed under microscopic control to secure that the removed specimens contained TM cells.
After sampling, the isolation was carried out by two experienced ophthalmologists in both preliminary NTM and GTM samples. Specifically, we removed all of the tunica pigmentosa and sclera under the optical microscope (40 magnificence) and only retained the translucent and meshwork-shaped tissue.
Every TM sample was divided into four equal amounts: one for DNA methylation analysis, one for embedding, one for protein homogenate, and one for RNA extraction. Histology examination was performed in three randomly selected samples (1 NTM and 2 GTM). The majority of TM cells in the specimens were determined by hematoxylin and eosin (H&E) staining (Figure S3B). Immediately after isolation, the samples were immersed in a corneal storage medium (Optisol; Chiron Vision, Irvine, CA, USA) at 4°C immediately and stored at –80°C until use.
The following NTM samples were used: NTM1 (52-year-old male), NTM2 (45-year-old male), NTM3 (64-year-old male), NTM4 (58-year-old male), NTM5 (58-year-old male), NTM6 (61-year-old female), NTM7 (45-year-old female), and NTM8 (64-year-old female). The average age of the NTM donors (n = 8), GTM donors (n = 8), and GTM validators (n = 90) was 55.9 years, 57.4 years, and 52.0 years, respectively (p > 0.05).
Genome-wide DNA methylation analysis
TM samples from the 8 POAG patients and 8 healthy donors were collected and used for genome-wide DNA methylation analysis as stated above. Illumina human methylation 850K chips (Illumina, San Diego, CA, USA) were used to examine the DNA methylation status. Total DNA was extracted using the QIAamp DNA Mini Kit (QIAGEN) and then subjected to bisulfite conversion using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). The bisulfite-converted DNA was loaded onto the Infinium Methylation EPIC BeadChips. The BeadChips were processed through a single nucleotide extension, followed by immunohistochemical staining. After the primer extension process, the chips were imaged on an Illumina iScan.
We processed all Illumina array raw data using the methylation module of Genome Studio version (v.)1.9 software. The criteria for identifying the disturbed methylation regions were |β-difference| > 0.2, and p values < 0.05. To assign biological meaning to the disrupted methylation regions, two forms of annotation clustering, Gene Ontology (GO) classifications and KEGG pathways, were employed. At least two genes had to be present in a GO term group or a KEGG pathway, and the p value had to be less than or equal to 0.05 for the genes to be considered.
BSP
For BSP, genomic DNA was isolated using the Genomic DNA Isolation Kit (QIAGEN, Valencia, CA, USA) and then subjected to bisulfite conversion using the EpiTect Fast DNA Bisulfite Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. The bisulfite-converted genomic DNA was used to measure the methylation level of five candidate genes with the methylation primers (details in Tables S5A and S5B). The amplified fragments were cloned into the pTG19-T vector (lot: GV6021; Beijing, China), and ten clones were randomly selected for BSP. Controls to assess the accuracy of the bisulfite conversion of the DNA were included in each run to ensure the fidelity of the measurements.
Reverse ChIP assay
Reverse ChIP was conducted as previously described.50 In brief, GTM cells were fixed, and the chromatin was solubilized. Twelve probes targeting different regions of the GDF7 gene (details in Table S6) were hybridized to the chromatin. The hybridized chromatin was captured using magnetic beads and then eluted. The captured proteins were identified by LC-MS/MS spectrometry. MS data were collected using the positive ion mode by selected reaction monitoring. Precursor ions were selected by the first quadrupole (Q1) and allowed to pass into the collision cell, which was a quadrupole in radio frequency (rf)-only mode (q). The second quadrupole (Q2) was set to allow only product ions of a specific mass-to-charge ratio (m/z) value.
ChIP-qPCR
ChIP-qPCR was performed essentially as described by Sinkkonen et al.51 In brief, TM cells were crosslinked with 1% formaldehyde while gently shaking for 15 min, and the DNA was sheared to fragments ranging from 200 to 400 bp by sonication. The genomic fragments were precipitated using antibodies against ETS1 (1:50; Cell Signaling Technology [CST], Danvers, MA, USA), Foxo1 (1:50; Abcam, Cambridge, MA, USA), and KDM3A (1:50; Abcam). The primers for the amplification of GDF7 were designed according to the four predicted binding sites detected in the reverse ChIP assay (details in Table S7). ChIP-qPCR was run in triplicate, and each ChIP reaction was repeated twice to confirm the reproducibility of results. The results were normalized to the input DNA amplifications.
Luciferase report assay
2.5 kb of the human GDF7 (NCBI: NC_000002.12) promoter sequence with all predicted binding sites was cloned upstream of the luciferase reporter. Human ETS1 cDNA plasmid was purchased from Sino Biological (PA, USA). Human ETS1 plasmid or siRNA of ETS1 (siETS1) was co-transfected with luciferase reporter plasmid into TM cells to test the interaction between the GDF7 promoter and ETS1 protein. pcDNA vectors without ETS1 open reading frame (ORF) or scramble siRNA were used as a negative control. Stable expression of the reporter construct, pGDF7-Luc, or the pGL-3 basic luciferase vector (Promega, Madison, WI, USA) served as a quality control. The Renilla reporter (RLuc; Promega) plasmid was used to normalize the transfection efficiency. Three independent experiments were performed.
Production and validation of the neutralizing antibody
For each immunization, 100 mL of the immunogen mixture, containing 30–60 mg of filter-sterilized rhGDF7 and 60 mg of poly I:C in PBS, was injected into both inner rear thighs of mice. The mice were immunized biweekly for four times. The mice spleens were collected 1 week after all immunizations and then fused with Sp2/0-Ag14 cells. Ten cell colonies were picked and cultured. The supernatants were harvested 1 week later, and the immunoglobulin G (IgG) was purified using protein G Sepharose (GE Healthcare, Chicago, IL, USA) and buffer exchanged with PBS. The neutralizing capability of the antibody was tested by regulating induced alkaline phosphatase production (Abcam, Cambridge, MA, USA) in ATDC5 cells.52,53 The neutralizing titer was tested in the ten strains, and the most efficient one, with half-maximum inhibitory concentration in 5 μg/mL, was used in a later experiment. The antibody purified from wild-type mice was taken as the control.
TM cell culture and specification
Tissue obtained from human donors <65 years of age provides an adequate number of TM cells with appropriate growth characteristics that enhance successful culture development.54 NTM cells were cultured from donor numbers (donor nos.) 3, 4, and 7, and GTM cells were cultured from donor Nos. 2, 3, and 6 (detailed information of patients listed in Table S1).
The iris and ciliary processes are gently removed from the anterior segment before isolation of the TM specimens. TM cells were isolated from the juxta-canalicular and corneoscleral regions of the NTM and GTM samples using the blunt isolation method as previously described.55,56
The cells were cultured at 37°C in 5% CO2 in TM Cell Medium (TMCM) (ScienCell, Carlsbad, CA, USA), supplemented with 2% (v/v) heat-inactivated fetal bovine serum (FBS; ScienCell), 1% TM Cell Growth Supplement (TMCGS; ScienCell), and 1% penicillin/streptomycin (P/S; ScienCell). All new primary cultures were treated with 0.1 μM dexamethasone (DEX; Sigma-Aldrich, St. Louis, MO, USA) for 4 days to validate the origin of cell strains,54 and only strains that had an increased expression of myocilin (a marker for human TM [HTM] cell identity) were used in this study (Figure S3C). Confluent TM cells were used between passages 1 and 5 for all experiments. Three normal and three glaucoma donor cell strains were used in every independent experiment.
Cell treatments
12 h prior to treatments, the cells were processed for serum starvation. After starvation, DAC (Sigma-Aldrich; 0.5 μM), rhGDF7 protein (Abcam; 5 ng/ml), nGDF7 (10 μg/mL), rhETS1 protein (Abcam; 5 ng/mL), DMOG (1 mM), or LDN (Selleck Chemicals, Houston, TX, USA; 5 nM) was added into the culture medium.
For siRNA transfection, the cells were cultured until 80% confluence. The siRNAs were mixed with Attractene transfection reagent (QIAGEN, Hilden, Germany) to a final concentration of 10 μg/mL, according to the manufacturer’s instructions. After incubation for 30 min, the siRNA mixtures were added to the culture medium. Transfection was conducted for 48 h to maximize the knockdown efficiency.
For plasmid transfection, the cells were cultured until 80% confluence. The plasmids were mixed with Attractene transfection reagent (QIAGEN) and OPTI-MEM (Gibco) to a final concentration of 1 μg/mL, according to the standard protocol. After incubation for 30 min, the plasmid mixtures were added to the culture medium. After 16 h transfection, the efficiency was visualized under a fluorescent microscope. Cells were harvested after 24 h transfection for the following assays. Three independent experiments were performed.
IF and histological staining
TM cells were seeded on coverslips (Millipore, Burlington, MA, USA), followed by fixation with 4% paraformaldehyde, permeabilization with 0.2% Triton X-100, and blocking with 1% bovine serum albumin. For whole-mount staining, TM samples were fixed with 10% neutral formalin for 24 h and then embedded in paraffin. The paraffin blocks were sliced into 4 μm-thick sections. The sections were deparaffinized, heat treated, and digested with protease K before staining.
The samples were incubated with primary antibodies at 4°C overnight, followed by a species-compatible secondary antibody for 1 h at room temperature. The sources and dilutions of antibodies are listed in Table S8. Cell nuclei were then labeled with 50 ng/mL 4′,6-diamidino-2-phenylindole (DAPI; CST) for 5 min. Images were visualized and captured by a Zeiss LSM 510 confocal laser-scanning microscope and processed by Adobe Photoshop CS8.
For histological staining, the TM sections and coverslips were examined by Masson’s trichrome (Accustain HT15; Sigma-Aldrich) and H&E staining, according to a standard procedure.57 Images were taken by Zeiss AxioObserver Z1. Ten fields were randomly checked in every slide.
Quantitative real-time PCR
Total RNA was extracted from tissues or cells using the TRIzol reagent (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instruction. The cDNA was synthesized using the PrimeScript RT Master Mix (TaKaRa, Kusatsu, Japan). The primers were listed in Table S9. Quantitative analysis was performed by quantitative real-time PCR using the SYBR Advantage qPCR Premix Master Mix (TaKaRa), according to a standard protocol. The target mRNA copy numbers were measured and normalized to that of β-actin. The comparative Cp method was used to evaluate the expression levels.
WB
Total protein was isolated from the tissues or cells with radioimmunoprecipitation assay (RIPA) lysis solution (Beyotime, China). Protein samples were electrophoresed on 4%–15% gradient polyacrylamide gels, according to a standard procedure. The expression levels of target proteins were normalized to β-actin using ImageJ software. The primary antibodies and dilutions were listed in Table S8.
ELISA
The tissue homogenates and cell supernatants were specifically prepared to measure the GDF7 level using the Human GDF7 ELISA Kit (Elabscience, Beijing, China). For tissue, it was rinsed with PBS for three times and immediate homogenization on ice, and the conditioned medium from TM cells should spin down at 4,000 × g for 40 min to remove the dead cells and debris before testing.
To measure the TET enzymatic activity, nuclei were extracted using the EpiQuik Nuclear Extraction Kit (Epigentek Group, Farmingdale, NY, USA) and then tested with the Epigenase 5mC Hydroxylase TET Activity/Inhibition Assay Kit (Epigentek Group). The absorbance was read at 450 nm with a reference wavelength of 655 nm. Three independent experiments were performed.
Husbandry statement of animals
All experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University and were performed in accordance with the Guide for the Care and Use of Laboratory Animals (Eighth Edition). Eight healthy adult rhesus monkeys (Macaca mulatta; three females and five males) with an initial body weight of 10.3 ± 3.1 kg and an initial age of 8.2 ± 1.2 years were purchased from Blue Island Biological Technology (Beijing, China). Each animal was individually caged and participated in an environmental enrichment program designed to encourage sensory engagement and social behaviors. The room temperature was maintained between 20°C and 24°C with the humidity between 40% and 70%. The animal rooms were set on a 12-h light and 12-h darkness cycle with scattered small light bulbs if necessary. Fresh air was ventilated into the animal room every 5−8 min. The animals were fed with monkey chow twice a day and supplemented with fresh fruits and vegetables once per day to ensure enough vitamin C intake. Routine veterinary physical examinations were performed on all animals.
Animal anesthesia and care after surgery
The veterinarian followed Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines to ensure appropriate handling for animal immobilization, sedation, and anesthesia. Abnormal postures, anorexia, vocalization, lethargy, and self-directed behaviors are indicators to evaluate pain or distress.
Anesthesia was first induced with ketamine hydrochloride (15 mg/kg, intramuscularly [i.m.], Ketalar 50; GuTian Pharmaceuticals, Beijing, China) and then maintained with inhalation anesthesia. All surgical procedures were performed under gaseous anesthesia (isoflurane 1%–2% in 1.0 l/min oxygen).58 The anesthesia depth was further confirmed by reflex tests. Animals’ vital life signs were monitored continuously with electrocardiogram, pulse oximeter, temperature, and end-tidal CO2 throughout the experiment.
Animals were covered by blankets to keep warm and closely monitored until revive. New toys, cage changes, or comforting foods (e.g., fruits, cookies, and candies) were employed to moderate stress without compromising the scientific aspects of the experiments. Monitoring of monkeys after surgery included daily clinical assessments by study staff and regular evaluations by veterinary staff. Animals were euthanized by overdosed sodium pentobarbital (∼200 mg/kg, intravenously [i.v.]) at the end of the experiment.
Ophthalmic examinations in animals
Before examination, 3.0% to 4.0% of isoflurane in 2.0 L/min oxygen was administered through a Bain coaxial system (Moduflex; Dispomed, Joliette, QC, Canada) connected to a mask for induction and maintenance with 2% isoflurane in 1.0 L/min oxygen. Monkeys were exposed to isoflurane through a mask for 5 min before topical proparacaine HCl (Alcaine, 0.5%; Alcon Laboratories, Ft. Worth, TX, USA). Then slit-lamp microscopy and photography (BX-900; Haag-Streit AG, Switzerland), gonioscopic examination (G-1 Trabeculum; Volk, USA), fundus photography (TRC-50DX retinal camera; Topcon, Tokyo, Japan), OCT (Spectralis OCT; Heidelberg Engineering, USA), and Schiotz tonography (YZ7A tonometer; 66 Vision Tech, China) were conducted according to standard procedures.
The AH facility (C) was measured in the supine position using a Schiotz tonometer with a standard 5.5-g weight over a period of 4 min. The initial reading of the Schiotz tonometer and reading after 4 min were recorded as Pt0 and Pt4, respectively. The outflow facility (C) can be calculated with the following formula:59
Ophthalmic examinations were performed every 2 weeks between 9:00 a.m. and 12:00 p.m. For measurement of IOP and AH outflow facility, each eye was measured 3 times, and the values were averaged.60 All of the examinations were conducted by the same two experienced technicians to minimize the technical variance. Isoflurane was stopped immediately after examination.58
Intracameral delivery of rhGDF7 and nGDF7
The eight monkeys were randomly divided into the following four groups: CON group nGDF7 group, rhGDF7 + nGDF7 group (rh + nGDF7), and rhGDF7 + CON antibody group (rhGDF7), with two monkeys in each group. All procedures were performed under general anesthesia, as stated above, followed with topical proparacaine HCl in all procedures involving contact with the cornea.
Under a stereoscopic microscope (M844; Carl Zeiss Meditec, Dublin, CA, USA), an insulin syringe was inserted into the anterior chamber of the eye through the limbus, taking care not to collapse the anterior chamber or damage Descemet’s membrane. After 0.1 mL of the AH was aspirated, the rhGDF7 solution (0.1 mL, 25 μg/mL) was delivered into the anterior chamber61 three times on the first days of weeks 0, 2, and 4. 10 weeks after the delivery of rhGDF7, 0.1 mL nGDF7 solution (0.5 mg/mL) was injected into the anterior chamber three times on the first days of weeks 10, 12, and 14. Balanced salt or NC antibody solution was used as control. Tobramycin ointment was applied to prevent bacterial infection.
Construction and validation of the ANN-based prediction model
Input variables included the regional methylation levels of six markers involved in the GDF7/BMPR2/Smad pathway (GDF7, BMPR2, Smad1, Smad5, Smad9, and Smad4) to predict four binary POAG-related outcomes (elevated IOP, attenuated RNFL thickness, high CDR, and VF defects) (detailed definitions in Table S10).
We constructed the neural network using the toolbox in MATLAB 2016b (MathWorks, Natick, MA, USA). The neural network algorithm is based on the systematic training of interlinked simple processing “elements” or artificial “neurons.”62 As every neuron was linked with many other neurons, the network offers an ability to represent both linear and nonlinear relationships directly from the data. Before building the model, the ANN was tuned using a simultaneous grid search for the optimal size of the hidden neurons. Regularization was used to combat ANN overfitting by augmenting the error function used for training with the squared magnitude of the weights used (more details could be referred to Arvind et al.63).
Eight-fold cross-validation was applied for training and testing. Specifically, for each fold, 14 samples were randomly selected for training, and the remaining two samples were used for validation. This procedure was repeated eight times. The performances were evaluated by receiver operating characteristic (ROC) curve values, AUC values, and indices of confusion matrix. The relative contribution of each methylation marker was also deciphered.64,65 All of the statistical tests were conducted using Statistical Software R (version 3.2.4).
Statistics and reproducibility
The results of the experiments presented in the figures were representative of at least three independent repetitions. All data were presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was performed, followed by the Bonferroni multiple comparison tests and two-way ANOVA using GraphPad Prism data analysis software (version 7.0; GraphPad Software). A p value less than 0.05 was considered statistically significant. The Pearson’s method was used to test the correlation between the clinical parameters and DNA methylation.
Acknowledgments
The sponsor of this study had no role in the design of the original study’s protocol; in the collection, analysis, and interpretation of the data; in the writing of the report; or in the decision to submit the manuscript for publication. The linguistic assistance provided by TopEdit LLC during the preparation of this manuscript is acknowledged. This study was supported by the National Natural Science Foundation of China (81470627, 81870658, and 81670897). P.W. and E.L. were supported by the International Program for PhD Candidates, Sun Yat-sen University.
Author contributions
P.W. provided the conception, design, and execution of experiments; data collection and interpretation; and manuscript writing. E.L. contributed computational modeling and interpretation and manuscript revision. Z.L. and Y.Z. executed the animal experiments and performed data collection and interpretation. W.S. approved the final manuscript and financial support. Y.Z. offered conception of experiments, approval of the final manuscript, and financial support.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.030.
Supplemental information
References
- 1.Tham Y.C., Li X., Wong T.Y., Quigley H.A., Aung T., Cheng C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Kwon Y.H., Fingert J.H., Kuehn M.H., Alward W.L. Primary open-angle glaucoma. N. Engl. J. Med. 2009;360:1113–1124. doi: 10.1056/NEJMra0804630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Braunger B.M., Fuchshofer R., Tamm E.R. The aqueous humor outflow pathways in glaucoma: A unifying concept of disease mechanisms and causative treatment. Eur. J. Pharm. Biopharm. 2015;95(Pt B):173–181. doi: 10.1016/j.ejpb.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 4.Tielsch J.M., Katz J., Sommer A., Quigley H.A., Javitt J.C. Family history and risk of primary open angle glaucoma. The Baltimore Eye Survey. Arch. Ophthalmol. 1994;112:69–73. doi: 10.1001/archopht.1994.01090130079022. [DOI] [PubMed] [Google Scholar]
- 5.Gong G., Kosoko-Lasaki S., Haynatzki G., Lynch H.T., Lynch J.A., Wilson M.R. Inherited, familial and sporadic primary open-angle glaucoma. J. Natl. Med. Assoc. 2007;99:559–563. [PMC free article] [PubMed] [Google Scholar]
- 6.Khawaja A.P., Cooke Bailey J.N., Wareham N.J., Scott R.A., Simcoe M., Igo R.P., Jr., Song Y.E., Wojciechowski R., Cheng C.Y., Khaw P.T., UK Biobank Eye and Vision Consortium. NEIGHBORHOOD Consortium Genome-wide analyses identify 68 new loci associated with intraocular pressure and improve risk prediction for primary open-angle glaucoma. Nat. Genet. 2018;50:778–782. doi: 10.1038/s41588-018-0126-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choquet H., Paylakhi S., Kneeland S.C., Thai K.K., Hoffmann T.J., Yin J., Kvale M.N., Banda Y., Tolman N.G., Williams P.A. A multiethnic genome-wide association study of primary open-angle glaucoma identifies novel risk loci. Nat. Commun. 2018;9:2278. doi: 10.1038/s41467-018-04555-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gordon M.O., Beiser J.A., Brandt J.D., Heuer D.K., Higginbotham E.J., Johnson C.A., Keltner J.L., Miller J.P., Parrish R.K., 2nd, Wilson M.R., Kass M.A. The Ocular Hypertension Treatment Study: baseline factors that predict the onset of primary open-angle glaucoma. Arch. Ophthalmol. 2002;120:714–720. doi: 10.1001/archopht.120.6.714. discussion 829–830. [DOI] [PubMed] [Google Scholar]
- 9.De Moraes C.G. Natural History of Normal-Tension Glaucoma with (Very) Low Intraocular Pressure. Ophthalmology. 2019;126:1117–1118. doi: 10.1016/j.ophtha.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 10.Wiggs J.L. The cell and molecular biology of complex forms of glaucoma: updates on genetic, environmental, and epigenetic risk factors. Invest. Ophthalmol. Vis. Sci. 2012;53:2467–2469. doi: 10.1167/iovs.12-9483e. [DOI] [PubMed] [Google Scholar]
- 11.Gauthier A.C., Liu J. Epigenetics and Signaling Pathways in Glaucoma. BioMed Res. Int. 2017;2017:5712341. doi: 10.1155/2017/5712341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bermudez J.Y., Webber H.C., Patel G.C., Liu X., Cheng Y.Q., Clark A.F., Mao W. HDAC Inhibitor-Mediated Epigenetic Regulation of Glaucoma-Associated TGFβ2 in the Trabecular Meshwork. Invest. Ophthalmol. Vis. Sci. 2016;57:3698–3707. doi: 10.1167/iovs16-19446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Byun H.M., Siegmund K.D., Pan F., Weisenberger D.J., Kanel G., Laird P.W., Yang A.S. Epigenetic profiling of somatic tissues from human autopsy specimens identifies tissue- and individual-specific DNA methylation patterns. Hum. Mol. Genet. 2009;18:4808–4817. doi: 10.1093/hmg/ddp445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rohen J.W., Lütjen-Drecoll E., Flügel C., Meyer M., Grierson I. Ultrastructure of the trabecular meshwork in untreated cases of primary open-angle glaucoma (POAG) Exp. Eye Res. 1993;56:683–692. doi: 10.1006/exer.1993.1085. [DOI] [PubMed] [Google Scholar]
- 15.Cordero-Espinoza L., Huch M. The balancing act of the liver: tissue regeneration versus fibrosis. J. Clin. Invest. 2018;128:85–96. doi: 10.1172/JCI93562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shu D.Y., Lovicu F.J. Myofibroblast transdifferentiation: The dark force in ocular wound healing and fibrosis. Prog. Retin. Eye Res. 2017;60:44–65. doi: 10.1016/j.preteyeres.2017.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hendrich B., Tweedie S. The methyl-CpG binding domain and the evolving role of DNA methylation in animals. Trends Genet. 2003;19:269–277. doi: 10.1016/S0168-9525(03)00080-5. [DOI] [PubMed] [Google Scholar]
- 18.Ando T., Nishimura M., Oka Y. Decitabine (5-Aza-2′-deoxycytidine) decreased DNA methylation and expression of MDR-1 gene in K562/ADM cells. Leukemia. 2000;14:1915–1920. doi: 10.1038/sj.leu.2401914. [DOI] [PubMed] [Google Scholar]
- 19.Blumenthal E.Z., Weinreb R.N. Assessment of the retinal nerve fiber layer in clinical trials of glaucoma neuroprotection. Surv. Ophthalmol. 2001;45(Suppl 3):S305–S312. doi: 10.1016/s0039-6257(01)00202-8. discussion S332–S334. [DOI] [PubMed] [Google Scholar]
- 20.Petraru D., Indrei A., Costin D., Mihalache G. [Morphopathological changes in glaucoma-induced trabecular meshwork] Rev. Med. Chir. Soc. Med. Nat. Iasi. 2011;115:894–898. [PubMed] [Google Scholar]
- 21.Belmares R., Raychaudhuri U., Maansson S., Clark A.F. Histological investigation of human glaucomatous eyes: Extracellular fibrotic changes and galectin 3 expression in the trabecular meshwork and optic nerve head. Clin. Anat. 2018;31:1031–1049. doi: 10.1002/ca.23263. [DOI] [PubMed] [Google Scholar]
- 22.Wecker T., Han H., Börner J., Grehn F., Schlunck G. Effects of TGF-β2 on cadherins and β-catenin in human trabecular meshwork cells. Invest. Ophthalmol. Vis. Sci. 2013;54:6456–6462. doi: 10.1167/iovs.13-12669. [DOI] [PubMed] [Google Scholar]
- 23.Wu X., Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat. Rev. Genet. 2017;18:517–534. doi: 10.1038/nrg.2017.33. [DOI] [PubMed] [Google Scholar]
- 24.Gomez-Puerto M.C., Iyengar P.V., García de Vinuesa A., Ten Dijke P., Sanchez-Duffhues G. Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 2019;247:9–20. doi: 10.1002/path.5170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wallace D.M., Murphy-Ullrich J.E., Downs J.C., O’Brien C.J. The role of matricellular proteins in glaucoma. Matrix Biol. 2014;37:174–182. doi: 10.1016/j.matbio.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 26.Stamer W.D., Braakman S.T., Zhou E.H., Ethier C.R., Fredberg J.J., Overby D.R., Johnson M. Biomechanics of Schlemm’s canal endothelium and intraocular pressure reduction. Prog. Retin. Eye Res. 2015;44:86–98. doi: 10.1016/j.preteyeres.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Primary open-angle glaucoma. Nat. Rev. Dis. Primers. 2016;2:16068. doi: 10.1038/nrdp.2016.68. [DOI] [PubMed] [Google Scholar]
- 28.Zhou B., Ge T., Zhou L., Jiang L., Zhu L., Yao P., Yu Q. Dimethyloxalyl Glycine Regulates the HIF-1 Signaling Pathway in Mesenchymal Stem Cells. Stem Cell Rev. Rep. 2020;16:702–710. doi: 10.1007/s12015-019-09947-7. [DOI] [PubMed] [Google Scholar]
- 29.Hirsch K., Taglauer E., Seedorf G., Callahan C., Mandell E., White C.W., Kourembanas S., Abman S.H. Perinatal Hypoxia-Inducible Factor Stabilization Preserves Lung Alveolar and Vascular Growth in Experimental Bronchopulmonary Dysplasia. Am. J. Respir. Crit. Care Med. 2020;202:1146–1158. doi: 10.1164/rccm.202003-0601OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Amouroux R., Nashun B., Shirane K., Nakagawa S., Hill P.W., D’Souza Z., Nakayama M., Matsuda M., Turp A., Ndjetehe E. De novo DNA methylation drives 5hmC accumulation in mouse zygotes. Nat. Cell Biol. 2016;18:225–233. doi: 10.1038/ncb3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang X., Yang J., Shi D., Cao Z. TET2 suppresses nasopharyngeal carcinoma progression by inhibiting glycolysis metabolism. Cancer Cell Int. 2020;20:363. doi: 10.1186/s12935-020-01456-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sardina J.L., Collombet S., Tian T.V., Gómez A., Di Stefano B., Berenguer C., Brumbaugh J., Stadhouders R., Segura-Morales C., Gut M. Transcription Factors Drive Tet2-Mediated Enhancer Demethylation to Reprogram Cell Fate. Cell Stem Cell. 2018;23:905–906. doi: 10.1016/j.stem.2018.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Good C.R., Madzo J., Patel B., Maegawa S., Engel N., Jelinek J., Issa J.J. A novel isoform of TET1 that lacks a CXXC domain is overexpressed in cancer. Nucleic Acids Res. 2017;45:8269–8281. doi: 10.1093/nar/gkx435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X., Hu C., Ferchen K., Nie J., Cui X., Chen C.H., Cheng L., Zuo Z., Seibel W., He C. Targeted inhibition of STAT/TET1 axis as a therapeutic strategy for acute myeloid leukemia. Nat. Commun. 2017;8:2099. doi: 10.1038/s41467-017-02290-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng J., Guo S., Chen S., Mastriano S.J., Liu C., D’Alessio A.C., Hysolli E., Guo Y., Yao H., Megyola C.M. An extensive network of TET2-targeting MicroRNAs regulates malignant hematopoiesis. Cell Rep. 2013;5:471–481. doi: 10.1016/j.celrep.2013.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl. Microbiol. Biotechnol. 2003;60:523–533. doi: 10.1007/s00253-002-1158-6. [DOI] [PubMed] [Google Scholar]
- 37.Vandemoortele G., Eyckerman S., Gevaert K. Pick a Tag and Explore the Functions of Your Pet Protein. Trends Biotechnol. 2019;37:1078–1090. doi: 10.1016/j.tibtech.2019.03.016. [DOI] [PubMed] [Google Scholar]
- 38.Roll J.D., Rivenbark A.G., Sandhu R., Parker J.S., Jones W.D., Carey L.A., Livasy C.A., Coleman W.B. Dysregulation of the epigenome in triple-negative breast cancers: basal-like and claudin-low breast cancers express aberrant DNA hypermethylation. Exp. Mol. Pathol. 2013;95:276–287. doi: 10.1016/j.yexmp.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Meng J., Sun B., Zhao X., Zhang D., Zhao X., Gu Q., Dong X., Zhao N., Liu P., Liu Y. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Mol. Cancer Ther. 2014;13:3107–3122. doi: 10.1158/1535-7163.MCT-13-1060. [DOI] [PubMed] [Google Scholar]
- 40.Donnenfeld E., Holland E. Dexamethasone Intracameral Drug-Delivery Suspension for Inflammation Associated with Cataract Surgery: A Randomized, Placebo-Controlled, Phase III Trial. Ophthalmology. 2018;125:799–806. doi: 10.1016/j.ophtha.2017.12.029. [DOI] [PubMed] [Google Scholar]
- 41.Ryman J.T., Meibohm B. Pharmacokinetics of Monoclonal Antibodies. CPT Pharmacometrics Syst. Pharmacol. 2017;6:576–588. doi: 10.1002/psp4.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinarello C.A., Simon A., van der Meer J.W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discov. 2012;11:633–652. doi: 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Clark A.F., Miggans S.T., Wilson K., Browder S., McCartney M.D. Cytoskeletal changes in cultured human glaucoma trabecular meshwork cells. J. Glaucoma. 1995;4:183–188. [PubMed] [Google Scholar]
- 44.Clark A.F., Wordinger R.J. The role of steroids in outflow resistance. Exp. Eye Res. 2009;88:752–759. doi: 10.1016/j.exer.2008.10.004. [DOI] [PubMed] [Google Scholar]
- 45.Hoare M.J., Grierson I., Brotchie D., Pollock N., Cracknell K., Clark A.F. Cross-linked actin networks (CLANs) in the trabecular meshwork of the normal and glaucomatous human eye in situ. Invest. Ophthalmol. Vis. Sci. 2009;50:1255–1263. doi: 10.1167/iovs.08-2706. [DOI] [PubMed] [Google Scholar]
- 46.Montecchi-Palmer M., Bermudez J.Y., Webber H.C., Patel G.C., Clark A.F., Mao W. TGFβ2 Induces the Formation of Cross-Linked Actin Networks (CLANs) in Human Trabecular Meshwork Cells Through the Smad and Non-Smad Dependent Pathways. Invest. Ophthalmol. Vis. Sci. 2017;58:1288–1295. doi: 10.1167/iovs.16-19672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Banes M.J., Culham L.E., Bunce C., Xing W., Viswanathan A., Garway-Heath D. Agreement between optometrists and ophthalmologists on clinical management decisions for patients with glaucoma. Br. J. Ophthalmol. 2006;90:579–585. doi: 10.1136/bjo.2005.082388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gyasi M., Amoako W., Adjuik M. Presentation patterns of primary open angle glaucomas in north eastern ghana. Ghana Med. J. 2010;44:25–30. doi: 10.4314/gmj.v44i1.68853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Saccà S.C., Pascotto A., Camicione P., Capris P., Izzotti A. Oxidative DNA damage in the human trabecular meshwork: clinical correlation in patients with primary open-angle glaucoma. Arch. Ophthalmol. 2005;123:458–463. doi: 10.1001/archopht.123.4.458. [DOI] [PubMed] [Google Scholar]
- 50.Rusk N. Reverse ChIP. Nat. Methods. 2009;6:187. [Google Scholar]
- 51.Sinkkonen L., Malinen M., Saavalainen K., Väisänen S., Carlberg C. Regulation of the human cyclin C gene via multiple vitamin D3-responsive regions in its promoter. Nucleic Acids Res. 2005;33:2440–2451. doi: 10.1093/nar/gki502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wadhwa M., Meager A., Dilger P., Bird C., Dolman C., Das R.G., Thorpe R. Neutralizing antibodies to granulocyte-macrophage colony-stimulating factor, interleukin-1alpha and interferon-alpha but not other cytokines in human immunoglobulin preparations. Immunology. 2000;99:113–123. doi: 10.1046/j.1365-2567.2000.00949.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fujii M., Takeda K., Imamura T., Aoki H., Sampath T.K., Enomoto S., Kawabata M., Kato M., Ichijo H., Miyazono K. Roles of bone morphogenetic protein type I receptors and Smad proteins in osteoblast and chondroblast differentiation. Mol. Biol. Cell. 1999;10:3801–3813. doi: 10.1091/mbc.10.11.3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Keller K.E., Bhattacharya S.K., Borrás T., Brunner T.M., Chansangpetch S., Clark A.F., Dismuke W.M., Du Y., Elliott M.H., Ethier C.R. Consensus recommendations for trabecular meshwork cell isolation, characterization and culture. Exp. Eye Res. 2018;171:164–173. doi: 10.1016/j.exer.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.McKee C.T., Wood J.A., Shah N.M., Fischer M.E., Reilly C.M., Murphy C.J., Russell P. The effect of biophysical attributes of the ocular trabecular meshwork associated with glaucoma on the cell response to therapeutic agents. Biomaterials. 2011;32:2417–2423. doi: 10.1016/j.biomaterials.2010.11.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rhee D.J., Tamm E.R., Russell P. Donor corneoscleral buttons: a new source of trabecular meshwork for research. Exp. Eye Res. 2003;77:749–756. doi: 10.1016/j.exer.2003.07.008. [DOI] [PubMed] [Google Scholar]
- 57.Fischer A.H., Jacobson K.A., Rose J., Zeller R. Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc. 2008;2008 doi: 10.1101/pdb.prot4986. pdb.prot4986. [DOI] [PubMed] [Google Scholar]
- 58.Authier S., Chaurand F., Legaspi M., Breault C., Troncy E. Comparison of three anesthetic protocols for intraduodenal drug administration using endoscopy in rhesus monkeys (Macaca mulatta) J. Am. Assoc. Lab. Anim. Sci. 2006;45:73–79. [PubMed] [Google Scholar]
- 59.Cao Y. [A method to identify the aqueous humor outflow facility (C value)] J. Pract. Ophthalmol. 1991:34–36. [Google Scholar]
- 60.Yan Z., Tian Z., Chen H., Deng S., Lin J., Liao H., Yang X., Ge J., Zhuo Y. Analysis of a method for establishing a model with more stable chronic glaucoma in rhesus monkeys. Exp. Eye Res. 2015;131:56–62. doi: 10.1016/j.exer.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 61.Polak S. In vitro to human in vivo translation - pharmacokinetics and pharmacodynamics of quinidine. ALTEX. 2013;30:309–318. doi: 10.14573/altex.2013.3.309. [DOI] [PubMed] [Google Scholar]
- 62.Kuhn M. GRIN Verlag GmbH; 2005. Manual for the Implementation of Neural Networks in MATLAB. pp. 2–8. [Google Scholar]
- 63.Arvind V., Kim J.S., Oermann E.K., Kaji D., Cho S.K. Predicting Surgical Complications in Adult Patients Undergoing Anterior Cervical Discectomy and Fusion Using Machine Learning. Neurospine. 2018;15:329–337. doi: 10.14245/ns.1836248.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Garson G.D. Interpreting neural-network connection weights. AI Expert. 1991;6:46–51. [Google Scholar]
- 65.Goh A.T.C. Back-propagation neural networks for modeling complex systems. AI Eng. 1995;9:143–151. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.