

Abstract
Introduction:
The occurrence of metal ions in biomolecules is required to exert vital cellular functions. Metal-containing biomolecules can be modulated by small-molecule inhibitors targeting their metal-moiety. As well, the discovery of cisplatin ushered the rational discovery of metal-containing-drugs. The use of both drug types exploiting metal–ligand interactions is well established to treat distinct pathologies. Therefore, characterizing and leveraging metal-coordinating drugs is a pivotal, yet challenging, part of medicinal chemistry.
Area covered:
Atomic-level simulations are increasingly employed to overcome the challenges met by traditional drug-discovery approaches and to complement wet-lab experiments in elucidating the mechanisms of drugs’ action. Multiscale simulations, allow deciphering the mechanism of metal-binding inhibitors and metallo-containing-drugs, enabling a reliable description of metal-complexes in their biological environment. In this compendium, the authors review selected applications exploiting the metal–ligand interactions by focusing on understanding the mechanism and design of (i) inhibitors targeting iron and zinc-enzymes, and (ii) ruthenium and gold-based anticancer agents targeting the nucleosome and aquaporin protein, respectively.
Expert opinion:
The showcased applications exemplify the current role and the potential of atomic-level simulations and reveal how their synergic use with experiments can contribute to uncover fundamental mechanistic facets and exploit metal–ligand interactions in medicinal chemistry.
Keywords: Metallodrug, metal-binding inhibitors, molecular dynamics, QM/MM, CYP450, ruthenium drug, breast cancer, prostate cancer, RAPTA, metallo-beta-lacatamases, aromatase
1. Introduction
Metal-containing biomolecules are involved in vital cellular processes, accounting for roughly 30–40% of the proteome [1]. These include metalloenzymes promoting complex biochemical reactions, metal-transporters that strictly preserve the homeostasis of critical metal ions [2], and metal-responsive transcriptional regulators, which modulate DNA-binding affinity and concomitant gene expression by binding to their cognate metal [3,4]. In addition to them, RNA enzymes also need divalent metal ions to stabilize their tertiary structure and promote the catalysis of nucleic acids [5,6].
Among the most abundant metal ions present in biomolecules are iron, copper, zinc, and magnesium. As an example, iron is contained within the heme moiety of cytochromes P450 (CYP450) and catalyzes the oxidative transformations of endogenous and exogenous compounds [7]. Some CYP450s also promote the biosynthesis of steroid hormones and, therefore, inhibitors targeting the Fe moiety are currently employed to fight diffused cancer types [8].
Due to their optimal redox potential, reactions mediated by copper enzymes promote fundamental biological processes (i.e. cellular respiration, iron oxidation, antioxidant defense) [9]. Yet, an excessive concentration of copper can possibly trigger cytotoxic cellular damages, as implicated in neurodegenerative disorders and cancer [10]. Deregulated copper metabolism, due to genetic abnormalities in Cu transporters, is responsible for Menkes’ and Wilson’s diseases [11]. In this respect, ligands that regulate mis-function of Cu(I) metabolism offer appealing opportunities to counteract these pathological states [12]. Zinc ions are the natural cofactors of a wide variety of enzymes such as (i) matrix metalloproteinase (MMP), responsible for protein degradation at the cell-extracellular matrix, typically targeted by anticancer compounds [13]; (ii) human carbonic anhydrase (hCA), promoting reversible hydration of carbon dioxide to bicarbonate [14], whose inhibitors exhibit several medical applications (i.e. diuretics, anticonvulsants, as anticancer agents/diagnostic tools for tumors, antiobesity agents); (iii) bacterial metallo-β-lactamases (MBL) enzymes that promote the degradation of β-lactam antibiotics. Their inhibitors are of critical importance to counteract resistance to commonly used β-lactam antibiotics [15,16].
Finally, magnesium-dependent proteins and ribozymes are widespread and promote the metabolism of nucleic acids in genome regulatory processes [17,18]. Among these, Mg2+-dependent pharmacological targets are (i) the flap endonuclease 1 (FEN1), an enzyme that removes the DNA and RNA flaps formed during replication and repair and is targeted by anticancer drugs [19,20]; (ii) HIV integrase, which inserts the viral genome into the host cell and is targeted by antiviral compounds [21]; (iii) the GTPase enzymes, which are critically linked with cellular differentiation, proliferation, division, and movement by regulating signal transduction. Their activity in many infiltrative (brain, ovarian, and melanoma) cancers can be inhibited by small-molecules binding to their Mg2+ ion cofactor [22].
The use of metal-containing drugs to treat patients affected by distinct pathologies is nowadays well established. As an example, metallodrugs can be effective anticancer agents [23]. Among these, cisplatin is currently the most widely used chemotherapeutic drug [24], being active in the treatment of 18 cancers, including testicular and ovarian carcinomas, lymphoma, melanoma, and neuroblastoma [25]. The activity of this drug and its derivatives (carboplatin, oxaliplatin, etc.) has been historically ascribed to the formation of DNA lesions that interfere with transcription, resulting in cellular apoptosis. Nowadays, it is however clear that its complex pharmacological profile is also due to its interactions with distinct proteins [26].
In spite of their successful clinical applications, the small size and the typical square planar geometry of platinum compounds only allow a poor site discrimination toward its biological target, resulting in severe toxicity and intrinsic or acquired resistance issues [27,28]. This prompted the development of novel chemotherapeutic strategies aimed at exploiting the potential of alternative metals such as ruthenium and gold compounds [29]. Ruthenium (Ru)-based compounds are promising candidates for their selective activity against specific cancer cell types and low toxicity [30]. The typical octahedral coordination sphere of ruthenium allows a higher degree of site selectivity, reducing the resulting toxicity. These properties led two ruthenium compounds – the anti-primary tumor indazolium trans-[tetrachloridobis (1 H-indazole) ruthenate(III)] (KP1019) and the anti-metastasis imidazolium trans- [tetrachloride (1 H-imidazole) (S-dimethylsulfoxide) ruthenate(III)] (NAMI-A) – in clinical trials, and later suspended for toxicity [30–32]. Again, gold (Au) complexes were object of intense research efforts due to their cytotoxic effects toward several cancers and their different pharmacological profiles as compared to Pt-drugs. Auranofin, an Au(I) compound originally used to treat rheumatic arthritis, was later approved for clinical trials against lung and ovarian carcinomas. The selenoprotein thioredoxin reductase is a well-documented target of auranofin. In addition, other Au(I) complexes were later demonstrated to bind different proteins, among which aquaporin-3 [33] and zinc finger proteins, such as poly(adenosine diphosphate ribose)polymerase-1 (PARP-1) [34].
Understanding the mechanism and designing of new drugs forming coordination bonds with metal ions pose serious challenges to structure-based drug discovery. Molecular docking simulations, traditionally used in medicinal chemistry, are unable to explicitly account for the intricate metal electronic structure [35,36]. Indeed, they cannot describe the formation/breaking of bonds of the metal-coordination sphere (i.e. the type and number of ligands coordinating to the metal center) and they cannot account for the readjustments of the metal electronic structure and charge density (polarization and charge transfer effects), occurring either during the binding of a drug to the meta-moiety of a biomolecule or while a metallodrug binds to its target biomolecule [37].
These limitations are shared by both main and transition metal elements [38,39]. Hence, a hierarchical computational approach is most commonly adopted in which docking calculations are instrumental to obtain the initial binding pose for (i) the inhibitor in proximity of the metal site to the target biomolecule or (ii) the metallodrug nearby the residues/nucleobases of the biological target. This binding pose is then relaxed with more advanced computational methods, traditionally used by computational chemists and biophysicists, such as all-atom molecular dynamics (MD) simulations at force field (FF) and electronic structure level [40].
FF-based MD simulations experience limitations for properly describing the electronic structure of metals or requires system-specific parameters, thus, making necessary the use of quantum mechanical (QM) methods. In this case, a hybrid quantum/classical (QM/MM) approach is employed that describe the metal and its coordination sphere (organic ligands and residues/nucleic acids) at a QM level, while treating the remainder of the system (the rest of the biomolecule in explicit solution, and, eventually, in a membrane mimic) with classical FF [41].
In this compendium, we review selected informative studies aimed at exemplifying the use of molecular simulations when dealing with transition metal elements, such as the biologically abundant iron and zinc atom embedded in metalloenzymes, or the ruthenium and gold ions contained in metallodrugs. In particular, we report how QM/MM MD simulations contributed to elucidate the reaction mechanisms of steroidogenic Fe-containing CYP450s and their inhibitors used in standard treatment regimens against distinct cancers [8], to study the binding of ligands to Zn-containing enzymes (MMP, hCA, MBL) [42], to dissect the mechanism of novel Ru-based anticancer drugs targeting the nucleosome [37], and the binding of Au(I) complexes to aquaporins [33].
This review highlights that understanding the mechanism of a metal-targeting or a metal-containing drug is not limited to the drug–ligand interactions or local drug-induced perturbations, but it can also be linked to a drug-induced allosteric modulation of the target, delineating different kinetic and thermodynamic properties. This information allows gathering precious information to rationalize and boost drug efficacy. This review also remarks that the interplay between experiments and computations is critical to clarify relevant biological questions at molecular level, assuming a prominent role in the rational design of drugs deploying metal–ligand interactions.
2. Computational methods
2.1. Force field-based MD
Molecular dynamics allows the solution of the classical equation of motion for a set of particles [43]. This provides a trajectory, which describes the time-evolution of each atom of a bimolecular system at finite temperature. In classical MD, the potential energy of the system is determined by empirical FFs, usually exhibiting the generic form of the potential energy reported in Eq. (1). FFs result from the sum of different contributions which mimic the molecular properties of the system (i.e. bonded terms, describing stretching, bending, torsional vibrational modes, and non-bonded terms, accounting for dispersion and electrostatic forces) [44,45].
| (1) |
AMBER [46], GROMOS [47], and CHARMM [48]. These are complemented for simulations of small molecules and cofactors by the Generalized AMBER FF (GAFF) [49] and CHARMM General FF (CgFF) [50].
Classical MD simulations are performed for time scales of tens-hundreds of μs, thus being impaired to directly monitor many biologically relevant phenomena. Nevertheless, by coupling classical MD with methods that enhance the sampling of the configurational space (i.e. enhanced sampling techniques) [51,52] or with non-equilibrium free-energy methods (i.e. free-energy perturbation, umbrella sampling, adaptive biasing force) [53], it becomes possible to study interesting biophysical phenomena taking place at longer time scales (μs-to-ms) [54,55].
2.2. Mixed quantum-classical (QM/MM) MD
FF-based methods can be combined to QM calculations, resulting in the so-called QM/MM approach. After its original formulation in the 70s [56], different QM/MM implementations have been proposed [57]. Here, we mostly refer to the fully Hamiltonian approaches included in the CPMD [58] and CP2K codes [59]. An efficient variation of this traditional scheme has been recently proposed [60]. This relies on a loose coupling of two highly parallelized codes such as the CPMD [61] and GROMACS [62], resulting in a higher computational efficiency.
In QM/MM studies of metallo-systems, the metal and its coordination sphere are treated at a higher level of accuracy (QM level), while the remainder of the system is described at the MM (FF) level of theory (Figure 1). Namely, for an inhibitor binding to the metalloenzyme, the QM region should comprise the enzyme’s metal center, the residues coordinating to it, and the inhibitor. In the case of a metallodrug binding an enzyme, the QM region includes the drug and the residues/nucleobases of the protein/nucleic acid directly binding the metal. In the general form of a hybrid QM/MM scheme, Eq. 2, the Hamiltonian of the system contains Hamiltonians for the quantum and classical systems and for the interacting part between the QM and MM regions :
| (2) |
where the ab initio Hamiltonian can be based on different QM approaches, spanning from semiempirical to ab initio Hartree-Fock or Density Functional Theory (DFT) methods. We remark that in the study of metallo-systems, the latter is most often the method of choice owing to its favorable scaling with the number of atoms and its reasonable accuracy to treat correlation effects [39].
Figure 1.
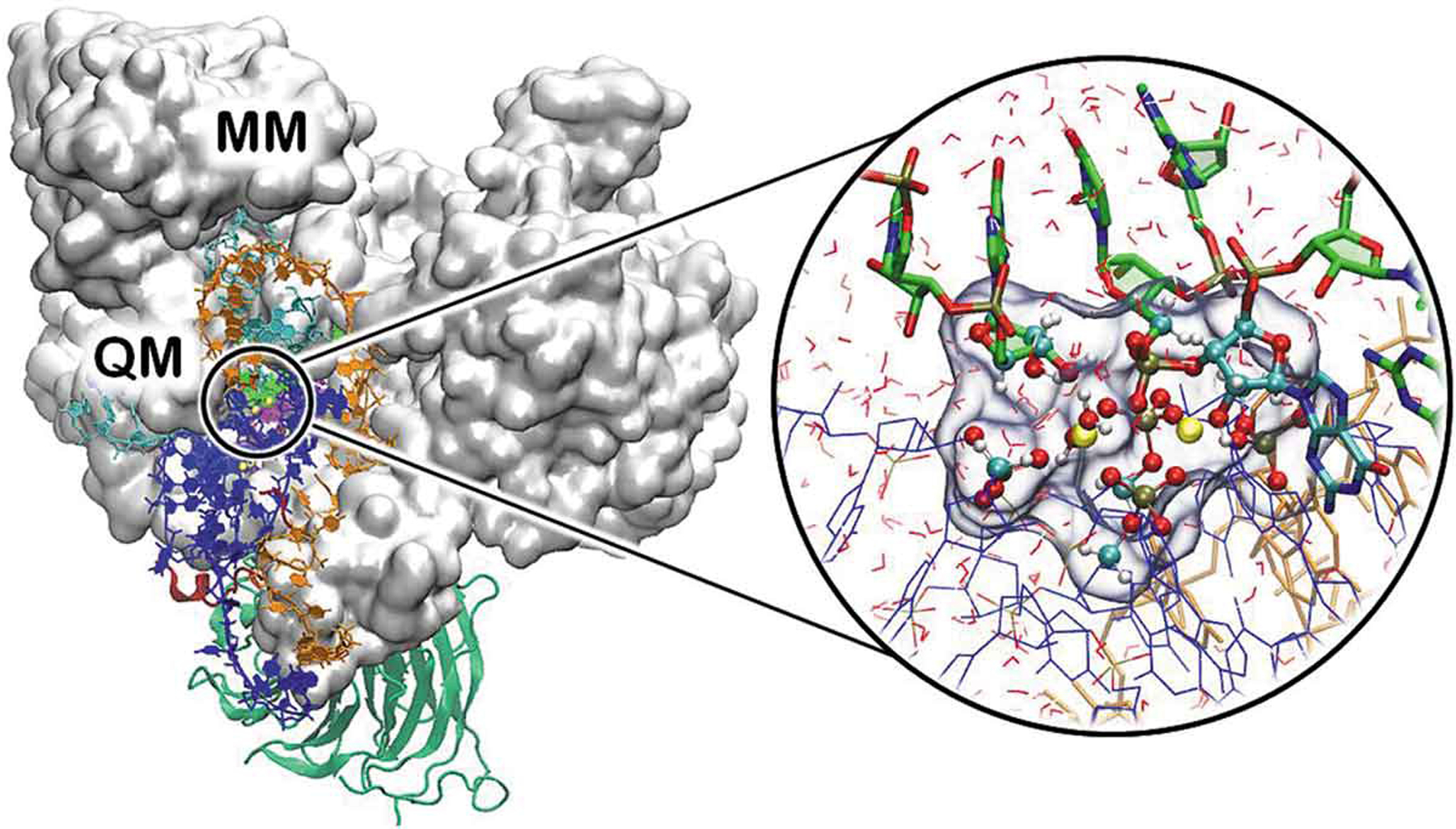
Representative QM/MM partitioning of a metal-containing biological system, showing the catalytic site of the spliceosome (total atoms 370,000 atoms). Proteins are shown with white surface and green new cartoons, distinct RNA strands are shown in blue, orange, cyan and green ribbons. The cycle on the right reports a close view of the QM region (highlighted with a transparent surface), composed by the Mg2+ ions (yellow), and the remaining RNA nucleobases and phosphate shown in licorice and ball and sticks and colored by atom name. The remaining part of the system, including RNA strands (shown as blue and orange ribbons), water molecules (shown as red sticks), protein and counter ions (not shown) are treated at the classical (MM) level. Adapted from Ref [94] with permission of Copyright © 2020, American Chemical Society.
QM/MM implementation has to devote particular care to the coupling between the QM and MM regions. This is described by the interaction Hamiltonian term, which accounts for both bonded and non-bonded interactions at the interface of the QM and MM regions. The description of the covalent bonds, split between the QM and MM regions, relies either on linking hydrogen atoms or on specially parameterized pseudo-atoms that saturate the valence of the terminal QM atoms. Furthermore, between the non-bonded interactions, the van der Waals terms are accounted at the classical FF level, while special care is needed for describing the electrostatic interactions. In the mechanical embedding scheme, the electrostatic interactions between the two partitions are either not described or are treated at the MM level. In the more rigorous and most commonly employed electrostatic embedding scheme, the electrostatic effects of the environment (MM portion) polarize the QM electronic charge density. Additionally, the interaction between MM point charges and QM electron density is incorporated in the as one-electron terms. Finally, in the polarized embedding scheme, the polarization effects of the QM region on the MM part are also considered toward a polarizable FF.
Since its first appearance [56], QM/MM approaches have been successfully applied to a growing number of drug-design [33,40,–40,63–67] and enzymatic reaction studies [68–81]. The QM/MM method, in combination with ab initio MD (i.e. through the Car-Parrinello and Born Oppenheimer approaches), has also been widely employed to study anticancer metallodrug–target interactions [40,41,82,83] and mechanistic studies of metalloenzyme catalysis [84–89]. Both the CPMD [90] and CP2K [91] codes are based on DFT and can be interfaced with distinct non-polarizable classical FFs. These continuous developments and code improvements enabled the study of huge cryo-EM structures accessible nowadays [92,93], with recent applications to biological systems of increasing size and complexity (reaching more than 370,000 atoms), such as the spliceosome and CRISPR-Cas9 [94–97].
3. Mechanism and design of metal-coordinating drugs within biomolecules
3.1. Drugs targeting metalloenzymes
3.1.1. Drugs targeting iron-containing enzymes
CYP450s are a wide family of enzymes involved in the metabolism of endogenous and exogenous substances [98,99]. CYP450s promote the biosynthesis of steroid hormones for which their de-regulated activity is linked to the onset of distinct diseases such as cancer [78,100]. Thanks to a specific catalytic scaffold, steroidogenic CYP450s promote complex biosynthetic processes with high precision and efficiency [8]. Their intricate catalytic functions are entwined with their environment, such as their membrane-associated nature, which affects the ligand channeling to/from the active site [101,102] and their interactions with specific redox partner, supplying the electrons needed for catalysis [103,104]. All these aspects are critical to understand and exploit at best CYP450s’ mechanism to devise inhibitors targeting the metal ions.
Among steroidogenic CYP450s, two enzymes have attracted particular interest for their implications in two diffused cancer types among the male and female populations. These are CYP19A1 (also referred to as aromatase, AR) and CYP17A1, which are in charge of the synthesis of estradiol and testosterone, the main adult female and male hormones, respectively.
CYP450 acts as monooxygenase enzymes which, by using molecular oxygen, insert one oxygen atom into their substrates (RH to ROH), while reducing the second oxygen to a water molecule (Eq. 3).
| (3) |
This occurs following a general mechanism (Figure 2A):
Figure 2.
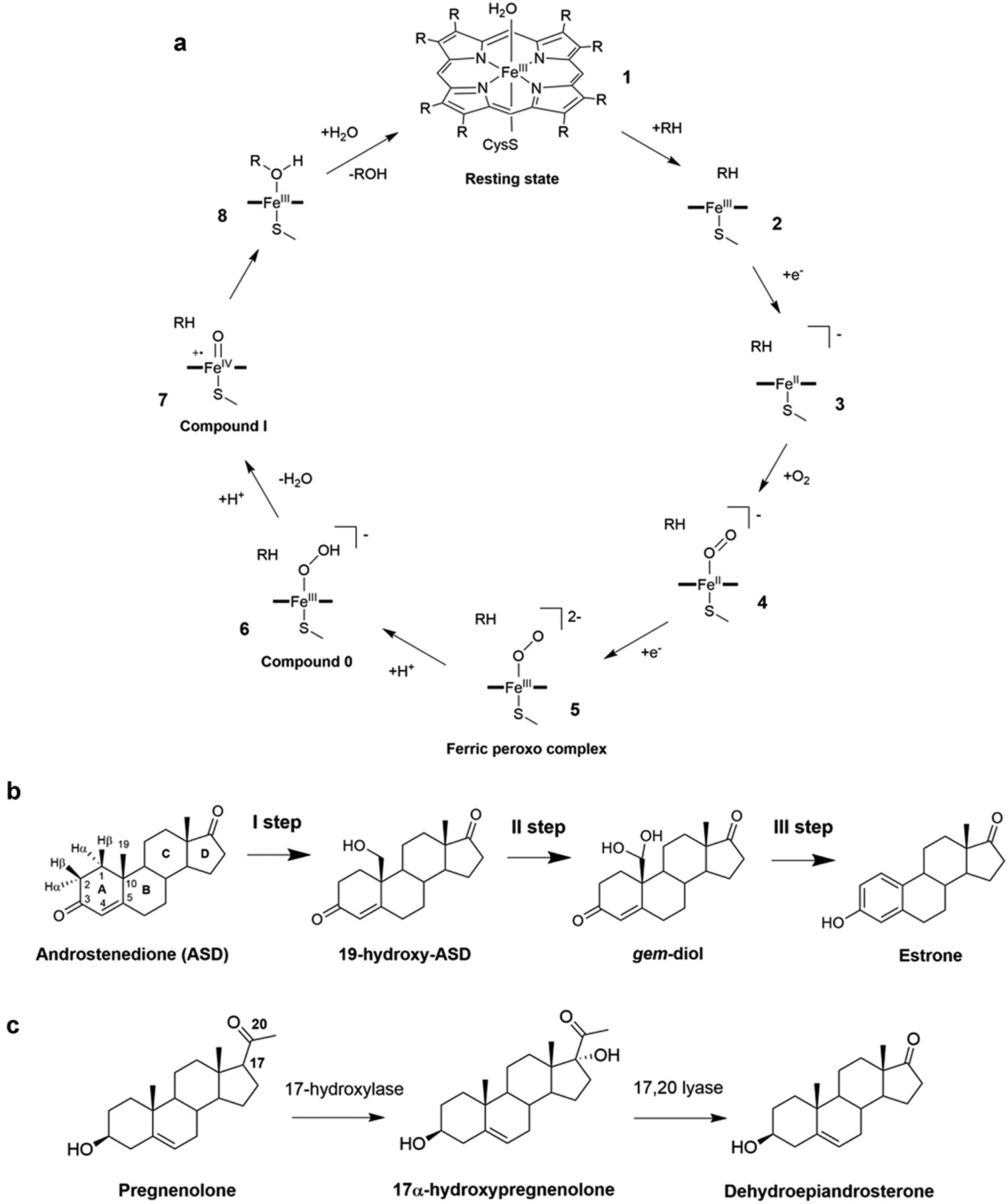
A) General CYP450 cycle and hormones conversion catalyzed by aromatase (AR) and CYP17A1 [77]. B) The three oxidation steps catalyzed by AR. C) 17α-hydroxylation and 17α-20-lyase reactions of pregnenolone as performed by CYP17A1.
In the resting state (1) CYP450s coordinate a water molecule to the iron of the heme moiety; (i) the substrate (RH) enters into the active site and water dissociates (2); (ii) the first electron transfer (ET) from the redox partner occurs, triggering the formation of a high spin ferrous complex (3); (iii) this species binds O2, resulting into oxy-ferrous complex (4); (iv) a second ET yields a ferric peroxo complex (5) and (v) after protonation this complex results into compound 0 (Cpd 0, 6); (vi) a proton transfer induces the formation of highly reactive iron-oxo species compound I (Cpd I), a FeIV-oxo porphyrin radical cation (7); (vii) in most CYP450s’ cycles, Cpd I performs substrate hydroxylation (8), yielding an alcohol, ROH, and ultimately restoring the heme resting state (1). The protons required for the reactions are supplied either from water molecules accessing the active site or a specific redox partner that furnishes the electrons.
3.1.2. Metal–ligand interactions in CYP19A1
Besides sharing these common features, the mechanism of substrate hydroxylation has specific differences in each CYP450 enzyme. This has been exhaustively addressed for the AR enzyme, which converts estrone and androstenedione to estradiol and estrogen, respectively (Figure 2B). The highly hydrophobic nature and substrate specificity of its active site have initially fostered the idea that a distinct mechanism may have been operative with respect to the general CYP450 catalysis. In particular, the last catalytic step, implicated in completing substrate aromatization, remained controversial and has been addressed in many computational studies [105–107]. After Cpd I was demonstrated to be the reactive species of final catalytic step, extensive classical and QM/MM metadynamic simulations supplied a comprehensive picture of AR’s enzymatic cycle (Figure 3A) [108]. This study disclosed that the most likely enzymatic path for the conversion of androstenedione to estrone occurs via (i) androstenedione enolization and Cpd 0 formation via a proton network mediated by Asp309 (Helmholtz’s free-energy barrier (ΔF#) = 11.4 ± 3.0 kcal/mol), (ii) subsequent formation of Cpd I, upon a rearrangement of the Asp309 sidechain (ΔF# = 9.1 ± 0.7 kcal/mol) and engagement of a proton network with Asp309 and Thr310. (iii) Two hydroxylation reactions next occur and are then followed by the conversion of 19,19-gem-diol is into estrone by Cpd I (ΔF#TS1 = 6.5/6.0 ± 1.3 kcal/mol [108].
Figure 3.
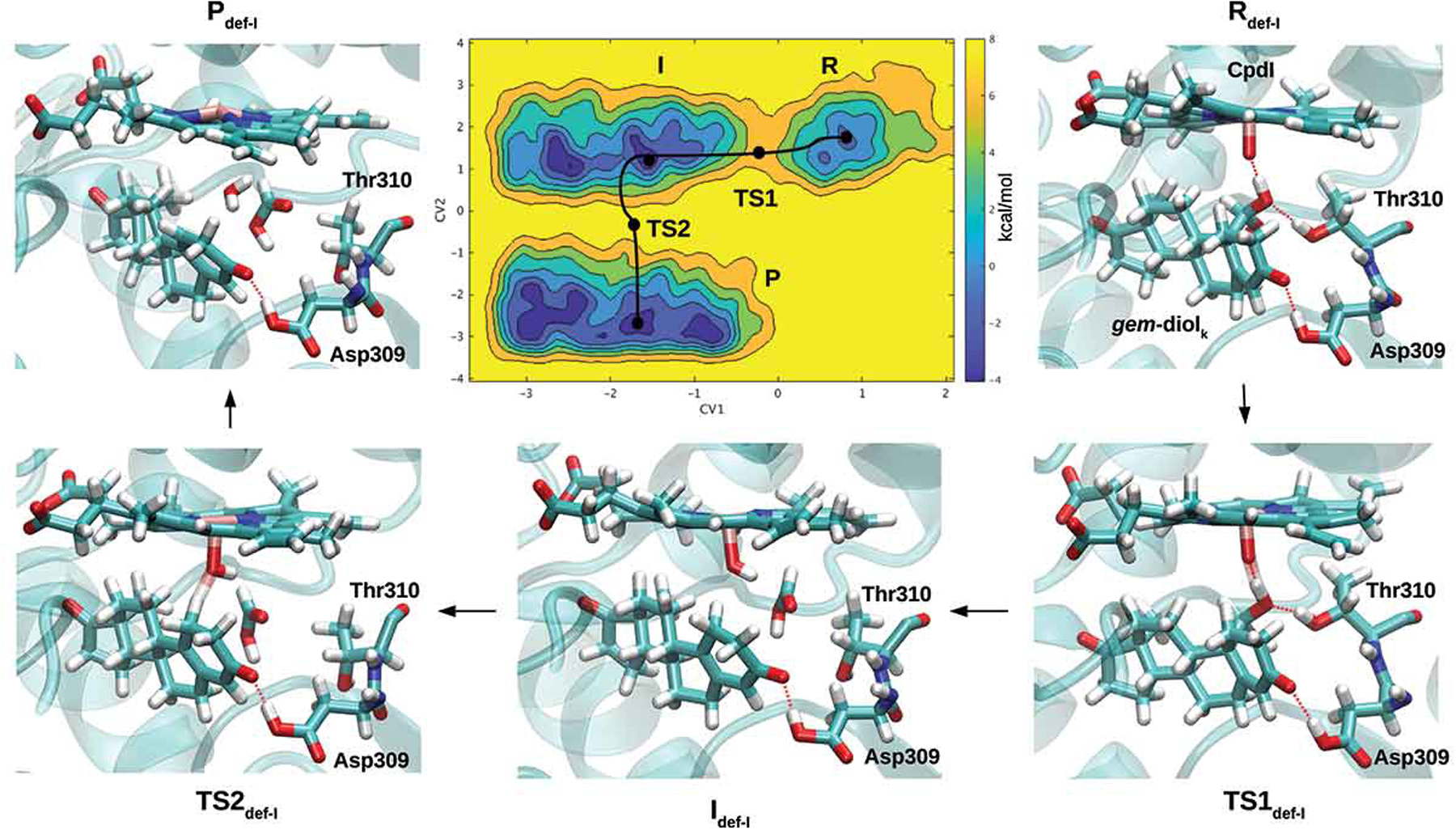
A) Free-energy surface for the aromatization (third catalytic) step, considering the enol form of androstenedione as a reagent [108]. Representative snapshots for the reagent (R), intermediate (I), product (P) and transition states (TSs) formed along the lowest energy path, are reported. Heme and the substrate are shown in a stick representation. Binding of B) letrozole and C) abiraterone to the Fe atom of CYP19A1 and CYP17A1, respectively, as predicted by QM/MM MD simulations [113] and X-ray crystallography (PDB 3RUK) [121]. Reproduced with permission from Ref [108], Copyright 2018 John Wiley & Sons.
Besides the key regulatory role that estrogens play in sexual and reproductive development, estrogens can also stimulate the development and growth of estrogen-dependent (ER+) breast cancer (BC), which currently is the most diffused BC type in post-menopausal woman. As such, AR is a pivotal target for standard pharmacological treatment against BC [109,110].
A clear understanding of the reaction mechanism by multiscale simulations is instrumental to design small-molecules that covalently inhibit the enzyme. This is notable in the steroidal inhibitor exemestane (EXE) [111] or for characterizing the metal–ligand interaction underlying the inhibition mechanism of non-steroidal inhibitors, which have so far eluded a structural characterization by experimental means [110]. The third-generation of AR inhibitors (AI) shares a common azole moiety, which is believed to coordinate the heme iron atom. QM/MM MD simulations have provided insights into the possible binding mode of letrozole, a-third generation AI, to the AR’s active site (Figure 3B) [112]. In addition, these inhibitors were used to estimate the binding free-energy and dissociation free-energy barrier of letrozole to/from the metal center [112]. This study showed that the dissociation free-energy cost associated with the cleavage of the covalent bond with the Fe atom is comparable to that for traveling among the egress channels. Therefore, both aspects have to be considered in order to ameliorate the kinetic properties of such drugs.
In addition, a recent study was aimed at identifying a novel class of non-steroidal inhibitors able to bind to the heme iron with their azole moiety [113] and to additionally target one allosteric site [114,115]. In this study, QM/MM MD simulations elucidated the key molecular traits underlying the measured trend of IC50 (half inhibitory concentration). Namely, this study showed that the binding geometries (bond lengths and angles) of all the newly developed inhibitors and letrozole are similar, in spite of their different potencies, suggesting that the structural deformation induced to the catalytic site is a key aspect to consider during the drug-design process (Figure 3B) [113]. Furthermore, it was observed that effective inhibitors active in the low nM range, such letrozole and exemestane, forms hydrogen (H)-bonds and engage optimal hydrophobic and π -stacking interactions that further boost their potency [113]. Ligand strain has already been indicated as an essential trait affecting the activity of inhibitors. Indeed, molecular simulations revealed that compounds exhibiting IC50s in the nM range undergo a protein-induced ligand strain smaller than 3 kcal/mol [116].
3.1.3. Metal–ligand interactions in CYP17A1
CYP17A1 promotes the biosynthesis of androgens [4] by merging two functions: 17-hydroxylase and 17,20 lyase, which are exploited to convert pregnenolone and progesterone to dehydroepiandrosterone and androstenedione [8] (Figure 2C). The 17-hydroxylation of pregnenolone and progesterone occurs via the canonical oxygen rebound mechanism detailed in Figure 2A, whereas the mechanism of the lyase step remains unclear. This may be promoted by Cpd I or by the nucleophilic attack of FeO2− on the steroid C20 atom [8]. The mechanistic intricacies of CYP17A1 have been addressed by DFT calculations, complemented by classical MD simulations. The first was employed to identify the structures of transition states and intermediates and their relative energies, while the latter were used to link the DFT-obtained geometries with the actual conformations of the protein framework. This study tackled the reaction mechanism of both the hydroxylase and lyase reactions considering Cpd I and peroxo anion as the reactants of the two steps, respectively. For the hydroxylase step, the reaction was observed to proceed with a Gibbs’s free-energy barrier (ΔG#) of 13–14 kcal/mol. Conversely, for the lyase step, two distinct mechanisms (stepwise or concerted) were discovered both having a ΔG# of 20 kcal/mol [117]. In addition, in line with previous findings of the AR enzymes, this study confirmed that the selectivity and specificity of the steroidogenic CYP450s are dictated by peculiar interactions between the substrate and the enzyme’s active site [117].
Prostate cancer (PC) is the second most common type of cancer in men and the fifth leading cause of death worldwide [118]. Several treatments are available against this disease. Most of the FDA approved drugs against PC are hormonal modulators targeting the androgen pathway such as gonadotropin-releasing agonists, and androgen-receptor blockers [119]. However, advanced PC ultimately develops resistance to current therapies, rapidly leading to castration-resistant prostate cancer (CRPC), where the androgens, synthesized by tumors and/or the adrenal glands, stimulate disease progression. Thus, the reduction/suppression of hormone levels in cancer cells remains a key pharmacological strategy against CRPC [120]. The use of CYP17A1 inhibitors, which prevent the conversion of cholesterol to testosterone, is the forefront therapy against CRPC. Abiraterone (ABI), the first CYP17A1 inhibitor approved in the United States [118], has a steroidal scaffold with a pyridin-3-yl moiety at position 17 that inhibits CYP17A1 by coordinating to the iron atom of the heme moiety [121] (Figure 3C). ABI is structurally similar to the substrates of other CYP450s involved in steroidogenesis, and interference can pose a liability in terms of side effects. Hence, designing and using non-steroidal scaffolds may lead to compounds that interact more selectively with CYP17A1. As such, a structure-based virtual screening approach to search compounds bearing azole or pyrazole moieties was done and complemented by QM(DFT)/MM simulations to assess the structural properties and compute the binding energy of the selected ligands. The success of the adopted protocol was confirmed by in vitro assays, demonstrating that two compounds selectively inhibited CYP17A1 with IC50s in the nM range [120]. The same protocol allowed optimizing these heme-Fe targeting ligands [120,122].
3.1.4. Metal–ligand interactions in Zn-containing enzymes
Many Zn-binding enzymes are the target of inhibitors directly binding to the metal moiety. Among them, the mechanism of matrix metalloproteinases (MMP), a family of Zn-dependent endopeptidases, which degrade the extracellular matrix has been intensively studied [76,123] due to their role in cell proliferation, adhesion, and migration. Therefore, these enzymes are implicated in cancer progression leading to metastasis. An articulated computational study investigated the binding of 28 hydroxamate inhibitors to the Zn-moiety of MMP9. In this study, the docking pose of these inhibitors has been initially scored with the traditional docking algorithm. Nevertheless, suitable binding poses have been selected on the basis of the Zn-ligand distance in order to select the pose, which enables the binding of the inhibitor to the metal moiety [124]. On a selection of suitable poses, QM/MM simulations were done to obtain the appropriate coordination geometries, and MD simulations were ultimately performed, while keeping constrained the distance and the angles defining the coordination geometry of the ligand to the metal moiety. This computational approach was instrumental to rationalize the observed inhibition constant and the different binding modes of the ligands [124].
Human carbonic anhydrase II (hCAII) is yet another Zn-dependent enzyme, which catalyzes the transformation of carbon dioxide into bicarbonate [75,125]. This is often used as a prototypical model system for medicinal chemistry applications.
Sulfonamide-carrying ligands are known to inhibit hCAII by directly binding to the catalytic Zn2+ ion of the protein, upon displacement of the fourth water/hydroxyl ligand. A recent computational study addressed the multistep hCAII–sulfonamide recognition process, identifying the structural features of the kinetically relevant intermediate along the binding pathway via enhanced sampling simulations at the classical FF level. On the identified free-energy minima, DFT calculations were performed to study the displacement of the hydroxyl ligands, after a proton transfer from the sulfonamide inhibitor to the hydroxyl ion. This allows the displacement of the resulting water molecule and the direct binding of the inhibitor to the metal moiety. This study revealed that the experimentally measured binding affinity of benzenesulfonamide to hCAII could be explained on the basis of the association rates, which is strongly connected with the hydrophobicity of the inhibitor substituents [42]. Moreover, QM/MM simulations have been used to refine the binding poses of a series of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamides directly binding to the zinc ion [126].
As well, Zn-dependent nucleases able to cleave single-strand DNA or RNA are object of research interest both for understanding their mechanism [127] and for their emerging role as the selective target of small-molecule inhibitors [128].
Finally, the metallo-β-lactamases (MBL) represent a large family of Zn-dependent enzymes involved in the cleavage of the β-lactam antibiotics. Several QM/MM MD studies in combination with free-energy methods have been done to assess the substrate cleavage mechanism [16,129–132]. QM/MM MD studies concerning the binding and the mechanism by which known inhibitors inactivate the enzyme include elucidated how a thiazolidinecarboxylic acid inhibitor inactivated the di zinc CcrA enzyme [129]. These studies were performed aiming at designing common inhibitors of all MBLs, in spite of the marked differences between the structure of MBLs’ active site, β-lactam specificity, and metal content [133].
In recent years, a series of small compounds like bisthiazolidines were shown to act as inhibitors of all MBL types, restoring the efficacy of currently used antibiotics against resistant bacterial strains producing different MBLs. This study offered precious insights for future MBL inhibitor design [134].
3.2. Metallodrugs targeting proteins and nucleic acids
3.2.1. Ru-containing drugs targeting the nucleosome core particle: DNA versus protein targeting strategies
Ru(II)-arene (RA) compounds represent a promising alternative to Pt-drugs (Figure 4). These complexes, originally introduced in the form of [Ru(II)(η6-arene)Cl(ethylene-diamine)] (RAED) and the [Ru(II)(η6-arene)Cl2(1,3,5-triaza-7-phosphoadamantane) (RAPTA) compounds, share a π-bonded arene ligand. It can be pictorially seen as the ‘seat of the stool,’ with a chelating ethylene-diamine (ED) or a monodentate phosphine 1,3,5-triaza-7-phosphadamantane (PTA) ligand occupying the remaining coordination sites (Figure 4(A, B)) [23]. The arene ligand provides a bulky hydrophobic surface, possibly fostering a high degree of complementarity and selectivity toward bimolecular targets [30]. The versatility of RA compounds stimulated the development of similar compounds, among which those bearing a p-cymene (RAPTA-C) or a toluene (RAPTA-T) ligand, exhibited selective antimetastatic activity in vitro and in vivo [135–137].
Figure 4.
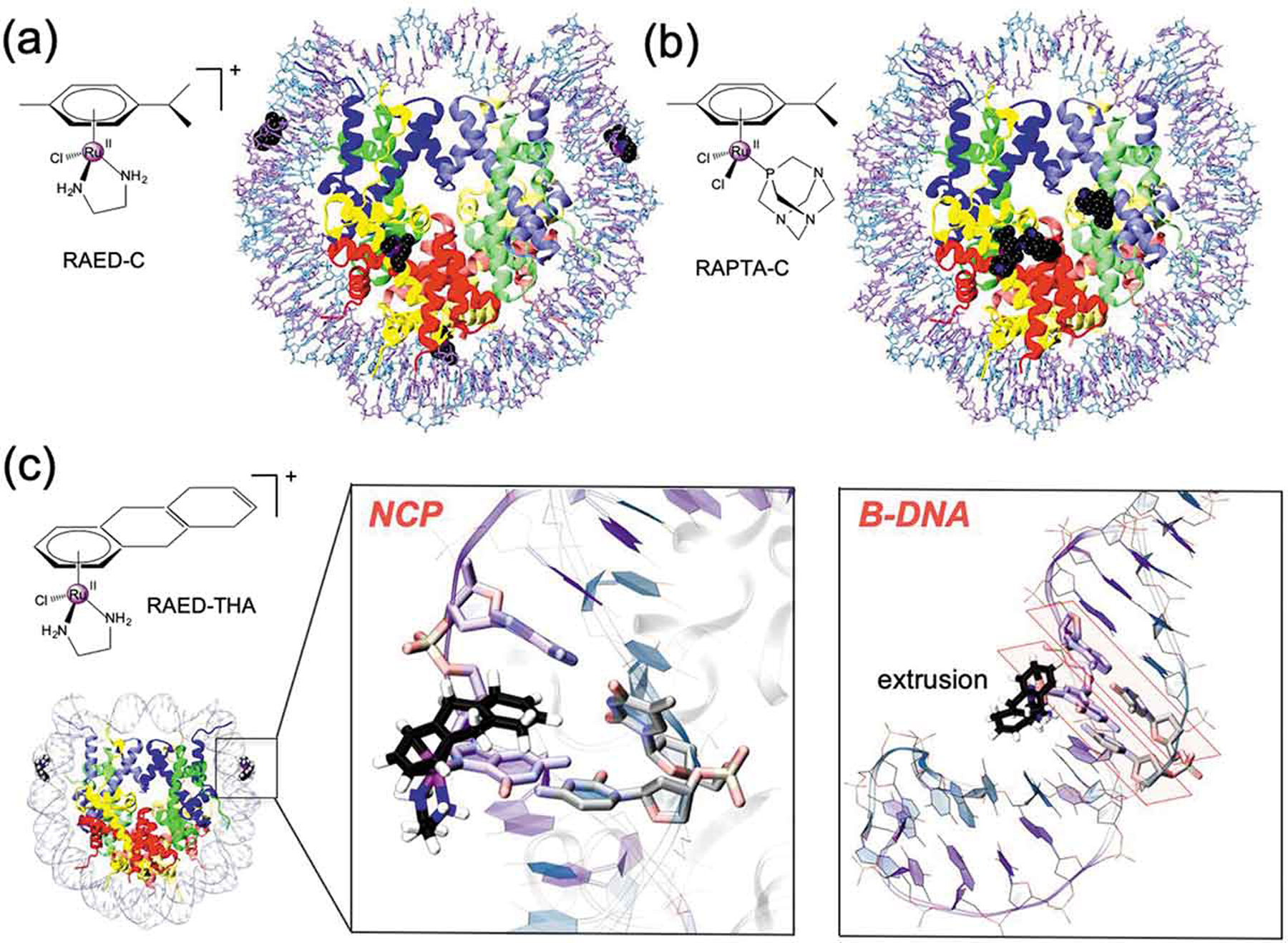
Chemical structure and binding mode of the RAED-C (A), RAPTA-C (B) and RAED-THA (C) at the Nucleosome Core Particle (NCP). RAED-C preferentially binds at DNA, while RAPTA-C selects protein histones. RAED-THA binds at the SHL ±1.5 sites of the DNA and engages ligand coordination with a guanine base (i.e. G ± 15). Within the nucleosomal DNA (close-up view), RAED-THA intercalates within the ±15/16 AG = CT step via a ‘mono-base stacking’ mechanism, which directly involves G ± 15. (B) Selected snapshots from classical MD simulations of a 14-mer DNA extracted from the RAED-THA/NCP model. After a ~ 20/30 ns MD, the THA is extruded from the double strand, which assumes a B-configuration. The typical base-stacking of DNA in B-configuration is highlighted in orange.
RA compounds have been demonstrated to bind to the nucleosome core particles (NCP)s [138,139]. Nucleosomes are the fundamental unit of chromatin and are composed of chromosomal DNA (about 145–147 base pairs (bp) long), wrapped around an octamer made by four core histone proteins (H3, H4, H2A, and H2B, Figure 4). The packaging of the genome into nucleosomes raises the intriguing possibility to form adducts between the drugs to the DNA and/or protein sites. These latter are particularly interesting, since the drugs binding at the histone may directly influence gene expression, opening novel opportunities for anticancer strategies [139,140].
Surprisingly, X-ray crystallography revealed that the chromatin-bound adducts in cancer cells treated with RAPTA-C are mostly associated with the NCP protein components, while RAED preferentially targets the guanine sites of the DNA components of chromatin (Figure 4(A, B)) [138,141,142]. When bound to the protein components, both RAPTA-C and RAED-C preferentially bind at glutamate sites of the histone components, while displaying a very different histone (RAPTA-C) vs. DNA (RAED-C) specificities. QM/MM MD simulations, complemented by free-energy methods, elucidated the molecular basis of this observed site selectivity. This was originating from both kinetic and thermodynamic factors due to the distinct size of the ligands present in the two drugs. Indeed, the steric constraints of the nucleosomal DNA, prevent the easy accommodation of the PTA ligand during the formation of the RAPTA-C-DNA adduct, also decreasing its thermodynamic stability. In contrast, the PTA ligands favor the binding of the drug at the histone sites owing to its special shape and hydrophobic complementarity with the histone proteins. In this scenario, the higher cytotoxicity of RAED-C may be related to its DNA lesion-forming proclivity, whereas the propensity of RAPTA-C to form protein adducts may be linked to its distinct therapeutic effect [138]. Building on the RAED compound, a novel antitumor compound [(η6-THA)Ru(ethylene-diamine)Cl][PF6] (THA = 5,8,9,10-tetrahydroanthracene; RAED-THA; Figure 4C), showing the highest cytotoxicity among the reported Ru-based agents, was identified [143]. This compound differs from RAED-C in the substitution of the THA group with p-cymene. RAED-THA cannot form bifunctional DNA cross-links. Its high cytotoxicity, comparable to that of cisplatin, has been attributed to the potential THA group ability to intercalate within the DNA base pairs.
Building on the X-ray crystallography revealing that RAED-THA attacks nucleosomal DNA at a pair of guanine sites establishing an unconventional ‘mono-base stacking,’ classical and QM/MM MD simulations enabled to compare its binding mode to both naked and nucleosomal DNA. These simulations confirmed a stable binding of RAED-THA by the coordination of Ru(II)-center to G15 and the p-stacking of the THA ligand with nucleosomal G ± 15 (Figure 4B). Due to the peculiar structural characteristics of the nucleosomal DNA [144], RAED-THA assumes an atypical, selective, and previously unknown semi-intercalative binding mode. Conversely, the simulations considering RAED-THA covalently bound to a 14-mer DNA section extracted from the RAEDTHA/NCP model experienced a rapid DNA (i.e. within the ~20/30 ns time scale) relaxation of the DNA double helix to the B-DNA conformation, with the THA group being extruded (Figure 4B) from ds DNA, while the adjacent bases returned to their canonical inter-base pair π-stacking [145]. These results further remark that the histone-induced deformation of the nucleosomal dsDNA is critical for stabilizing the newly reported semi-intercalation mechanism of RAED-THA [145].
3.2.2. Synergistic effects of Ru(II) and Au(I) compounds
Gold-based compounds have increasingly attracted interest of medicinal chemists for their potential therapeutic applications [146]. Among these, auranofin [(1-thio-b-d-glucopyranosyl) (triethylphosphine) Au(I) 2,3,4,6-tetraacetate], a clinically approved drug for the treatment of rheumatoid arthritis, has raised the interest of medicinal chemistry community for its therapeutic application in a wide number of diseases including cancer, neurodegenerative disorders, HIV, parasitic and bacterial infections [147]. The antitumor activity of gold compounds is known to be ‘DNA-independent,’ being its main target thioredoxin reductase, a mitochondrial enzyme, which is overexpressed in most cancer cells [148]. In vivo, this drug is metabolized to tetraacetylthioglucose and the bioactive fragment [Au(PEt3)]+ (AUF – Figure 5A), with the first fragment being excreted, the cationic AUF species can coordinate His imidazole nitrogens and Cys thiol sulfurs of biomolecules.
Figure 5.
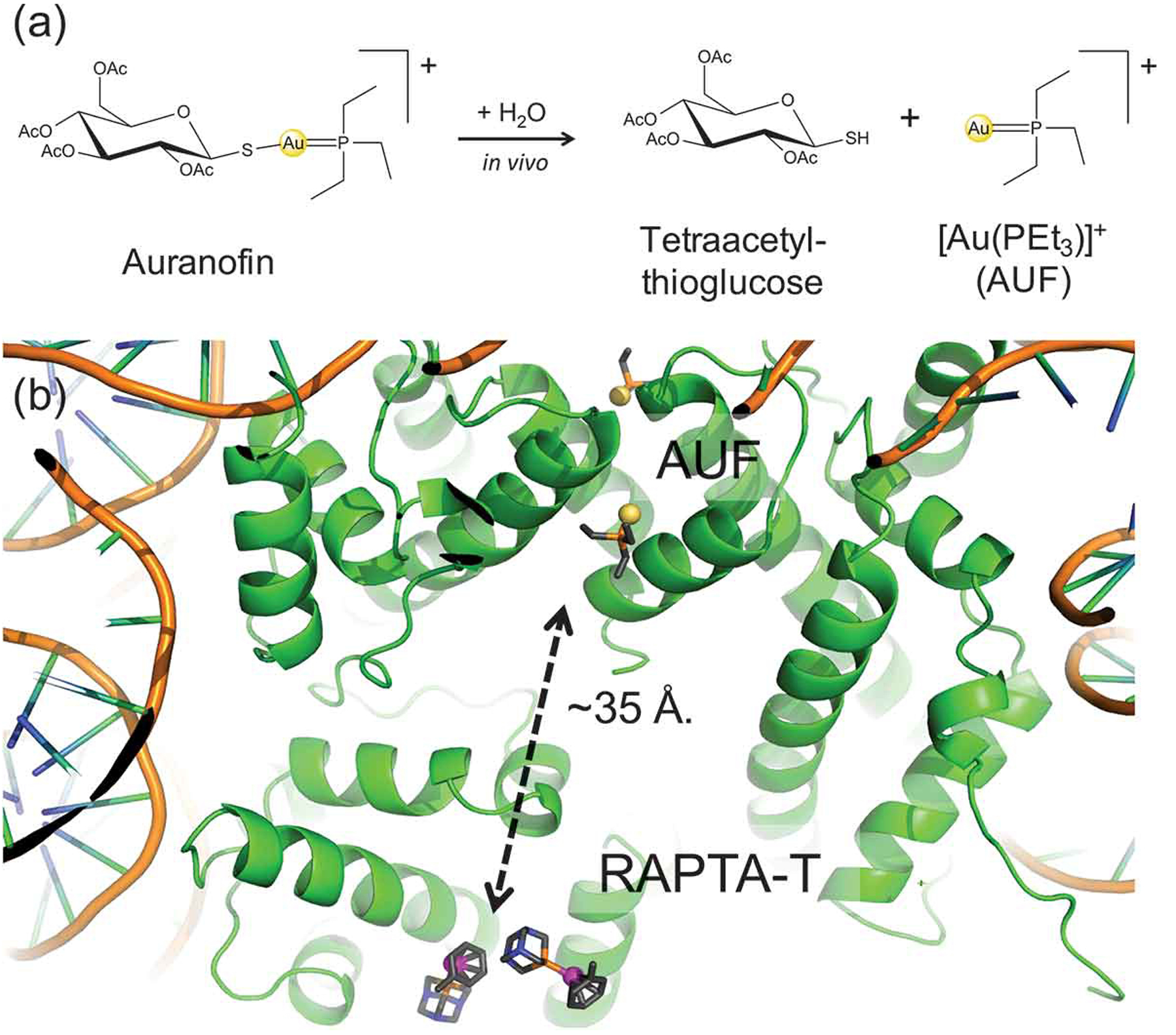
(A) Chemical structure and in vivo metabolic pathway of auranofin. Auranofin is a pro-drug, which is metabolized leading to tetraacetylthioglucose and to the bioactive fragment [Au(PEt3)]+ (AUF). (B) X-ray structure of the NCP displaying the binding of Auranofin (AUF) and RAPTA-T at distal histone sites, separated by ~35 Å.
The importance of auranofin in this context is due to the recent discovery of its synergistic cytotoxic activity with RAPTA-T (with an η6-toluene ring) on ovarian cancer cells, along with a ~ 3 fold increased uptake of auranofin into nucleosomes when simultaneously applied with RAPTA-T. Crystallographic studies on the NCP in complex with RAPTA-T and AUF showed that the AUF fragment binds at two symmetry-related histidine residues (H113/H113ʹ) located at ~35 Å distance away from the two adjacent RAPTA-T binding sites [149]. Extensive MD simulations (considering the apo NCP, the NCP/RAPTA-T and NCP/AUF adducts and the NCP in complex with both drugs) contributed to shed light on the synergistic mechanism of the two drugs at the molecular level. These simulations allowed discriminating the differences in the structural and dynamic properties induced by simultaneous binding of both drugs. Indeed, a cross-correlation analysis showed that a cross-talk between the two metals binding sites was taking place, thanks to a series of subtle conformational changes occurring within the protein framework that are transmitted via the coupled motions of the histones.
We remark that the binding of RAPTA compounds at the protein histones occurs at the specific binding sites of chromatin factors. These latter are enzymes determining the degree of chromatin compaction by binding NCPs [140]. Thus, the RAPTA compounds directly interfere with the epigenetic mechanisms of gene expression. In light of this evidence, the synergistic action of RAPTA-T and auranofin might be explained by a mechanism of sequestration of specific binding sites in which the NCP is made inaccessible for the epigenetic machineries [149]. This is opening new research lines attempting to fully exploit the allosteric nature of the NCP [150,151] through the cross-link of allosteric sites [152].
3.2.3. Au(III)-drugs targeting aquaporins
Au(III) compounds have been also object of research interest [146]. Recently, [Au(III)(phen)Cl2]Cl (phen = 1,10-phenanth roline)], AuPhen, was identified as a selective and potent inhibitor of human aquaporin-3 (AQP3). This is an aquaglyceroporin family member allowing permeation of water and small uncharged solutes such as glycerol. Remarkably, AQP3 overexpression is linked to cancer development and obesity [153]. A computational workflow, composed of docking, FF-based MD, and QM/MM calculations, was applied to Auphen and other similar Au(III) complexes to predict their binding mode to AQP3’s channel. Interestingly, Au(III) binding to Cys40 induces thorough conformational changes of the aquaglyceroporin channel, ultimately leading to pore shrinkage and inhibition of substrate permeation [33].
Furthermore, MD simulations of Hg2+ ions (benchmark inhibitors of all AQPs) binding to AQP3 have shown metal-induced pore closure of the channel, upon Hg2+ binding to Cys40, triggering a protein conformational change rather than the steric blockage of AQP3 channel by the metal inhibitor [154].
Finally, a multi-level theoretical approach, relying on classical MD and QM/MM studies, was used to investigate both the interaction with the physiological medium and the binding at the extracellular pore of AQP3 of the Au(III) complex [Au(N^N)Cl2][PF6] (N^N = 2,2ʹ-bipyridine, Aubipy). This study clarified the importance of the initial formation of non-covalent Au(III) complex/AQP3 adduct to compensate the thermodynamic and kinetic barriers linked with the production of the final covalent adduct [155].
4. Conclusions
The limits of in silico drug-discovery strategies to properly account for metal–ligand interactions have prevented their applications to the inorganic medicinal chemistry research field, where metal–ligand interactions have to be accurately monitored and described. The exponential increase of computational power occurred in the last decades, together with the promise of modern (quantum) computers, is expected to markedly boost the use of accurate, yet computationally intensive, all-atom simulations in drug-discovery programs in the near future. To show the potential of the synergistic use of massive classical and QM/MM MD simulations in unraveling the mechanism of action, rationalizing the potency and the underlying binding specificity of metal-binding drugs, we have presented a selection of informative showcase applications. In particular, this compendium focuses on the role of molecular simulations in decrypting the metal-mediated catalytic mechanism of steroidogenic CYP450s and exploiting the Fe-ligand interactions to inhibit their function in cancer. Again, few applications of computer simulations in studying the mechanism or the binding of inhibitors to distinct members of the Zn-containing enzymes are also reported. The studies discussed clearly exemplifies that the action, mechanism, and specificity of metal-targeting drugs do not only depend on the coordination geometries while being most often entwined with the biological environment. This should be explicitly considered to optimize inhibitors’ potency and specificity [156]. Complementarily, in the field of metallodrugs, our selected examples show how molecular simulations can contribute to elucidate key aspects of the targeting characteristics of Ru and Au-anticancer agents, such as the binding preferences, the topological dependence of their binding mode and allosteric regulations of drugs targeting distinct binding sites.
Besides showing the predictive power of multiscale simulations, this review also highlights how the synergism between experiments and all-atom simulations can contribute to enrich and expand our comprehension of inorganic medicinal chemistry, elucidating mechanistic features, which are often inaccessible to experiments and molecular simulations taken singularly. This synergism is also expected to contribute to pinpointing off-target interactions and their associated toxicity mechanism and the synergism/interference mechanism of different drugs. This information will contribute to design and develop the next generation of clinically applicable drugs or drug-combinations exploiting metal–ligand interactions.
5. Expert opinion
The use of metal–ligand interactions in medicine to treat patients with different pathologies is well established. Nowadays, the mechanism of action is increasingly used to drive the drug-discovery process to optimize the properties and the efficacy of drugs exploiting metal–ligand interactions [64].
In this scenario, the rapid and constant increase in accuracy and computational affordance of atomic-level simulations provide computational inorganic chemists with powerful tools to gain direct views on the mechanism of action of two categories of drugs: those directly binding the metal moiety of metal-enzymes (metal-interacting drugs) or those containing non-bio-abundant metals (metal-containing drugs). Recent advances in free energy and enhanced sampling methods, the refinements of FFs, the development of specific computational platforms to run all-atom simulations have partially overcome traditional shortcomings of classical and QM/MM MD simulations (i.e. short time-scales, size-limit, and predictability problems).
More importantly, the growing body of data gathered from experimental and computational means enables computers to learn from data, paving toward machine learning (ML) approaches, a burgeoning research field in drug discovery and chemistry [157,158]. Its impact has been so far more limited in drug-discovery of metal-coordinating drugs. Indeed, metal–ligand interactions are very sensitive to atom identity, exhibit coordination, and environment-dependence of bonds. These properties need to be captured by complex and sophisticated descriptors and functions, therefore, posing a challenge for ML [159]. Additionally, available training data sets are small, owing to the high computational cost of simulation of transition metal compounds with very accurate methods. This has a strong impact on the predictive power of ML [159] in this research area. In spite of these limitations, we expect that ML will hit on inorganic medicinal chemistry. On one hand, ML is expected to foster the development of cost-effective and more accurate description of the system either based on quantum mechanics (by contributing to the development of more accurate exchange-correlation functional, orbital-free density functional theory, many-body expansions) or based on molecular mechanics (by contributing to develop more accurate and versatile classical FFs) suitable to describe the full range of inorganic chemical bonding [160]. We expect that these advances will contribute to further bridge pharmaceutical companies and academic institutions.
Novel research areas representing an opportunity for computational studies of metallo-ligand/target interactions include nuclear medicine. Radiometals can be applied for both imaging and therapy, especially in the oncology field [161]. Selective tumor targeting is required in radiotherapy to avoid compromising the health of surrounding tissues. Therefore, the rational design of radiopharmaceutical ligands, along with a detailed comprehension of their transport routes and mechanisms, is required to ensure an optimal drug performance in terms of stability of the radiometal compound and selective targeting properties [162].
The structural biology research is currently routinely accessing increasingly large biological macromolecular machineries at (or near) atomic resolution, thanks to single-particle cryo-EM. We envision that in forthcoming years, this technique will enable to obtain structures of new pharmacologically relevant metal-dependent machineries amenable to structure-based design studies [93,97]. Extending the metallodrug interaction context from the single protein target to complex macromolecular aggregates of proteins and/or nucleic acids will further advance our mechanistic understanding of metal-containing or metal-binding drugs. Irrespective of the methodological advances achieved, computational drug-design efforts will have to be supported, integrated, and complemented by experimental data, as exemplified by the successfully showcased studies presented in this compendium.
Article highlights.
Small-molecule inhibitors can establish metal-coordination bonds to impair the function of pharmacologically relevant metalloenzymes.
Transition metals find application mainly as anticancer agents exerting their cytotoxic action by coordinating to nucleic acids and/or proteins.
Multiscale simulations can rationalize the potency of metal-targeting inhibitors and unveil the mechanism of metalloenzymes, thereby boosting the efficacy inhibitor’s potency.
Classical and quantum-classical simulations enable prediction and assessment of the binding mode, mechanism, and the targeting proclivity of metallodrugs.
This review highlights the current role and challenges faced by all-atom simulations, envisioning their permeation in the drug-discovery workflow to target and exploit metal ions.
This box summarizes the key points contained in the article.
Funding
A Spinello was supported by a FIRC-AIRC ‘Mario e Valeria Rindi’ fellowship while A Magistrato was financially supported through the project ‘Finding personalized therapies with in silico and in vitro strategies’ (ARES) CUP: D93D19000020007 POR FESR 2014 2020-1.3.b, Friuli Venezia Giulia. G Palermo meanwhile has received grant support from the National Science Foundation under grant No. CHE-1,905,374.
Declaration of interest
AM thanks the financial support of the project ‘Finding personalized therapies with in silico and in vitro strategies’ (ARES) CUP: D93D19000020007 POR FESR 2014 2020 - 1.3.b - Friuli Venezia Giulia. AS was supported by a FIRC-AIRC ‘Mario e Valeria Rindi’ fellowship for Italy. AM thank the CINECA Italian Super Computing Center for computational resources. GP thanks the National Science Foundation for supporting her research grant under Grant No. CHE-1905374. GP acknowledges funding from the National Science Foundation (NSF) and the National Institute of Health (NIH). Specifically, this work was supported by the NSF under Grant No. CHE-1905374, and partially by the NIH under Grant No. R01 EY027440. GP also acknowledges funding for computer time by the Extreme Science and Engineering Discovery Environment (XSEDE) via grant TG- MCB160059.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Footnotes
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Putignano V, Rosato A, Banci L, et al. MetalPDB in 2018: a database of metal sites in biological macromolecular structures. Nucleic Acids Res. 2018;4:459–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Magistrato A, Pavlin M, Qasem Z, et al. Copper trafficking in eukaryotic systems: current knowledge from experimental and computational efforts. Curr Opin Struct Biol. 2019;58:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sala D, Musiani F, Rosato A. Application of molecular dynamics to the investigation of metalloproteins involved in metal homeostasis. Eur J Inorg Chem. 2018;43:4661–4677. [Google Scholar]
- 4.Musiani F, Bertosa B, Magistrato A, et al. Computational study of the DNA-binding protein helicobacter pylori NikR: the Role of Ni(2 +). J Chem Theory Comput. 2010;6:3503–3515. [DOI] [PubMed] [Google Scholar]
- 5.Sgrignani J, Magistrato A. The structural role of Mg2+ ions in a class I RNA polymerase ribozyme: a molecular simulation study. J Phys Chem B. 2012. February 23;116:2259–2268. [DOI] [PubMed] [Google Scholar]
- 6.Casalino L, Magistrato A. Structural, dynamical and catalytic interplay between Mg2+ ions and RNA. Vices and virtues of atomistic simulations. Inorg Chim Acta. 2016;452:73–81. [Google Scholar]
- 7.Dubey KD, Shaik S. Cytochrome P450—The wonderful nanomachine revealed through dynamic simulations of the catalytic cycle. Acc Chem Res. 2019;52:389–399. [DOI] [PubMed] [Google Scholar]; •• This review presents a summary of current understanding on the mechanism of CYP450
- 8.Spinello A, Ritacco I, Magistrato A. The catalytic mechanism of steroidogenic cytochromes P450 from all-atom simulations: entwinement with membrane environment, redox partners, and post-transcriptional regulation. Catalysts. 2019;9:81. [Google Scholar]; • This review presents a summary of current understanding on the mechanism of steroidogenic CYP450
- 9.Lutsenko S Human copper homeostasis: a network of interconnected pathways. Curr Opin Chem Biol. 2010;14:2011–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spinello A, Bonsignore R, Barone G, et al. Metal ions and metal complexes in Alzheimer’s disease. Curr Pharm Des. 2016;22:3996–4010. [DOI] [PubMed] [Google Scholar]
- 11.Qasem Z, Pavlin M, Ritacco I, et al. The pivotal role of MBD4–ATP7B in the human Cu(I) excretion path as revealed by EPR experiments and all-atom simulations. Metallomics. 2019;11:1288–1297. [DOI] [PubMed] [Google Scholar]
- 12.Wang J, Luo C, Shan C, et al. Inhibition of human copper trafficking by a small molecule significantly attenuates cancer cell proliferation. Nat Chem. 2015;7:968–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Winer A, Adams S, Mignatti P. Matrix metalloproteinase inhibitors in cancer therapy: turning past failures into future successes. Mol Cancer Ther. 2018;17:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Supuran CT. Carbonic anhydrases-an overview. Curr Pharm Des. 2008;14:603–614. [DOI] [PubMed] [Google Scholar]
- 15.Lopez C, Ayala JA, Bonomo RA, et al. Protein determinants of dissemination and host specificity of metallo-beta-lactamases. Nat Commun. 2019;10:3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Simona F, Magistrato A, Vera DM, et al. Protonation state and substrate binding to B2 metallo-beta-lactamase CphA from Aeromonas hydrofila. Proteins: Struct., Func., Bioinfo 2007;69:595–605. [DOI] [PubMed] [Google Scholar]
- 17.Palermo G, Casalino L, Magistrato A, et al. Understanding the mechanistic basis of non-coding RNA through molecular dynamics simulations. J Struct Biol. 2019;206:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Casalino L, Magistrato A. Unraveling the molecular mechanism of Pre-mRNA splicing from multi-scale simulations. Front Mol Biosci. 2019. August 6;6(62):1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Exell JC, Thompson MJ, Finger LD, et al. Cellularly active N-hydroxyurea FEN1 inhibitors block substrate entry to the active site. Nat Chem Biol. 2016;12:815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sgrignani J, Magistrato A. QM/MM MD simulations on the enzymatic pathway of the human flap endonuclease (hFEN1) Elucidating common cleavage pathways to RNase H enzymes. ACS Catal. 2015;5:3864–3875. [Google Scholar]
- 21.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4:236–248. [DOI] [PubMed] [Google Scholar]
- 22.Maldonado MDM, Dharmawardhane S. Targeting Rac and Cdc42 GTPases in cancer. Cancer Res. 2018;78:3101–3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sava G, Bergamo A, Dyson P. Metal-based antitumour drugs in the post-genomic era: what comes next? Dalton Trans. 2011;40:9069–9075. [DOI] [PubMed] [Google Scholar]
- 24.Jung Y, Lippard SJ. Direct cellular responses to platinum-induced DNA damage. Chem Rev. 2007;107:1387–1407. [DOI] [PubMed] [Google Scholar]
- 25.Kelland L The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer. 2007;7:573–584. [DOI] [PubMed] [Google Scholar]
- 26.Casini A, Reedijk J. Interactions of anticancer Pt compounds with proteins: an overlooked topic in medicinal inorganic chemistry. Chem Sci. 2012;3:3135–3144. [Google Scholar]
- 27.Wu B, Davey GE, Nazarov AA, et al. Specific DNA structural attributes modulate platinum anticancer drug site selection and cross-link generation. Nucleic Acids Res. 2011;39:8200–8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Spiegel K, Magistrato A, Carloni P, et al. Azole-bridged diplatinum anticancer compounds. Modulating DNA flexibility to escape repair mechanism and avoid cross resistance. J Phys Chem B. 2007;111:11873–11876. [DOI] [PubMed] [Google Scholar]
- 29.Spinello A, Magistrato A. An omics perspective to the molecular mechanisms of anticancer metallo-drugs in the computational microscope era. Expert Opin Drug Discov. 2017;12:813–825. [DOI] [PubMed] [Google Scholar]
- 30.Murray BS, Dyson PJ. Recent progress in the development of organometallics for the treatment of cancer. Curr Opin Chem Biol. 2019;56:28–34. [DOI] [PubMed] [Google Scholar]; • This review summarizes current advances in the development of anticancer organometallic drugs
- 31.Vargiu AV, Robertazzi A, Magistrato A, et al. The hydrolysis mechanism of the anticancer ruthenium drugs NAMI-A and ICR investigated by DFT-PCM calculations. J Phys Chem B. 2008;112:4401–4409. [DOI] [PubMed] [Google Scholar]
- 32.Alessio E, Messori L. NAMI-A and KP1019/1339, two iconic ruthenium anticancer drug candidates face-to-face: a case story in medicinal inorganic chemistry. Molecules. 2019;24:1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de Almeida A, Mosca AF, Wragg D, et al. The mechanism of aquaporin inhibition by gold compounds elucidated by biophysical and computational methods. Chem Commun. 2017;53:3830–3833. [DOI] [PubMed] [Google Scholar]
- 34.Jacques A, Lebrun C, Casini A, et al. Reactivity of Cys4 zinc finger domains with gold(III) complexes: insights into the formation of “gold fingers”. Inorg Chem. 2015;54:4104–4113. [DOI] [PubMed] [Google Scholar]
- 35.Riccardi L, Genna V, De Vivo M. Metal–ligand interactions in drug design. Nat Rev Chem. 2018;26:100–112. [Google Scholar]; • This review reports current advances and limitations in treating metal-ligand interactions for drug design
- 36.Sgrignani J, Magistrato A. First-principles modeling of biological systems and structure-based drug-design. Curr Comp Aid Drug Des. 2013;9:15–34. [PubMed] [Google Scholar]
- 37.Palermo G, Magistrato A, Riedel T, et al. Fighting cancer with transition metal complexes: from naked DNA to protein and chromatin targeting strategies. ChemMedChem. 2016;11:1199–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This review summariezes QM/MM and MD computational studies of different binding drugs to treat cancer
- 38.Casalino L, Palermo G, Abdurakhmonova N, et al. Development of site-specific Mg2+-RNA force field parameters: a dream or reality? Guidelines from combined molecular dynamics and quantum mechanics simulations. J Chem Theory Comput. 2017;1:340–352. [DOI] [PubMed] [Google Scholar]
- 39.Vidossich P, Magistrato A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules. 2014;4:616–645. [DOI] [PMC free article] [PubMed] [Google Scholar]; • This review summarizes QM/MM MD computational studies of metal binding enzymes
- 40.Vargiu AV, Magistrato A. Atomistic-level portrayal of drug-DNA Interplay: a history of courtships and meetings revealed by molecular simulations. ChemMedChem. 2014;9:1966–1981. [DOI] [PubMed] [Google Scholar]
- 41.Spiegel K, Magistrato A. Modeling anticancer drug-DNA interactions via mixed QM/MM molecular dynamics simulations. Org Biomol Chem. 2006;4:2507–2517. [DOI] [PubMed] [Google Scholar]
- 42.Gaspari R, Rechlin C, Heine A, et al. Kinetic and structural insights into the mechanism of binding of sulfonamides to human carbonic anhydrase by computational and experimental studies. J Med Chem. 2016;12:4245–4256. [DOI] [PubMed] [Google Scholar]
- 43.Karplus M, McCammon JA. Molecular dynamics simulations of biomolecules. Nat Struct Biol. 2002;9:646–652. [DOI] [PubMed] [Google Scholar]
- 44.Hollingsworth SA, Dror RO. Molecular dynamics simulation for all. Neuron. 2018;99:1129–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
- 46.Case TAD DA, Cheatham TE, Simmerling CL III, et al. AMBER 12. San Francisco: University of California; 2012. [Google Scholar]
- 47.Christen M, Hunenberger PH, Bakowies D, et al. The GROMOS software for biomolecular simulation: GROMOS05. J Comput Chem. 2005;26:1719–1751. [DOI] [PubMed] [Google Scholar]
- 48.MacKerell AD, Bashford D, Bellott M, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 49.Wang J, Wang W, Kollman PA, et al. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol. Graph Mod 2006;25:247–260. [DOI] [PubMed] [Google Scholar]
- 50.Vanommeslaeghe K, Hatcher E, Acharya C, et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J Comput Chem. 2010;31:671–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci USA. 2002;99:12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miao Y, Feher VA, McCammon JA. Gaussian accelerated molecular dynamics: unconstrained enhanced sampling and free energy calculation. J Chem Theory Comput. 2015;11:3584–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Baron R, McCammon JA. Molecular recognition and ligand association. Annu Rev Phys Chem. 2013;64:151–175. [DOI] [PubMed] [Google Scholar]
- 54.Franco D, Vargiu AV, Magistrato A. Ru[(bpy)(2)(dppz)](2)(+) and Rh [(bpy)(2)(chrysi)](3)(+) targeting double strand DNA: the shape of the intercalating ligand tunes the free energy landscape of deintercalation. Inorg Chem. 2014;53:7999–8008. [DOI] [PubMed] [Google Scholar]
- 55.Granata D, Camilloni C, Vendruscolo M, et al. Characterization of the free-energy landscapes of proteins by NMR-guided metadynamics. Proc Natl Acad Sci USA. 2013;110:6817–6822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Warshel A, Levitt M. Theoretical studies of enzymic reactions: dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103:227–249. [DOI] [PubMed] [Google Scholar]
- 57.Brunk E, Rothlisberger U. Mixed quantum mechanical/molecular mechanical molecular dynamics simulations of biological systems in ground and electronically excited states. Chem Rev. 2015;115:6217–6263. [DOI] [PubMed] [Google Scholar]; •• This review reports and extesive survey of QM/MM metodology and its applications
- 58.Laio A, VandeVondele J, Rothlisberger UA. Hamiltonian electrostatic coupling scheme for hybrid Car–Parrinello molecular dynamics simulations. J Chem Phys. 2002;116:6941. [Google Scholar]
- 59.Laino T, Mohamed F, Laio A, et al. An efficient real space multigrid QM/MM electrostatic coupling. J Chem Theory Comput. 2005;1:1176–1184. [DOI] [PubMed] [Google Scholar]
- 60.Olsen JMH, Bolnykh V, Meloni S, et al. MiMiC: a novel framework for multiscale modeling in computational chemistry. J Chem Theory Comput. 2019;15:3810–3823. [DOI] [PubMed] [Google Scholar]
- 61.Car R, Parrinello M. Unified approach for molecular dynamics and density-functional theory. Phys Rev Lett. 1985;55:2471–2474. [DOI] [PubMed] [Google Scholar]
- 62.Van Der Spoel D, Lindahl E, Hess B, et al. GROMACS: fast, flexible, and free. J Comput Chem. 2005;26:1701–1718. [DOI] [PubMed] [Google Scholar]
- 63.Spiegel K, Rothlisberger U, Carloni P. Cisplatin binding to DNA oligomers from hybrid car-parrinello/molecular dynamics simulations. J Phys Chem B. 2004;108:2699–2707. [Google Scholar]
- 64.Janos P, Spinello A, Magistrato A. All-atom simulations to studying metallo-drugs/target interactions. Curr Opin Chem Biol. 2020;61:1–8. [DOI] [PubMed] [Google Scholar]; •• This review reports the state of the art of all-atom simulations to study the interactions between metallo-drugs and their biological target
- 65.Calandrini V, Rossetti G, Arnesano F, et al. Computational metallomics of the anticancer drug cisplatin. J Inorg Biochem. 2015;153:231–238. [DOI] [PubMed] [Google Scholar]
- 66.Biancardi A, Burgalassi A, Terenzi A, et al. A theoretical and experimental investigation of the spectroscopic properties of a DNA intercalator salphen type ZnII complex. Chem Eur J. 2014;20:7439–7447. [DOI] [PubMed] [Google Scholar]
- 67.Vargiu AV, Ruggerone P, Magistrato A, et al. Anthramycin-DNA binding explored by molecular simulations. J Phys Chem B. 2006;110:24687–24695. [DOI] [PubMed] [Google Scholar]
- 68.Biarnes X, Ardevol A, Iglesias-Fernandez J, et al. Catalytic itinerary in 1,3-1,4-beta-glucanase unraveled by QM/MM metadynamics. charge is not yet fully developed at the oxocarbenium ion-like transition state. J Am Chem Soc. 2011;133:20301–20309. [DOI] [PubMed] [Google Scholar]
- 69.Campomanes P, Neri M, Horta BAC, et al. Origin of the spectral shifts among the early intermediates of the rhodopsin photocycle. J Am Chem Soc. 2014;136:3842–3851. [DOI] [PubMed] [Google Scholar]
- 70.De Vivo M, Dal Peraro M, Klein ML. Phosphodiester cleavage in ribonuclease H occurs via an associative two-metal-aided catalytic mechanism. J Am Chem Soc. 2008;130:10955–10962. 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ganguly A, Thaplyal P, Rosta E, et al. Hammes-Schiffer S Quantum mechanical/molecular mechanical free energy simulations of the self-cleavage reaction in the hepatitis delta virus ribozyme. J Am Chem Soc. 2014;136:1483–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Otyepka M, Banáš P, Magistrato A, et al. Second step of hydrolytic dehalogenation in haloalkane dehalogenase investigated by QM/MM methods.. Proteins: Struct., Func., Bioinfo 2008;70:707–717. [DOI] [PubMed] [Google Scholar]
- 73.Carloni P, Rothlisberger U, Parrinello M. The role and perspective of ab initio molecular dynamics in the study of biological systems. Acc Chem Res. 2002;35:455–464. [DOI] [PubMed] [Google Scholar]
- 74.Palermo G, Campomanes P, Cavalli A, et al. Anandamide hydrolysis in FAAH reveals a dual strategy for efficient enzyme-assisted amide bond cleavage via nitrogen inversion. J Phys Chem B. 2015;119:789–801. [DOI] [PubMed] [Google Scholar]
- 75.Riccardi D, Yang S, Cui Q. Proton transfer function of carbonic anhydrase: insights from QM/MM simulations. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics. 2010;1084:342–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Feliciano GT, Roque da Silva AJ. Unravelling the reaction mechanism of matrix metalloproteinase 3 using QM/MM calculations. J Mol Struct Theochem. 2015;1091:125–132. [Google Scholar]
- 77.Shaik S, Kumar D, de Visser SP, et al. Theoretical perspective on the structure and mechanism of cytochrome P450 enzymes. Chem Rev. 2005;105:2279–2328. [DOI] [PubMed] [Google Scholar]
- 78.Su H, Wang B, Shaik S. Quantum-mechanical/molecular-mechanical studies of CYP11A1-catalyzed biosynthesis of pregnenolone from cholesterol reveal a C–C bond cleavage reaction that occurs by a compound i-mediated electron transfer. J Am Chem Soc. 2019;141:20079–20088. [DOI] [PubMed] [Google Scholar]
- 79.Sérgio FS, Ribeiro AJM, Neves RPP, et al. Application of quantum mechanics/molecular mechanics methods in the study of enzymatic reaction mechanisms. Wiley Interdiscip Rev Comput Mol Sci. 2016;7:e1281. [Google Scholar]
- 80.Raich L, Nin-Hill A, Ardevol A, et al. Enzymatic cleavage of glycosidic bonds: strategies on how to set up and control a QM/MM metadynamics simulation. Methods Enzymol. 2016;577:159–183. [DOI] [PubMed] [Google Scholar]
- 81.Ferreira P, Cerqueira N, Fernandes PA, et al. Catalytic mechanism of human aldehyde oxidase. ACS Catal. 2020;10:9276–9286. [Google Scholar]
- 82.Brunk E, Ashari N, Athri P, et al. Pushing the frontiers of first-principles based computer simulations of chemical and biological systems. Chimia. 2011;65:667–671. [DOI] [PubMed] [Google Scholar]
- 83.Vargiu AV, Magistrato A. Detecting DNA mismatches with metallo-insertors: a molecular simulation study. Inorg Chem. 2012;51:2046–2057. [DOI] [PubMed] [Google Scholar]
- 84.Casalino L, Palermo G, Rothlisberger U, et al. Who activates the nucleophile in ribozyme catalysis? An answer from the splicing mechanism of group II introns. J Am Chem Soc. 2016;138:10374–10377. [DOI] [PubMed] [Google Scholar]
- 85.Palermo G, Stenta M, Cavalli A, et al. Molecular simulations highlight the role of metals in catalysis and inhibition of type II topoisomerase. J Chem Theory Comput. 2013;9:857–862. [DOI] [PubMed] [Google Scholar]
- 86.Blomberg MRA, Borowski T, Himo F, et al. Quantum chemical studies of mechanisms for metalloenzymes. Chem Rev. 2014;114:3601–3658. [DOI] [PubMed] [Google Scholar]
- 87.Cao L, Ryde U. N2H2 binding to the nitrogenase FeMo cluster studied by QM/MM methods. J Biol Inorg Chem. 2020;25:521–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang B, Cao Z, Rovira C, et al. Fenton-derived OH radicals enable the MPnS enzyme to convert 2-hydroxyethylphosphonate to methylphosphonate: insights from Ab Initio QM/MM MD simulations. J Am Chem Soc. 2019;23:9284–9291. [DOI] [PubMed] [Google Scholar]
- 89.Wang B, Walton PH, Rovira C. Molecular mechanisms of oxygen activation and hydrogen peroxide formation in lytic polysaccharide monooxygenases. ACS Catal. 2019;9:4958–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Parrinello M, Andreoni W, Curioni A. CPMD. Armonk, New York and Stuttgart, Germany: IBM Corporation and Max-Planck Institut; 2000. [Google Scholar]
- 91.Kühne TD, Iannuzzi M, Del Ben M, et al. CP2K: an electronic structure and molecular dynamics software package - Quickstep: efficient and accurate electronic structure calculations. J Chem Phys. 2020;152:194103. [DOI] [PubMed] [Google Scholar]
- 92.Palermo G, Sugita Y, Wriggers W, et al. Frontiers in cryoEM modeling. J Chem Inf Model. 2019;59:3091–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Borisek J, Saltalamacchia A, Spinello A, et al. Exploiting cryo-EM structural information and all-atom simulations to decrypt the molecular mechanism of splicing modulators. J Chem Inf Model. 2019;60:2510–2521. [DOI] [PubMed] [Google Scholar]
- 94.Borišek J, Magistrato A. All-atom simulations decrypt the molecular terms of RNA catalysis in the exon-ligation step of the spliceosome. ACS Catal. 2020;10:5328–5334. [Google Scholar]; •• This study reports one of the largest QM/MM study of a protein directed metallo-ribozyme such as the splicesome
- 95.Structure Palermo G. and dynamics of the CRISPR-Cas9 catalytic complex. J Chem Inf Model. 2019;59:2394–2406. [DOI] [PubMed] [Google Scholar]
- 96.Casalino L, Nierzwicki Ł, Jinek M, et al. Catalytic mechanism of non-target DNA cleavage in CRISPR-Cas9 revealed by Ab Initio molecular dynamics. ACS Catal. 2020;10:13596–13605. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study reports the first QM/MM study of CRISPR-Cas9, another example of the largest metalloproteins studied through ab-initio MD
- 97.Casalino L, Palermo G, Spinello A, et al. All-atom simulations disentangle the functional dynamics underlying gene maturation in the intron lariat spliceosome. Proc Natl Acad Sci USA. 2018;115:6584–6589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Shaik S, Cohen S, Wang Y, et al. P450 enzymes: their structure, reactivity, and selectivity modeled by QM/MM calculations. Chem Rev. 2010;110:949–1017. [DOI] [PubMed] [Google Scholar]; •• This review reports an extensive description of the catalytic mechanism of CYP450
- 99.Ali HS, Henchman RH, de Visser SP. Cross-linking of aromatic phenolate groups by cytochrome P450 enzymes: a model for the biosynthesis of vancomycin by OxyB. Org Biomol Chem. 2020;18:4610–4618. [DOI] [PubMed] [Google Scholar]
- 100.Spinello A, Ritacco I, Magistrato A. Recent advances in computational design of potent aromatase inhibitors: open-eye on endocrine-resistant breast cancers. Expert Opin Drug Discov. 2019;14:1065–1076. [DOI] [PubMed] [Google Scholar]
- 101.Sgrignani J, Magistrato A. Influence of the membrane lipophilic environment on the structure and on the substrate access/egress routes of the human aromatase enzyme. A computational study. J Chem Inf Model. 2012;52:1595–1606. [DOI] [PubMed] [Google Scholar]
- 102.Šrejber M, Navrátilová V, Paloncýová M, et al. Membrane-attached mammalian cytochromes P450: an overview of the membrane’s effects on structure, drug binding, and interactions with redox partners. J Inorg. Biochem 2018;183:117–136. [DOI] [PubMed] [Google Scholar]
- 103.Ritacco I, Saltalamacchia A, Spinello A, et al. All-atom simulations disclose how cytochrome reductase reshapes the substrate access/egress routes of its partner CYP450s. J Phys Chem Lett. 2020;11:1189–1193. [DOI] [PubMed] [Google Scholar]
- 104.Ritacco I, Spinello A, Ippoliti E, et al. The post-translational regulation of CYP450s metabolism as revealed by all-atoms simulations of the aromatase enzyme. J Chem Inf Model. 2019;59:2930–2940. [DOI] [PubMed] [Google Scholar]
- 105.Hackett JC, Brueggemeier RW, Hadad CM. The final catalytic step of cytochrome P450 aromatase: a density functional theory study. J Am Chem Soc. 2005;127:5224–5237. [DOI] [PubMed] [Google Scholar]
- 106.Sgrignani J, Iannuzzi M, Magistrato A. Role of water in the puzzling mechanism of the final aromatization step promoted by the human aromatase enzyme. insights from QM/MM MD simulations. J Chem Inf Model. 2015;55:2218–2226. [DOI] [PubMed] [Google Scholar]
- 107.Xu K, Wang Y, Hirao H. Estrogen formation via H-abstraction from the O-H bond of gem-diol by compound I in the reaction of CYP19A1: mechanistic scenario derived from multiscale QM/MM calculations. ACS Catal. 2015;5:4175–4179. [Google Scholar]; • A recent mechanistic study on the steroidogenic human aromatase enzyme
- 108.Spinello A, Pavlin M, Casalino L, et al. A dehydrogenase dual hydrogen abstraction mechanism promotes estrogen biosynthesis: can we expand the functional annotation of the aromatase enzyme? Chem Eur J. 2018;24:10840–10849. [DOI] [PubMed] [Google Scholar]; •• A recent and complete mechanistic study of the catalytic mechanism of human aromatase
- 109.Ferreira Almeida C, Oliveira A, Ramos MJ, et al. Estrogen receptor-positive (ER+) breast cancer treatment: are multi-target compounds the next promising approach? Biochem Pharmacol. 2020;177:113989. [DOI] [PubMed] [Google Scholar]
- 110.Ghosh D, Lo J, Egbuta C. Recent progress in the discovery of next generation inhibitors of aromatase from the structure-function perspective. J Med Chem. 2016;59:5131–5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Viciano I, Marti S. Theoretical study of the mechanism of exemestane hydroxylation catalyzed by human aromatase enzyme. J Phys Chem B. 2016;120:3331–3343. [DOI] [PubMed] [Google Scholar]
- 112.Magistrato A, Sgrignani J, Krause R, et al. Single or multiple access channels to the CYP450s active site? An answer from free energy simulations of the human aromatase enzyme. J Phys Chem Lett 2017;8:2036–2042. [DOI] [PubMed] [Google Scholar]; • Study of the binding of Fe-targeting inhibitor and its dissociation free energy cost from the aromatase enzyme
- 113.Caciolla J, Spinello A, Martini S, et al. Targeting orthosteric and allosteric pockets of aromatase via dual-mode novel azole inhibitors. ACS Med Chem Lett. 2020;11:732–739. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• A recent study elucidating the difficulties of describing inhibitor-iron interactions in drug design
- 114.Sgrignani J, Bon M, Colombo G, et al. Computational approaches elucidate the allosteric mechanism of human aromatase inhibition: a novel possible route to Small-molecule regulation of CYP450s activities? J Chem Inf Model. 2014;54:2856–2868. [DOI] [PubMed] [Google Scholar]
- 115.Spinello A, Martini S, Berti F, et al. Rational design of allosteric modulators of the aromatase enzyme: an unprecedented therapeutic strategy to fight breast cancer. Eur J Med Chem. 2019;168:253–262. [DOI] [PubMed] [Google Scholar]
- 116.Bostrom J, Norrby PO, Liljefors T. Conformational energy penalties of protein-bound ligands. J Comput Aided Mol Des. 1998;12:383–396. [DOI] [PubMed] [Google Scholar]
- 117.Bonomo S, Jorgensen FS, Olsen L. Mechanism of cytochrome P450 17A1-catalyzed hydroxylase and lyase reactions. J Chem Inf Model. 2017;57:1123–1133. [DOI] [PubMed] [Google Scholar]
- 118.Gomez L, Kovac JR, Lamb DJ. CYP17A1 inhibitors in castration-resistant prostate cancer. Steroids. 2015;95:80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Khemlina G, Ikeda S, Kurzrock R. Molecular landscape of prostate cancer: implications for current clinical trials. Cancer Treat Rev. 2015;41:761–766. [DOI] [PubMed] [Google Scholar]
- 120.Bonomo S, Hansen CH, Petrunak EM, et al. Promising tools in prostate cancer research: selective non-steroidal cytochrome P450 17A1 inhibitors. Sci Rep. 2016;6:29468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482:116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Larsen M, Hansen CH, Rasmussen TB, et al. Structure-based optimisation of non-steroidal cytochrome P450 17A1 inhibitors. Chem Commun. 2017;53:3118–3121. [DOI] [PubMed] [Google Scholar]
- 123.Vasilevskaya T, Khrenova MG, Nemukhin AV, et al. Mechanism of proteolysis in matrix metalloproteinase-2 revealed by QM/MM modeling. J Comput Chem. 2015;36:1621–1630. [DOI] [PubMed] [Google Scholar]
- 124.Khandelwal A, Lukacova V, Comez D, et al. A combination of docking, QM/MM methods, and MD simulation for binding affinity estimation of metalloprotein ligands. J Med Chem. 2005;48:5437–5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Piazzetta P, Marino T, Russo N. Promiscuous ability of human carbonic anhydrase: QM and QM/MM investigation of carbon dioxide and carbodiimide hydration. Inorg Chem. 2014;53:3488–3493. [DOI] [PubMed] [Google Scholar]
- 126.Samanta PN, Das KK. Prediction of binding modes and affinities of 4-substituted-2,3,5,6-tetrafluorobenzenesulfonamide inhibitors to the carbonic anhydrase receptor by docking and ONIOM calculations. J Mol Graph Mod. 2016;63:38–48. [DOI] [PubMed] [Google Scholar]
- 127.Liao RZ, Yu JG, Himo F. Phosphate mono- and diesterase activities of the trinuclear zinc enzyme nuclease P1—insights from quantum chemical calculations. Inorg Chem. 2010;49:6883–6888. [DOI] [PubMed] [Google Scholar]
- 128.Swale C, Bougdour A, Gnahoui-David A, et al. Metal-captured inhibition of pre-mRNA processing activity by CPSF3 controls cryptosporidium infection. Sci Transl Med. 2019;11:eaax7161. [DOI] [PubMed] [Google Scholar]
- 129.Park H, Brothers EN, Merz KM Jr. Hybrid QM/MM and DFT investigations of the catalytic mechanism and inhibition of the dinuclear zinc metallo-beta-lactamase CcrA from Bacteroides fragilis. J Am Chem Soc. 2005;127:4232–4241. [DOI] [PubMed] [Google Scholar]
- 130.Dal Peraro M, Vila AJ, Carloni P, et al. Role of zinc content on the catalytic efficiency of B1 metallo beta-lactamases. J Am Chem Soc. 2007;129:2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Zhu K, Lu J, Liang Z, et al. A quantum mechanics/molecular mechanics study on the hydrolysis mechanism of New Delhi metallo-beta-lactamase-1. J Comput Aided Mol Des. 2013;27:247–256. [DOI] [PubMed] [Google Scholar]
- 132.Simona F, Magistrato A, Dal Peraro M, et al. Common mechanistic features among metallo-beta-lactamases: a computational study of Aeromonas hydrophila CphA enzyme. J Biol Chem. 2009;284:28164–28171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Lisa MN, Palacios AR, Aitha M, et al. A general reaction mechanism for carbapenem hydrolysis by mononuclear and binuclear metallo-beta-lactamases. Nat Commun. 2017;8:538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hinchliffe P, Gonzalez MM, Mojica MF, et al. Cross-class metallo-beta-lactamase inhibition by bisthiazolidines reveals multiple binding modes. Proc Natl Acad Sci USA. 2016;113:E3745–E3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nowak-Sliwinska P, van Beijnum JR, Casini A, et al. Organometallic ruthenium(II) arene compounds with antiangiogenic activity. J Med Chem. 2011;54:3895–3902. [DOI] [PubMed] [Google Scholar]
- 136.Scolaro C, Bergamo A, Brescacin L, et al. In vitro and in vivo evaluation of ruthenium(II)-arene PTA complexes. J Med Chem. 2005;48:4161–4171. [DOI] [PubMed] [Google Scholar]
- 137.Wolters DA, Stefanopoulou M, Dyson PJ, et al. Combination of metallomics and proteomics to study the effects of the metallo-drug RAPTA-T on human cancer cells. Metallomics. 2012;4: 1185–1196. 201. [DOI] [PubMed] [Google Scholar]
- 138.Adhireksan Z, Davey GE, Campomanes P, et al. Ligand substitutions between ruthenium-cymene compounds can control protein versus DNA targeting and anticancer activity. Nat Commun. 2014;5:3462.. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This article report a combined experimental and computational study rationalizing the preferential binding mode of ruthenium ligand
- 139.Wu B, Ong MS, Groessl M, et al. A ruthenium antimetastasis agent forms specific histone protein adducts in the nucleosome core. Chem Eur J. 2011;17:3562–3566. [DOI] [PubMed] [Google Scholar]
- 140.McGinty RK, Tan S. Nucleosome structure and function. Chem Rev. 2015;115:2255–2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Novakova O, Chen HM, Vrana O, et al. DNA interactions of mono-functional organometallic ruthenium(II) antitumor complexes in cell-free media. Biochemistry. 2003;42:11544–11554. [DOI] [PubMed] [Google Scholar]
- 142.Novakova O, Kasparkova J, Bursova V, et al. Conformation of DNA modified by monofunctional Ru(II) arene complexes: recognition by DNA binding proteins and repair. Relationship to cytotoxicity. Chem Biol. 2005;12:121–129. [DOI] [PubMed] [Google Scholar]
- 143.Noffke AL, Habtemariam A, Pizarro AM, et al. Designing organometallic compounds for catalysis and therapy. Chem Commun. 2012;48:5219–5246. [DOI] [PubMed] [Google Scholar]
- 144.Davey CA, Richmond TJ. DNA-dependent divalent cation binding in the nucleosome core particle. P Natl Acad Sci USA. 2002;99:11169–11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ma Z, Palermo G, Adhireksan Z, et al. An organometallic compound displays a unique one-stranded intercalation mode that is DNA topology-dependent. Angew Chem Int Ed. 2016;128:7441–7444.. [DOI] [PubMed] [Google Scholar]; •• This elucidate the topological dependence of the binding mode of a metal-based ligand
- 146.Nobili S, Mini E, Landini I, et al. Gold compounds as anticancer agents: chemistry, cellular pharmacology, and preclinical studies. Med Res Rev. 2010;30:550–580. [DOI] [PubMed] [Google Scholar]
- 147.Tejman-Yarden N, Miyamoto Y, Leitsch D, et al. A reprofiled drug, auranofin, is effective against metronidazole-resistant giardia lamblia. Antimicrob Agents Ch. 2013;57:2029–2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Vergara E, Casini A, Sorrentino F, et al. Anticancer Therapeutics that target selenoenzymes: synthesis, characterization, in vitro cytotoxi-city, and thioredoxin reductase inhibition of a series of gold(i) complexes containing hydrophilic phosphine ligands. Chemmed chem. 2010;5:96–102. [DOI] [PubMed] [Google Scholar]
- 149.Adhireksan Z, Palermo G, Riedel T, et al. Allosteric cross-talk in chromatin can mediate drug-drug synergy. Nat Commun. 2017;8:14860.. [DOI] [PMC free article] [PubMed] [Google Scholar]; •• This study elucidates the mechanism allosteric cross-talk of two metallo drugs
- 150.Bowerman S, Hickok RJ, Wereszczynski J. Unique dynamics in asymmetric macroH2A-H2A hybrid nucleosomes result in increased complex stability. J Phys Chem B. 2019;123:419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bowerman S, Wereszczynski J. Effects of macroh2a and H2A.Z on nucleosome dynamics as elucidated by molecular dynamics simulations. Biophys J. 2016;110:327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Batchelor LK, De Falco L, von Erlach T, et al. Crosslinking allosteric sites on the nucleosome. Angew Chem Int Ed Engl. 2019;58:15660–15664. [DOI] [PubMed] [Google Scholar]
- 153.Serna A, Galan-Cobo A, Rodrigues C, et al. Functional inhibition of aquaporin-3 with a gold-based compound induces blockage of cell proliferation. J Cell Physiol. 2014;229:1787–1801. [DOI] [PubMed] [Google Scholar]
- 154.Spinello A, de Almeida A, Casini A, et al. The inhibition of glycerol permeation through aquaglyceroporin-3 induced by mercury (II): A molecular dynamics study. J Inorg Biochem. 2016;60:78–84. [DOI] [PubMed] [Google Scholar]
- 155.Graziani V, Marrone A, Re N, et al. Multi-level theoretical study to disclose the binding mechanisms of gold (III)–bipyridyl compounds as selective aquaglyceroporin inhibitors. Chem Eur J. 2017;23:138 02–13813. [DOI] [PubMed] [Google Scholar]
- 156.Sgrignani J, Casalino L, Doro F, et al. Can multiscale simulations unravel the function of metallo-enzymes to improve knowledge-based drug discovery? Future Med Chem. 2019;11:771–791. [DOI] [PubMed] [Google Scholar]
- 157.Lavecchia A Machine-learning approaches in drug discovery: methods and applications. Drug Discovery Today. 2015;20:318–331. [DOI] [PubMed] [Google Scholar]; • This review elucidates the role of machine learning in drug discovery
- 158.Mater AC, Coote ML. Deep learning in chemistry. J Chem Info Mod. 2019;59:2545–2559. [DOI] [PubMed] [Google Scholar]; • This review elucidates the role of machine learning in chemistry
- 159.Janet JP, Kulik HJ. Resolving Transition metal chemical space: feature selection for machine learning and structure-property relationships. J Phys Chem A. 2017;121:8939–8954. [DOI] [PubMed] [Google Scholar]
- 160.Nandy A, Duan C, Janet JP, et al. Strategies and software for machine learning accelerated discovery in transition metal chemistry. Ind Eng Chem Res. 2018;57:13973–13986. [Google Scholar]
- 161.Kostelnik TI, Orvig C. Radioactive Main Group and Rare Earth Metals for Imaging and Therapy. Chem Rev. 2019;119:902–956. [DOI] [PubMed] [Google Scholar]
- 162.Boros E, Packard AB. Radioactive transition metals for imaging and therapy. Chem Rev. 2019;119:870–901. [DOI] [PubMed] [Google Scholar]; • This review report the applications of transition metal in radio imaging and therapy