

Abstract
Glypican-3 (GPC3) is a well-characterized hepatocellular carcinoma (HCC)-associated antigen, yet anti-GPC3 therapies have achieved only minimal clinical progress. CD47 is a ubiquitously expressed innate immune checkpoint that promotes evasion of tumors from immune surveillance. Given both the specific expression of GPC3 in HCC and the known phagocytosis inhibitory effect of CD47 in liver cancer, we hypothesized that a bispecific antibody (BsAb) that co-engages with GPC3 and CD47 may offer excellent antitumor efficacy with minimal toxicity. Here, we generated a novel BsAb: GPC3/CD47 biAb. With the use of both in vitro and in vivo assays, we found that GPC3/CD47 biAb exerts strong antitumor activity preferentially against dual antigen-expressing tumor cells. In hCD47/human signal regulatory protein alpha (hCD47/hSIRPα) humanized mice, GPC3/CD47 biAb had an extended serum half-life without causing systemic toxicity. Importantly, GPC3/CD47 biAb induced enhanced Fc-mediated effector functions to dual antigen-expressing HCC cells in vitro, and both macrophages and neutrophils are required for its strong efficacy against xenograft HCC tumors. Notably, GPC3/CD47 biAb outperformed monotherapies and a combination therapy with anti-CD47 and anti-GPC3 monoclonal antibodies (mAbs) in a xenograft HCC model. Our study illustrates a strategy for improving HCC treatment by boosting innate immune responses and presents new insights to inform antibody design for the future development of innovative immune therapies.
Keywords: hepatocellular carcinoma, GPC3, CD47, bispecific antibody, cancer immunotherapy
Graphical abstract
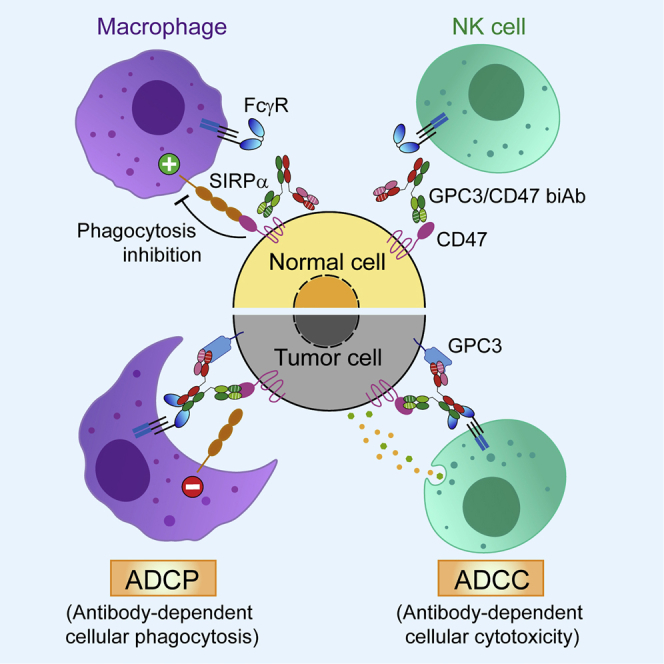
Du and colleagues generated a bispecific antibody that co-engages hepatocellular carcinoma (HCC)-associated GPC3 and CD47, an innate-immune checkpoint. This bispecific antibody promotes macrophage phagocytosis by blocking CD47’s “don’t-eat-me” signal. Experiments with dual antigen-expressing HCC mouse models showed that the bispecific antibody enhances innate-immune responses and outperforms a two-antibody combination therapy.
Introduction
Hepatocellular carcinoma (HCC) is the sixth-most common carcinoma worldwide and the third leading cause of cancer-related death.1 For patients with de novo or recurrent advanced HCC, the treatment options are limited. Currently, only two multikinase inhibitors—sorafenib and lenvatinib—have been approved by the US Food and Drug Administration (FDA) as first-line systemic treatments for advanced HCC.2 However, their treatment benefits and response rates are often modest. Tumor antigen-targeting antibody-based and immune-modulating antibody-based immunotherapies represent emerging approaches that may improve HCC treatment outcomes.
Glypican-3 (GPC3) is one of the best-characterized HCC-associated antigens. It is a member of the GPC family; these are heparin sulfate proteoglycans anchored to cell membranes by a glycosylphosphatidylinositol (GPI) anchor.3 Many studies have confirmed that GPC3 is specifically upregulated in HCC, with minimal or no expression in normal (and even cirrhotic) liver tissues,4 making it an excellent tumor-specific target for an antibody therapy. However, a humanized anti-GPC3 antibody, GC33, did not confer a clinical benefit in a phase II clinical trial as a monotherapy, potentially owing to suboptimal dosing and/or an unfavorable immune environment.5
One strategy for modulating immune responses is use of T cell-engaging bispecific antibodies (T-BsAbs) that combine two different antigen-binding specificities within a single molecule; such antibodies engage T cells with one arm and engage a tumor antigen with the other arm. There are T-BsAbs that redirect T cells to HCC cells (by binding to both an epitope of CD3 on T cells and to GPC3 on tumor cells) that are currently under preclinical or clinical development.6 In addition to harnessing T cell immune responses to fight malignant cells, increasing evidence is underscoring the impacts of the innate immune system in cancer treatment. For example, studies have reported macrophage recruitment in GPC3-positive HCC patients.7 Phagocytosis of cancer cells is the major mechanism through which macrophages mediate antitumor activity. Therefore, we speculated that harnessing macrophages may be a promising strategy to improve antitumor efficacy against this type of HCC.
CD47 is an inhibitory innate immune checkpoint.8 It interacts with its receptor signal regulatory protein alpha (SIRPα) on myeloid cells (especially on macrophages) and confers a “don’t eat me” signal so that cancer cells can evade immune surveillance.9 Therefore, blockade of the interaction between CD47 and SIRPα with antibodies targeting CD47 represents a promising strategy to enhance the phagocytic clearance of cancer cells. The expression of CD47 is upregulated in many solid tumors, including HCC,10,11 and anti-CD47 antibodies can inhibit HCC tumor growth.12,13 However, CD47-targeted antibodies that are currently under clinical development are cleared rapidly and lead to hemotoxicity, owing largely to the expression of CD47 on normal cells and especially on red blood cells (RBCs).14
Given the specific expression of GPC3 in HCC and considering the problematically widespread expression of CD47, we here speculated that a BsAb, which could co-engage with GPC3 and CD47, may offer both enhanced antitumor efficacy and reduced toxicity. After generating and demonstrating the basic specificity and binding performance of our BsAb (GPC3/CD47 biAb), we conducted a series of in vitro and in vivo studies to assess its safety and antitumor performance. With the use of the hCD47/hSIRPα humanized mouse model that we established, GPC3/CD47 biAb demonstrated reduced systemic toxicity and extended serum half-life (t1/2). Importantly, experiments with a xenograft HCC model showed that GPC3/CD47 biAb exhibited Fc-dependent antitumor efficacy that outperformed both monotherapies and a two-antibody combination therapy. We also confirmed that both macrophages and neutrophils are required for GPC3/CD47 biAb’s antitumor effects. These results together illustrate a BsAb strategy to safely boost innate immune responses and confer efficient antitumor activity against HCC.
Results
Generation of BsAbs and demonstration of GPC3/CD47 biAb’s greatly improved specificity for dual antigen-expressing cells
After characterizing the binding affinity, epitopes, and antitumor activities of a variety of anti-GPC3 or anti-CD47 monoclonal antibodies (mAbs), we adopted two antibodies—the GC3315 and the hCD47-targeting mAb (BC18)—to generate the GPC3/CD47 biAb (Figure S1A). BC18, obtained through screening of our phage display naive antibody library (single-chain fragment of variable domain [scFv]),16 effectively blocks the interaction between CD47 and its receptor SIRPα (Figure S1B). Both GC33 and BC18 have nanomolar range affinities for their own antigens, as measured by surface plasmon resonance (SPR) analysis (Figure S1C). Another two BsAbs (control [Ctrl]/CD47 biAb or GPC3/Ctrl biAb), respectively, for CD47 or GPC3 were generated as Ctrls (Figure S1A). All biAbs were produced via transient transfection of HEK293F cells and were then purified via protein A affinity chromatography (Figure S1D). Subsequently, SPR-based analysis demonstrated the dual specificity of the purified and properly assembled GPC3/CD47 biAb (Figure 1A) and confirmed the respective specificities of the two Ctrl biAbs (Figure S1E).
Figure 1.

Binding ability and selectivity analysis of the GPC3/CD47 biAb
(A) Binding curve of the GPC3/CD47 biAb with the GPC3 or CD47 antigens as measured by SPR-based bridge assays (depicted in the schematic). The blue and magenta circles, respectively, denote GPC3 and CD47. R1: binding response of the GPC3/CD47 biAb to the immobilized GPC3. R2: binding response of CD47 to the GPC3/CD47 biAb. (B) The binding ability of each antibody (Ab) to Raji-WT and Raji-GPC3H cells was detected by flow cytometry. (C) The binding selectivity of each antibody was measured by competitive flow cytometry. Raji-WT cells (red contour) mixed with Raji-GPC3H cells (black contour) at a ratio of 1:1 were incubated with 0.2 μg/mL of each antibody prior to staining with FITC-conjugated anti-human (h)IgG secondary antibody. The binding selectivity was determined based on the fluorescence intensity for FITC in each cell population. (D) GPC3/CD47 biAb competes with SIRPα for binding to Raji-GPC3H cells, exerting stronger competitive ability than Raji-WT. Raji-WT or Raji-GPC3H cells were incubated with 50 nM of biotinylated SIRPα-mFc fusion protein in the presence of anti-CD47 mAb, control (Ctrl)/CD47 biAb, and GPC3/CD47 biAb (10 μg/mL). The binding of SIRPα to each cell type was assessed based on the fluorescence intensity of FITC-conjugated streptavidin. See also Figure S1.
The widespread expression of CD47 on normal tissue cells may result in decreased antibody bioavailability to tumor cells, thus jeopardizing the treatment efficacy of anti-CD47 antibodies (the so-called “antigen sink” phenomenon).17 We used an engineered GPC3+CD47+ double-positive cell (Raji-GPC3H) (Figure S1F) to examine if GPC3/CD47 biAb possesses selective binding properties for dual antigen-expressing cells as designed. Supporting that our dual-targeting strategy confers improved specificity, we found that GPC3/CD47 biAb had much stronger binding for double-positive Raji-GPC3H cells than for single antigen-expressing Raji-wild-type (WT) cells (Figure 1B). Furthermore, we designed an in vitro experiment to mimic an antigen sink scenario where both double-positive and single-positive cells exist. Specifically, we labeled Raji cells with deep red dye and mixed them with Raji-GPC3H cells at a 1:1 ratio prior to incubation with each antibody. During fluorescence-activated cell sorting (FACS) analysis, cells were initially gated according to cell type, followed by analysis of antibody binding. GPC3/CD47 biAb exerts preferential binding for Raji-GPC3H over Raji cells; no such specificity was evident for the aforementioned Ctrl anti-CD47 mAb or the Ctrl/CD47 biAb (Figure 1C).
We next tested the SIRPα/CD47 blockade activity of GPC3/CD47 biAb: competitive FACS analysis showed that compared to anti-CD47 mAb and Ctrl/CD47 biAb—which each blocked the binding of SIRPα to CD47-expressing Raji-WT cells and Raji-GPC3H cells to the same extent—GPC3/CD47 biAb exerts stronger blockade activity for Raji-GPC3H cells than for unmodified Raji cells (Figure 1D). Collectively, these results indicate that GPC3/CD47 biAb readily achieves preferential binding to, and effective SIRPα/CD47 blockade of, dual antigen-expressing tumor cells.
GPC3/CD47 biAb has a superior safety profile and extended serum t1/2 in hCD47/hSIRPα gene-modified mice compared with anti-CD47 mAb
Severe side effects represent one of the major concerns for anti-CD47 antibody treatment.17 To evaluate the hematologic safety of our GPC3/CD47 biAb, we first performed in vitro agglutination assays with hRBCs. Anti-CD47 mAb caused hemagglutination as expected, whereas GPC3/CD47 biAb did not induce hemagglutination. In line with this, no hemagglutination occurred with Ctrl/CD47 biAb, indicating that binding to CD47, with only one antibody arm, avoids induction of RBC agglutination (Figure 2A).
Figure 2.

GPC3/CD47 biAb has a good safety profile and has an extended serum half-life in hCD47/hSIRPα humanized mice
(A) Human red blood cell (hRBC) agglutination induced by anti-CD47 antibodies. Non-agglutinated RBCs sediment and form a red dot in the bottom of the well. (B−D) RBC counts, body temperature change, and pharmacokinetic analyses after a single administration for each antibody (10 mg/kg). RBC counts in untreated mice (NT) were measured for use as the baseline Ctrl value (B). Body temperature was recorded 24 h before antibody administration and at 45 min and 3, 8, and 24 h after antibody administration (C). Serum antibody concentration versus time profile is shown in (D). All data are shown as the mean ± SD. See also Figure S2.
To assess the safety profile of GPC3/CD47 biAb in vivo, we generated hCd47/hSirpα double-gene-modified mice (hCD47/hSIRPα humanized mice); specifically, the exons encoding the extracellular domains of murine Cd47 and Sirpα genes were replaced with human counterparts (Figures S2A and S2B). No obvious signs of hematologic toxicity were observed for the biAbs, whereas anti-CD47 mAb induced acute RBC depletion within 1 h in the humanized mice (Figures 2B and S2C). Additionally, anti-CD47 mAb caused a significant drop in body temperature; no such drop occurred upon treatment with either the GPC3/CD47 or Ctrl/CD47 biAbs (Figure 2C).
CD47-mediated rapid drug clearance is also a concern for clinical applications.17 We therefore performed a single-dose pharmacokinetic (PK) study with the aforementioned humanized mice. Similar to Ctrl/CD47 biAb, GPC3/CD47 biAb showed an extended serum t1/2 (∼13.4 days) compared to the parental anti-CD47 mAb (t1/2 ∼ 2.4 days) (Figure 2D). These extensions likely result from reduced antibody binding to normal somatic cells that express CD47 (antigen sink). Collectively, these results support that GPC3/CD47 biAb is apparently safer than anti-CD47 mAb and has a favorable PK profile in vivo.
GPC3/CD47 biAb exerts enhanced Fc-mediated functions and selective growth suppression against dual antigen-expressing tumors
Antibody-dependent cellular cytotoxicity (ADCC) and antibody-dependent cellular phagocytosis (ADCP) mediated by the Fc domain are two functions elicited by antibodies during cancer therapy.18 In light of our finding that GPC3/CD47 biAb can preferentially bind to dual antigen-expressing tumor cells (Figure 1C), we investigated GPC3/CD47 biAb’s ability to induce ADCC and ADCP against Raji-GPC3H cells. For the ADCC assays, we used a reporter system in which engineered Jurkat T lymphocyte cells were used as effector cells (Figure S3A). We also knocked out (KO) CD47 expression to overcome a background toxicity issue caused by CD47 expression in Jurkat cells (Jurkat-CD16A-CD47KO) (Figures S3B and S3C). Compared to anti-CD47 mAb, which induced equally strong ADCC against Raji-GPC3H and Raji cells, GPC3/CD47 biAb induced stronger ADCC against Raji-GPC3H cells than Raji cells, doing so in a dose-dependent manner (Figure 3A).
Figure 3.

GPC3/CD47 biAb is highly efficacious against dual antigen-expressing tumor cells both in vitro and in vivo
(A) GPC3/CD47 biAb induced a stronger ADCC effector function against Raji-GPC3H cells than against Raji-WT cells. ADCC activity was measured using a bioluminescence-based reporter system. This experiment was performed in triplicate with Jurkat-CD16A-CD47KO cells at an effector:target (E:T) ratio of 6:1. Relative luminescence units (RLU) were plotted against the log of antibody concentration. (B) GPC3/CD47 biAb induced specific phagocytosis of Raji-GPC3H cells. Raji-WT (red) and Raji-GPC3H (green) target cells were initially mixed in a 1:1 ratio before being combined with mouse effector macrophages (BMDMs, white) at an E:T ratio of 1:2 in the presence of the indicated antibodies. The fluorescently labeled tumor cells engulfed by the mouse BMDMs were evaluated via fluorescence microscopy (scale bar, 50 μm; triplicate measurement), and the phagocytosis percentage was determined by calculating the number of engulfed cells per 100 total macrophages. The differences in phagocytosis percentage between the two types of target cells were analyzed with Holm-Sidak multiple t tests (∗∗p < 0.01). (C) Schematic diagram illustrating tumor inoculation (subcutaneously [s.c.]) and antibody treatment (i.p.) schedules in a mouse model with bilateral xenograft tumors (Raji-WT and Raji-GPC3H cells). Treatment began 8 days after tumor cell inoculation (at which point, both tumors had reached approximately 80–200 mm3 in size). (D) Growth curves of the bilateral Raji-WT and Raji-GPC3H tumors for each treatment group (n = 5/group). Tumor volumes were measured by caliper, and the data are shown as the mean ± SEM (∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 2-way ANOVA followed by Tukey’s multiple comparisons test). See also Figure S3.
We then analyzed biAb-induced ADCP of Raji-GPC3H cells upon co-incubation with macrophages in vitro. In this experiment, bone marrow-derived macrophages (BMDMs) expressing hSIRPα from the humanized mice were used as effector cells, whereas a mixture of Raji-GPC3H and Raji cells was used as target cells. Fluorescence microscopy and quantification of phagocytosis showed that GPC3/CD47 biAb induced preferential phagocytosis of Raji-GPC3H cells (Figure 3B). In contrast, the anti-CD47 mAb exhibited no selectivity: it induced phagocytosis of both types of cells.
To examine the antitumor efficacy of GPC3/CD47 biAb against dual antigen-expressing tumors during in vivo treatment, we used a bilateral mouse model wherein Raji or Raji-GPC3H cells were, respectively, implanted in the right or left flank of non-obese diabetic (NOD)-severe combined immunodeficiency (SCID) mice (Figure 3C). Encouragingly, and in contrast to both anti-CD47 mAb and Ctrl/CD47 biAb—which each suppressed Raji-GPC3H and Raji tumor progression at similar levels—GPC3/CD47 biAb’s tumor-suppressing effects were obviously more pronounced for Raji-GPC3H than for Raji cells (Figure 3D). Anti-GPC3 mAb did not suppress Raji-GPC3H tumor progression, likely owing to its weak effector functions. Collectively, these in vitro and in vivo results show that GPC3/CD47 biAb exerts enhanced Fc-mediated functions and selective growth suppression against dual antigen-expressing tumors.
GPC3/CD47 biAb outperforms monotherapies and an anti-CD47 and anti-GPC3 mAb combination therapy in a xenograft HCC model
To test the performance of GPC3/CD47 biAb on HCC progression in vivo, we used a GPC3 and CD47 double-positive hHCC cell line (Hep3B) in a xenograft mouse model. After confirming that Hep3B cells express both GPC3 and CD47 at moderate levels (Figure S3D), we established a Hep3B-Luc23 cell line stably expressing luciferase to enable measurement of tumor growth using in vivo bioluminescence imaging.19 NOD-SCID mice were subcutaneously injected with these Hep3B-Luc23 cells and were then randomized into four groups with similar mean tumor bioluminescence intensities prior to receiving antibody treatment twice each week for 3 weeks.
GPC3/CD47 biAb conferred significantly enhanced tumor suppression as compared to either anti-GPC3 mAb or anti-CD47 mAb, although it should be noted that each of these mAb monotherapies also inhibited Hep3B tumor growth. Bioluminescence imaging revealed complete eradication of the tumors in three of the five GPC3/CD47 biAb group mice, whereas no tumor eradication was detected for mice of the mAbs monotherapy groups (Figures 4A−4C). With the use of a similar dosing regimen, we also compared the effects of each antibody on the overall survival of tumor-bearing mice. Although both anti-CD47 mAb and anti-GPC3 mAb significantly increased the overall survival compared to PBS Ctrls, all mice eventually died due to tumor progression. In contrast, GPC3/CD47 biAb elicited robust antitumor responses, resulting in complete response (CR) in 50% of mice (3 out of 6) for 2 months after the end of the treatment (Figure S4A). GPC3/CD47 biAb treatment extended median survival to 88 days compared to a median survival of 65 days in the anti-CD47 mAb group and 68.5 days in the anti-GPC3 mAb group (Figure S4B). We next compared the antitumor performance of GPC3/CD47 biAb against a combination therapy comprising anti-GPC3 mAb and anti-CD47 mAb. The monitoring of tumor growth over time by measuring tumor volumes and bioluminescence intensities showed that GPC3/CD47 biAb conferred superior antitumor effects compared to the combination therapy (Figures 4D−4F). Taken together, these results provide a powerful demonstration of the antitumor activity of GPC3/CD47 biAb against GPC3 and CD47 double-positive HCC tumors.
Figure 4.

GPC3/CD47 biAb exhibits excellent antitumor activity in a hHCC xenograft model
(A−C) Hep3B-Luc23 cells were subcutaneously inoculated into NOD-SCID mice. Mice were divided into four groups (n = 5/group) with similar mean tumor bioluminescence intensities, followed by treatment with PBS (Ctrl) or the indicated antibodies (10 mg/kg) two times per week for 3 weeks. Tumor growth was measured by bioluminescence (A and B) and by caliper (C). (D−F) Tumor-bearing mice were prepared and divided into three groups (n = 5/group) as described in (A). These mice were treated with PBS, GPC3/CD47 biAb (10 mg/kg), or a combination of anti-GPC3 mAb and anti-CD47 mAb (5 mg/kg for each). Tumor growth was measured by bioluminescence (D and E) and by caliper (F). All tumor volumes are shown as the mean ± SEM (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 2-way ANOVA followed by Tukey’s multiple comparisons test). See also Figure S4.
Macrophages and neutrophils are involved in GPC3/CD47 biAb’s Fc-dependent antitumor activities in vivo
To investigate GPC3/CD47 biAb’s mechanism of action for treatment of HCC, we first examined Fc-mediated effector functions against Hep3B cells. In vitro ADCP experiments showed that GPC3/CD47 biAb induced significant phagocytosis of Hep3B cells. We also found that anti-GPC3 mAb did not induce phagocytosis of Hep3B cells, whereas genetic KO of CD47 from Hep3B cells (Hep3B-CD47KO) restored the phagocytic activity of this anti-GPC3 mAb (Figures 5A and 5B), a finding that confirms previous reports about CD47 expression on tumor cells conferring resistance to antibody-induced phagocytosis.20 In in vitro ADCC assays, GPC3/CD47 biAb induced dose-dependent cytotoxic effects against Hep3B cells at a level comparable to or higher than the anti-CD47 or anti-GPC3 mAbs (Figure 5C).
Figure 5.

GPC3/CD47 biAb exerts Fc-dependent antitumor activity mediated by macrophages and neutrophils
(A and B) GPC3/CD47 biAb induced significant phagocytosis of both Hep3B and Hep3B-CD47KO cells. Hep3B or Hep3B-CD47KO cells (green) were incubated with mouse BMDMs (red) in the presence of 4 μg/mL of the indicated antibodies for 2 h (scale bar, 50 μm). The E:T ratio was 2.5:1. Tumor cells engulfed by mouse BMDMs (A) and the phagocytosis percentage (B) were analyzed as described in Figure 3B. The white arrowheads indicate representative images of tumor cells engulfed by macrophages. (C) ADCC activity of each antibody against Hep3B cells. Hep3B cells were incubated with Jurkat-CD16A-CD47KO effector cells at an E:T ratio of 6:1 for 8 h. (D) In vivo antitumor activities of GPC3/CD47 biAb and its Fc variant. GPC3/CD47-DANG is an Fc variant of GPC3/CD47 biAb with abolished binding activity to all classes of FcγRs. (E) Time-lapse in vivo NIRF imaging of Hep3B tumor-bearing mice. The tumors are indicated with white circles. (F) Representative ex vivo NIRF images of dissected organs (top) and the organ-to-tumor average radiant fluorescence ratio at 48 h after antibody administration (bottom). The ratio differences between the two groups were analyzed with Holm-Sidak multiple t tests (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001). (G) In vivo tumor growth after GPC3/CD47 biAb treatment with or without the depletion of either macrophage (clodronate liposome) or neutrophils (anti-Ly6G). All tumor volumes were measured by caliper and are shown as the mean ± SEM (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001; 2-way ANOVA followed by Tukey’s multiple comparisons test). See also Figures S5 and S6.
To further assess Fc-mediated effector functions in vivo, we generated a GPC3/CD47 biAb variant (GPC3/CD47-DANG) bearing two mutations (D265A and N297G) in its Fc region that are known to abolish the binding of Fc regions with all classes of FcγRs.21 Prior to conducting mouse studies, we successfully confirmed that GPC3/CD47-DANG exhibited no binding activity for mouse FcγR (mFcγRs) yet retained its capacity to bind to Hep3B cells, doing so with similar affinity as GPC3/CD47 biAb (Figures S5A and S5B). In experiments using the aforementioned Hep3B-Luc xenograft model, we found that GPC3/CD47-DANG conferred no antitumor effects (Figure 5D), indicating that Fc-mediated effector functions are required for biAb’s antitumor activity. With the use of whole-body near-infrared fluorescence (NIRF) imaging, we also assessed the distribution of GPC3/CD47 biAb in tumor-bearing mice. GPC3/CD47 biAb exhibited increased tumor accumulation over time as compared to the isotype Ctrl antibody (Figure 5E). To measure the accumulation of each antibody in different organs, the fluorescence signals from freshly dissected tumors and organs were quantified. In contrast to the apparently nonspecific distribution of the isotype Ctrl antibody, the significantly decreased organ-to-tumor fluorescence ratios in GPC3/CD47 biAb-treated mice supported its specific accumulation at the tumor site (Figure 5F).
We next sought to identify the immune cell types that contribute to the antitumor effects of GPC3/CD47 biAb. Given the immunocompromising immune background of NOD-SCID mice, we used an immune cell depletion method to assess the contribution of the model’s three major innate immune cell subsets: macrophages, neutrophils, and natural killer (NK) cells. The depletion efficiencies were validated to be over 90% for all three immune cell subsets (Figure S6A). Depletion of either macrophages or neutrophils from mice led to significant reductions in GPC3/CD47 biAb’s antitumor effects, whereas depletion of NK cells did not cause any effect (Figures 5G and S6B). This result supports that both macrophages and neutrophils functionally contribute to biAb’s antitumor effects.
In light of previous studies showing that GPC3 overexpression in HCC may recruit M2-polarized immunosuppressive macrophages—an outcome thought to limit the treatment efficacy of antibodies22,23—we further analyzed the infiltrated macrophages after GPC3/CD47 biAb treatment. Compared to the vehicle Ctrl, FACS analysis revealed that the frequency of infiltrated macrophages was significantly elevated in mice treated with GPC3/CD47 biAb (Figures S6C and S6D). This result was further supported by results from an immunofluorescence microscopy analysis (Figure S6E). Additionally, GPC3/CD47 biAb treatment also appeared to cause an increased M1-like/M2-like ratio, a finding suggesting that GPC3/CD47 biAb can somehow bias intratumoral macrophages toward a proinflammatory status (Figure S6F). Viewed together, these results from NOD-SCID xenograft tumor models establish that GPC3/CD47 biAb’s antitumor activity requires both its Fc-mediated effector functions and the involvement of macrophages and neutrophils.
Discussion
Despite numerous advances in cancer therapy in recent years, there is still a large proportion of patients with advanced HCC who do not respond to first-line treatment or to immune checkpoint inhibitors. Therefore, development of a treatment strategy that exploits distinct mechanisms would likely greatly benefit HCC patients. BsAbs represent an emerging class of antibody therapies that have been explored for treating a variety of tumor types.24 In the present study, we generated a BsAb that binds to GPC3 and CD47 to specifically target HCC tumor cells, and we assessed the PK and potential adverse effects of hCD47-targeting BsAbs in hCD47/hSIRPα double humanized mice. GPC3/CD47 biAb has an attractive t1/2 and displayed no hematological toxicity. We also exploited a bilateral mouse model to empirically establish the preferential activity of GPC3/CD47 biAb for tumor cells over normal cells. Collectively, the safety profile, long t1/2, and preferential antitumor activity of GPC3/CD47 biAb highlight its apparent advantages compared to combinational therapies that employ an anti-CD47 mAb.
Recall that our in vitro ADCP experiments showed how anti-GPC3 mAb exerts significant phagocytosis of Hep3B after CD47 KO. This result corroborates previous studies reporting that CD47 expression on tumor cells can hamper the pro-phagocytic potential of tumor antigen-targeted therapeutic antibodies.20,25 Therefore, our work illustrates how the exploitation of the CD47 blockade can add an additional pro-phagocytic antitumor response for the treatment of CD47-positive HCC. Tumor-associated macrophages (TAMs) display a continuum of different polarization states between antitumorigenic M1 and pro-tumorigenic M2 phenotypes. Previous studies showed that elevated M2 macrophage levels in tumors are associated with poor prognosis for HCC.26,27 In addition to activating macrophage phagocytosis, GPC3/CD47 biAb also improved the ratio of M1/M2 macrophages. Although it remains unknown whether such a modulatory effect of GPC3/CD47 biAb is relevant to its enhanced antitumor activity, it is plausible that this circumstance may be additionally favorable for HCC patients.
In contrast to the well-studied antitumor activities of macrophages, there is relatively limited evidence that neutrophils mediate antitumor effects. Although neutrophils, which express FcRs, represent the most abundant population of circulating cytotoxic effector cells in humans,28 their exact mechanism(s) for killing antibody-opsonized cancer cells remain elusive.29 Intriguingly, our results support that neutrophils are required for the antitumor effects of GPC3/CD47 biAb in NOD-SCID mice. Similar to our findings, previous studies have shown that selective depletion of neutrophils significantly reduces the protective activity of an anti-CD52 mAb30 and an anti-CD20 mAb.31 Blockade of the CD47-SIRPα interaction has been shown to augment antibody-mediated tumor cell killing by neutrophils.32,33 Interestingly, there are also reports of neutrophil involvement in HCC progression.34,35 Thus, it will be interesting to examine whether and how neutrophils contribute to GPC3/CD47 biAb’s antitumor effects in HCC patients.
When considering the antitumor activity of GPC3/CD47 biAb in HCC patients, hepatic NK cells cannot be ignored. These cells, which account for up to 50% of total hepatic lymphocytes,36 are involved in the inhibition of viral infection as well as liver tumorigenesis.37 It is notable that many studies have observed impaired cytotoxicity and decreased infiltration of NK cells as HCC progresses.38 This reduction in NK cells in tumors would apparently limit the antitumor efficacy of ADCC-mediating antibodies. Previous studies have demonstrated that the antitumor efficacy of ADCC-mediating therapeutic antibodies is affected by polymorphisms in FcγRIIIa (158V/F) expressed on NK cells.39 Moreover, it is notable that more than 85% of the human population are known to carry a low-affinity FcγRIIIa-158F allotype. The failure of a phase II clinical trial of anti-GPC3 mAb (GC33) in patients with advanced HCC can perhaps be attributed to suboptimal ADCC activity, and selection of patients with high-affinity FcγRIIIa allotypes or high expression levels of GPC3 and/or FcγRIIIa may promote a high ADCC effect, which could ultimately improve outcomes.5,40 Taking this into consideration and recalling GPC3/CD47 biAb’s greatly enhanced ADCC activity as compared to the parental anti-GPC3 mAb against Hep3B cells (which express moderate levels of GPC3), we anticipate that an ability of GPC3/CD47 biAb to elicit enhanced ADCC of NK cells may promote its overall efficacy for treating a broader range of HCC patients (i.e., patients having heterogeneous GPC3 expression profiles). Although the NK-dysfunctional NOD-SCID mouse model of our study precluded the unambiguous confirmation of NK cells’ involvement in GPC3/CD47 biAb’s antitumor effects, close monitoring of NK cell activity in future preclinical and clinical trials of this antibody (including combination therapies) will very likely deepen our understanding of how ADCC activity can promote NK-mediated tumor killing.
It should be emphasized that other host immune cells, such as T cells, also contribute significantly in antitumor protection. Several studies have demonstrated therapeutic efficacy of GPC3-targeting chimeric antigen receptor (CAR) T cells and T-BsAbs in HCC xenograft models,41, 42, 43 but these presently meet numerous challenges in clinical practice, especially for solid tumors. It is noteworthy that CD47-blocking antibodies can harness antigen-presenting cells (macrophages or dendritic cells) and promote tumor antigen cross-presentation to T cells, which lead to durable adaptive antitumor responses.44 Thus, we assume that our GPC3/CD47 biAb may also exploit T cell antitumor activity, and its HCC therapeutic efficacy could potentially be further maximized when deployed as a combination agent alongside other T cell-stimulating immune checkpoint inhibitors or chemotherapeutics. Future studies administering GPC3/CD47 biAb together with T cell immune checkpoint inhibitors, such as anti-PD-1 or anti-CTLA-4 mAbs in immune-competent models, are warranted to address their respective contributions.
It is interesting to note that, in addition to exhibiting much stronger antitumor activity than mAbs alone, treatment with GPC3/CD47 biAb conferred superior antitumor benefit over a combination therapy comprising two antibodies that separately recognize the GPC3 and CD47 targets. This observation supports that the binding of two antigens using a BsAb may offer improved clinical value through some form of synergism. In sum, we here developed a BsAb and demonstrated its safe and efficient antitumor activity for HCC. Ultimately, as we learn more about the specific mechanisms of GPC3/CD47 biAb in treating HCC, we may uncover more general trends that can support the development of BsAbs to boost innate immune responses for innovative therapeutic strategies to further improve cancer treatment outcomes.
Materials and methods
Cell lines
FreeStyle 293F cells were cultured following the manufacturer’s instructions (Thermo Fisher Scientific). Raji cells were from the Cell Bank of Type Culture Collection, Chinese Academy of Sciences. The Raji-GPC3H cell line (expressing hGPC3) was generated via electroporation, followed by G418 selection and single-cell clone isolation. The hHCC cell line Hep3B was generously provided by Dr. Fengmin Lu (Peking University Health Science Center, Beijing, China). The Hep3B-Luc23 cell line was constructed as previously described.19 The CD47 gene KO cell lines Hep3B-CD47KO and Jurkat-CD16A-CD47KO were generated with CRISPR-Cas9 technology. Raji, Raji-GPC3H, and Jurkat-CD16A-CD47KO cells were cultured with RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS). Hep3B, Hep3B-Luc23, and Hep3B-CD47KO cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% FBS.
BsAb construction
The variable heavy chain (VH) and variable light chain (VL) gene sequences of GC33 were synthesized by GenScript. Anti-hCD47 antibody BC18 was selected from our human nonimmune scFv phage display antibody library,16 and the genes of its VH and VL were sequenced. The coding sequences of the VH and VL of GC33 and BC18 were subcloned, respectively, into a human immunoglobulin 1 (hIgG1) heavy chain (HC) expression vector and a light chain (LC) expression vector.
To construct BsAbs, we modified the original wide-type HC and LC expression vectors (blank) using previously reported KiHs and CrossMab technologies.45, 46, 47 Briefly, several mutations were introduced into the constant heavy chain 3 (CH3) of the HC via site-directed mutagenesis to generate the “knob chain” (T366W, S354C) and the “hole chain” (Y349C, T366S, L368A, Y407V). The CH1 of the hole chain and the constant light chain (CL) of the LC were interchanged to, respectively, generate the paired crossover HC (CL-hole chain) and LC (CH1-LC). The VH and VL fragments of GC33, BC18, and a Ctrl antibody were then cloned into the knob chain, CL-hole chain, or CH1-LC expression vectors as needed.
SPR analysis
All SPR analyses were performed on a Biacore T200 instrument (Biacore, GE Healthcare). To measure the binding affinity of each mAb to its own antigen, GC33 or BC18 (isotype hIgG1) was first captured using a protein A/G (Pierce, Thermo Fisher Scientific) immobilized to a CM5 sensor chip; the analytes (hGPC3-ΔHS or hCD47 proteins; see Supplemental Materials and Methods) were then injected over each flow cell at serially diluted concentrations. The association rates (Kon), dissociation rates (Koff), and affinity constants (KD) were calculated using BiacoreT200 evaluation software. Kinetics analyses of GPC3/CD47 biAb or GPC3/CD47-DANG variant binding to mFcγRs were performed as described above (here the analytes were serial diluted mFcγRs).
To demonstrate simultaneous binding of GPC3/CD47 biAb, hGPC3-ΔHS was covalently attached to a CM5 sensor chip using an amine coupling kit (Biacore) at a surface density of ∼600 response units (RUs). GPC3/CD47 biAb was injected at 20 μg/mL with a dissociation phase of 80 s, and 1 mM hCD47-mFc fusion protein was then injected with a dissociation phase of 120 s, followed by regeneration with 3 M MgCl2.
FACS-based binding and blocking assays
To compare binding abilities of each antibody to single antigen- or dual antigen-expressing cells, Raji or Raji-GPC3H cells (5 × 105 per well in 96-well plates) were incubated with 0.4 μg/mL or 2 μg/mL of test antibodies for 30 min at 4°C in FACS buffer (0.5% BSA/PBS). Antibody-bound cells were then washed and incubated with fluorescein isothiocyanate (FITC)-conjugated goat anti-hIgG secondary antibody (Sigma-Aldrich) for 20 min at 4°C. These samples were analyzed with a FACSAria II instrument (BD Biosciences), and data were processed using FCS Express version (v.)4 (De Novo Software).
To assess the binding selectivity of these antibodies, Raji cells were labeled with CellTrack Deep Red, according to the manufacturer’s protocol (Thermo Fisher Scientific), followed by mixing with unstained Raji-GPC3H cells at a 1:1 ratio. The mixed cells (1 × 106 per well) were then incubated with 0.2 μg/mL of each antibody for 30 min at 4°C and analyzed as described above.
To assess the abilities of all anti-CD47 antibodies to block the interaction between CD47 and SIRPα, Raji or Raji-GPC3H cells were incubated with 50 nM of biotinylated SIRPα-mFc in the presence of different anti-CD47 antibodies (10 μg/mL) at 4°C for 30 min. Streptavidin-FITC (Sigma-Aldrich) was then added after washing to measure the binding of SIRPα to each type of cells.
RBC agglutination assay
Human whole blood collected from healthy donors was washed and resuspended with PBS. 3 million RBCs in PBS were plated per well in polypropylene 96-well plates (Corning) and incubated with serially diluted antibodies for 1 h at room temperature.
Generation of hCD47/hSIRPα humanized mice
All animal experiments were conducted following the National Guidelines for Housing and Care of Laboratory Animals in China and performed under the approved Institutional Animal Care and Use Committee (IACUC) protocols at National Institute of Biological Sciences, Beijing. The humanized mice were generated by CRISPR-Cas9-mediated gene-editing technology.48 The humanized Sirpα mice were developed by replacing exon 2 of the mouse Sirpα gene that encodes the extracellular domain with the hSirpα exon counterpart. Similarly, the humanized Cd47 mice were developed by replacing exon 2 of the mouse Cd47 gene that encodes the extracellular domain with the hCd47 exon counterpart. The hCd47/hSirpα double-gene humanized mice were developed by mating the two types of humanized mice together. The homozygous humanized mice were used in this study.
PK and hematologic toxicity studies in mice
A single-dose PK study was carried out in hCD47/hSIRPα humanized mice. 8- to 10-week-old humanized mice (n = 3 per group) were intraperitoneally (i.p.) injected with 10 mg/kg of each antibody. Blood samples were collected at different time points, and serum concentrations of each human antibody were measured with a hIgG ELISA quantitation kit (Bethyl Laboratories). The evaluation of the PK data was conducted with WinNonlin software. 6- to 8-week-old humanized mice (n = 4–6 per group) were i.p. injected with 10 mg/kg of each antibody. Blood samples were collected from orbital sinus 45 min or 2.5 h after antibody administration, and the RBC counts were measured using the ADVIA 2120i instrument at Beijing BrightShines.
ADCC reporter bioassay
The ADCC reporter gene bioassay was performed according to a previous report.49 Target cells (Raji, Raji-GPC3H, or Hep3B) were seeded at 1.5 × 104 into each well of a 96-well solid white polystyrene microplate (Corning). 3-fold serially diluted anti-GPC3 mAbs, anti-CD47 mAbs, or GPC3/CD47 biAbs, as well as Jurkat-CD16A-CD47KO effector cells, were added; the final effector-to-target cell ratio (E:T) was 6:1. Luciferase activity was measured using a luminescent substrate (Promega Bright-Glo) after incubation for approximately 8 h at 37°C, 5% CO2.
ADCP assays
Mouse BMDMs were used as effector cells in ADCP assays. To prepare BMDMs, mouse bone marrow cells were collected from the femurs and tibia of hCD47/hSIRPα humanized mice and induced by DMEM, supplemented with 15% L929 (secreting granulocyte macrophage-colony-stimulating factor [GM-CSF]) cell-culture medium for 3 days. The differentiated BMDMs were labeled with a 1:200 dilution of anti-mouse F4/80-Alexa Fluor647 (Thermo Fisher Scientific; clone BM8) prior to incubation with target cells.
For the competitive ADCP experiment, Raji and Raji-GPC3H cells were used as the target cells; these were stained with CellTrace Yellow or carboxyfluorescein succinimidyl ester (CFSE), according to the manufacturer’s protocol (Thermo Fisher Scientific). The fluorescently labeled target cells were mixed at 1:1 and plated at a density of 4 × 105 cells/well. For the HCC cell line ADCP experiment, Hep3B or Hep3B-CD47KO cells were stained with CFSE and plated at a density of 8 × 104 cells/well. Each target cell was incubated with 4 μg/mL of each antibody at room temperature (RT) for 10 min before being added to the differentiated and labeled BMDMs (∼2 × 105 cells/well) at 37°C for 2 h. Phagocytosis of fluorescent-labeled target cells by Alexa Fluor647-labeled BMDMs was recorded using a Nikon A1R confocal microscope. Prior to microscopy imaging, unphagocytosed cells were washed. Statistical analyses (multiple Student’s t tests or one-way ANOVA) were implemented in GraphPad Prism.
In vivo HCC xenograft model
5 × 106 Hep3B-Luc23 cells were injected subcutaneously into the right flank of 6- to 8-week-old, NOD-SCID mice. Mice were randomized into groups (n = 5 per group) based on equivalent mean tumor volumes and received 10 mg/kg of each antibody (or its variants) via i.p. injection twice a week for 3 weeks. In vivo tumor bioluminescence intensities of tumor-bearing mice were measured using an IVIS Lumina III In Vivo Imaging System (PerkinElmer) after i.p. injection of 15 mg/kg D-luciferin (PerkinElmer). Tumor dimensions were measured with an electronic caliper, and tumor volume was calculated using the formula (L × W2)/2, where L and W are the largest and smallest measured diameters, respectively. All mice were euthanized by CO2 if their tumor size exceeded 2,000 mm3 or at the end of the study.
For in vivo NIRF imaging, GPC3/CD47 biAb and an isotype Ctrl antibody were conjugated with Cy7 NHS ester (GE Healthcare). Hep3B tumor-bearing mice were randomized into two groups (n = 3 per group), based on equivalent mean tumor volumes, and injected with 8 mg/kg of each fluorescently labeled antibody. Mice were imaged 12, 24, 36, and 48 h after antibody administration, with excitation and emission wavelengths of 745 and 800 nm. Organs were dissected and imaged 48 h after antibody administration.
For depletion of macrophages, NOD-SCID mice were i.p. injected with 200 μL clodronate liposome (FormuMax), 3 days prior to initiation of GPC3/CD47 biAb treatment, followed by additional injection (100 μL) once a week until the end of the study. For depletion of neutrophils, NOD-SCID mice were i.p. injected with 200 μg anti-Ly6G antibody (1A8 clone; BioXCell) every 5 days.
Statistical analysis
Statistical significance between different experimental groups was analyzed by Student’s t test, one-way ANOVA, or two-way ANOVA with Tukey’s test (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). All statistical analyses and graph preparation were performed using GraphPad Prism.
Acknowledgments
We thank F. Yang, X. Lv, Y. Yang, and X. Tian in the Sui lab for their technical assistance. We would also like to thank the NIBS Biological Resource Center for DNA sequencing, the NIBS Animal Facility for its help in animal handling and care, and the NIBS imaging facility for assistance with the microscope experiment. This work was supported by grants from the Ministry of Science and Technology of the People's Republic of China (973 program #2012CB837600 to J.S.), Beijing Municipal Science and Technology Commission, and Beijing Key Laboratory of Pathogen Invasion and Immune Defense (Z171100002217064 to J.S.).
Author contributions
K.D. and J.S. conceptualized this study, interpreted the results, and drafted the manuscript. K.D., Y. Li, J.L., W.C., Z.W., and Y.L uo performed experiments. Y.Q. and F.W. provided Jurkat T effector cells and transgenic mice. K.D. prepared figures. J.S. supervised the study. All authors commented on the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.01.006.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Vogel A., Saborowski A. Current strategies for the treatment of intermediate and advanced hepatocellular carcinoma. Cancer Treat. Rev. 2020;82:101946. doi: 10.1016/j.ctrv.2019.101946. [DOI] [PubMed] [Google Scholar]
- 3.Iozzo R.V., Schaefer L. Proteoglycan form and function: A comprehensive nomenclature of proteoglycans. Matrix Biol. 2015;42:11–55. doi: 10.1016/j.matbio.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo M., Zhang H., Zheng J., Liu Y. Glypican-3: A New Target for Diagnosis and Treatment of Hepatocellular Carcinoma. J. Cancer. 2020;11:2008–2021. doi: 10.7150/jca.39972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abou-Alfa G.K., Puig O., Daniele B., Kudo M., Merle P., Park J.W., Ross P., Peron J.M., Ebert O., Chan S. Randomized phase II placebo controlled study of codrituzumab in previously treated patients with advanced hepatocellular carcinoma. J. Hepatol. 2016;65:289–295. doi: 10.1016/j.jhep.2016.04.004. [DOI] [PubMed] [Google Scholar]
- 6.Hoseini S.S., Cheung N.V. Immunotherapy of hepatocellular carcinoma using chimeric antigen receptors and bispecific antibodies. Cancer Lett. 2017;399:44–52. doi: 10.1016/j.canlet.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Takai H., Kato A., Kato C., Watanabe T., Matsubara K., Suzuki M., Kataoka H. The expression profile of glypican-3 and its relation to macrophage population in human hepatocellular carcinoma. Liver Int. 2009;29:1056–1064. doi: 10.1111/j.1478-3231.2008.01968.x. [DOI] [PubMed] [Google Scholar]
- 8.Matlung H.L., Szilagyi K., Barclay N.A., van den Berg T.K. The CD47-SIRPα signaling axis as an innate immune checkpoint in cancer. Immunol. Rev. 2017;276:145–164. doi: 10.1111/imr.12527. [DOI] [PubMed] [Google Scholar]
- 9.Barclay A.N., Van den Berg T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: structure, function, and therapeutic target. Annu. Rev. Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 10.Willingham S.B., Volkmer J.-P., Gentles A.J., Sahoo D., Dalerba P., Mitra S.S., Wang J., Contreras-Trujillo H., Martin R., Cohen J.D. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA. 2012;109:6662–6667. doi: 10.1073/pnas.1121623109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheung C.H., Lee K.W., Ng I.O.-L. Vol. 71. 2011. Abstract 2453: CD47 is a novel therapeutic target for hepatocellular carcinoma; p. 2453. (Proceedings of the 102nd Annual Meeting of the American Association for Cancer Research). [Google Scholar]
- 12.Lo J., Lau E.Y.T., So F.T.Y., Lu P., Chan V.S.F., Cheung V.C.H., Ching R.H.H., Cheng B.Y.L., Ma M.K.F., Ng I.O.L., Lee T.K.W. Anti-CD47 antibody suppresses tumour growth and augments the effect of chemotherapy treatment in hepatocellular carcinoma. Liver Int. 2016;36:737–745. doi: 10.1111/liv.12963. [DOI] [PubMed] [Google Scholar]
- 13.Xiao Z., Chung H., Banan B., Manning P.T., Ott K.C., Lin S., Capoccia B.J., Subramanian V., Hiebsch R.R., Upadhya G.A. Antibody mediated therapy targeting CD47 inhibits tumor progression of hepatocellular carcinoma. Cancer Lett. 2015;360:302–309. doi: 10.1016/j.canlet.2015.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sikic B.I., Lakhani N., Patnaik A., Shah S.A., Chandana S.R., Rasco D., Colevas A.D., O’Rourke T., Narayanan S., Papadopoulos K. First-in-Human, First-in-Class Phase I Trial of the Anti-CD47 Antibody Hu5F9-G4 in Patients With Advanced Cancers. J. Clin. Oncol. 2019;37:946–953. doi: 10.1200/JCO.18.02018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ishiguro T., Sugimoto M., Kinoshita Y., Miyazaki Y., Nakano K., Tsunoda H., Sugo I., Ohizumi I., Aburatani H., Hamakubo T. Anti-glypican 3 antibody as a potential antitumor agent for human liver cancer. Cancer Res. 2008;68:9832–9838. doi: 10.1158/0008-5472.CAN-08-1973. [DOI] [PubMed] [Google Scholar]
- 16.Li D., He W., Liu X., Zheng S., Qi Y., Li H., Mao F., Liu J., Sun Y., Pan L. A potent human neutralizing antibody Fc-dependently reduces established HBV infections. eLife. 2017;6:e26738. doi: 10.7554/eLife.26738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu J., Wang L., Zhao F., Tseng S., Narayanan C., Shura L., Willingham S., Howard M., Prohaska S., Volkmer J. Pre-Clinical Development of a Humanized Anti-CD47 Antibody with Anti-Cancer Therapeutic Potential. PLoS ONE. 2015;10:e0137345. doi: 10.1371/journal.pone.0137345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weiner L.M., Surana R., Wang S. Monoclonal antibodies: versatile platforms for cancer immunotherapy. Nat. Rev. Immunol. 2010;10:317–327. doi: 10.1038/nri2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu H., Zheng S., Hou X., Liu X., Du K., Lv X., Li Y., Yang F., Li W., Sui J. Novel Abs targeting the N-terminus of fibroblast growth factor 19 inhibit hepatocellular carcinoma growth without bile-acid-related side-effects. Cancer Sci. 2020;111:1750–1760. doi: 10.1111/cas.14353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chao M.P., Alizadeh A.A., Tang C., Myklebust J.H., Varghese B., Gill S., Jan M., Cha A.C., Chan C.K., Tan B.T. Anti-CD47 antibody synergizes with rituximab to promote phagocytosis and eradicate non-Hodgkin lymphoma. Cell. 2010;142:699–713. doi: 10.1016/j.cell.2010.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lo M., Kim H.S., Tong R.K., Bainbridge T.W., Vernes J.M., Zhang Y., Lin Y.L., Chung S., Dennis M.S., Zuchero Y.J. Effector-attenuating Substitutions That Maintain Antibody Stability and Reduce Toxicity in Mice. J. Biol. Chem. 2017;292:3900–3908. doi: 10.1074/jbc.M116.767749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mano Y., Aishima S., Fujita N., Tanaka Y., Kubo Y., Motomura T., Taketomi A., Shirabe K., Maehara Y., Oda Y. Tumor-associated macrophage promotes tumor progression via STAT3 signaling in hepatocellular carcinoma. Pathobiology. 2013;80:146–154. doi: 10.1159/000346196. [DOI] [PubMed] [Google Scholar]
- 23.Takai H., Ashihara M., Ishiguro T., Terashima H., Watanabe T., Kato A., Suzuki M. Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer Biol. Ther. 2009;8:2329–2338. doi: 10.4161/cbt.8.24.9985. [DOI] [PubMed] [Google Scholar]
- 24.Labrijn A.F., Janmaat M.L., Reichert J.M., Parren P.W.H.I. Bispecific antibodies: a mechanistic review of the pipeline. Nat. Rev. Drug Discov. 2019;18:585–608. doi: 10.1038/s41573-019-0028-1. [DOI] [PubMed] [Google Scholar]
- 25.Buatois V., Johnson Z., Salgado-Pires S., Papaioannou A., Hatterer E., Chauchet X., Richard F., Barba L., Daubeuf B., Cons L. Preclinical Development of a Bispecific Antibody that Safely and Effectively Targets CD19 and CD47 for the Treatment of B-Cell Lymphoma and Leukemia. Mol. Cancer Ther. 2018;17:1739–1751. doi: 10.1158/1535-7163.MCT-17-1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yeung O.W., Lo C.M., Ling C.C., Qi X., Geng W., Li C.X., Ng K.T., Forbes S.J., Guan X.Y., Poon R.T. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. J. Hepatol. 2015;62:607–616. doi: 10.1016/j.jhep.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 27.Dong P., Ma L., Liu L., Zhao G., Zhang S., Dong L., Xue R., Chen S. CD86+/CD206+, Diametrically Polarized Tumor-Associated Macrophages, Predict Hepatocellular Carcinoma Patient Prognosis. Int. J. Mol. Sci. 2016;17:320. doi: 10.3390/ijms17030320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mestas J., Hughes C.C.W. Of mice and not men: differences between mouse and human immunology. J. Immunol. 2004;172:2731–2738. doi: 10.4049/jimmunol.172.5.2731. [DOI] [PubMed] [Google Scholar]
- 29.van Egmond M., Bakema J.E. Neutrophils as effector cells for antibody-based immunotherapy of cancer. Semin. Cancer Biol. 2013;23:190–199. doi: 10.1016/j.semcancer.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Siders W.M., Shields J., Garron C., Hu Y., Boutin P., Shankara S., Weber W., Roberts B., Kaplan J.M. Involvement of neutrophils and natural killer cells in the anti-tumor activity of alemtuzumab in xenograft tumor models. Leuk. Lymphoma. 2010;51:1293–1304. doi: 10.3109/10428191003777963. [DOI] [PubMed] [Google Scholar]
- 31.Hernandez-Ilizaliturri F.J., Jupudy V., Ostberg J., Oflazoglu E., Huberman A., Repasky E., Czuczman M.S. Neutrophils contribute to the biological antitumor activity of rituximab in a non-Hodgkin’s lymphoma severe combined immunodeficiency mouse model. Clin. Cancer Res. 2003;9:5866–5873. [PubMed] [Google Scholar]
- 32.Zhao X.W., van Beek E.M., Schornagel K., Van der Maaden H., Van Houdt M., Otten M.A., Finetti P., Van Egmond M., Matozaki T., Kraal G. CD47-signal regulatory protein-α (SIRPα) interactions form a barrier for antibody-mediated tumor cell destruction. Proc. Natl. Acad. Sci. USA. 2011;108:18342–18347. doi: 10.1073/pnas.1106550108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ring N.G., Herndler-Brandstetter D., Weiskopf K., Shan L., Volkmer J.P., George B.M., Lietzenmayer M., McKenna K.M., Naik T.J., McCarty A. Anti-SIRPα antibody immunotherapy enhances neutrophil and macrophage antitumor activity. Proc. Natl. Acad. Sci. USA. 2017;114:E10578–E10585. doi: 10.1073/pnas.1710877114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilson C.L., Jurk D., Fullard N., Banks P., Page A., Luli S., Elsharkawy A.M., Gieling R.G., Chakraborty J.B., Fox C. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun. 2015;6:6818. doi: 10.1038/ncomms7818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou S.-L., Zhou Z.-J., Hu Z.-Q., Huang X.-W., Wang Z., Chen E.-B., Fan J., Cao Y., Dai Z., Zhou J. Tumor-Associated Neutrophils Recruit Macrophages and T-Regulatory Cells to Promote Progression of Hepatocellular Carcinoma and Resistance to Sorafenib. Gastroenterology. 2016;150:1646–1658.e17. doi: 10.1053/j.gastro.2016.02.040. [DOI] [PubMed] [Google Scholar]
- 36.Mikulak J., Bruni E., Oriolo F., Di Vito C., Mavilio D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019;10:946. doi: 10.3389/fimmu.2019.00946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tian Z., Chen Y., Gao B. Natural killer cells in liver disease. Hepatology. 2013;57:1654–1662. doi: 10.1002/hep.26115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sung P.S., Jang J.W. Natural Killer Cell Dysfunction in Hepatocellular Carcinoma: Pathogenesis and Clinical Implications. Int. J. Mol. Sci. 2018;19:3648. doi: 10.3390/ijms19113648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellor J.D., Brown M.P., Irving H.R., Zalcberg J.R., Dobrovic A. A critical review of the role of Fc gamma receptor polymorphisms in the response to monoclonal antibodies in cancer. J. Hematol. Oncol. 2013;6:1. doi: 10.1186/1756-8722-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen G., Chen Y.C., Reis B., Belousov A., Jukofsky L., Rossin C., Muehlig A., Xu C., Essioux L., Ohtomo T. Combining expression of GPC3 in tumors and CD16 on NK cells from peripheral blood to identify patients responding to codrituzumab. Oncotarget. 2018;9:10436–10444. doi: 10.18632/oncotarget.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gao H., Li K., Tu H., Pan X., Jiang H., Shi B., Kong J., Wang H., Yang S., Gu J., Li Z. Development of T cells redirected to glypican-3 for the treatment of hepatocellular carcinoma. Clin. Cancer Res. 2014;20:6418–6428. doi: 10.1158/1078-0432.CCR-14-1170. [DOI] [PubMed] [Google Scholar]
- 42.Li D., Li N., Zhang Y.-F., Fu H., Feng M., Schneider D., Su L., Wu X., Zhou J., Mackay S. Persistent Polyfunctional Chimeric Antigen Receptor T Cells That Target Glypican 3 Eliminate Orthotopic Hepatocellular Carcinomas in Mice. Gastroenterology. 2020;158:2250–2265.e20. doi: 10.1053/j.gastro.2020.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu L., Yang X., Huang N., Lang Q.L., He Q.L., Jian-Hua W., Liang-Peng G. A novel targeted GPC3/CD3 bispecific antibody for the treatment hepatocellular carcinoma. Cancer Biol. Ther. 2020;21:597–603. doi: 10.1080/15384047.2020.1743158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Veillette A., Chen J. SIRPα-CD47 Immune Checkpoint Blockade in Anticancer Therapy. Trends Immunol. 2018;39:173–184. doi: 10.1016/j.it.2017.12.005. [DOI] [PubMed] [Google Scholar]
- 45.Ridgway J.B., Presta L.G., Carter P. ‘Knobs-into-holes’ engineering of antibody CH3 domains for heavy chain heterodimerization. Protein Eng. 1996;9:617–621. doi: 10.1093/protein/9.7.617. [DOI] [PubMed] [Google Scholar]
- 46.Merchant A.M., Zhu Z., Yuan J.Q., Goddard A., Adams C.W., Presta L.G., Carter P. An efficient route to human bispecific IgG. Nat. Biotechnol. 1998;16:677–681. doi: 10.1038/nbt0798-677. [DOI] [PubMed] [Google Scholar]
- 47.Schaefer W., Regula J.T., Bähner M., Schanzer J., Croasdale R., Dürr H., Gassner C., Georges G., Kettenberger H., Imhof-Jung S. Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies. Proc. Natl. Acad. Sci. USA. 2011;108:11187–11192. doi: 10.1073/pnas.1019002108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wefers B., Bashir S., Rossius J., Wurst W., Kühn R. Gene editing in mouse zygotes using the CRISPR/Cas9 system. Methods. 2017;121-122:55–67. doi: 10.1016/j.ymeth.2017.02.008. [DOI] [PubMed] [Google Scholar]
- 49.Parekh B.S., Berger E., Sibley S., Cahya S., Xiao L., LaCerte M.A., Vaillancourt P., Wooden S., Gately D. Development and validation of an antibody-dependent cell-mediated cytotoxicity-reporter gene assay. MAbs. 2012;4:310–318. doi: 10.4161/mabs.19873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.