

Abstract
Cancer-associated lymphatic endothelial cells (LECs) are an active barrier to the effector arm of the anti-tumor immune response; however, it remains unclear how LECs become immunosuppressive in the tumor microenvironment (TME). Exosomal microRNAs (miRNAs) have recently been implicated in intercellular crosstalk within the TME. Here, we report a mechanistic model via which cervical cancer-secreted, exosome-encapsulated microRNA (miR)-1468-5p promotes lymphatic PD-L1 upregulation and lymphangiogenesis to impair T cell immunity. Subsequently, exosomal miR-1468-5p epigenetically activates the JAK2/STAT3 pathway in LECs by directly targeting homeobox containing 1 (HMBOX1) in the SOCS1 promoter, activating an immunosuppressive program that allows cancer cells to escape anti-cancer immunity. Furthermore, clinical data reveal that high serum exosomal miR-1468-5p levels correlate with TME immunosuppressive status and poor prognosis in cervical cancer (CCa) patients. Taken together, our results suggest that cancer-secreted exosomal miR-1468-5p instructs LECs to form an integrated immunosuppressive TME component and may be a prognostic biomarker and therapeutic target for CCa.
Graphical abstract
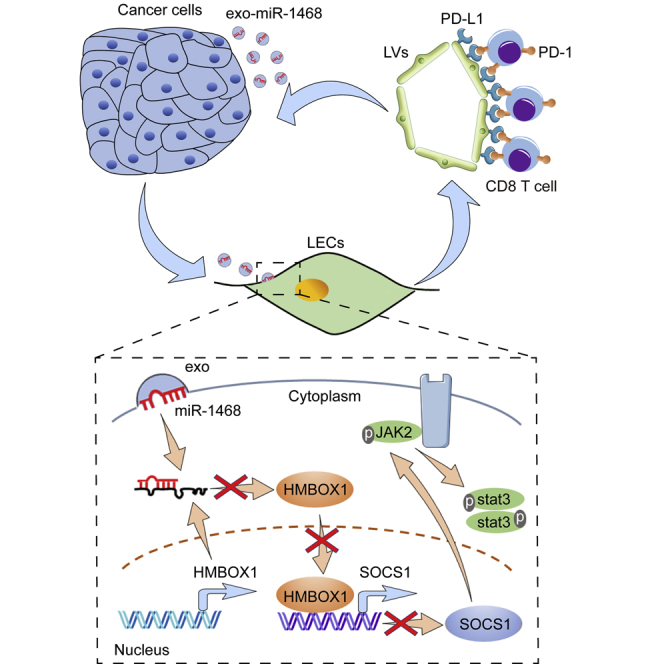
Our study elucidated a mechanistic model via which cervical cancer-secreted, exosome-encapsulated miR-1468-5p promotes lymphatic PD-L1 upregulation and lymphangiogenesis to impair T cell immunity. Moreover, clinical data revealed that high serum exosomal miR-1468-5p levels correlate with TME immunosuppressive status and poor prognosis in cervical cancer patients.
Introduction
Although the majority of patients with early-stage cervical cancer (CCa) achieve good recovery through surgical treatment and concurrent chemoradiotherapy, those with recurrent and metastatic CCa are rarely treated effectively.1 The modification of T cell function by immune checkpoint blockade has recently been reported as a novel strategy for improving the clinical outcomes of these patients;2 however, there are several issues with regard to T cell-mediated immunotherapies that have yet to be solved.3 For instance, the ratio of responders has been limited so far, and the precise mechanism of action is not yet clear. The tumor microenvironment (TME) has been strongly implicated in determining immunotherapeutic outcomes.4 Cancer-associated lymphatic endothelial cells (LECs) are one of many cell types that promote a suppressive TME and help the tumor escape host immunity.5 An improved understanding of the immunosuppressive mechanisms of cancer-associated LECs should therefore enable the rational development of immunotherapeutic strategies.
PD-L1, also known as B7-H1 or CD274, promotes T cell dysfunction by binding to the programmed death 1 (PD-1) receptor.6 Many human cancers express high PD-L1 protein levels, which correlate with poor prognosis in some cases.7, 8, 9 In addition to PD-L1 expression by tumor cells, recent mouse and human studies have highlighted the role of host hematopoietic cells in PD-L1-dependent T cell exhaustion,10, 11, 12 indicating that the TME contributes to effector T cell exhaustion. However, little is currently known about the expressional and functional significance of nonhematopoietic PD-L1 in the stroma of human tumors in situ.
Cancer involves a dynamic interaction between cells in the TME,13 with cancer-secreted exosomes known to play a role across many levels of intercellular crosstalk.14, 15, 16 Exosomes transfer biomolecules between adjacent or distant cells by traveling through interstitial spaces and the blood.17 Cancer-secreted exosomes have been implicated in angiogenesis,18 vascular permeability,19 cancer-associated fibroblast activation,16 and neutrophil recruitment in the metastatic niche,20 with many of these functions attributed to the post-transcriptional regulation of gene expression in niche cells by exosome-encapsulated microRNAs (miRNAs). In this study, we aimed to investigate whether cancer-secreted exosomal miRNAs are involved in the immunosuppressive reprogramming of LECs in the TME to evade host immunity.
Results
PD-L1+ lymphatics correlate with PD-1+ CD8+ T cells in human CCa peritumoral stroma
As the main immune checkpoint, PD-L1 plays a critical role in shaping the immunosuppressive TME,21 whereas cancer-associated LECs have been reported to help tumor cells evade cytotoxic T cell responses.22 Therefore, we investigated whether cancer-associated, LEC-derived PD-L1 functionally suppresses CD8+ T cell immunity in the TME by examining PD-L1 expression on LECs and CD8+ T cells in CCa (n = 102) and normal control (n = 67) tissues (Figure 1A). In all samples, CD8+ T cell, PD-1+ cell, and lymphatic density, as well as PD-L1 expression, were higher in CCa stroma than in normal stroma (Figures 1B−1E). Moreover, PD-L1 was mainly expressed on stromal cells, especially on lymphatics (Figures 1A and 1G), whereas most infiltrating CD8+ T cells accumulated in peritumoral CCa stroma but not normal stroma and were positive for PD-1 (Figures 1A and 1F). Notably, the majority of PD-1+ CD8+ T cells in the peritumoral stroma were in close contact with PD-L1+ lymphatics (Figure 1A), with statistical analysis revealing a positive association between PD-L1+ lymphatics and PD-1+ CD8+ T cells in peritumoral CCa stroma but not normal stroma (p < 0.0001, R = 0.6097 and p = 0.3091, R = 0.1262, respectively; Pearson’s correlation coefficients; Figure 1H). Taken together, these results suggest that cancer-associated LECs may promote tumor progression by impairing T cell immunity via PD-L1.
Figure 1.

PD-L1+ lymphatics correlate with PD-1+ CD8+ T cells in human cervical cancer (CCa) peritumoral stroma
(A) Representative micrographs of CD8, PD-1, lymphatics (D240 positive), and PD-L1 immunofluorescence staining in sections of CCa and healthy controls. Scale bar, 20 μm. (B−G) Quantification of CD8 (B), PD-1 (C), lymphatics (D), PD-L1 (E), PD-1+ CD8 (F), and PD-L1+ lymphatics (G) expression. (H) Correlation analysis between PD-1+ CD8 and PD-L1+ lymphatics in CCa and healthy specimens. E, epithelium; T, tumor. Error bars represent the mean ± SD of three independent experiments. ∗∗∗p < 0.001.
High PD-L1+ lymphatic and PD-1+ CD8+ T cell infiltration correlate with poor CCa prognosis
Based on our observation that PD-L1 and PD-1 were expressed highly in the lymphatics and CD8+ T cells of CCa tissues, respectively, we hypothesized that the presence of PD-L1+ lymphatics and PD-1+ CD8+ T cells would have adverse effects on survival. Statistical analysis revealed a negative correlation between PD-1+ CD8+ T cells (integrated optical density [IOD]/area) and overall survival (OS; p = 0.0023, R = −0.3068, Pearson’s correlation coefficients) and disease-free survival (DFS; p = 0.0126, R = −0.2649, Pearson’s correlation coefficients; Figure 2A). As expected, there was also a negative correlation between PD-L1+ lymphatics and OS (p < 0.0001, R = −0.4122, Pearson’s correlation coefficients) and DFS (p = 0.0004, R = −0.3512, Pearson’s correlation coefficients) in the same CCa group (Figure 2B).
Figure 2.

High PD-L1+ lymphatic and PD-1+ CD8+ T cell infiltration correlate with poor CCa prognosis
(A and B) Correlation of PD-1+ CD8+ T cells (A) and PD-L1+ lymphatics (B) to overall survival (OS) and disease-free survival (DFS). (C−E) The OS and DFS of CCa patients with lower versus higher expression of PD-1+ CD8+ T cells (C), or PD-L1+ lymphatics (D), or PD-1+ CD8+ T cells plus PD-L1+ lymphatics (E) were estimated using Kaplan-Meier curves. The median expression was used as the cut-off value. ∗∗∗p < 0.001.
A total of 102 CCa patients underwent curative resection with available follow-up data and were thus divided into six groups according to the median (IOD)/area value for PD-L1+ lymphatics and PD-1+ CD8+ T cells. As shown in Figure 2C, patients with a higher PD-1+ CD8+ T cell density had poorer OS (p = 0.0015, log-rank test) and DFS (p = 0.0075, log-rank test). Similarly, patients with a higher PD-L1+ lymphatic density displayed shorter OS (p < 0.0001, log-rank test) and DFS (p = 0.0004, log-rank test) (Figure 2D). Comprehensive analysis revealed that patients with both higher PD-1+ CD8+ T cell and PD-L1+ lymphatic densities had significantly shorter OS (p < 0.0001, log-rank test) and DFS (p = 0.0002, log-rank test; Figure 2E). These findings demonstrate that PD-1+ CD8+ T cells and PD-L1+ lymphatics have prognostic value and thus might serve as a promising predictor of survival in CCa.
Cancer-secreted exosomes reprogram human dermal LECs (HDLECs) to suppress CD8+ T cell immunity
Clinical evidence has revealed that cancer-associated LECs actively participate in tumor progression; thus, it is crucial to improve our understanding of cancer-associated LEC reprogramming by cancer cells. Since exosomes are an important method via which tumors induce systemic changes, exosomes may rationally participate in the reprogramming of cancer-associated LECs. Then we choose HDLECs to represent normal LECs in our model because of its low PD-L1 expression. To observe the transfer of cancer-secreted exosomes from cancer cells to HDLECs, hCEp (human cervical epithelium cell line) and Siha (CCa cell line) cells were transduced with a lentiviral plasmid encoding the CD63-green fluorescent protein (GFP) fusion protein, a common exosomal marker (Figure S1). With the use of a semipermeable Transwell membrane, GFP-exosome-producing hCEp (hCEpGFP-exo) or Siha (SihaGFP-exo) cells were co-cultured with phalloidin-labeled HDLECs (1:1) for 0, 1, 4, or 24 h to measure the labeled exosomes. We found that HDLECs showed a time-dependent uptake of exosomes in both hCEpGFP-exo and SihaGFP-exo groups (Figure 3A), suggesting exosomes could be transferred into recipient cells independent of their origin.
Figure 3.

Cancer-secreted exosomes reprogram HDLECs to suppress CD8+ T cell immunity
(A) Confocal imaging showed the transfer of GFP-labeled exosomes (green) to phalloidin-labeled HDLECs (red) at the indicated time point using Transwell chamber. (B and C) Exosomes secreted by Siha and hCEp were detected by transmission electron microscopy (TEM) (B) and NanoSight analysis (C). Scale bar, 100 nm. (D) Western blot for characteristic proteins of exosomes compared with conditioned media (CM) from Siha and hCEp. (E and F) Flow cytometry analysis of PD-L1 expression (E) and tube-formation assay (F) in HDLECs treated with PBS, hCEp-exo, or Siha-exo. Scale bar, 10 μm. (G−J) Flow cytometry analysis of IFN-γ (G), CD69 (H), PD-1 (I), and Annexin V (J) expression on CD8+ T cells co-cultured with HDLECs treated with PBS, hCEp-exo, or Siha-exo. (K and L) Quantification of PD-L1 expression (K) and tube formation (L) in HDLECs with indicated treatment. (M−P) Quantification of IFN-γ (M), CD69 (N), PD-1 (O), and Annexin V (P) expression on CD8+ T cells co-cultured with HDLECs with indicated treatment. The numeric values under the western blot bands represent the protein relative expression (baseline value 1.00). Error bars represent the mean ± SD of three independent experiments. ∗∗∗p < 0.001.
To explore the biological function of different exosomes, they were isolated and purified from the conditioned media (CM) of the normal hCEp and the Siha by standard ultracentrifugation. The typical cup-shaped morphology, 30−150 nm size, and number of the isolated exosomes were detected by transmission electron microscopy (TEM) and NanoSight analysis (Figures 3B and 3C), with western blot verifying that the isolated particles were exosomes by detecting TSG101, CD63, and heat shock protein 70 (HSP70) (Figure 3D). Then, the purified exosomes were added to HDLECs for 48 h. The results showed that PD-L1 expression and tube formation were significantly stimulated in HDLECs by Siha-exo but not hCEp-exo (Figures 3E, 3F, 3K, and 3L).
To further evaluate the effect of exo-treated HDLECs on T cell immunity, we contact co-cultured, activated human CD8+ T cells with exo-treated HDLECs. The percentage of CD69+ (activated) and interferon (IFN)-γ+ CD8+ (effector) T cells was significantly lower after incubation with Siha-exo-treated HDLECs than with hCEp-exo-treated HDLECs, yet the percentage of PD-1+ (inhibitory) and Annexin V+ (apoptotic) CD8+ T cells was dramatically higher (Figures 3G−3J and 3M−3P).
Cancer-secreted exosomal microRNA (miR)-1468-5p reprograms HDLECs to suppress CD8+ T cell immunity
Next, we explored the mechanism via which cancer-secreted exosomes reprogram HDLECs. Many miRNAs are selectively encapsulated in exosomes and have important roles in cell−cell communication;23 thus, we hypothesized that cancer-derived exosomal miRNAs reprogram HDLECs to suppress CD8+ T cell immunity. To identify the specific miRNAs involved, hCEp cells, hCEp-exo, Siha cells, and Siha-exo were sequenced using miRNA microarrays (Figure 4A), and 16 upregulated miRNAs (fold change > 2) that were selectively enriched in Siha-exo were validated further (Figure 4B). Only miR-1468-5p clearly promoted PD-L1 expression in HDLECs (Figure 4C), with qRT-PCR analysis verifying that miR-1468-5p was more abundant in CCa-exo than in CCa cells and hCEp-exo (Figure 4D). To confirm that CCa-secreted miR-1468-5p could be transferred to HDLECs via exosomes, we measured miR-1468-5p levels in HDLECs treated with CCa-exo, observing increased cellular miR-1468-5p levels in recipient HDLECs following CCa-exo treatment (Figure 4E). Moreover, flow cytometry analysis and tube-formation assay both showed that miR-1468-5p mimics also promoted PD-L1 expression and tube formation in HDLECs (Figures S2, 4F, and 4G).
Figure 4.

Cancer-secreted exosomal miR-1468-5p reprograms HDLECs to suppress CD8+ T cell immunity
(A) Microarray analysis of exosomal and cellular miRNAs from hCEp and Siha were presented in a heatmap. (B) Overlapping results of upregulated miRNAs in indicated groups. (C) qRT-PCR analysis of PD-L1 expression in HDLECs transfected with indicated mimics. (D) qRT-PCR analysis of miR-1468-5p expression in indicated cells and paired exosomes. (E) qRT-PCR analysis of miR-1468-5p expression in HDLECs treated with PBS or indicated exosomes. (F and G) Flow cytometry analysis of PD-L1 expression (F) and quantification of tube formation (G) in HDLECs transfected with miR-1468-5p mimics or negative control (NC). (H and I) Flow cytometry analysis of PD-L1 expression (H) and quantification of tube formation (I) in HDLECs treated with hCEp-exo or Siha-exo in the presence of miR-1468-5p inhibitors or NC. Scale bar, 10 μm. (J−M) Flow cytometry analysis of IFN-γ (J), CD69 (K), PD-1 (L), and Annexin V (M) expression on CD8+ T cells co-cultured with HDLECs treated with hCEp-exo or Siha-exo in the presence of miR-1468-5p inhibitors or NC. Error bars represent the mean ± SD of three independent experiments. ∗∗∗p < 0.001.
To confirm that the transcriptional activation of endogenous miR-1468-5p in HDLECs did not activate lymphatic reprogramming, we established miR-1468-5p-knockdown HDLECs (HDLECs/anti-1468-5p) and Siha cells (Siha/anti-1468-5p) by selectively silencing miR-1468-5p using a lentiviral vector. Interestingly, miR-1468-5p was significantly downregulated in the Siha/anti-1468-5p cells but not the HDLECs/anti-1468-5p cells, suggesting that endogenous miR-1468-5p expression was extremely low in HDLECs (Figure S3A). Next, we evaluated the effects of Siha-exo on HDLEs/anti-1468-5p or negative control (NC) cells by flow cytometry analysis and tube-formation assays. Siha-exo promoted PD-L1 expression and tube formation in HDLECs/anti-1468-5p more than hCEp-exo, consistent with the results obtained in HDLECs/anti-NC (Figures S3B−S3D). Taken together, these results suggest that cancer cells reprogram HDLECs via the transfer of exosomal miR-1468-5p.
To confirm the role of miR-1468-5p, exo-treated HDLECs were treated with miR-1468-5p inhibitors. Consistent with the results above, miR-1468-5p inhibitors abolished the effect of Siha-exo on HDLECs (Figures 4H and 4I) and reversed the immunosuppression of activated CD8+ T cells by Siha-exo-treated HDLECs (Figures 4J−4M). Collectively, these findings reveal that cancer-secreted exosomal miR-1468-5p reprograms HDLECs to suppress CD8+ T cell immunity.
Cancer-secreted exosomal miR-1468-5p enhances tumor-specific T cell immunity suppression to assist tumor growth in vivo
Immunosuppression in the TME leads to CD8+ T cell exhaustion, which can promote immune escape and greatly reduce the efficacy of cancer immunotherapy;24 however, PD-L1 antibodies can block the interaction between PD-L1 and its cognate receptors to enhance the cytotoxic activity of antitumor T cells.25 To test the effect of exosomal miR-1468-5p-mediated HDLECs on T cell immunity in vivo, we generated allogenic T cell lines specific to Siha/anti-1468-5p cells with low PD-L1 expression (Figure S4A) by extended culture with primary human CD8+ T cells and transplanted the cells into B-NDG mice via intravenous tail injection (Figure S4B). The next day, Siha/anti-1468-5p cells were subcutaneously implanted alone or together with HDLECs treated with Siha/anti-1468-5p-exo or Siha/anti-NC-exo (Figure 5A), with the same human leukocyte antigen (HLA)-A24 typing detected in Siha/anti-1468-5p, HDLECs, and CD8+ T cells by flow cytometry (Figure S4C). α-PD-L1 was intraperitoneally injected 2 days after implantation and every 3 days afterward. Compared to Siha/anti-1468-5p alone, Siha/anti-1468-5p plus Siha/anti-NC-exo-treated HDLECs, but not those treated with Siha/anti-1468-5p-exo, displayed progressive tumor growth that was significantly reversed by α-PD-L1 (Figure 5B). These data indicate that exosomal miR-1468-5p-treated HDLEC PD-L1 signals are immunopathological in vivo.
Figure 5.

Cancer-secreted exosomal miR-1468-5p enhances tumor-specific T cell immunity suppression to assist tumor growth in vivo
(A) Schematic illustration of animal model. (B) Tumor growth curves in indicated groups. (C) Representative micrographs of CD8, PD-1, lymphatics, and PD-L1 immunofluorescence staining in sections of xenograft tumors. Scale bar, 20 μm. (D) Quantification of CD8, PD-1, lymphatics, PD-L1, PD-1+ CD8, and PD-L1+ lymphatics expression in indicated groups. Error bars represent the mean ± SD of three independent experiments. ∗∗p < 0.01; ∗∗∗p < 0.001.
To further confirm the role of PD-L1+ HDLECs in CD8+ T cell exhaustion, we examined CD8+ cell infiltration, PD-1 expression, lymphatic density, and PD-L1 expression by multiple immunofluorescent staining in xenograft tumors. As expected, PD-1 expression, lymphatic density, and PD-L1 expression were much higher in the Siha/anti-1468-5p plus Siha/anti-NC-exo-treated HDLECs group than in either the Siha/anti-1468-5p-alone group or Siha/anti-1468-5p plus Siha/anti-1468-5p-exo-treated HDLEC group, whereas CD8+ T cell density was much lower (Figures 5C and 5D). Despite the reduced number of CD8+ T cells, the PD-1+ CD8+ T cell density was higher when in close contact with PD-L1+ lymphatics (Figures 5C and 5D), consistent with our previous clinical observation. Moreover, α-PD-L1 was able to neutralize the effect of exosomal miR-1468-5p-induced CD8+ T cell immunosuppression in the tumor environment but not affect the lymphatic density (Figures 5C and 5D), suggesting that exosomal miR-1468-5p-treated HDLECs reduce tumor-specific T cell immunity in vivo via PD-L1 signaling and thereby contribute to tumor growth.
miR-1468-5p directly targets homeobox containing 1 (HMBOX1) in HDLECs to suppress CD8+ T cell immunity
To identify miR-1468-5p targets in HDLECs, we used two bioinformatics tools (miRWalk and miRDB) to predict seven common miR-1468-5p target genes with binding scores > 0.85 (Figure 6A). Among these, only HMBOX1 was verified as significantly downregulated at the mRNA level and downregulated at the protein level in HDLECs treated with miR-1468-5p mimics (Figures 6B and 6C). The miR-1468-5p and full-length HMBOX1 sequences were then aligned, demonstrating a HMBOX1 coding sequence to be a potential miR-1468-5p target (Figure 6D). Wild-type (WT) and mutated-type (MT) miR-1468-5p binding sites were then cloned into luciferase vectors, with luciferase activity significantly lower in HDLECs treated with the WT vector in the presence of miR-1468-5p mimics (Figure 6E).
Figure 6.

miR-1468-5p directly targets HMBOX1 in HDLECs to suppress CD8+ T cell immunity
(A) Target gene prediction of miR-1468-5p with two bioinformatics tools. (B) qRT-PCR analysis of predicted genes expression in HDLECs transfected with miR-1468-5p mimics or NC. (C) Western blot of HMBOX1 expression in HDLECs transfected with miR-1468-5p mimics or NC. (D) The wild type (WT) and a mutated type (MT) of binding site between miR-1468-5p and HMBOX1. (E) Relative luciferase activity of HDLECs with indicated treatments. (F and G) Flow cytometry analysis of PD-L1 expression (F) and quantification of tube formation (G) in HDLECs transfected with siHMBOX1 or siRNA. (H and I) Flow cytometry analysis of PD-L1 expression (H) and quantification of tube formation (I) in HDLECs treated with miR-1468-5p mimics or NC in the presence of HMBOX1 or vector. Scale bar, 10 μm. (J−M) Flow cytometry analysis of IFN-γ (J), CD69 (K), PD-1 (L), and Annexin V (M) expression on CD8+ T cells co-cultured with HDLECs treated with miR-1468-5p mimics or NC in the presence of HMBOX1 or vector. The numeric values under the western blot bands represent the protein relative expression (the indicated protein/GAPDH). Error bars represent the mean ± SD of three independent experiments. ∗p < 0.05; ∗∗∗p < 0.001.
To determine the role of HMBOX1 in lymphatic reprogramming, we knocked down HMBOX1 expression using small interfering (si)RNAs in HDLECs and confirmed the effect by qRT-PCR analysis (Figure S5A). Flow cytometry analysis and tube-formation assays showed that siHMBOX1-treated HDLECs displayed increased PD-L1 expression and tube formation (Figures S5B, 6F, and 6G), whereas HMBOX1 overexpression (Figure S5C) neutralized the effects of miR-1468-5p on lymphatic reprogramming (Figures 6H and 6I) and reversed CD8+ T cell immunosuppression induced by miR-1468-5p-treated HDLECs (Figures 6J−6M). Taken together, these data indicate that HMBOX1 is a direct downstream target of miR-1468-5p and may mediate lymphatic reprogramming.
miR-1468-5p reprograms HDLECs via the HMBOX1-SOCS1-JAK2/STAT3 axis
Since the JAK2/STAT3 pathway is associated with PD-L1 expression and lymphogenesis,26,27 we detected JAK2/STAT3 pathway activation in our experimental model. As shown in Figures 7A−7C, miR-1468-5p mimics promoted phosphorylated JAK2/STAT3 expression and JAK2/STAT3 pathway activation in HDLECs, whereas similar results were obtained when HDLECs were treated with Siha-exo but not hCEp-exo. More importantly, the JAK/STAT pathway inhibitor tofacitinib was able to neutralize the effect of exosomal miR-1468-5p-induced CD8+ T cell immunosuppression in vivo (Figures S6A and S6B). These results suggest that miR-1468-3p reprograms HDLECs by mediating JAK2/STAT3 pathway activation.
Figure 7.

miR-1468-5p reprograms HDLECs via the HMBOX1-SOCS1-JAK2/STAT3 axis
(A) Western blot of phosphorylated and total JAK2/STA3 expression in HDLECs treated with miR-1468-5p mimics or indicated exosomes. (B and C) Relative luciferase activity of JAK2 (B) and STAT3 (C) pathway in HDLECs treated with miR-1468-5p mimics or indicated exosomes. (D) Western blot of indicated proteins in HDLECs with indicated treatment. (E and F) Schematic outlines of the predicted binding of HMBOX1 to the SOCS1 promoter (E) and the putative binding sites (F) in the promoter. (G) Relative luciferase activity of the SOCS1 promoter in HMBOX1 overexpression or knockdown HDLECs. (H) ChIP assays of the enrichment of HMBOX1 on indicated HMBOX1 binding sites (HBSs) in the promoter region of SOCS1 relative to IgG. The numeric values under the western blot bands represent the protein relative expression (the indicated protein/GAPDH). Error bars represent the mean ± SD of three independent experiments. ∗p < 0.05; ∗∗∗p < 0.001.
The SOCS family is known to negatively regulate the JAK2/STAT3 pathway;28 therefore, we investigated the relationship between HMBOX1 and key SOCS family proteins. Western blot analysis showed that HMBOX1 knockdown decreased SOCS1 expression to activate the JAK2/STAT3 pathway in hCEp-exo-treated HDLECs, whereas HMBOX1 overexpression increased SOCS1 expression to suppress the JAK2/STAT3 pathway in Siha-exo-treated HDLECs (Figure 7D). However, SOCS2 and SOCS3 protein expression was not significantly affected by HMBOX1 knockdown or overexpression in HDLECs, indicating that HMBOX1 mediates JAK2/STAT3 pathway activation via SOCS1, not SOCS2 or SOCS3.
Bioinformatics analysis has predicted four HMBOX1 binding sites (HBSs) in the SOCS1 promoter region based on the JASPAR database (Figures 7E and 7F).29 Luciferase reporter assays revealed increased SOCS1 promoter-driven luciferase activity in HMBOX1-overexpressing cells but decreased activity in HMBOX1 knockdown cells (Figure 7G). Moreover, chromatin immunoprecipitation (ChIP) assay revealed that HMBOX1 bound to HBS1 and HBS4 in the SOCS1 promoter region (Figure 7H), indicating that HMBOX1 transcriptionally upregulates SOCS1 expression. These results strongly suggest that miR-1468-5p suppresses HMBOX1-SOCS1 expression to activate the JAK2/STAT3 pathway in HDLEC reprogramming.
Serum exosomal miR-1468-5p is associated with clinicopathological progression in CCa
Accumulating evidence has shown that exosomal miRNAs secreted by CCa cells can be detected in serum and that these serum exosomal miRNAs may be promising biomarkers for early CCa diagnosis.30 Therefore, it is important to clarify the clinical significance of exosomal miR-1468-5p in CCa. In this study, we examined serum exosomes collected from paired CCa patients and controls by TEM and NanoSight analysis (Figures 8A and 8B) and detected TSG101, CD63, and HSP70 protein levels to confirm that the isolated particles were exosomes (Figure 8C). Next, we explored whether exosomal miR-1468-5p was clinically relevant to CCa, with qRT-PCR analysis showing that serum exosomal miR-1468-5p expression was elevated in CCa patients compared to the controls (Figure 8D). Moreover, statistical analysis revealed a positive correlation between serum exosomal miR-1468 expression and PD-1+ CD8+ T cell infiltration (p < 0.0001, R = 0.4993, Pearson’s correlation coefficients) and PD-L1+ lymphatics density from the same patients (p < 0.0001, R = 0.5469, Pearson’s correlation coefficients) (Figures 8E and 8F), suggesting that exosomal miR-1468-5p plays a crucial role in miR-1468-5p-related lymphatic immunosuppression during CCa progression. Moreover, Kaplan-Meier analysis showed that high serum exosomal miR-1468 levels are associated with a shorter OS (p = 0.0026, log-rank test) and DFS (p = 0.0053, log-rank test) in CCa (Figures 8G and 8H). Taken together, our data suggest that serum exosomal miR-1468-5p may be a diagnostic biomarker and therapeutic target for CCa.
Figure 8.

Serum exosomal miR-1468-5p is associated with clinicopathological progression in CCa
(A−C) Serum exosomes isolated from the same healthy and CCa subjects were detected by TEM (A), NanoSight analysis (B), and western blot (C). Scale bar, 100 nm. (D) qRT-PCR analysis of exosomal miR-1468-5p in healthy and CCa serums. (E and F) Correlation of serum exosomal miR-1468-5p to PD-1+ CD8+ T cells (E) and PD-L1+ lymphatics (F) from the same CCa patients. (G and H) The OS (G) and DFS (H) of CCa patients with lower versus higher expression of serum exosomal miR-1468-5p were estimated using Kaplan-Meier curves. The median expression was used as the cut-off value. (I) Proposed schematic diagram of cancer-secreted exosomal miR-1468-5p-reprogramming HDLECs to suppress CD8+ T cell immunity for CCa progression. The numeric values under the western blot bands represent the protein relative expression (baseline value 1.00). Error bars represent the mean ± SD of three independent experiments. ∗∗∗p < 0.001.
In summary, the results of this study show that cancer-secreted exosomal miR-1468-5p promotes lymphatic PD-L1 expression and lymphangiogenesis to impair CD8+ T cell immunity by suppressing HMBOX1-SOCS1 expression and activating JAK2/STAT3 signaling, thus enabling the immune escape of cancer cells (Figure 8I). Our findings also indicate that cancer cell-secreted exosomal miR-1468-5p has an important role in intercellular communication that generates an immunosuppressive TME to promote CCa progression.
Discussion
Immune checkpoint blockade, including α-PD-L1 antibodies, has achieved promising clinical responses.31 Whereas the majority of responders are patients with PD-L1+ tumor cells, PD-L1− patients also respond to treatment,32 indicating that additional anatomical locations contribute to patient responses and should be explored further. A recent study highlighted the role of host and particularly hematopoietic PD-L1 expression in cancer-associated T cell exhaustion,33 whereas nonhematopoietic PD-L1 expression has been shown to contribute to immunopathology during chronic viral infection,34 and cancer-associated, LEC-derived PD-L1 expression suppresses T cell activation in vitro.35 In this study, we found that the majority of PD-L1+ lymphatics were in close contact with PD-1+ CD8+ T cells in peritumoral stroma, suggesting that cancer-associated lymphatics may impair CD8+ T cell immunity via PD-L1 signaling. Moreover, patients with both higher PD-L1+ lymphatic and PD-1+ CD8+ T cell density displayed shorter OS and DFS in CCa, suggesting that PD-L1+ lymphatics and PD-1+ CD8+ T cells may have prognostic value for CCa survival. Several studies have specifically demonstrated that whereas PD-L1 on tumor cells was largely dispensable for the response to checkpoint blockade, PD-L1 in host cells was essential for this response.36 Consistently, our data showed that blocking PD-L1 expression on cancer-associated LECs using a specific antibody contributed to tumor control in vivo, suggesting that PD-L1 suppression on cancer-associated LECs may be an important immunotherapeutic strategy for CCa.
The TME is a dynamic system involving complex interactions that are responsible for tumor progression.37 Exosomes, which mediate intercellular crosstalk within the TME, have been the focus of recent studies since they can transfer a prototype message reflecting the genetic and epigenetic alterations in donor cells to recipient cells without these features.38 Onco-miRNAs with oncogenic mutations are found in cancer-secreted exosomes and may participate in the transfer of a cancer prototype to noncancerous cells.39, 40, 41 Herein, we screened the ability of the novel miR-1468-5p, which is enriched in CCa-secreted exosomes, to promote lymphatic PD-L1 expression and lymphangiogenesis in order to impair CD8+ T cell immunity. High serum exosome miR-1468-5p expression was positively correlated with PD-L1+ lymphatics and PD-1+ CD8+ T cells, as well as poor OS and DFS, indicating that exosomal miR-1468-5p could be a molecular target for clinical intervention in patients with CCa.
Altered immune status in tumor stroma has emerged as an important aspect of cancer-associated immune reprogramming.22 The reprogramming of cancer-mediated LECs is correlated with increased immune suppression in progressing tumors.42 Thus, an intervention with LEC reprogramming in the TME could be a promising therapeutic strategy for improving CCa prognosis. In this study, we found that cancer-secreted exosomal miR-1468-5p was a potent target for diminishing LEC reprogramming in CCa and may be an attractive option for cancer treatment. We demonstrated that knocking down exosomal miR-1468-5p expression in CCa cells by RNAi inhibited lymphatic PD-L1 expression, and lymphangiogenesis to promote CD8+ T cell antitumor immune responses thus may be an approach for suppressing tumor progression. In addition, human papillomavirus (HPV) induced somatic mutations and a multitude of neoantigens, which played a crucial role in the suppressive TME of CCa and could lead to notable alterations among checkpoint-related genes, such as PD-L1, CTLA-4 and PD-1.43 Specifically, PD-L1 showed a positive correlation with E6/E7 oncoprotein from major oncogenic HPV genotypes.44 It would be important to explore the underlying mechanisms of the HPV E6/E7 oncoprotein leading to miR-1468-5p overexpression in CCa, which as an ongoing direction in our lab, may reveal additional strategies for immunotherapeutic intervention.
Another key finding of this study was that cancer-secreted exosomal miR-1468-5p downregulated HMBOX1-SOCS1 expression to epigenetically activate JAK2/STAT3 signaling in LECs. HMBOX1 is a member of the homeobox gene family that was screened from a pancreatic cDNA library.45 Most homeobox genes, including HMBOX1, are transcription factors that regulate genes associated with embryonic development and cell differentiation,46 with a study revealing that HMBOX1 is a key factor in the differentiation of bone marrow-derived stroma cells into endothelial cells.47 Herein, we demonstrated that CCa cell-secreted exosomal miR-1468-5p reduced HMBOX1 expression in LECs, whereas HMBOX1 overexpression was able to reverse the immunosuppressive phenotype of LECs on CD8+ T cells induced by miR-1468-5p. Emerging evidence has suggested that JAK2/STAT3 signaling pathway activation contributes to PD-L1 expression and lymphangiogenesis;26,27 therefore, we examined whether JAK2/STAT3 signaling was a downstream effector of the miR-1468-5p-HMBOX1 axis. Exosomes with high miR-1468-5p and miR-1468-5p mimic expression activated JAK2/STAT3 signaling in LECs and could be stimulated and reversed by HMBOX1 knockdown or overexpression, respectively. More importantly, the JAK/STAT pathway inhibitor tofacitinib was able to neutralize the effect of exosomal miR-1468-5p-induced CD8+ T cell immunosuppression in vivo. The SOCS family includes JAK kinase binding proteins that can inhibit JAK2/STAT3 activation directly by binding the JAK2 activation loop via its SH2 domain and targeting JAK2 for ubiquitination and proteasomal degradation via its COOH-terminal homology domain.48 We found that the HMBOX1-SOCS1 axis mediated JAK2/STAT3 signaling activation by targeting SOCS1 rather than SOCS2 or SOCS3, whereas HMBOX1 could directly bind the SOCS1 promoter to mediate JAK2/STAT3 activation.
In conclusion, our findings provide evidence of a lymphatic immunosuppression-dependent tumor progression mechanism in which CCa cell-secreted exosomal miR-1468-5p promotes lymphatic PD-L1 upregulation and lymphangiogenesis to impair CD8+ T cell immunity by transcriptionally suppressing HMBOX1-SOCS1 expression and activating JAK2/STAT3 signaling. We also found that miR-1468-5p was overexpressed in the serum-exo of patients with CCa and positively correlated with both PD-L1+ lymphatics and PD-1+ CD8+ T cells and thus is clinically relevant to CCa prognosis. Our study not only identifies a crucial mechanism of exosomal miRNA-mediated intercellular communication between CCa cells and the TME to promote tumor progression but also suggests a potential noninvasive diagnostic approach and therapeutic strategy for patients with CCa.
Materials and methods
Clinical samples and study approval
Clinical tissue and blood samples were obtained from 102 patients with CCa who had not undergone preoperative radiotherapy or chemotherapy and 67 patients with uterine leiomyoma at the Department of Gynecological Oncology of Nanfang Hospital of Southern Medical University (Guangzhou, China) and Tongji Hospital of Huazhong University of Science and Technology (Wuhan, China) between 2007 and 2016. Pathological diagnosis was performed preoperatively and confirmed postoperatively. Patients without CCa were confirmed as eligible using a ThinPrep cytologic test (TCT) combined with HPV-DNA detection. All experiments were conducted in accordance with and approved by the World Medical Association Declaration of Helsinki and Institutional Research Ethics Committee involving Human Subjects of Southern Medical University and Huazhong University of Science and Technology. Informed consent was obtained from all participants prior to sample collection.
Cell lines and cell culture
The human CCa cell lines Siha, Caski, HeLa, C33A, ME180, and MS751 were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured according to the respective guidelines. hCEp cells (#7060) and HDLECs (#2010) were purchased from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in cervical epithelial cell medium (CerEpiCM; Cat. #7601; ScienCell) and endothelial cell medium (ECM; Cat. #1001; ScienCell), respectively, in a humidified incubator with 5% CO2 at 37°C. CCa cell lines were used within ten passages, whereas hCEp cells and HDLECs were used within six passages.
Exosome purification and characterization
Exosomes were purified from CCa cell-derived supernatant or clinical serum samples by ultracentrifugation as described previously.49 Cell culture supernatant was collected after 48 h and centrifuged at 500 × g for 10 min at 4°C, followed by 10,000 × g for 30 min at 4°C. Serum was diluted with an equal volume of PBS and centrifuged at 2,000 × g for 30 min at 4°C, followed by 12,000 × g for 45 min at 4°C. The supernatant was then passed through a 0.22-μm filter (Millipore, Burlington, MA, USA), ultracentrifuged at 110,000 × g for 70 min at 4°C, and washed in PBS using the same ultracentrifugation conditions. Pelleted exosomes were resuspended in 100 μL of PBS and subjected to several experiments, including morphological identification by TEM (H-7500; Hitachi, Tokyo, Japan); nanoparticle tracking analysis using a NanoSight NS300 (Malvern Instruments, UK); and RNA extraction, western blot, and cell treatment. For RNA extraction, 1 × 1011 of exosomes were first incubated with 50 U of RNase If (M0243; New England Biolabs, Ipswich, MA, USA) at 37°C for 30 min and inactivated at 75°C for 5 min. RNA was extracted using a miRNeasy Mini Kit (QIAGEN, Dusseldorf, Germany). For cell treatment, 1 × 1011 of exosomes (equivalent to 2 μg protein) was added to 1 × 105 recipient cells for 48 h.
Exosome uptake assay
For the exosome uptake experiments, hCEp and Siha cells were transduced with a lentiviral plasmid expressing a CD63-GFP fusion protein (GeneChem, Shanghai, China). hCEpGFP-exo or SihaGFP-exo cells were co-cultured with HDLECs (1:1) for 0, 1, 4, or 24 h using a semipermeable Transwell membrane. The HDLECs were then labeled with phalloidin and imaged by confocal microscopy.
Stable lentiviral transfection
Lenti-GFP vectors containing an miR-1468-5p overexpression sequence (miR-1468-5p) and its NC RNA (miR-NC) or containing an anti-1468-5p and its NC vector (anti-NC) were purchased from GeneChem (Shanghai, China). hCEp and Siha cells were transfected with lenti-miR-1468-5p overexpression or knockdown vectors, respectively. Polyclonal cells with GFP signals were purified for further experiments using a fluorescence-activated cell sorting flow cytometer.
Transient transfection with oligonucleotides and plasmids
miR-7704, miR-5787, miR-127-3p, miR-126-5p, miR-4492, miR-142-5p, miR-4488, miR-3656, miR-1468-5p, miR-126-3p, miR-5096, miR-199-3p, miR-409-3p, miR-223-3p, miR-4466, and miR-144-5p mimics or miR-NC and anti-1468-5p or anti-NC were designed and cloned by RiboBio (Guangzhou, China). The HMBOX1 coding sequence (without 3′ UTR) was cloned into the pCDNA3.1(+) vector (Thermo Fisher Scientific, Waltham, MA, USA), whereas an empty vector was used as a blank control. siHMBOX1 and its NC (siRNA) were designed and synthesized by GenePharma (Shanghai, China). Cells were transfected with miR mimics, miR inhibitors, siHMBOX1, and pCDNA3.1(+)-HMBOX1 using Lipofectamine 2000 Reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s protocol. RNA extraction, western blot, and in vitro assays were performed on cells 48 h after transfection. The siHMBOX1 and siRNA sequences are shown in Table S1.
RNA isolation and qRT-PCR
Total RNA was extracted from samples using TRIzol reagent (Thermo Fisher Scientific, Waltham, MA, USA), according to the manufacturer’s instructions. Reverse transcription was performed using a Mir-X miRNA First-Strand Synthesis Kit for miRNAs (TaKaRa, Dalian, China) or PrimeScript RT Master Mix for general genes (TaKaRa, Dalian, China). qRT-PCR was conducted using SYBR Premix Ex Taq (TaKaRa, Dalian, China) on an Applied Biosystems 7500 Fast Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA). Specific primer sets for miR-7704, miR-5787, miR-127-3p, miR-126-5p, miR-4492, miR-142-5p, miR-4488, miR-3656, miR-1468-5p, miR-126-3p, miR-5096, miR-199-3p, miR-409-3p, miR-223-3p, miR-4466, miR-144-5p, and U6 were purchased from RiboBio. miRNA and mRNA expression was normalized to U6 and glyceraldehyde 3-phosphate dehydrogenase (GAPDH), respectively. The primer sequences are shown in Table S1.
Western blot
Exosomes or cells were lysed with radioimmunoprecipitation assay (RIPA; Thermo Fisher Scientific, Waltham, MA, USA) buffer containing a complete protease inhibitor tablet (Roche, Mannheim, Germany). The lysates were cleared by centrifugation at 14,000 × g for 20 min and the supernatant fraction protein concentrations detected using a Pierce Bicinchoninic Acid (BCA) Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA). Next, 30 μg of lysate was electrophoresed by 10% SDS-PAGE, electro-transferred onto immobilon polyvinylidene membranes (Sigma-Aldrich, St. Louis, MO, USA), blocked for 1 h, and incubated overnight at 4°C with the following primary antibodies: TSG101 (1:1,000, ab125011), CD63 (1:1,000, ab216130), HSP70 (1:1,000, ab2787), SOCS1 (1 μg/mL, ab62584), SOCS2 (1 μg/mL, ab3692), and SOCS3 (1 μg/mL, ab16030; Abcam, Cambridge, UK); JAK2 (1:1,000, #3230), pJAK2 (1:1,000, #4406), STAT3 (1:1,000, #30835), pSTAT3 (1:1,000, #9145), and GAPDH (1:1,000, #2118; Cell Signaling Technology [CST], Danvers, MA, USA); and HMBOX1 (1:500, PA5-21558; Thermo Fisher Scientific, Waltham, MA, USA). Horseradish peroxidase (HRP)-linked anti-mouse (1:2000, ab6728) and anti-rabbit (1:2000, ab6721; Abcam, Cambridge, UK) immunoglobulin G (IgG) were used as secondary antibodies.
Multiplexed immunofluorescence staining
Multiplexed immunofluorescence staining was performed using an Opal 7-Color Fluorescence Immunohistochemistry (IHC) Kit (PerkinElmer, Waltham, MA, USA), according to the manufacturer’s protocol with the following primary antibodies: CD8 (1:800, ab93278; Abcam), PD-1 (1:800, ab137132; Abcam), D240 (1:500, M361929-2; Dako, Glostrup, Denmark), and PD-L1 (5 μg/mL, ab205921; Abcam). After deparaffinization, the sections were microwaved in antigen retrieval buffer for 45 s at 100°C, washed and blocked for 10 min at 25°C, and incubated with each primary antibody. Next, the slides were incubated with an HRP-broad spectrum SuperPicture Polymer Detection Kit (Thermo Fisher Scientific, Waltham, MA, USA) and incubated with Opal fluorochromes (Opal520, Opal570, Opal 620, and Opal 690), diluted 1:150 in amplification buffer (all provided by the Opal 7-Color Fluorescence IHC Kit) for 10 min at 25°C. Finally, the slides were microwave treated with AR6 buffer, incubated with a working 4′,6-diamidino-2-phenylindole (DAPI) solution (provided in the Opal 7-Color Fluorescence IHC Kit) for 5 min at 25°C, and mounted with ProLong Diamond Antifade Mounting Medium (Life Technologies). Images were obtained using a Zeiss LSM 880 Confocal Laser-Scanning Microscope (Jena, Germany).
Human-activated CD8+ T cell preparation
Peripheral blood mononuclear cells (PBMCs) were isolated from HLA-A24 healthy donors using Ficoll-Paque Plus (GE Healthcare, Chicago, IL, USA). CD8+ T cells were isolated from the PBMCs by magnetic bead purification using a Human CD8+ T Cell Enrichment Kit (STEMCELL Technologies, Vancouver, BC, Canada). HLA-A24 phenotype and purity (>90%) were checked by flow cytometry using anti-HLA-A24 (human) monoclonal antibody (mAb)-phycoerythrin (PE) (K0208-5; MBL International) and eF450-labeled antibodies against CD8 (48-0087-42; Thermo Fisher Scientific, Waltham, MA, USA). CD8+ T cells were grown in complete RPMI-1640 medium plus interleukin (IL)-2 (10 IU/mL) and activated (1 × 105 cells) by stimulation with plates coated with 2.5 μg/mL of anti-CD3 (OKT3; Thermo Fisher Scientific, Waltham, MA, USA) and 2 μg/mL of anti-CD28 (10F3; Thermo Fisher Scientific, Waltham, MA, USA) in vitro.
In vitro CD8+ T cell immune suppression
HDLECs (1 × 105) were pretreated with indicated exosomes for 48 h and washed twice with PBS for 3 min. To detect PD-L1 expression, surface staining was performed on HDLECs using PE-conjugated PD-L1 (329706; BioLegend, San Diego, CA, USA). Then, conditioned HDLECs were co-cultured with activated CD8+ T cells (1 × 105) to evaluate their immunity. To assess their cytotoxic activity, surface or intracellular CD8+ T cell staining was performed after 3 days of co-culture using the following anti-human or control antibodies: fluorescein isothiocyanate (FITC)-conjugated anti-CD69 (11-0699-42; Thermo Fisher Scientific, Waltham, MA, USA), PE-conjugated anti-IFN-γ (12-7319-42; Thermo Fisher Scientific, Waltham, MA, USA), allophycocyanin (APC)-conjugated α-PD-1 (17-2799-42; Thermo Fisher Scientific, Waltham, MA, USA), or fluorochrome-conjugated antibodies with a control IgG isotype. To analyze CD8+ T cell apoptosis, the proportion of apoptotic CD8+ T cells was examined using an Annexin V-APC Apoptosis Detection Kit (KGF004; KeyGEN BioTech, Nanjing, China) after 7 days of co-culture. Flow cytometry was performed by the Department of Immunology, School of Basic Medical Sciences, Southern Medical University, using a fluorescence-activated cell sorting (FACS) LSRFortessa (BD Biosciences, Franklin Lakes, NJ, USA), and data were analyzed using FlowJo VX software (Tree Star, Ashland, OR, USA).
In vivo xenograft model
Female B-NDG (non-obese diabetic [NOD]-PrkdcscidIL-2rgtm1/Bcgen) mice (5 weeks old) were purchased from Biocytogen (Beijing, China) and housed in an Association for Assessment and Accreditation of Laboratory Animal Care-licensed facility under sterile and standardized environmental conditions. The mice received autoclaved food and bedding and acidified drinking water ad libitum. All animal studies were approved by the Institutional Animal Research Ethics Committee of Guangzhou Medical University.
Human PBMCs were obtained as described above and were enriched for CD8+ T cells by T cell enrichment (STEMCELL Technologies, Vancouver, BC, Canada), according to the manufacturer’s instructions. Tumor-reactive CD8+ T cells were expanded by culture with mitomycin C-treated Siha cells for 10 days in RPMI 1640, supplemented with 10% fetal bovine serum (FBS) and IL-2. Tumor-reactive CD8+ T cells (2 × 106) were implanted into B-NDG mice by tail-vein injection. The next day, Siha/anti-1468-5p alone (1 × 105) with low PD-L1 expression or mixed with conditioned HDLECs (1 × 105) was subcutaneously implanted into the left flank of B-NDG mice (n = 6 per group), whereas α-PD-L1 (5 μg/g, MEDI4736; AstraZeneca, London, UK) or tofacitinib (3 μg/g, HY-40354; MedChemExpress [MCE], NJ, USA) was administered intraperitoneally on days 3, 6, 9, 12, 15, 18, and 21. Tumor size (cubic millimeters) was measured every 5 days and calculated as follows: volume = (width)2 × length/2. Xenograft tumors were harvested on day 25.
Tube-formation assay
Tube-formation assays were performed as described previously.50 Briefly, conditioned HDLECs (1.5 × 104) were diluted with 100 μL of serum-free RPMI 1640 in each well of a 96-well culture plate precoated with 50 μL of basement membrane matrix (BD Biosciences, Franklin Lakes, NJ, USA). The plate was then incubated at 37°C for 6 h and tube formation visualized under an inverted microscope (CX41; Olympus, Tokyo, Japan). The length of tube structures was analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
Luciferase activity assay
To analyze the targeting of HMBOX1 by miR-1468-5p, the putative miR-1468-5p complementary site in the 3′ UTR of HMBOX1 or its mutant sequence was cloned into the pmiR-RB-REPORT vector (RiboBio, Guangzhou, China). The resultant pmiR-RB-REPORT-HMBOX1-3′ UTR-WT, pmiR-RB-REPORT-HMBOX1-3′ UTR-MT, or pmiR-RB-REPORT control vectors were co-transfected into HLDECs with miR-1468-5p mimics or a NC. To determine the luciferase activity of the JAK2/STAT3 pathway, HDLECs transfected with luciferase constructs containing JAK2 (MV-h11386; Applied Biological Materials, Vancouver, BC, Canada) or STAT3 (#79730; BPS Bioscience, San Diego, CA, USA) were treated with miR-1468 mimics or the indicated exosomes. To analyze the targeting of the SOCS1 promoter by HMBOX1, HDLECs with HMBOX1 overexpression or knockdown were co-transfected with SOCS1 promoter plasmids containing firefly luciferase reporters with an internal control pRL-TK containing a full-length Renilla luciferase gene. Luciferase activity was detected using a Dual Luciferase Kit (Promega, Madison, WI, USA), according to the manufacturer’s instructions.
ChIP assays
Cells were cross linked with 1% formaldehyde and quenched in glycine solution and ChIP assays performed using an Enzymatic ChIP Kit (#9003; CST, Danvers, MA, USA), according to the manufacturer’s protocol. Anti-HMBOX1 antibodies (16123-1-AP; Proteintech, Chicago, IL, USA) and normal IgG (Millipore, Burlington, MA, USA) were used for immunoprecipitation, whereas ChIP-enriched DNA samples were quantified by qPCR to determine HBSs in the SOCS1 promoter region. Data were shown as relative enrichment normalized to control IgG. The primer sequences used for ChIP-qPCR are presented in Table S1.
miRNA microarray
Next generation sequencing of exosomal miRNAs and cellular miRNAs from hCEp and Siha was established using Illumina GAIIx (Illumina, San Diego, CA, USA). qRT-PCR validation was performed using TaqMan and SYBR Green assays. Data analysis was performed by South China Institute of Biomedicine (Guangzhou, China). Raw data were submitted to the National Center for Biotechnology Information (NCBI: GSE143339).
Statistical analysis
SPSS V.13.0 software (IBM, Armonk, NY, USA) was used for statistical analysis. Differences were considered statistically significant with p <0.05. All experiments were performed in triplicate, and quantitative data were presented as the mean ± SD. The statistical significance of differences between samples with non-normal distribution was identified using Mann-Whitney U tests, whereas two-tailed Student’s t tests was used for parametric variables, and the χ2 test was used for nonparametric variables. The comparisons of means among groups were analyzed by one-way analysis of variance (ANOVA), and the Dunn’s multiple comparison test was further used to determine significant differences between groups. OS and DFS were evaluated using the Kaplan-Meier method.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 82073165, 81672589, 81971341, and 81902621).
Author contributions
S.W., W. Wang, and C.Z. conceived and designed the experiments. C.Z., W. Wei, and J.M. carried out experiments. C.Z., W. Wei, and Y.Y. drafted the manuscript. L.L., Y.Z., and Z.W. contributed to the sample collection and interpretation of the data. J.M., X.C., and L.H. performed the statistical analysis.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.034.
Contributor Information
Wei Wang, Email: smugowwang@126.com.
Sha Wu, Email: shawu99@outlook.com.
Supplemental information
References
- 1.Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- 2.Borcoman E., Le Tourneau C. Pembrolizumab in cervical cancer: latest evidence and clinical usefulness. Ther. Adv. Med. Oncol. 2017;9:431–439. doi: 10.1177/1758834017708742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shekarian T., Sivado E., Jallas A.C., Depil S., Kielbassa J., Janoueix-Lerosey I., Hutter G., Goutagny N., Bergeron C., Viari A. Repurposing rotavirus vaccines for intratumoral immunotherapy can overcome resistance to immune checkpoint blockade. Sci. Transl. Med. 2019;11:eaat5025. doi: 10.1126/scitranslmed.aat5025. [DOI] [PubMed] [Google Scholar]
- 4.Liao W., Overman M.J., Boutin A.T., Shang X., Zhao D., Dey P., Li J., Wang G., Lan Z., Li J. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell. 2019;35:559–572.e7. doi: 10.1016/j.ccell.2019.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bordry N., Broggi M.A.S., de Jonge K., Schaeuble K., Gannon P.O., Foukas P.G., Danenberg E., Romano E., Baumgaertner P., Fankhauser M. Lymphatic vessel density is associated with CD8+ T cell infiltration and immunosuppressive factors in human melanoma. OncoImmunology. 2018;7:e1462878. doi: 10.1080/2162402X.2018.1462878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong H., Strome S.E., Salomao D.R., Tamura H., Hirano F., Flies D.B., Roche P.C., Lu J., Zhu G., Tamada K. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- 7.Heeren A.M., Punt S., Bleeker M.C., Gaarenstroom K.N., van der Velden J., Kenter G.G., de Gruijl T.D., Jordanova E.S. Prognostic effect of different PD-L1 expression patterns in squamous cell carcinoma and adenocarcinoma of the cervix. Mod. Pathol. 2016;29:753–763. doi: 10.1038/modpathol.2016.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pawelczyk K., Piotrowska A., Ciesielska U., Jablonska K., Gletzel-Plucinska N., Grzegrzolka J., Podhorska-Okolow M., Dziegiel P., Nowinska K. Role of PD-L1 Expression in Non-Small Cell Lung Cancer and Their Prognostic Significance according to Clinicopathological Factors and Diagnostic Markers. Int. J. Mol. Sci. 2019;20:824. doi: 10.3390/ijms20040824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matikas A., Zerdes I., Lövrot J., Richard F., Sotiriou C., Bergh J., Valachis A., Foukakis T. Prognostic Implications of PD-L1 Expression in Breast Cancer: Systematic Review and Meta-analysis of Immunohistochemistry and Pooled Analysis of Transcriptomic Data. Clin. Cancer Res. 2019;25:5717–5726. doi: 10.1158/1078-0432.CCR-19-1131. [DOI] [PubMed] [Google Scholar]
- 10.Lin H., Wei S., Hurt E.M., Green M.D., Zhao L., Vatan L., Szeliga W., Herbst R., Harms P.W., Fecher L.A. Host expression of PD-L1 determines efficacy of PD-L1 pathway blockade-mediated tumor regression. J. Clin. Invest. 2018;128:805–815. doi: 10.1172/JCI96113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Herbst R.S., Soria J.C., Kowanetz M., Fine G.D., Hamid O., Gordon M.S., Sosman J.A., McDermott D.F., Powderly J.D., Gettinger S.N. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuang D.M., Zhao Q., Peng C., Xu J., Zhang J.P., Wu C., Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. J. Exp. Med. 2009;206:1327–1337. doi: 10.1084/jem.20082173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barcellos-Hoff M.H., Lyden D., Wang T.C. The evolution of the cancer niche during multistage carcinogenesis. Nat. Rev. Cancer. 2013;13:511–518. doi: 10.1038/nrc3536. [DOI] [PubMed] [Google Scholar]
- 14.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9:654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 15.Yan W., Wu X., Zhou W., Fong M.Y., Cao M., Liu J., Liu X., Chen C.H., Fadare O., Pizzo D.P. Cancer-cell-secreted exosomal miR-105 promotes tumour growth through the MYC-dependent metabolic reprogramming of stromal cells. Nat. Cell Biol. 2018;20:597–609. doi: 10.1038/s41556-018-0083-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang T., Lv H., Lv G., Li T., Wang C., Han Q., Yu L., Su B., Guo L., Huang S. Tumor-derived exosomal miR-1247-3p induces cancer-associated fibroblast activation to foster lung metastasis of liver cancer. Nat. Commun. 2018;9:191. doi: 10.1038/s41467-017-02583-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou W., Fong M.Y., Min Y., Somlo G., Liu L., Palomares M.R., Yu Y., Chow A., O’Connor S.T., Chin A.R. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. 2014;25:501–515. doi: 10.1016/j.ccr.2014.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bai M., Li J., Yang H., Zhang H., Zhou Z., Deng T., Zhu K., Ning T., Fan Q., Ying G., Ba Y. miR-135b Delivered by Gastric Tumor Exosomes Inhibits FOXO1 Expression in Endothelial Cells and Promotes Angiogenesis. Mol. Ther. 2019;27:1772–1783. doi: 10.1016/j.ymthe.2019.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19.Zeng Z., Li Y., Pan Y., Lan X., Song F., Sun J., Zhou K., Liu X., Ren X., Wang F. Cancer-derived exosomal miR-25-3p promotes pre-metastatic niche formation by inducing vascular permeability and angiogenesis. Nat. Commun. 2018;9:5395. doi: 10.1038/s41467-018-07810-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y., Gu Y., Han Y., Zhang Q., Jiang Z., Zhang X., Huang B., Xu X., Zheng J., Cao X. Tumor Exosomal RNAs Promote Lung Pre-metastatic Niche Formation by Activating Alveolar Epithelial TLR3 to Recruit Neutrophils. Cancer Cell. 2016;30:243–256. doi: 10.1016/j.ccell.2016.06.021. [DOI] [PubMed] [Google Scholar]
- 21.Watanabe R., Shirai T., Namkoong H., Zhang H., Berry G.J., Wallis B.B., Schaefgen B., Harrison D.G., Tremmel J.A., Giacomini J.C. Pyruvate controls the checkpoint inhibitor PD-L1 and suppresses T cell immunity. J. Clin. Invest. 2017;127:2725–2738. doi: 10.1172/JCI92167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lund A.W., Duraes F.V., Hirosue S., Raghavan V.R., Nembrini C., Thomas S.N., Issa A., Hugues S., Swartz M.A. VEGF-C promotes immune tolerance in B16 melanomas and cross-presentation of tumor antigen by lymph node lymphatics. Cell Rep. 2012;1:191–199. doi: 10.1016/j.celrep.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 23.Hannafon B.N., Trigoso Y.D., Calloway C.L., Zhao Y.D., Lum D.H., Welm A.L., Zhao Z.J., Blick K.E., Dooley W.C., Ding W.Q. Plasma exosome microRNAs are indicative of breast cancer. Breast Cancer Res. 2016;18:90. doi: 10.1186/s13058-016-0753-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mascaux C., Angelova M., Vasaturo A., Beane J., Hijazi K., Anthoine G., Buttard B., Rothe F., Willard-Gallo K., Haller A. Immune evasion before tumour invasion in early lung squamous carcinogenesis. Nature. 2019;571:570–575. doi: 10.1038/s41586-019-1330-0. [DOI] [PubMed] [Google Scholar]
- 25.Stewart R., Morrow M., Hammond S.A., Mulgrew K., Marcus D., Poon E., Watkins A., Mullins S., Chodorge M., Andrews J. Identification and Characterization of MEDI4736, an Antagonistic Anti-PD-L1 Monoclonal Antibody. Cancer Immunol. Res. 2015;3:1052–1062. doi: 10.1158/2326-6066.CIR-14-0191. [DOI] [PubMed] [Google Scholar]
- 26.Green M.R., Monti S., Rodig S.J., Juszczynski P., Currie T., O’Donnell E., Chapuy B., Takeyama K., Neuberg D., Golub T.R. Integrative analysis reveals selective 9p24.1 amplification, increased PD-1 ligand expression, and further induction via JAK2 in nodular sclerosing Hodgkin lymphoma and primary mediastinal large B-cell lymphoma. Blood. 2010;116:3268–3277. doi: 10.1182/blood-2010-05-282780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lien M.Y., Tsai H.C., Chang A.C., Tsai M.H., Hua C.H., Wang S.W., Tang C.H. Chemokine CCL4 Induces Vascular Endothelial Growth Factor C Expression and Lymphangiogenesis by miR-195-3p in Oral Squamous Cell Carcinoma. Front. Immunol. 2018;9:412. doi: 10.3389/fimmu.2018.00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Durham G.A., Williams J.J.L., Nasim M.T., Palmer T.M. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol. Sci. 2019;40:298–308. doi: 10.1016/j.tips.2019.03.001. [DOI] [PubMed] [Google Scholar]
- 29.Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., van der Lee R., Bessy A., Chèneby J., Kulkarni S.R., Tan G. JASPAR 2018: update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018;46(D1):D1284. doi: 10.1093/nar/gkx1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou C.F., Ma J., Huang L., Yi H.Y., Zhang Y.M., Wu X.G., Yan R.M., Liang L., Zhong M., Yu Y.H. Cervical squamous cell carcinoma-secreted exosomal miR-221-3p promotes lymphangiogenesis and lymphatic metastasis by targeting VASH1. Oncogene. 2019;38:1256–1268. doi: 10.1038/s41388-018-0511-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frenel J.S., Le Tourneau C., O’Neil B., Ott P.A., Piha-Paul S.A., Gomez-Roca C., van Brummelen E.M.J., Rugo H.S., Thomas S., Saraf S. Safety and Efficacy of Pembrolizumab in Advanced, Programmed Death Ligand 1-Positive Cervical Cancer: Results From the Phase Ib KEYNOTE-028 Trial. J. Clin. Oncol. 2017;35:4035–4041. doi: 10.1200/JCO.2017.74.5471. [DOI] [PubMed] [Google Scholar]
- 32.Ribas A., Hamid O., Daud A., Hodi F.S., Wolchok J.D., Kefford R., Joshua A.M., Patnaik A., Hwu W.J., Weber J.S. Association of Pembrolizumab With Tumor Response and Survival Among Patients With Advanced Melanoma. JAMA. 2016;315:1600–1609. doi: 10.1001/jama.2016.4059. [DOI] [PubMed] [Google Scholar]
- 33.Liu J., Fan L., Yu H., Zhang J., He Y., Feng D., Wang F., Li X., Liu Q., Li Y. Endoplasmic Reticulum Stress Causes Liver Cancer Cells to Release Exosomal miR-23a-3p and Up-regulate Programmed Death Ligand 1 Expression in Macrophages. Hepatology. 2019;70:241–258. doi: 10.1002/hep.30607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mueller S.N., Vanguri V.K., Ha S.J., West E.E., Keir M.E., Glickman J.N., Sharpe A.H., Ahmed R. PD-L1 has distinct functions in hematopoietic and nonhematopoietic cells in regulating T cell responses during chronic infection in mice. J. Clin. Invest. 2010;120:2508–2515. doi: 10.1172/JCI40040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dieterich L.C., Ikenberg K., Cetintas T., Kapaklikaya K., Hutmacher C., Detmar M. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front. Immunol. 2017;8:66. doi: 10.3389/fimmu.2017.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang H., Liang Y., Anders R.A., Taube J.M., Qiu X., Mulgaonkar A., Liu X., Harrington S.M., Guo J., Xin Y. PD-L1 on host cells is essential for PD-L1 blockade-mediated tumor regression. J. Clin. Invest. 2018;128:580–588. doi: 10.1172/JCI96061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whiteside T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Invest. 2016;126:1216–1223. doi: 10.1172/JCI81136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kalluri R. The biology and function of exosomes in cancer. J. Clin. Invest. 2016;126:1208–1215. doi: 10.1172/JCI81135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cao L.Q., Yang X.W., Chen Y.B., Zhang D.W., Jiang X.F., Xue P. Exosomal miR-21 regulates the TETs/PTENp1/PTEN pathway to promote hepatocellular carcinoma growth. Mol. Cancer. 2019;18:148. doi: 10.1186/s12943-019-1075-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Felicetti F., De Feo A., Coscia C., Puglisi R., Pedini F., Pasquini L., Bellenghi M., Errico M.C., Pagani E., Carè A. Exosome-mediated transfer of miR-222 is sufficient to increase tumor malignancy in melanoma. J. Transl. Med. 2016;14:56. doi: 10.1186/s12967-016-0811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Casadei L., Calore F., Creighton C.J., Guescini M., Batte K., Iwenofu O.H., Zewdu A., Braggio D.A., Bill K.L., Fadda P. Exosome-Derived miR-25-3p and miR-92a-3p Stimulate Liposarcoma Progression. Cancer Res. 2017;77:3846–3856. doi: 10.1158/0008-5472.CAN-16-2984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Balsat C., Blacher S., Herfs M., Van de Velde M., Signolle N., Sauthier P., Pottier C., Gofflot S., De Cuypere M., Delvenne P. A specific immune and lymphatic profile characterizes the pre-metastatic state of the sentinel lymph node in patients with early cervical cancer. OncoImmunology. 2017;6:e1265718. doi: 10.1080/2162402X.2016.1265718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qin Y., Ekmekcioglu S., Forget M.A., Szekvolgyi L., Hwu P., Grimm E.A., Jazaeri A.A., Roszik J. Cervical Cancer Neoantigen Landscape and Immune Activity is Associated with Human Papillomavirus Master Regulators. Front. Immunol. 2017;8:689. doi: 10.3389/fimmu.2017.00689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mezache L., Paniccia B., Nyinawabera A., Nuovo G.J. Enhanced expression of PD L1 in cervical intraepithelial neoplasia and cervical cancers. Mod. Pathol. 2015;28:1594–1602. doi: 10.1038/modpathol.2015.108. [DOI] [PubMed] [Google Scholar]
- 45.Chen S., Saiyin H., Zeng X., Xi J., Liu X., Li X., Yu L. Isolation and functional analysis of human HMBOX1, a homeobox containing protein with transcriptional repressor activity. Cytogenet. Genome Res. 2006;114:131–136. doi: 10.1159/000093328. [DOI] [PubMed] [Google Scholar]
- 46.Wu L., Zhang C., Zhang J. HMBOX1 negatively regulates NK cell functions by suppressing the NKG2D/DAP10 signaling pathway. Cell. Mol. Immunol. 2011;8:433–440. doi: 10.1038/cmi.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Su L., Zhao H., Sun C., Zhao B., Zhao J., Zhang S., Su H., Miao J. Role of Hmbox1 in endothelial differentiation of bone-marrow stromal cells by a small molecule. ACS Chem. Biol. 2010;5:1035–1043. doi: 10.1021/cb100153r. [DOI] [PubMed] [Google Scholar]
- 48.Zeng Y.T., Liu X.F., Yang W.T., Zheng P.S. REX1 promotes EMT-induced cell metastasis by activating the JAK2/STAT3-signaling pathway by targeting SOCS1 in cervical cancer. Oncogene. 2019;38:6940–6957. doi: 10.1038/s41388-019-0906-3. [DOI] [PubMed] [Google Scholar]
- 49.Wu X.G., Zhou C.F., Zhang Y.M., Yan R.M., Wei W.F., Chen X.J., Yi H.Y., Liang L.J., Fan L.S., Liang L. Cancer-derived exosomal miR-221-3p promotes angiogenesis by targeting THBS2 in cervical squamous cell carcinoma. Angiogenesis. 2019;22:397–410. doi: 10.1007/s10456-019-09665-1. [DOI] [PubMed] [Google Scholar]
- 50.Liu D., Li L., Zhang X.X., Wan D.Y., Xi B.X., Hu Z., Ding W.C., Zhu D., Wang X.L., Wang W. SIX1 promotes tumor lymphangiogenesis by coordinating TGFβ signals that increase expression of VEGF-C. Cancer Res. 2014;74:5597–5607. doi: 10.1158/0008-5472.CAN-13-3598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.