

Abstract
Ongoing clinical trials for treatment of beta-globinopathies by gene therapy involve the transfer of the beta-globin gene, which requires integration of three to four copies per genome in most target cells. This high proviral load may increase genome toxicity, potentially limiting the safety of this therapy and relegating its use to total body myeloablation. We hypothesized that introducing an additional hypersensitive site from the locus control region, the complete sequence of the second intron of the beta-globin gene, and the ankyrin insulator may enhance beta-globin expression. We identified a construct, ALS20, that synthesized significantly higher adult hemoglobin levels than those of other constructs currently used in clinical trials. These findings were confirmed in erythroblastic cell lines and in primary cells isolated from sickle cell disease patients. Bone marrow transplantation studies in beta-thalassemia mice revealed that ALS20 was curative at less than one copy per genome. Injection of human CD34+ cells transduced with ALS20 led to safe, long-term, and high polyclonal engraftment in xenograft experiments. Successful treatment of beta-globinopathies with ALS20 could potentially be achieved at less than two copies per genome, minimizing the risk of cytotoxic events and lowering the intensity of myeloablation.
Keywords: gene addition, gene therapy, beta-globinopathies, lentiviral vectors, hematopoietic stem cells
Graphical abstract
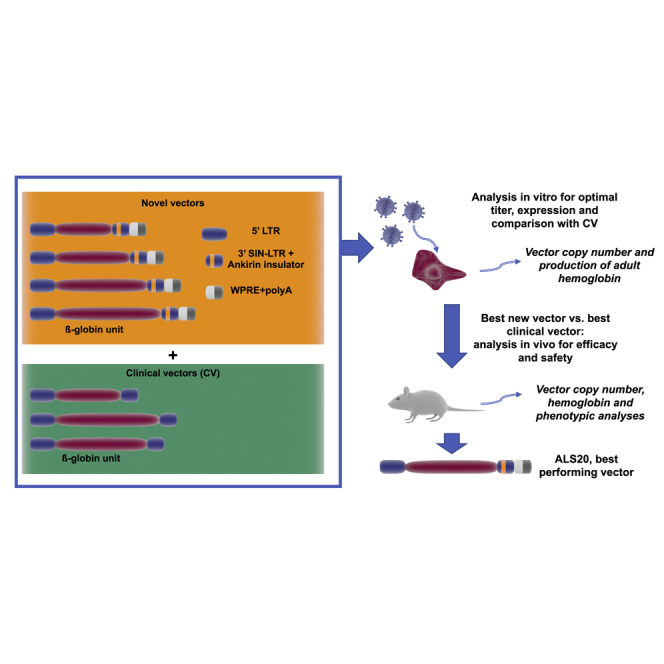
Increase of beta-globin expression by gene addition is critical to successfully curing patients with beta-thalassemia and sickle cell disease. Here, Breda and colleagues report genomic features that maximize transgene expression at low genome integration rates, preserving efficacy and safety and potentially reducing the burden of autologous bone marrow transplantation.
Introduction
Gene therapy holds potential for the treatment of patients affected by beta-globinopathies. However, initial clinical trials run by Bluebird Bio in beta-thalassemia patients reported relatively low integration of the BB305 vector (with a vector copy number [VCN] < 2) and suboptimal hemoglobin (Hb) synthesis.1 Similarly, BB305 exhibited insufficient expression of vector-derived Hb for treatment of sickle cell disease (SCD), and it only ameliorated SCD in one out of seven children affected by the disease.2,3 Nonetheless, transduction efficiency was improved in the most recent phase III trials, reaching VCN ∼3–4, and transfusion independence was reported in four out of five patients with non-beta0beta0 genotypes and in three out of four patients with severe beta0beta0 or IVS1-110 genotypes.4,5 Furthermore, this improved protocol led to 5.3 g/dL of curative Hb and a reduction in vaso-occlusive events in SCD patients.6 The use of a different lentiviral vector (GLOBE) led to transfusion discontinuation in three out of four pediatric patients with beta0beta0 or severe beta+ genotypes; however, this was not the case for adult patients.7 Finally, a trial at the Memorial Sloan Kettering Cancer Center with the TNS93.55 lentiviral vector failed due to low engraftment levels of transduced hematopoietic stem cells (HSCs).8 Although the recent trials with BB305 treatment improved patient outcomes with a VCN > 2, an increased integration rate may impair the success of gene therapy by increasing genome toxicity9,10 and ruling out lower myeloablation requirements and lower cost of viral transductions.
The lentiviral vectors used in all of these trials contained the human beta-globin gene along with its promoter, hypersensitive site (HS)2, HS3, and HS4 of the locus control region (LCR), and a truncated version (Δ) of intron 2 (Δ 374 bp and Δ 593 bp).11, 12, 13, 14, 15, 16, 17 The main modifications included insertion of the deleted sequence of intron 2, which was not included in oncoretroviral vectors because it decreased viral titer and production. For this reason, the region was also excluded from lentiviral vectors; however, it may contribute to sustained beta-globin expression because it contains an OCT-1 binding site and an enhancer, which were shown to augment beta-globin expression in transgenic mice.18 Therefore, we hypothesize that inclusion of the full intron as well as other genomic elements, such as HS1 of the LCR and the ankyrin element, which directs position-independent, uniform expression of the beta-globin gene and other transgenes, will enhance beta-globin expression.11,19, 20, 21, 22, 23 In addition, we further modified the location of the woodchuck post-regulatory element (WPRE) in our vectors to preserve titer and reduce size of the integration cassette, as indicated in detail in the Results section.
Using a variety of in vitro and in vivo screening approaches, we identified a new vector, ALS20, that expresses significantly higher transgenic Hb levels compared with other vectors that are currently used to treat beta-globinopathies. This finding has many potential benefits for the successful treatment of patients affected by beta-globinopathies, including a low VCN and genomic integrations, a minimized chance of clonal expansion and tumorigenesis, a reduced cost of therapy, as well as a potentially reduced need for myeloablation.
Results
ALS20 expresses significantly high levels of Hb in HUDEP cells and in patient-derived CD34+ erythroid cells
We generated novel lentiviral vectors (ALS16–ALS20) by combining different genomic elements, including a long or short version of the beta-globin promoter, a portion of HS1, HS2, HS3, and HS4 of the LCR, the full coding sequence, and a 3′ beta-globin enhancer (Figures 1 and S1). These vectors also included the insulator element from the human ankyrin gene. Inclusion of this element in all of our constructs was supported by a previous study that showed a marked increase in Hb expression in mice transduced with a parental vector, AnkT9W11 (Figure 1). As a benchmark, we generated vectors similar to those that have already been used in clinical trials: CV-I12 (for BB305 or Zynteglo, ClinicalTrials.gov: NTC03207009 and NTC02906202; recently approved by the European Medicines Agency), CV-II24 (for GLOBE; ClinicalTrials.gov: NTC02453477), and CV-III13 (for TNS9.3.55; ClinicalTrials.gov: NTC01639690). All these vectors carried the T87Q modification of the beta-globin gene14 for uniform comparison and identification of the beta-globin transgene.
Figure 1.

Maps of AnkT9W, AnkT9W-derived ALS16–20 vectors, and clinical vectors (CVs) engineered in other laboratories
Viral features—LTR, long terminal repeat; SD, splice donor; SA, splice acceptor; RRE, reverse responsive element; sinLTR, self-inactivating long terminal repeat. Transgene features—e+, β-globin gene enhancer; βp, β-globin gene promoter; HS, hypersensitive site; 3′ UTR, 3′ untranslated region of the β-globin gene. β-globin gene exons and introns are indicated by red and blue segments, respectively. The length of the segments from the β-globin gene promoter to the end of the enhancer and the mini LCR are indicated under each segment. T87Q indicates a threonine to glutamine substitution at amino acid 87. Miscellaneous features—ankyrin, human ankyrin gene insulator element. The region indicated as a Δ+ comprises a 374-bp DNA sequence that was reinstated to reproduce the wild-type intron II (850 bp) in all ALS vectors. Note, WPRE, woodchuck hepatitis post-transcriptional regulatory element; pA, bovine growth hormone polyadenylation sequence, which are not included in the diagrams but are present in these constructs downstream of the sinLTR (see Figure S1 for reference). More specifically, five main modifications of the AnkT9W vector were performed to make ALS17. First, we replaced the truncated intravenous 2 (IVS2) sequence with the full endogenous IVS2 sequence. Second, we removed the woodchuck post-regulatory element (WPRE) from the vector integration sequence and placed it directly after the 3′ LTR. Third, we placed a strong bovine growth hormone poly(A) signal after the WPRE. Fourth, we altered the β-globin gene to express β-globin with the T87Q modification (threonine to glutamine substitution at amino acid 87) instead of endogenous β-globin. Last, we extended the hypersensitive site (HS)4 (1,157 bp) and removed a short polylinker from the 3′ LTR flanking the ankyrin sequence. For ALS16, we replaced the ALS17 LCR with that of the CV-I vector, as previously published. In ALS18, we swapped the 500-bp promoter from ALS16 with a shorter version (~200 bp) to asses if the shorter β-globin promoter was equally efficient in transcribing the β-globin gene. ALS19 and ALS20 have the same elements as ALS16 and also include the core element of HS1 (556 bp), as previously reported25 in either sense or antisense orientation.
Using CRISPR/Cas9 technology, we modified the human umbilical-cord-blood-derived erythroid progenitor (HUDEP-2) cell line and generated a new erythroid cell line, HUDEP M#13, which completely expresses a mutant form of human hemoglobin (HbM) (Figure S2A). Sequencing analysis indicated that clone M#13 contained one allele with a deletion of codon 6 (Glu6), which leads to a corresponding protein one amino acid shorter (146 aa) than the canonic beta-globin chain (Figure S2B). The second allele carried a single-nucleotide deletion, which results in a premature termination codon (PTC) and no globin chain synthesis (Figure S2B). HbM is distinguishable from adult hemoglobin A (HbA) that is produced by any of the proposed therapeutic lentiviral vectors (Figure S2C). We performed a dose-response analysis of HUDEP M#13 cells that were treated with each vector by correlating the HbA levels with the corresponding VCN.11 In summary, ALS20 was among the best performing vectors and produced 157%, 84%, and 40% more HbA than CV-II, CV-III, and CV-I, respectively (Figures 2A and 2B). To corroborate these findings, we performed parallel assessments using ALS20, ALS17, and CV-I in primary human ErPCs, which were isolated from patients with SCD (Figures 2C and 2D).26 We excluded from this set of experiments CV-II and III, which reported significantly lower expression of beta-globin in HUDEP M#13 cells. Additionally, while no significant HbA% difference was detected at VCN = 1 between ALS20 and ALS19 in HUDEP M#13 cells, we pursued further experiment with ALS20 only, because the orientation of HS1 in this construct was the preferred one (antisense, see supplementary methods, under “plasmid generation” for details). In SCD cells, ALS20 produced an average of 21% HbA at a VCN = 1 (p < 0.001), whereas CV-I produced an average of 14.9% HbA at a VCN = 1 (p < 0.001). These results confirmed a 40% increase in HbA production with ALS20 compared with CV-I, as shown in HUDEP M#13 cells. Due to the lower dose-response values and consistently lower titer (data not shown), most likely due to increase length of the transgenic cassette compared with ALS20, ALS17 was not used in subsequent experiments.
Figure 2.

Hemoglobin A synthesis in HUDEP M#13 cells, primary human sickle cell disease cells, and in beta0 beta0 erythroblasts after transduction with beta-globin lentiviral vectors
(A) Representative graphics of hemoglobin separation obtained by cation exchange liquid chromatography in untreated (top) and ALS-treated (bottom) HUDEP M#13 cells at day 7 after differentiation, according to the protocol described in the “Human primary cell isolation, propagation, transduction, and analyses” section in the Supplemental materials and methods. (B) Dose-response analyses obtained plotting integration levels (vector copy number [VCN], x axes) against adult hemoglobin (HbA) percentages (y axes) to establish a dose-response effect in cells treated with CV-I and CV-II versus ALS16–20 within a VCN range between 0.25 and 3 in M#13 cells. (C) Representative graphics of hemoglobin separation obtained by cation exchange liquid chromatography in untreated and ALS-treated sickle cell disease (SCD) primary erythroblasts after differentiation. Primary cell culture conditions and infection are described in the “Human primary cell isolation, propagation, transduction, and analyses” section in the Supplemental materials and methods. (D) Dose-response analyses obtained plotting integration levels (VCN, x axes) against HbA percentages (y axes) in cells treated with CV-I versus ALS17 and ALS20 within a VCN range between 0.25 and 3 in primary SCD erythroblasts isolated from patients and differentiated in vitro (n = 4). At the bottom of each plot in (B) and (D), linear regression analyses for comparison of HbA levels are indicated by black arrows. Results from mixed-effects linear regression models are reported as difference with 95% confidence interval (CI) at a VCN = 1. In (B), for CV-I, p = 0.1875; for CV-II, p = 0.0055; for CV-III, p = 0.0002; for ALS16, p = 0.0024; for ALS17, p = 0.0018; for ALS18, p = 0.0003; for ALS19, p = 0.019; and for ALS20, p < 0.0001. In (D), for CV-I and ALS20, p < 0.001; for ALS17, p = 0.345. (E) Representative graphics of hemoglobin separation obtained by cation exchange liquid chromatography in untreated and ALS-treated beta0beta0 primary erythroblasts after differentiation. (F) Dose-response analyses obtained plotting integration levels (VCN, x axes) against HbA percentages (y axes) to establish a dose-response effect in three independent experiments with three different beta0beta0 erythroblast progenitor cell samples treated with ALS20, p < 0.001. In (B) and (D), each data point represents an average of three independent transductions, whereas in (F), each data point represents a single transduction. Hemoglobin peaks—HbF, fetal hemoglobin; mutant Hb and MutAcHb, mutant form of hemoglobin containing a shorter aminoacidic sequence (146 aa) (produced by HUDEP M#13) and its acetylated form; HbA and AcHbA, adult hemoglobin and its acetylated form; HbA2, secondary adult hemoglobin (α2δ2); HbS and AcHbS, sickle hemoglobin and its acetylated form; α-aggr, α-globin aggregates.
We also performed a dose-response analysis using ALS20 in beta0beta0 ErPCs (Figures 2E and 2F). Untreated cells did not produce HbA, and the mean fetal hemoglobin (HbF) value was 82% (Figure 2E), with the HbA2 variant making up the remainder of the Hb produced. Cells treated with ALS20 showed a significant correlation between an increased HbA percentage and VCN (p < 0.001; Figure 2F). On average, one copy of ALS20 produced 42% of the entire HbA (p < 0.001), which was 52% of the HbA levels produced in ErPCs from asymptomatic carriers.11 This suggests that in beta0beta0 HSCs, two copies per genome of ALS20 could significantly improve anemia and reduce transfusion dependency.
ALS20 viral genome and transgenic cassette exhibit integrity and clonal abundance after integration
We removed the WPRE from the vector integrating sequence and placed it directly after the 3′ long terminal repeat (LTR) as shown in Figure 1. Since the HIV polyadenylation signal in the SIN-3′-LTR leaks,27 allowing 3′-viral RNA readthrough, a strong and accurate bovine growth hormone poly(A) signal (bGHpoly(A))28 was added after the WPRE. The rationale for this design was to maintain WPRE in the viral RNA to preserve high viral titers by maximizing the amount of retrotranscribed sequence while minimizing potential recombination and integration of the WPRE into the host genome.29 Furthermore, inclusion of bGHpoly(A) aimed to limit inclusion of non-functional RNA sequence derived from the plasmid downstream of the viral vector.
To verify transgenic molecule integrity in viral preparations, we sequenced cDNA obtained from the viral genomic RNA of ALS20 and aligned the number of reads from the viral sequences with the consensus sequence (Figure S3A). From these analyses, 97,278 reads aligned to the viral cassette within the reference plasmid sequence and no high abundance mismatches were observed in the coding sequence or regulatory regions. Appendix SA shows the predicted sequence. Less than 0.2% of reads aligned to the plasmid backbone, indicating minimal plasmid DNA contamination, as expected with research-grade viral preparations. These findings were confirmed by droplet digital (dd)PCR that was performed with the same cDNA template used for sequencing (Figures S3B and S3C). The ddPCR results indicated that a high proportion of the viral molecules included the complete cassette, ending with the WPRE followed by the bGHpolyA.
To demonstrate that intron 2 did not undergo recombination, we amplified the region between the beta-globin promoter and GAG codon, which included the entire transgenic cassette, in ALS20-tranduced human CD34+ cells and erythroid progenitor cells (ErPCs). We confirmed that intron 2 was integrated intact (Figure 3A) and that no cryptic splicing occurred in the entire sequence between the LTRs of the viral molecule (Figures 3B). The primer locations in the viral cassette are schematized in Figure 3C.
Figure 3.

Viral genome and transgenic cassette stability after integration and polyclonality
(A) Representation of amplicons obtained after semiquantitative PCR of genomic DNA isolated from primary beta0beta0 erythroblasts, human CD34+ cells transduced with ALS20, and from control ALS20 plasmid and proviral cDNA using the Tr1A and Tr2A primer set, which amplifies a 3.63-kb transgenic region within ALS20. (B) Representation of amplicons obtained after semiquantitative PCR of genomic DNA isolated from primary beta0beta0 erythroblasts, human CD34+ cells transduced with ALS20, and from control ALS20 plasmid using the Int1A and Int2A primer set, which amplifies the 7.6-kb cassette that is included between the viral long terminal repeats. (C) Schematic representation of the viral components and location of the primers designed to amplify the regions obtained in (A) and (B). (D and E) Representative heatmaps summarizing the distribution sites of integration relative to the genomic features or mapped sites of epigenetic modifications or bound proteins, respectively, in human hematopoietic stem cells. The distribution of lentiviral vector integration sites were compared with the distribution of random sites using the receiver operating characteristic (ROC) curve method.30 The columns indicate the sample analyzed. Data from samples transduced with ALS20, ALS17, and CV-I (three biological replicas per vector) were analyzed with samples from patients treated safely in clinical trial for Wiskott-Aldrich syndrome (WAS) and chronic granulomatous disease (CGD). (D) The rows indicate comparisons to genomic feature annotations that are mapped on the human genome (hg38). Associations are quantified as ROC areas, which compare experimental integration site sets to computationally generated random distributions. Values are color coded on each tile and range between 0 (negatively associated; blue) and 1 (positively associated; red). The most appropriate chromosomal interval for comparison is not known in advance, so multiple intervals were used. p values were calculated using the Wald test using a chi-square distribution; no correction for multiple comparisons was applied. p values are marked on the heatmap tiles: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. RefSeq.counts indicate comparisons based on the RefSeq gene numbers in the indicated intervals; gene.width indicates the width of genes containing integration sites; general.width indicates the width of intergenic regions that host integration sites; start.dist shows comparisons based on distances to transcription start sites; and boundary.dist indicates comparisons based on distances to gene boundaries. (E) Similar to what is presented in (D) but with comparisons of integration site distributions and random distributions relative to mapped epigenetic marks and sites of bound proteins in human hematopoietic stem cells. All comparisons used 10-kb intervals.
We assessed the genomic integration distributions of ALS20, ALS17, and CV-I in human CD34+ cells according to a transduction protocol that recapitulates clinical trial standard operating procedures. Data from samples transduced with ALS20, ALS17, and CV-I were analyzed with samples from patients treated safely in clinical trials. These samples were obtained from post-transduction, pre-infusion samples of lentiviral vector-transduced human HSCs in two successful clinical trials for Wiskott-Aldrich syndrome (WAS) and chronic granulomatous disease (CGD).31,32 The distribution of lentiviral vector integration sites were compared with the distribution of random sites using the receiver operating characteristic (ROC) curve method.30
Through the use of various parameters, our analysis confirmed that none of the samples exhibited preferential or skewed genomic integrations. We also compared the vector integration site distributions and mapped chromosomal features or epigenetic modifications in the cells (Figures 3D and 3E). Distributions diverged from random controls (asterisks on tiles), but the patterns generally followed those seen for CV-I and lentiviral integration in successful human gene therapy trials to treat WAS and CGD. Integration mostly occurred in or near transcriptional units or features that correlated with active transcription, similar to what was observed in many other studies with HIV and lentiviral vectors.33 Data comparisons for the ASL17 and ASL20 vectors showed a slight but significantly reduced integration frequency near cancer-associated genes compared with the results of trials for lentiviral vectors that were deemed safe (Table S1). Thus, the integration site distributions seen in our ASL17 and ASL20 pre-infusion products resemble those observed with the CV-I vector and other previous trials with safe outcomes.
ALS20 ameliorates anemia in beta-thalassemia mice with different levels of disease severity
We compared the ability of ALS20 to correct anemia to that of CV-I in beta-thalassemia mice with different levels of disease severity. We used the Hbbth3/+ and Hbbth3/th3 mouse models, which mimic the milder non-beta0beta0 and the more severe beta0beta0 forms of human thalassemia, respectively.
Hbbth3/+ mice treated with ALS20 showed significantly higher Hb concentrations (14.5 ± 2.7 g/dL), red blood cell (RBC) counts (10.7 ± 2, 106/mL), and hematocrit (HCT) (48.3% ± 9.4%), with lower reticulocyte counts (5% ± 2.4%) and RDW-CV (17.7% ± 2.5%) compared with the untreated Hbbth3/+ control mice, in which these same parameters were 7.4 ± 0.6 g/dL, 6.4 ± 0.5 106/mL, 26% ± 2%, 28.1% ± 7.3%, and 32.8% ± 3.8%, respectively (Figure 4A). All parameters in ALS20-treated Hbbth3/+ mice were comparable to those detected in healthy control Hbb+/+ chimeras. In comparison to ALS20-treated mice, mice treated with CV-I exhibited significantly lower Hb concentrations (11.9 ± 1.8 g/dL), RBC counts (8.7 ± 1.4, 106/mL), and HCT (39% ± 7.3%), at VCN ∼1. Similar differences in Hb levels were also detected in Hbbth3/+ mice treated ALS20 and CV-I (Hb = 11.74 ± 1.37 g/dL and 10.16 ± 1.48 g/dL, respectively) with lower and comparable integration (VCN = 0.43; Figure S4A). CV-I-treated mice had higher RDW-CV (218.8 ± 23) compared with healthy control Hbb+/+ chimeras, in which the RDW-CV was 148 ± 14.7. Consequently, the curves that defined Hb concentrations, RBC counts, and HCT over VCN of all animals treated with ALS20 converged to normal values (light blue bars) at a lower VCN compared with those of mice treated with CV-I (Figure 4B).
Figure 4.

Dose-response analysis for viral copy number and hemoglobin concentration and complete blood count assessment of experimental mice
(A) Hemoglobin (Hb) concentration (top left), hematocrit (HCT, %; top middle), red blood cell (RBC) count (top right), reticulocyte count (%; bottom left), and RBC distribution width coefficient of variation (RDW-CV; bottom right) in Hbb+/+ (n = 3, 100% donor chimerism), Hbbth3/+ (n = 22, 96.3% donor chimerism), and in Hbbth3/+ mice treated with ALS20-T87Q (VCN = 1 ± 0.1) or CV-I (VCN = 1 ± 0.2) (n = 9, 95.5% donor chimerism, and 5, 93% donor chimerism for ALS20 and CV-I-treated groups, respectively). Groups were tested by non-parametric ANOVA analyses. Asterisks indicate p values that refer to ALS20, while pound sign indicates p values that refer to CV-I. ∗ or #p ≤ 0.05; ##p ≤ 0.005; and ∗∗∗∗p < 0.0001. (B) Hb concentration (top left), HCT (%; top middle), RBC count (top right), reticulocyte count (%; bottom left), and RDW (bottom right) are plotted against the VCN in all ALS20- or CV-I-treated Hbbth3/+ mice. Blue horizontal bars represent values in wild-type Hbbt+/+ mice. R squared for each trendline are indicated in red for ALS20 and in black for CV-I.
ALS20-treated Hbbth3/+ mice displayed significantly decreased erythroid expansion and an increased proportion of mature erythroid cells in their bone marrow and spleens along with the absence of splenomegaly compared with untreated Hbbth3/+ mice (Figures 5, S4B, and S4C). The distribution of splenic erythroid cells in ALS20-treated Hbbth3/+ mice was comparable to that of control Hbb+/+ mice (Figures 5A and 5B). At a VCN as low as 0.4, ALS20-treated Hbbth3/+ mice showed splenic recovery with 86% of mature RBCs (Figure 5B). Whereas CV-I-treated Hbbth3/+ mice showed a normalized splenic erythroid cell proportion only at a VCN > 1, indicating that the mice with a VCN < 1 were still compensating for anemia by producing a larger proportion of ErPCs. This was further confirmed by splenic architecture analyses (Figure S5A and S5B), which showed that at a VCN as low as 0.22, ALS20-treated Hbbth3/+ mice displayed an improved distribution of splenic red and white pulp, whereas CV-I-treated Hbbth3/+ mice did not until a VCN > 1 was reached. Furthermore, ALS20-treated Hbbth3/+ chimeras (VCN ≥ 0.22) did not show iron accumulation in the liver and kidneys, as observed in untreated Hbbth3/+ chimeras (Figures S6A and S6B).
Figure 5.

Spleen phenotype correction in ALS20- and CV-I-treated primary Hbbth3/+ chimeras
(A) Flow cytometry assessment of the proportion of Ter119+ erythroid cells (hematopoietic progenitors - > RBC) and their distribution by CD44+ and forward scatter (FCS) in the spleens of Hbbth3/+ and Hbb+/+ control mice. (B) Flow cytometry assessment of the spleens of Hbbth3/+ mice that were treated with either ALS20 or CV-I. (C) Ratio of spleen over body weight (left). (D) Proportion of R1 (pro-erythroblasts), R2 (basophilic), R3 (polychromatic), R4 (orthochromatic and reticulocytes), and R5 (mature erythrocytes) in the spleen of Hbb+/+ (n = 3), Hbbth3/+ (n = 21), and in Hbbth3/+ mice treated with ALS20-T87Q (VCN = 1 ± 0.1) or CV-I (VCN = 1 ± 0.2) (n = 7 and 6 for ALS20 and CV-I treated groups, respectively). Groups were tested by non-parametric ANOVA and multiple comparison analyses. Q1 through 4 gates represent live cells gated for CD71 and Ter119 markers. Asterisks indicate p values referred to ALS20, while pound signs indicate p values referred to CV-I. #p ≤ 0.05; ## or ∗∗p ≤ 0.005; ∗∗∗p ≤ 0.0005, and ∗∗∗∗p < 0.0001.
We assessed the long-term expression of the beta-globin transgene in secondary chimeras that were generated with bone marrow harvested from primary ALS20-treated Hbbth3/+ chimeras. In the secondary chimeras, we measured the proportion of immature/mature erythroid cells in the spleens and bone marrow (Figures 6A and 6B), complete blood cell counts (Figure 6C), spleen size (Figure 6D), and the proportion of the chimeric Hb products (Figure 6E). In one cohort (secondary transplant A; Figure 6C, left), all parameters, including the chimeric Hb expression levels, were comparable to that of the primary chimeras, confirming that sustained amelioration of the anemia phenotype was achieved with ALS20. In another cohort (secondary transplant B; Figure 6C, right), the hematological parameters were greatly improved but somewhat blunted, due to significantly lower donor chimerism (45.65% ± 9.64%) compared with the primary chimera (85%).
Figure 6.

Maintained beta-thalassemia phenotype correction in secondary ALS20 Hbbth3/+ chimeras
(A and B) Flow cytometry assessment of Ter119+ erythroid cells (hematopoietic progenitors - > RBC) and their distribution by CD44+ and forward scatter (FCS) in the spleens and bone marrow, respectively, of a primary ALS20 Hbbth3/+ chimera (left column) and three secondary chimeras (right, other three columns) that were generated with the bone marrow harvested from a primary chimera. (C) Values of VCN, chimeric hemoglobin % (resulting from the association of two human beta-globin chains and two mouse alpha-globin chains), and hematopoietic values from complete blood cell counts of the secondary chimeras (n = 3 for each group) from two independent transplants normalized by values of primary chimeras (indicated by the red line at 1, for individual primary chimeras), with a 1:3 ratio between primary and secondary chimeras analyzed in two independent transplants (transplant A and B). (D) Images of spleens and relative spleen/body weight ratios in untreated Hbbth3/+ chimera, primary ALS20 treated Hbbth3/+ chimera, and secondary ALS20 treated Hbbth3/+ chimeras. (E) Chromatographic separation of chimeric hemoglobin and mouse hemoglobin (endogenous mouse α2β2) of mice represented in (A) and (B).
Chimeras injected with untreated Hbbth3/th3 fetal liver cells (FLCs) were euthanized 7–8 weeks after bone marrow transplantation due to extremely severe anemia (mean Hb concentrations 3.7 g/dL), whereas treated chimeras survived and were harvested at the anticipated endpoint (5–6 months after bone marrow transplantation). Notably, the ALS20-treated mouse (VCN = 2.9, Hb 15.9 g/dL) exhibited much more robust phenotype improvement, as shown by the bone marrow and spleen erythroid cell distributions, splenic size reduction, and normalization of hematological parameters, compared with the CV-I-treated mouse with a higher VCN = 6 and lower Hb concentration (8.55 g/dL) (Figures S7A and S7B; Table S2). Nonetheless, both of these mice only expressed chimeric Hb (Figure S7C). All together, the comparative data in bone marrow chimeras using Hbbth3/+ HSC and Hbbth3/th3 FLC cells indicate that ALS20 maintained its superiority in producing curative Hb as observed in vitro using HUDEP M#13- and SCD-derived CD34+ cells. To evaluate long-term amelioration in the beta0beta0 mouse model, we generated secondary chimeras with primary ALS20-treated Hbbth3/th3 bone marrow (VCN = 2.9). At 5 months after the bone marrow transplantation, these mice exhibited similar donor chimerism, VCN, hematological parameters, as well as spleen/body weight ratio and spleen composition (not shown), to those of the primary chimera (Figure S7D), which confirmed stable and long-term expression of the transgene in the beta0beta0 mouse model.
ALS20-transduced human CD34+ cells show safe and long-term engraftment in xenograft experiments
We transduced healthy donor CD34+ human HSCs with ALS20 and tested cell products (fresh or in culture) in immunocompromised NSG mice. After 4 months, the mice were harvested for further analyses. Overall examinations for toxicity included complete pathological assessments, full necropsies with macroscopic post-mortem examinations, and analysis of the hematopoietic tissues, including the bone marrow and spleens. These analyses confirmed the absence of proliferative disorders or neoplastic transformation, indicating that engraftment of ALS20-transduced human CD34+ cells was safe and that there were no remarkable differences between mice injected with ASL20-transduced or control cells (Appendix SB). Human donor chimerism and the proportion of human HSCs were comparable among the experimental arms. In fact, the proportions of human CD45+ over mouse CD45+ cells were 1.08 ± 0.96, 0.8 ± 0.65, and 0.38 ± 0.21, and the proportions of human CD34+ over human CD45+ cells were 0.11 ± 0.02, 0.07 ± 0.03, and 0.08 ± 0.02 in mice injected with freshly thawed, untreated, or ALS20-treated cells, respectively (Figure 7A). The average VCN in ALS20-treated mice was 0.8 ± 0.56 (Figure 7B).
Figure 7.

Long-term engraftment of ALS20-transduced human CD34+ cells in immunocompromised mice
(A) Flow cytometry assessment of human CD45+ cells over mouse CD45+ cells (top) and of human CD34+ cells within the human CD45+ cells in bone marrow harvested 4 months after bone marrow transplantation (bottom). (B) VCN in bone marrow of NSG mice (n = 8) injected with human CD34+ cells transduced with ALS20 and collected after 4 months. (C and D) Comparison of the distributions of lentiviral vector integration sites in bone marrow specimens from the 8 animals injected with ALS20 (from A and B, coded with p and a number) relative to similar data from human stem cell gene therapy studies with safe outcomes for 6 peripheral blood mononuclear cell (PBMC) samples, 6 months after gene therapy for WAS and CGD. Samples (in columns) are compared to genomic features and epigenetic marks (in rows), respectively, as in Figure 3.
Integration site distributions were quantified by ligation-mediated PCR and compared with those of historic controls.31,32 These distributions diverged from random controls, and integration predominately occurred in transcriptional units and near features associated with active transcription (Figures 7C and 7D). Integration frequency near cancer-associated genes was assessed to ensure that transduced cell populations were not evolving by clonal expansion due to insertional mutagenesis, which was suggested by the results of an earlier study on the treatment of beta-thalassemia by lentiviral vector transduction.34,35 No global integration frequency increase was observed in or near cancer-associated genes when compared with the integration patterns observed in successful gene therapy trials to treat WAS and CGD (Table S3). Clone sizes varied in the samples, but no obvious signs of genotoxicity were observed when cell expansion was assessed in regard to vector integration near cancer-associated genes, which was observed in at least 50% of the clonal populations.
Discussion
Our previous study as well as clinical trials clearly showed that a VCN > 2 was required for successful lentiviral vector use to ameliorate the phenotype of patients with the beta0beta0 genotype compared to the non-beta0beta0 genotype.1,7,11 Consistent with these findings, recent clinical trials conducted by Bluebird Bio indicated that a VCN of 3–4 was required for curative outcomes in patients with either SCD or the beta0beta0 genotype. Based on reported data, one copy of BB305, which is similar to CV-I, makes an average of 6.9 g/dL of HbA.36 Our goal was to achieve higher Hb levels, while possibly reducing the VCN below that used in current trials. Our data from HUDEP M#13 cells and SCD patient samples showed that one copy of ALS20 had the potential to make 40% more HbA than CV-I, translating to at least 9 g/dL HbA per VCN. Therefore, our findings have a greater clinical impact on patients, especially those who require higher Hb production to become transfusion independent. Here, we showed that two ALS20 copies were sufficient to elevate HbA levels in beta0beta0 primary cells to the levels observed in asymptomatic carrier primary cells. However, we believe that this projection is somewhat conservative and that the dose-response trend may be underestimated due to high HbF expression, which has been previously reported to be associated with the culture system.11,26
Compared to humans, busulfan-based conditioning in mice is not ideal because it does not always achieve complete myeloablation. Despite this intrinsic limitation, we assessed the ability of ALS20 to ameliorate the phenotype of Hbbth3/+ mice not only at a low VCN, but also under limited chimerism conditions. The Hbbth3/+ mice exhibited ∼35% improvement in Hb synthesis with ALS20 treatment compared with CV-I treatment. This increase in Hb synthesis, with a reduced VCN as low as 0.22, resulted in significant amelioration of ineffective erythropoiesis, which was demonstrated by comparing the relative abundance of erythroid populations and by the absence of splenomegaly in the ALS20-treated mice. In the beta0beta0 mice, we observed dramatic recovery of physiological erythropoietic features and complete reversal of splenomegaly with ALS20 treatment at a VCN of 2.9, whereas CV-I treatment at a VCN > 6 failed to achieve complete reversal of anemia and splenomegaly. Although the quantity of this data is limited, a secondary transplantation from ALS20-treated mice showed long-term stability of transgene expression with consistent improvement in erythropoiesis and splenomegaly. Furthermore, this data likely underestimates the efficacy of ALS20 that may be achieved with complete myeloablation and pancellular reconstitution, which are common in a conventional bone marrow transplantation regimen.
The use of a relatively low VCN and/or partial chimerism could offer major advantages. One advantage is a possible reduction in genome toxicity. Genome toxicity is likely higher at a VCN ≥ 3,9,10 which is required in many current clinical trials. A second advantage is that if relatively low VCNs are effective in restoring normal erythropoiesis, the conditioning regimens for patients could be lowered. This would make gene therapy more appealing and would potentially result in a higher number of patients willing to undergo the procedures. An additional advantage is that lower integration requirements would result in successful transduction with lower viral loads, which may significantly reduce the financial burden of gene therapy, making it more affordable.
Compared to current trials that aim to reactivate HbF by gene addition or genome editing (NCT04208529 and NCT03745287), our approach may offer the advantage of normalizing anemia with high beta-globin expression in less cells at a relatively low VCN. Furthermore, a powerful vector may help to develop non-toxic myeloablative regimens and in vivo delivery methods because lower integration and/or a reduction in the number of corrected cells are expected under such conditions.
On a safety standpoint, ALS20 viral particles did not contain unwanted genomic RNA byproducts derived from cryptic splicing events, which was confirmed by detection of the full-length viral molecules in ALS20-transduced primary patient cells and human HSCs. Additionally, pre-infusion, ALS20-transduced human CD34+ cells displayed an integration pattern comparable to those of historical pre-infusion products used in previous clinical trials that successfully treated WAS and CGD.31,32 We reproduced this same pattern in long-term xenograft experiments, in which NSG mice were injected with ALS20-treated human HSCs. These mice exhibited normal physiology without any proliferative disorders or neoplastic transformation compared with mice injected with untreated cells. Considering the body of evidence presented in this work, we believe that ALS20 is an outstanding candidate for the successful treatment of beta-globinopathies. Given its improved capability to increase Hb synthesis compared with other vectors used in clinical trials, its use may reduce the requirement for high viral integration, with a VCN as low as 2, for effective treatment. This may not only result in a safer therapy with a reduced probability of genome toxicity and milder conditioning requirements, but it may also improve the efficacy and offer a competitive product for the gene-therapy market.
Materials and methods
Viral genome stability analysis
Research-grade ALS20 viral particles were lysed for RNA isolation and subsequent reverse transcription, and the cDNA was sequenced. Specifically, the DNA library was prepared using the Illumina Nextera XT DNA library prep kit, according to the manufacturer’s protocol. Vector cDNA was fragmented and tagged by the Nextera method, and Agencourt AMPure XP magnetic beads were used for PCR purification. PicoGreen and Qubit were used for DNA quantification, and fragment size was determined by Agilent TapeStation. The DNA library was denatured and loaded on an Illumina V2 500-cycle flow cell on a MiSeq system sequencer at a concentration of 8 pM with 15% PhiX spike-in. Raw demultiplexed sequencing reads were processed using the Sunbeam pipeline (v2.1).37 Reads were filtered for quality, and adapters were trimmed. Then, reads were aligned to the reference vector sequence, and the results were viewed and analyzed using IGV 2.7.0 (Integrative Genomics Viewer), which was downloaded from the IGV website (https://www.broadinstitute.org/igv).
Evaluation of integrity and recombination potential of the viral expression cassette in the host genome
Integrity of the transgenic viral expression cassette was assessed using genomic DNA from CD34+ or ErPCs that were previously transduced with ALS20. PCR primers were designed to amplify the transgenic cassette, which was entirely encompassed between the 5′ and 3′ LTRs of the vector. An expand long template PCR system (Roche, Basel, Switzerland) and 10 mM deoxyribonucleotide triphosphate (dNTP) mix (Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA) were used. ALS20 plasmid and its proviral cDNA were used as positive controls. PCR products were run on a 1.2% agarose gel.
Bone marrow transplantation
C57BL/6-Hbbth3/+ or wild-type (WT) CD45.2 mice were bred at the Children’s Hospital of Philadelphia (CHOP) facility. Lin− HSCs were isolated from the bone marrow of donor mice (8–10 weeks old) via immunomagnetic separation using a mouse lineage cell depletion kit (Miltenyi Biotec, Auburn, CA, USA). Lin− cells were transduced overnight at a viral multiplicity of infection (MOI) of 50–100 in StemSpan Serum Free Expansion Medium (SFEM) culture medium (StemCell Technologies, Vancouver, Canada) supplemented with 50 ng/mL recombinant murine stem cell factor (SCF), 10 ng/mL recombinant murine interleukin-6 (IL-6), 6 ng/mL IL-3 (PeproTech, Rocky Hill, NJ, USA), 200 mM L-glutamine, 100 U/mL penicillin/streptomycin (Gibco, Thermo Fisher Scientific), and 8 ng/mL polybrene. A minimum of 300,000 viable Lin− donor cells in phosphate-buffered saline (PBS) (Gibco, Thermo Fisher Scientific) were intravenously injected into pre-conditioned Hbbth3/+ recipient mice (8–10 weeks old) of the opposite gender. Sub-lethal myeloablation was carried out by daily intraperitoneal injections of busulfan (Otsuka America Pharmaceutical, Rockville, MD, USA) at a total dose of 70 mg/kg over a duration of 4 days. Hbbth3/th3 FLCs were obtained as previously described,15 transduced as previously described for Lin− HSCs, and intravenously injected into lethally irradiated Pepcb/BoyJ CD45.1 recipient mice (8–10 weeks old; The Jackson Laboratory, Bar Harbor, ME, USA).
Xenograft
We transduced healthy donor CD34+ human HSCs (STEMCELL Technologies, #70002.4) with ALS20. The cells were kept in culture for the pre-stimulation (24 h) and infection periods (24 h), and the transduced or mock-treated cells were then injected into immunocompromised NOD scid gamma (NSG) mice. A third group of animals were instead injected with freshly thawed cells. NSG recipient mice were pre-conditioned with an intravenous injection of prescription-grade busulfan (3 mg/kg; Sagent Pharmaceuticals, Schaumburg, Illinois, USA). After 24 h, the recipient mice were intravenously injected with 150,000–300,000 CD34+ cells suspended in 110 mL of 2% fetal bovine serum (FBS) in PBS.
Integration site distribution analysis
Integration site distribution analysis was performed using the INSPIIRED pipeline, as previously described.38,39 Clonal abundance was quantified using the sonic abundance method.40 Associations between integration site distribution and genomic features were quantified using the ROC curve method.30 Cancer-associated genes were quantified by comparison to the allOnco list, which compiles known cancer-associated genes in humans and their homologs in other organisms.41
Institutional Review Board (IRB) regulation
Patients were recruited and samples were obtained according to the Declaration of Helsinki, following the approval by the Children’s Hospital of Philadelphia IRB (#15-012123). All subjects gave their informed consent prior to inclusion in the study.
Institutional Animal Care and Use Committee (IACUC) regulation
All bone marrow transplantation studies in mice were approved by the IACUC (protocol #1173) at the CHOP. Reporting of animal studies have been provided in accordance with ARRIVE guidelines. Studies in the immunocompromised animals were done at the Stem Cell and Xenograft Core Facility at the University of Pennsylvania, and all procedures were conducted under IACUC protocol #803506.
Data Sharing Statement
For original data, please contact L.B. at bredal@email.chop.edu. The pathology assessment in NSG mice can be found in Appendix SB. All integration site data were deposited in the NCBI sequence read archive (SRA) with accession ID for NC: PRJNA649778, available at https://www.ncbi.nlm.nih.gov/sra/PRJNA649778.
Acknowledgments
This work was supported by the Comprehensive Center for the Cure of Sickle Cell Disease and other Red Cell Disorders (CuRED) – Frontier Program at the Children’s Hospital of Philadelphia (CHOP), the Commonwealth Universal Research Enhancement Program (CURE) – Department of Health of Pennsylvania, and the Associazione Veneta per la Lotta alla Talassemia (AVLT) of Rovigo, Italy. We would like to thank the caregivers, patients, and their families at the Comprehensive Thalassemia Center and the Comprehensive Sickle Cell Center at CHOP. We would also like to thank Gregory and Jami Davis, Gerald and Elizabeth Rorer, donors who wish to remain anonymous, generous supporters of the 2018 CHOP Daisy Days campaign and The Runway, Patrick and Devaki Jean for their leadership through CHOP’s Sickle Cell Parent Network, and Megan Fredericks at the CHOP Foundation for their constant support. This paper is dedicated to the memory of Kwame Ohene-Frempong and his family.
Author contributions
L.B. and S.R. designed the experiments; V.G., K.S.-W., and J.L.K. provided patient specimens; X.S. and G.A.D.-D. provided technical and analytical support with the xenograft work; E.R., C.A.A., J.C.T., and N.H. performed the pathology assessments and analyses of the xenograft work; L.B., V.G., N.T., D.J., A.D., C.C., Y.I., O.A., A.D., J.E., and T.D.R. performed the experiments; L.B., S.R., J.L.K., and F.D.B. wrote the initial draft of the manuscript; Y.N. and R.K. provided the parental HUDEP-2 cell line. L.B., S.R., and F.D.B. oversaw all experiments and data interpretation; all authors contributed to the revision of the manuscript.
Declaration of interests
S.R. serves in the scientific advisory board of Disc Medicine, IONIS Pharmaceuticals, and Meira GTx. S.R. is a consultant for BVF Partners LP; Cambridge Healthcare Research; Celgene Corp.; First Manhattan Co.; FORMA Therapeutics; Ghost Tree Capital; Incyte Corp.; Keros Therapeutics, Inc.; Noble Insight; Protagonist Therapeutics; Rallybio, LLC; Sanofi Aventis U.S., Inc.; Slingshot Insight; Techspert.io; venBio Select LLC; and Catenion. S.R. has stock options in Meira GTx.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2020.12.036.
Contributor Information
Laura Breda, Email: bredal@email.chop.edu.
Stefano Rivella, Email: rivellas@email.chop.edu.
Supplemental information
Document S1. Supplemental materials and methods, Figures S1–S7, and Appendix A
Document S2. Tables S1–S5
Document S3. Appendix B
Document S4. Article plus supplemental information
References
- 1.Thompson A.A., Kwiatkowski J., Rasko J., Hongeng S., Schiller G.J., Anurathapan U., Cavazzana M., Ho J., von Kalle C., Kletzel M. Lentiglobin Gene Therapy for Transfusion-Dependent β-Thalassemia: Update from the Northstar Hgb-204 Phase 1/2 Clinical Study. Blood. 2016;128:1175. [Google Scholar]
- 2.Ribeil J.A., Hacein-Bey-Abina S., Payen E., Magnani A., Semeraro M., Magrin E., Caccavelli L., Neven B., Bourget P., El Nemer W. Gene Therapy in a Patient with Sickle Cell Disease. N. Engl. J. Med. 2017;376:848–855. doi: 10.1056/NEJMoa1609677. [DOI] [PubMed] [Google Scholar]
- 3.Kanter J., Walters M.C., Hsieh M.M., Krishnamurti L., Kwiatkowski J., Kamble R.T., von Kalle C., Kuypers F.A., Cavazzana M., Leboulch P. Interim Results from a Phase 1/2 Clinical Study of Lentiglobin Gene Therapy for Severe Sickle Cell Disease. Blood. 2016;128:1176. [Google Scholar]
- 4.Thompson A.A., Walters M.C., Kwiatkowski J.L., Hongeng S., Porter J.B., Sauer M.G., Thrasher A.J., Thuret I., Elliot H., Tao G. Northstar-2: Updated Safety and Efficacy Analysis of Lentiglobin Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia and Non-β0/β0 Genotypes. Blood. 2019;134(Suppl. 1):3543. [Google Scholar]
- 5.Lal A., Locatelli F., Kwiatkowski J.L., Kulozik A.E., Yannaki E., Porter J.B., Thuret I., Sauer M.G., Elliot H., Chen Y. Northstar-3: Interim Results from a Phase 3 Study Evaluating Lentiglobin Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia and Either a β0 or IVS-I-110 Mutation at Both Alleles of the HBB Gene. Blood. 2019;134(Suppl. 1):815. [Google Scholar]
- 6.Kanter J., Tisdale J.F., Mapara M.Y., Kwiatkowski J.L., Krishnamurti L., Schmidt M., Miller A.L., Pierciey F.J., Jr., Huang W., Ribeil J.-A. Resolution of Sickle Cell Disease Manifestations in Patients Treated with Lentiglobin Gene Therapy: Updated Results from the Phase 1/2 Hgb-206 Group C Study. Blood. 2019;134(Suppl. 1):990. [Google Scholar]
- 7.Marktel S., Scaramuzza S., Cicalese M.P., Giglio F., Galimberti S., Lidonnici M.R., Calbi V., Assanelli A., Bernardo M.E., Rossi C. Intrabone hematopoietic stem cell gene therapy for adult and pediatric patients affected by transfusion-dependent ß-thalassemia. Nat. Med. 2019;25:234–241. doi: 10.1038/s41591-018-0301-6. [DOI] [PubMed] [Google Scholar]
- 8.Mansilla-Soto J., Riviere I., Boulad F., Sadelain M. Cell and Gene Therapy for the Beta-Thalassemias: Advances and Prospects. Hum. Gene Ther. 2016;27:295–304. doi: 10.1089/hum.2016.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baum C., Düllmann J., Li Z., Fehse B., Meyer J., Williams D.A., von Kalle C. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood. 2003;101:2099–2114. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- 10.Woods N.B., Muessig A., Schmidt M., Flygare J., Olsson K., Salmon P., Trono D., von Kalle C., Karlsson S. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101:1284–1289. doi: 10.1182/blood-2002-07-2238. [DOI] [PubMed] [Google Scholar]
- 11.Breda L., Casu C., Gardenghi S., Bianchi N., Cartegni L., Narla M., Yazdanbakhsh K., Musso M., Manwani D., Little J. Therapeutic hemoglobin levels after gene transfer in β-thalassemia mice and in hematopoietic cells of β-thalassemia and sickle cells disease patients. PLoS ONE. 2012;7:e32345. doi: 10.1371/journal.pone.0032345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Negre O., Eggimann A.V., Beuzard Y., Ribeil J.A., Bourget P., Borwornpinyo S., Hongeng S., Hacein-Bey S., Cavazzana M., Leboulch P., Payen E. Gene Therapy of the β-Hemoglobinopathies by Lentiviral Transfer of the β(A(T87Q))-Globin Gene. Hum. Gene Ther. 2016;27:148–165. doi: 10.1089/hum.2016.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.May C., Rivella S., Callegari J., Heller G., Gaensler K.M., Luzzatto L., Sadelain M. Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human beta-globin. Nature. 2000;406:82–86. doi: 10.1038/35017565. [DOI] [PubMed] [Google Scholar]
- 14.Pawliuk R., Westerman K.A., Fabry M.E., Payen E., Tighe R., Bouhassira E.E., Acharya S.A., Ellis J., London I.M., Eaves C.J. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science. 2001;294:2368–2371. doi: 10.1126/science.1065806. [DOI] [PubMed] [Google Scholar]
- 15.Rivella S., May C., Chadburn A., Rivière I., Sadelain M. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood. 2003;101:2932–2939. doi: 10.1182/blood-2002-10-3305. [DOI] [PubMed] [Google Scholar]
- 16.Arumugam P.I., Scholes J., Perelman N., Xia P., Yee J.K., Malik P. Improved human beta-globin expression from self-inactivating lentiviral vectors carrying the chicken hypersensitive site-4 (cHS4) insulator element. Mol. Ther. 2007;15:1863–1871. doi: 10.1038/sj.mt.6300259. [DOI] [PubMed] [Google Scholar]
- 17.Roselli E.A., Mezzadra R., Frittoli M.C., Maruggi G., Biral E., Mavilio F., Mastropietro F., Amato A., Tonon G., Refaldi C. Correction of beta-thalassemia major by gene transfer in haematopoietic progenitors of pediatric patients. EMBO Mol. Med. 2010;2:315–328. doi: 10.1002/emmm.201000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buzina A., Lo M.Y., Moffett A., Hotta A., Fussner E., Bharadwaj R.R., Pasceri P., Garcia-Martinez J.V., Bazett-Jones D.P., Ellis J. Beta-globin LCR and intron elements cooperate and direct spatial reorganization for gene therapy. PLoS Genet. 2008;4:e1000051. doi: 10.1371/journal.pgen.1000051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pasceri P., Pannell D., Wu X., Ellis J. Full activity from human beta-globin locus control region transgenes requires 5‘HS1, distal beta-globin promoter, and 3’ beta-globin sequences. Blood. 1998;92:653–663. [PubMed] [Google Scholar]
- 20.Lisowski L., Sadelain M. Locus control region elements HS1 and HS4 enhance the therapeutic efficacy of globin gene transfer in beta-thalassemic mice. Blood. 2007;110:4175–4178. doi: 10.1182/blood-2007-08-108647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gallagher P.G., Romana M., Tse W.T., Lux S.E., Forget B.G. The human ankyrin-1 gene is selectively transcribed in erythroid cell lines despite the presence of a housekeeping-like promoter. Blood. 2000;96:1136–1143. [PubMed] [Google Scholar]
- 22.Romero Z., Campo-Fernandez B., Wherley J., Kaufman M.L., Urbinati F., Cooper A.R., Hoban M.D., Baldwin K.M., Lumaquin D., Wang X. The human ankyrin 1 promoter insulator sustains gene expression in a β-globin lentiviral vector in hematopoietic stem cells. Mol. Ther. Methods Clin. Dev. 2015;2:15012. doi: 10.1038/mtm.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leboulch P., Huang G.M., Humphries R.K., Oh Y.H., Eaves C.J., Tuan D.Y., London I.M. Mutagenesis of retroviral vectors transducing human beta-globin gene and beta-globin locus control region derivatives results in stable transmission of an active transcriptional structure. EMBO J. 1994;13:3065–3076. doi: 10.1002/j.1460-2075.1994.tb06605.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pasceri P., Pannell D., Wu X., Ellis J. 5'HS1 and the distal beta-globin promoter functionally interact in single copy beta-globin transgenic mice. Ann. N Y Acad. Sci. 1998;850:377–381. doi: 10.1111/j.1749-6632.1998.tb10497.x. [DOI] [PubMed] [Google Scholar]
- 25.Miccio A., Cesari R., Lotti F., Rossi C., Sanvito F., Ponzoni M., Routledge S.J., Chow C.M., Antoniou M.N., Ferrari G. In vivo selection of genetically modified erythroblastic progenitors leads to long-term correction of beta-thalassemia. Proc. Natl. Acad. Sci. USA. 2008;105:10547–10552. doi: 10.1073/pnas.0711666105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Breda L., Motta I., Lourenco S., Gemmo C., Deng W., Rupon J.W., Abdulmalik O.Y., Manwani D., Blobel G.A., Rivella S. Forced chromatin looping raises fetal hemoglobin in adult sickle cells to higher levels than pharmacologic inducers. Blood. 2016;128:1139–1143. doi: 10.1182/blood-2016-01-691089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaiss A.K., Son S., Chang L.J. RNA 3′ readthrough of oncoretrovirus and lentivirus: implications for vector safety and efficacy. J. Virol. 2002;76:7209–7219. doi: 10.1128/JVI.76.14.7209-7219.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goodwin E.C., Rottman F.M. The 3′-flanking sequence of the bovine growth hormone gene contains novel elements required for efficient and accurate polyadenylation. J. Biol. Chem. 1992;267:16330–16334. [PubMed] [Google Scholar]
- 29.Zufferey R., Donello J.E., Trono D., Hope T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999;73:2886–2892. doi: 10.1128/jvi.73.4.2886-2892.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Berry C., Hannenhalli S., Leipzig J., Bushman F.D. Selection of target sites for mobile DNA integration in the human genome. PLoS Comput. Biol. 2006;2:e157. doi: 10.1371/journal.pcbi.0020157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hacein-Bey Abina S., Gaspar H.B., Blondeau J., Caccavelli L., Charrier S., Buckland K., Picard C., Six E., Himoudi N., Gilmour K. Outcomes following gene therapy in patients with severe Wiskott-Aldrich syndrome. JAMA. 2015;313:1550–1563. doi: 10.1001/jama.2015.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kohn D.B., Booth C., Kang E.M., Pai S.Y., Shaw K.L., Santilli G., Armant M., Buckland K.F., Choi U., De Ravin S.S., Net4CGD consortium Lentiviral gene therapy for X-linked chronic granulomatous disease. Nat. Med. 2020;26:200–206. doi: 10.1038/s41591-019-0735-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schröder A.R., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110:521–529. doi: 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- 34.Cavazzana-Calvo M., Payen E., Negre O., Wang G., Hehir K., Fusil F., Down J., Denaro M., Brady T., Westerman K. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bushman F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020;28:352–356. doi: 10.1016/j.ymthe.2019.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson A.A., Walters M.C., Kwiatkowski J., Rasko J.E.J., Ribeil J.A., Hongeng S., Magrin E., Schiller G.J., Payen E., Semeraro M. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018;378:1479–1493. doi: 10.1056/NEJMoa1705342. [DOI] [PubMed] [Google Scholar]
- 37.Clarke E.L., Taylor L.J., Zhao C., Connell A., Lee J.J., Fett B., Bushman F.D., Bittinger K. Sunbeam: an extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome. 2019;7:46. doi: 10.1186/s40168-019-0658-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sherman E., Nobles C., Berry C.C., Six E., Wu Y., Dryga A., Malani N., Male F., Reddy S., Bailey A. INSPIIRED: A Pipeline for Quantitative Analysis of Sites of New DNA Integration in Cellular Genomes. Mol. Ther. Methods Clin. Dev. 2016;4:39–49. doi: 10.1016/j.omtm.2016.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berry C.C., Nobles C., Six E., Wu Y., Malani N., Sherman E., Dryga A., Everett J.K., Male F., Bailey A. INSPIIRED: Quantification and Visualization Tools for Analyzing Integration Site Distributions. Mol. Ther. Methods Clin. Dev. 2016;4:17–26. doi: 10.1016/j.omtm.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berry C.C., Gillet N.A., Melamed A., Gormley N., Bangham C.R., Bushman F.D. Estimating abundances of retroviral insertion sites from DNA fragment length data. Bioinformatics. 2012;28:755–762. doi: 10.1093/bioinformatics/bts004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sadelain M., Papapetrou E.P., Bushman F.D. Safe harbours for the integration of new DNA in the human genome. Nat. Rev. Cancer. 2011;12:51–58. doi: 10.1038/nrc3179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Document S1. Supplemental materials and methods, Figures S1–S7, and Appendix A
Document S2. Tables S1–S5
Document S3. Appendix B
Document S4. Article plus supplemental information