

Summary
Haemophilia A is a congenital bleeding disorder characterised by recurrent haemorrhages into the major joints. Haemophilic arthropathy is a well-established outcome of recurrent joint bleeding; however, it is clear that multiple factors determine the extent and severity of its occurrence. We sought to identify genetic factors related to abnormalities in range of motion (ROM) in the knees, ankles and elbows in a cohort of children and adolescents with haemophilia A not treated primarily with regular prophylaxis. Using data from the Haemophilia Growth and Development Study, we examined associations between 13,342 genetic markers and ROM scores measured at six-month intervals for up to seven years. As a first step, ordered logistic regression models were fit for each joint separately. A subset of SNP markers showing significant effects (p<0.01) on the right and left sides for at least two joints were included in a full model fit using a multivariate generalised linear mixed model assuming an ordinal response. The models contained all ROM scores obtained at all visits. Twenty-five markers analysed in the full model showed either increased or decreased risk of ROM abnormalities at the p<0.001 level. Several genes identified at either the first or second stage of the analysis have been associated with arthritis in a variety of large studies. Our results support the likelihood that risk for haemophilic arthropathy is associated with genetic factors, the identification of which holds promise for further advancing the individualisation of treatment.
Keywords: Genetics, haemophilia, haemarthrosis, joint range of motion, arthritis
Introduction
The moderate and severe forms of both haemophilia A and B demonstrate frequent haemorrhages into the major joints of the limbs, specifically the knees, ankles and elbows (1–3). While there is a broad variation in age at onset, these are seen to occur prior to 24 months of age, when the child initially becomes actively mobile (4–6). The pathophysiologic changes arising from these recurrent bleeds result in the clinical hallmark of the disease, specifically haemophilic arthropathy, which is responsible for the significant morbidity, pain and decreased quality of life that is so well documented in these individuals (7).
Research has shown that blood within a joint produces a cascade of events that results in joint inflammation which continues until blood is no longer present within the impacted joint (8–12). The observed clinical changes and tissue inflammatory pathology show similarities to those seen in other degenerative joint diseases such as rheumatoid arthritis (13, 14). All anatomic structures in and around the joint are affected, including the joint synovium, cartilage, epiphyseal structures as well as the bones around the impacted joint. Although the specific pathophysiologic processes underlying this arthropathy have not been elucidated, documented changes include the degradation of heme with deposition of iron, infiltration of neutrophils, macrophages and lymphoid cells, swelling of the joint tissues with fluid deposition, angiogenesis and proliferation of soft tissue structures including the synovium (11, 12).
There is insufficient knowledge regarding the pathogenesis of haemophilic arthropathy. In vitro and animal research has identified two major processes associated with the development of this complication. The first is the sequence of events mediated by biologic response modifiers that lead to soft tissue and synovial proliferation (15, 16). The second is the degenerative changes that impact the articular cartilage (17). The role of iron, derived from red cells as a consequence of the intra-articular haemorrhages, has received focus (11).
While research has demonstrated that blood in the joint is the key event in triggering the pathophysiology of haemophilic arthropathy, it is apparent that other factors underlie the extent and severity of the processes that produce the ultimate outcomes. Clearly, the amount of blood within the joint as well as the frequency of subsequent bleeding into the same joint are major underlying events for the development of haemophilic arthropathy. On this basis, the current major therapeutic target is to prevent bleeding into the major joints of children and adults with haemophilia and this approach has resulted in considerable forward movement in limiting joint damage (18–20). Even under careful preventative treatment schedules, however, both clinical and radiologic pathologic joint changes have been noted during the first few years of life. This research has also shown that there is clinically significant variability in the phenotypic expression of joint pathology (18, 21, 22). Consequently, it is clear that further knowledge is needed to understand the basic molecular changes within the joints that are set in motion by a bleeding event and this knowledge will be extended by elucidating the mechanisms that underpin the observed phenotypic variability.
We used clinical data collected from participants in the multicentre Haemophilia Growth and Development Study (HGDS) to explore genetic factors associated with the development of joint disease in children and adolescents with haemophilia A. Genetics data were generated as part of the Haemophilia Inhibitor Genetics Study Combined Cohort (23) (HIGS), in which the HGDS participated.
Methods
Study population
The HGDS is a longitudinal cohort investigation conducted in 14 US haemophilia treatment centres (HTC). Details of recruitment of the HGDS cohort have been reported by Hilgartner (24). In summary, a census was completed at participating centres for every individual receiving care in that centre. Eligibility criteria included age between 6 and 19 years, at least one visit to the HTC within the two years prior to the census, use of 100 U of clotting factor or more per kilogram of body weight or receipt of nine or more clotting factor infusions during that period. Between 1989 and 1990, a total of 333 children and adolescents, 274 of whom had haemophilia A, were enrolled and followed prospectively at six-month intervals for up to seven years. The enrolled cohort was representative of the total population identified in the census with regard to type and severity of haemophilia, number of infusions in the two years prior to enrolment, prevalence of human immunodeficiency virus (HIV) infection, and racial and ethnic origin. At entry, and throughout the period of study, subjects were predominantly treated on demand with treatment given episodically at the onset of bleeding. During follow-up, some participants were reported to have received prophylaxis for surgical coverage, during periods of increased activity, during involvement in sports, or if they were having increased bleeding in a joint. Some were likely on prophylaxis for longer periods. The collection of information on regimen was not sufficiently systematic, however, to permit classification of type of treatment prior to enrolment or at each study visit. Those members of the HGDS cohort with haemophilia A and DNA samples available (N=265, 97%) were included in the current analysis. Of these, 231, 87%, were unrelated. The human-subjects committees of collaborating institutions approved the HGDS, informed consent was obtained from parents or legal guardians, and informed consent or assent was obtained from all participants, in compliance with the human-experimentation guidelines of the US Department of Health and Human Services.
Clinical and laboratory data
Demographic data including date of birth, race and ethnicity were collected. Severe haemophilia was defined as FVIII activity <0.01 IU/ml, moderate as 0.01–0.05 IU/ml, and mild as >0.05–0.40 IU/ ml. For this analysis, an inhibitor was defined as a current or history of a titre ≥ 1 BU measured at the local laboratory.
Joint dysfunction was assessed by measurement of range-of-motion (ROM) for elbows, knees, and ankles at entry and six-month intervals throughout follow-up by study personnel centrally trained in the administration of an evaluation using a goniometer to measure joint ROM. Normal values for the study were established by consensus prior to the initiation of the research. A five-point scale was used with 1=normal, and 2–5 indicating degrees of abnormality. A 5 degree extension defect at the elbow, for example, would receive a score of 2; whereas a score of 5 would indicate virtual loss of range of motion at the elbow, or virtual loss of dorsiflexion and plantar flexion of the ankle. Quality assurance was maintained for joint assessment throughout the study. Personnel were newly trained or participated in refresher activities, as appropriate, to ensure consistent outcomes.
F8 mutation typing
The presence or absence of a F8 inversion mutation on intron 1 or 22 (inversion/no inversion) had been determined for 58% of the HGDS cohort during regular follow-up (25). The remaining HGDS samples were mutation typed using the methods of Oldenburg (26).
Genotyping
An Illumina iSelect array was used to genotype 14,626 single nucleotide polymorphisms (SNPs) from a set of 1,081 genes. The genes — chiefly immune response and immune modifier genes, and cytokines, cytokine receptors, chemokines, chemokine receptors, immune and inflammatory pathway genes, and HLA genes were selected from a literature review of inflammatory and immune genes and pathway public databases. SNPs were selected from a region spanning 5 kb upstream and 1 kp downstream of the target genes using data from the International HapMap Project. We first selected all known or putative functional coding region (nonsynonymous, insertion, deletions, frameshift) and regulatory SNPs. Functional status was vetted using information from NCBI, PupaSuite (http: //pupasuite.bioinfo.cipf.es/) and SNPEffect (http: //snpeffect.vib.be/index.php). SNPs that are reported to affect amino acid composition, or occurred in splice sites, exonic splicing enhancer or exonic splicing silencer, or regulatory regions were selected as long as they met minimum Illumina design requirements. Using these as the first seeds, we added tagging SNPs (r2>0.8) equally spaced across the targeted gene region. Because of the paucity of exomic SNPs, most of the SNPs were located on introns or non-coding regions. Genotypes that exhibited significant deviation from Hardy-Weinberg equilibrium (p<0.001) or that exhibited missingness greater than 20% were removed.
Statistical analysis
The genetics analysis was conducted in two steps.
Independent, single joint models (Step 1)
Due to the complexity of the data — both longitudinal and containing multiple observations per subject at each time point — the first set of analyses were conducted in separate models by specific joint — the independent or single joint models. These were fit using the ROM data for the knee, elbow and ankle joints on both the left and right sides. As noted, the ROM was measured on a 1–5 scale with 1 representing a normal score and 5 being the farthest from normal. Given the distribution of the response variable, an ordered logistic regression model was fitted for each joint to estimate the odds of an association between a specific SNP and an increase in ROM score. The covariates in the model include severity of haemophilia, current or history of an inhibitor (yes/no), presence of an inversion mutation on intron 1 or 22 (yes/no), age at entry, visit number, and SNP. Body mass index (BMI) and HIV status were not significant predictors of joint outcome and were not included in the models. As genetics data were not available for HGDS participants with factor IX deficiency, type of haemophilia could not be used as a covariate in this study. The SNPs were modelled as additive effects with the count of population defined minor alleles for each individual. Population substructure due to ancestry differences among subjects was adjusted for using eigenvectors obtained from a principal components analysis of ancestry informative markers on the Illumina array (EIGENSOFT software [27]). The results from each model estimated the effect of the covariates on the odds of limiting the ROM of a specific joint.
All joints at all visits – full models (Step 2)
Next, to address how a given SNP may affect the overall odds of joint destruction, full models were fit with all joints at all visits for a subset of the SNP markers. The full model utilised the relationship between all joints and estimated the odds of association between a given SNP and a monotonic increase in ROM score for an individual. A SNP was used in the full model if it had an association with ROM measurement with a p-value less than 0.01 on both right and left sides for two of the three joints tested in the single joint models. The full models were fit using a multivariate generalised linear mixed model assuming an ordinal response. Random intercepts were fit for individuals, random slopes for visit number and the fixed covariates used in the individual models. The models were fit using a Markov Chain Monte Carlo (MCMC) technique with 10,000 iterations per SNP.
Results
There were 265 children and adolescents with six outcomes and up to 15 visits included in the analysis. The demographic and haemophilia-related characteristics of the study group are shown in ▶Table 1. The cohort was predominately white and the age at study entry ranged from seven to 19 years. The majority of participants had severe haemophilia (75.1%) and approximately 20% had a current or history of an inhibitor.
Table 1:
Demographics and haemophilia-related characteristics.
| Race/ethnicity (N, %) | |
| White | 195 (73.6 %) |
| Hispanic | 42 (15.9 %) |
| Black | 24 (9.1 %) |
| Other | 4 (1.5 %) |
| Severity (N, %) | |
| Mild | 16 (6.0 %) |
| Moderate | 50 (18.9 %) |
| Severe | 199 (75.1 %) |
| Current or history of inhibitor (N, %) | |
| Yes | 52 (19.6) |
| No | 213 (80.4) |
| Age at study entry (years) | |
| Mean (SD) | 12.3 (3.33) |
| Median | 12.2 |
| Range | 7.0–19.1 |
| Inversion mutation (N, %) | |
| Yes | 98 (38.7 %) |
| No | 155 (61.3 %) |
| Number of study visits | |
| Mean (SD) | 10.4 (3.76) |
| Median | 11 |
| Range | 1–15 |
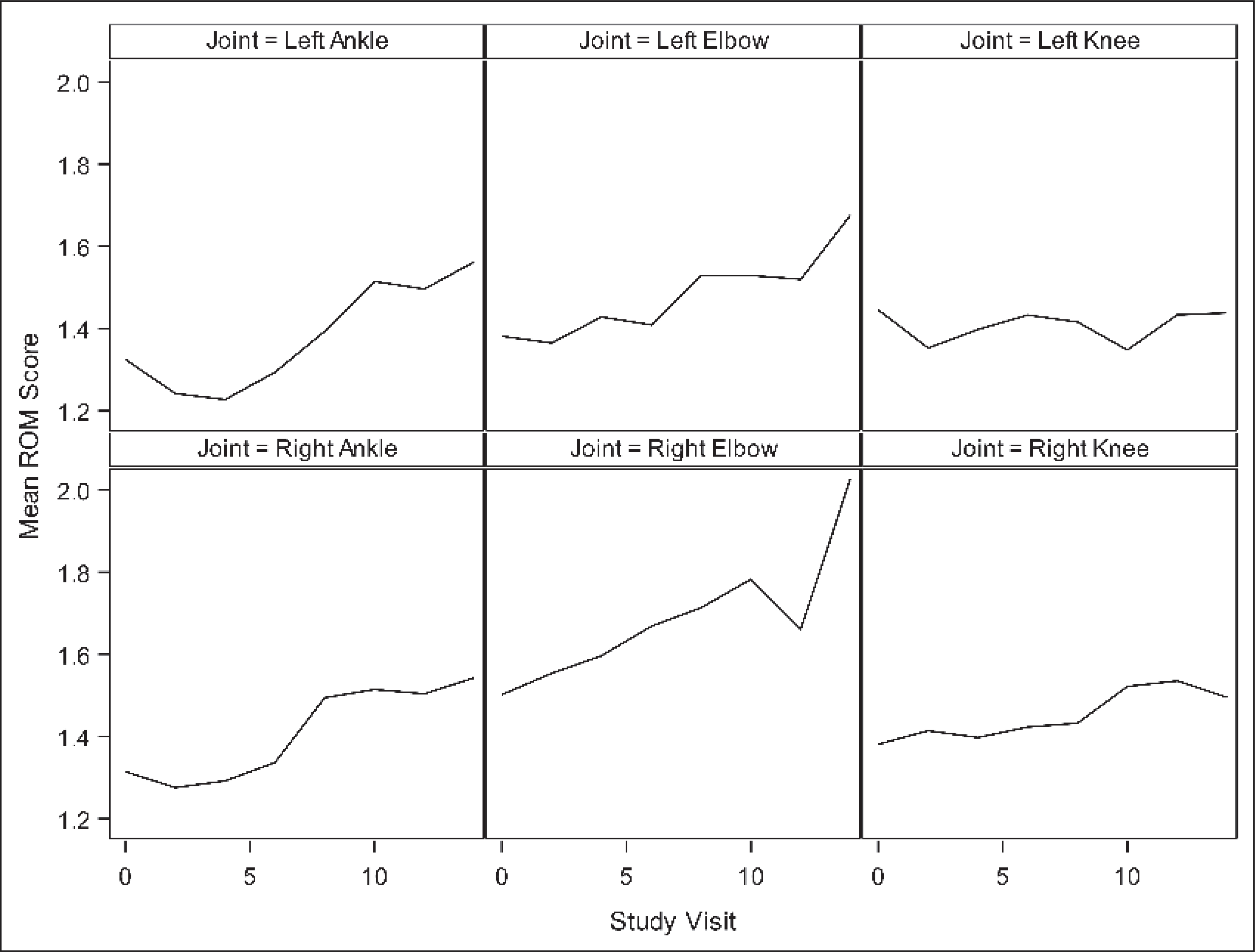
The incidence and severity of ROM abnormalities increased over the course of the study. At study entry, 55% of participants had an abnormal ROM in at least one joint with an average of 1.2 joints affected (▶Table 2). At the end of the study, 72% of participants had an abnormal ROM in at least one joint with an average of 1.8 joints affected. ROM scores also increased over the study period (▶Figure 1).
Table 2:
Range-of-motion scores for each joint and number of joints affected at study entry.
| Right elbow | |
| Mean (SD) | 1.5 (1.0) |
| Median | 1 |
| Range | 1–5 |
| Left elbow | |
| Mean (SD) | 1.4 (0.9) |
| Median | 1 |
| Range | 1–5 |
| Right knee | |
| Mean (SD) | 1.4 (0.9) |
| Median | 1 |
| Range | 1–5 |
| Left knee | |
| Mean (SD) | 1.4 (1.0) |
| Median | 1 |
| Range | 1–5 |
| Right ankle | |
| Mean (SD) | 1.3 (0.7) |
| Median | 1 |
| Range | 1–5 |
| Left ankle | |
| Mean (SD) | 1.3 (0.7) |
| Median | 1 |
| Range | 1–5 |
| Number of affected joints | |
| Mean (SD) | 1.2 (1.0) |
| Median | 1 |
| Range | 0–6 |
Figure 1: Mean ROM score by joint and study visit (0=baseline) over the follow-up period.
Of 14,626 SNPs on the Illumina panel, 13,952 (95%) were successfully genotyped. A total of 13,342 passed the selection criteria and Hardy-Weinberg equilibrium testing (▶Figure 2). The single joint models were fit for each of these SNP markers in the first step of the analysis.
Figure 2: Flow diagram illustrating the steps in the analysis.
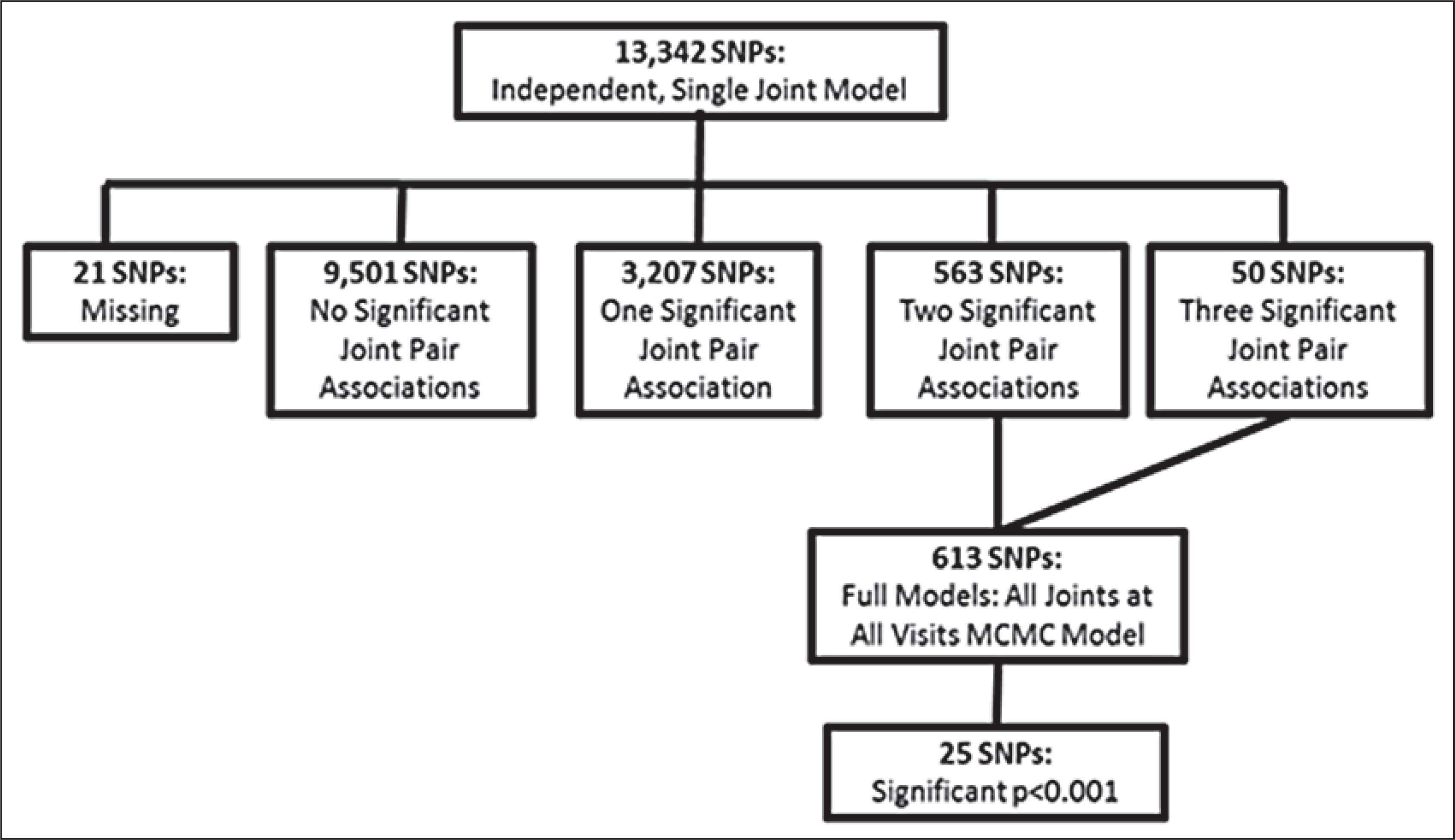
Step 1 (N=13,342 SNPs): independent, single joint models. Step 2 (N=613 SNPs): full models — all joints at all visits.
From the results of the single joint models, 613 SNP markers were shown to have a p-value <0.01 for both left and right sides on at least two of the three joints. The subset of 613 SNPs was then fit in the full model with all joint outcomes at all visits. Of the total models run, 609 converged and provided estimates for the SNP markers. The average odds ratio from the MCMC was reported for each SNP model. A number of SNPs resulted in p-values less than 0.001 (n=25) with a substantial peak in chromosome 2 near the BCL2L11 gene (▶Table 3). Six SNPs were located on the BCL2L11 gene; however, the average pairwise D’ between the markers near the gene is 0.93 with a standard deviation of 0.15. The high degree of linkage disequilibrium (LD) would provide similar associations for all markers within the region. Eight of these SNPs from the BCL2L11 gene included on the panel did not pass the independent model threshold and, therefore, were not tested with the MCMC model. Similarly, the gene CARD10 had several significant markers, again likely due to the linkage between SNPs — an average LD of 0.69 with a standard deviation of 0.10. There were, however, nine other SNPs that did not pass the independent model threshold for CARD10 and were not tested with the MCMC model. Given the linkage between SNPs and the number of SNPs that did not show significance in the independent models, BCL2L11 or CARD10 may not have higher likelihood of association compared to other genes on the list.
Table 3:
Most significant SNPs from the full model with all joint outcomes at all visits.
| SNP | CHR | Type | Gene | Mean OR | MAF | CI | P |
|---|---|---|---|---|---|---|---|
| rs10204044 | 2 | intron | BCL2L11 | 0.644 | 0.354 | (0.513,0.8) | 0.0001 |
| rs2066845 | 16 | coding-nonsynonymous | NOD2 | 0.000 | 0.002 | (0,0.081) | 0.0001 |
| rs2241842 | 2 | locus-region | BCL2L11 | 1.447 | 0.432 | (1.171,1.776) | 0.0001 |
| rs3115630 | 6 | locus-region | HLA-G | 2.133 | 0.062 | (1.44,3.128) | 0.0001 |
| rs3761704 | 2 | intron | BCL2L11 | 1.736 | 0.151 | (1.323,2.28) | 0.0001 |
| rs742153 | 22 | intron | CARD10 | 1.649 | 0.145 | (1.242,2.15) | 0.0001 |
| rs7625958 | 3 | intron | PTPRG | 1.604 | 0.355 | (1.313,2.001) | 0.0001 |
| rs13210247 | 6 | intron | TRAF3IP2 | 1.849 | 0.081 | (1.333,2.604) | 0.0002 |
| rs686952 | 2 | intron | BCL2L11 | 0.592 | 0.317 | (0.442,0.775) | 0.0002 |
| rs724710 | 2 | coding-synonymous | BCL2L11 | 0.683 | 0.357 | (0.553,0.848) | 0.0002 |
| rs10172990 | 2 | intron | TACR1 | 1.691 | 0.151 | (1.318,2.189) | 0.0004 |
| rs6441977 | 3 | coding-nonsynonymous | CCRL2 | 1.656 | 0.126 | (1.225,2.226) | 0.0004 |
| rs10491067 | 10 | intron | BLNK | 0.656 | 0.251 | (0.511,0.84) | 0.0004 |
| rs738304 | 22 | intron | CARD10 | 0.638 | 0.281 | (0.502,0.793) | 0.0004 |
| rs10100561 | 8 | intron | LYN | 1.433 | 0.261 | (1.144,1.776) | 0.0006 |
| rs11466655 | 4 | coding-nonsynonymous | TLR10 | 5.117 | 0.011 | (1.978,13.295) | 0.0006 |
| rs2075642 | 19 | intron | PVRL2 | 1.477 | 0.245 | (1.198,1.826) | 0.0006 |
| rs932327 | 22 | intron | CARD10 | 1.629 | 0.234 | (1.234,2.131) | 0.0006 |
| rs11674517 | 2 | intron | BCL2L11 | 1.468 | 0.415 | (1.207,1.784) | 0.0008 |
| rs4938807 | 11 | intron | ARHGEF12 | 0.661 | 0.194 | (0.507,0.843) | 0.0008 |
| rs11230550 | 11 | intron | CD6 | 1.644 | 0.106 | (1.227,2.211) | 0.001 |
| rs12320259 | 12 | intron | LRP6 | 1.582 | 0.200 | (1.186,2.122) | 0.001 |
| rs2228059 | 10 | coding-nonsynonymous | IL15RA | 1.372 | 0.460 | (1.139,1.655) | 0.001 |
| rs2273346 | 1 | coding-nonsynonymous | MASP2 | 2.349 | 0.038 | (1.428,3.924) | 0.001 |
| rs1986027 | 5 | intron | CSF1R | 0.483 | 0.066 | (0.315,0.757) | 0.001 |
CHR=chromosome. MAF=minor allele frequency.
A nonsynonymous SNP rs2066845 in the NOD2 gene had the estimate with the highest magnitude of association: 4.4×10−4 (3.75×10−6, 0.081). However, there was only one individual in the study population who carried the minor allele. The missense SNP rs11466655 in the TLR10 gene showed the next highest magnitude, but similar to NOD2, the minor allele was carried by only six individuals in the study population. For both genes, the results indicate a potentially strong association with arthropathy, but given the low frequency of the variant alleles in the study population, further evaluations are required.
Estimates of the effects of several covariates were obtained from the complex models, including change in ROM scores associated with an increase of one year in age (▶Table 4). While the change in risk from a single visit was small, odds ratio (OR) of 1.07 (confidence interval [CI] 1.05, 1.08), the estimated risk after several visits was substantially greater. For example, the risk after seven visits, or half the maximum, was 1.55 (CI 1.43, 1.68) and increased to 2.39 (CI 2.17, 3.03) by the 15th visit. The effect at the average age at enrolment, 12, was 1.66 (CI 1.33, 2.10) times higher risk when compared to the youngest subjects in the study, age seven. This risk rose to 3.37 (CI 1.98, 5.75) times higher for the oldest subjects in the study, enrolment age of 19, compared to the youngest. The effect of severity was also evaluated, showing a 1.77 (CI 1.4, 2.22) fold increase in risk for those with moderate compared to mild haemophilia and a 1.96 (CI 1.83, 2.10) fold increase in risk for those with severe haemophilia compared to those with moderate disease. While there was an association between presence of an inversion mutation and severity of haemophilia, inclusion of both covariates added information to the model. For those with a current or history of an inhibitor, the average OR was 1.75 (CI 1.19, 2.58), indicating an increased risk of having a higher ROM score compared to those without an inhibitor.
Table 4:
Covariates used in the models – their odds ratios and 95 % confidence intervals (CI).
| Covariates | Mean Odds | 95 % CI |
|---|---|---|
| Severity of haemophilia | ||
| Moderate* | 1.77 | (1.4, 2.22) |
| Severe* | 3.46 | (2.57, 4.67) |
| Current or history of an inhibitor | 1.75 | (1.19, 2.58) |
| Presence of inversion mutation | 1.15 | (0.85, 1.57) |
| Age** | 1.11 | (1.06, 1.16) |
Compared to mild.
Risk associated with increase of one year.
Discussion
By combining experience from two large, multicenter studies, i.e. HGDS and the HIGS Combined Cohort, we analysed 14,626 SNPs from a set of 1,081 genes to explore whether genes or pathways involved in inflammation and immune modulation may be associated with the occurrence of haemophilic arthropathy. The subjects were children or adolescents predominantly treated on demand with treatment given episodically at the onset of bleeding. We have shown that the incidence and severity of ROM abnormalities increased over the course of the study. Hence, the joint damage seen among those in the HGDS cohort may reflect the natural course of haemophilic arthropathy, as regular prophylaxis had not been used. The finding of associations between SNPs and affected/not affected joints may well reflect the genetic impact on development of arthropathy. To our knowledge, there are no previous reports on genetic factors and haemophilic arthropathy in humans. It is gratifying, however, that our study findings have replicated the observations of others. Focusing on studies of arthritis, several genes have previously been associated with joint outcomes. The genes HLA-G and TRAF3IP2 have been reported as associated with arthritis (28, 29) and both of these are in our list of top significant SNPs. Several other genes including IL12B, REL, PAX5 and CD247 have also been shown to be associated with arthritis in studies with large numbers of subjects (29–35). These genes do not appear among our list of top markers, but met the criteria for testing with the MCMC model, and warrant further study. A number of SNPs (n=25) were strongly associated with arthropathy in the full statistical model, either with a high degree of ROM abnormalities or protection against joint disease.
The most significant strength of our study is that a relatively large cohort of young people with haemophilia was followed prospectively for seven years at six-months intervals. They were largely treated on demand; therefore, the intervention in the natural course of development of arthropathy observed with regular prophylactic treatment did not influence the results. It cannot be ruled out that time from symptoms of bleeding to treatment likely differed among individuals, thereby varying the effect on haemorrhagic blood volume and intraarticular pressure, both factors which may be of importance for development of arthropathy. The SNPs chosen were the same as those examined in the HIGS Combined Cohort study (23). These were a group of SNPs selected because they occurred within or near genes with immune function and it is quite possible that other important SNPs, such as might be found using genome wide screening, may have been overlooked. On the other hand, a number of markers were found to be highly significantly related to ROM scores.
It is important to document that detailed evaluation of haemophilic arthropathy was not a primary or even a secondary objective of the HGDS. Thus, the extent to which major joint dysfunction was assessed and documented was not as comprehensive as other outcomes such as physical growth, viral markers, and clinical progression of HIV and hepatitis C. Nonetheless, ROM was measured by study-trained staff specialised in haemophilia and haemophilic arthropathy, utilising a clinically meaningful agreed upon 5-point scale which likely assures good quality of these ”real life” data. However, a lack of stringent measurement of joint disease likely introduced a degree of variability that limited the sensitivity of our analysis. We saw an opportunity to explore possible associations between a large set of genetic markers and degree of ROM deficit accumulating over time. There are other weaknesses of the study including the fact that detailed treatment data such as age of first bleeding episode, annual bleeding rate, treatment intensity, and use of home treatment are lacking. Nor was there availability of data with which to estimate frequency or intensity of physiotherapy. Without a subclinical/clinical bleeding event, a genetic susceptibility for arthropathy would not likely have an impact. A patient would not bleed simply because he has a specific genotype for arthropathy. While the lack of bleeding history may be a weakness, it is known that all subjects eligible for enrolment in the HGDS had experienced bleeding to some degree, thereby placing them at risk for joint damage. In the best case scenario, the identification of genetic factors that predict the development of haemophilic arthropathy would be done in a cohort of patients having the same frequency of bleeding events with the condition of the joints measured at specific time points and ages. Clearly, that study design would be quite difficult to implement.
While some of the genetic factors identified in our study may be related to bleeding phenotype rather than susceptibility to arthropathy, a landmark study in small children with severe haemophilia (18) clearly documented that the number of clinical haemarthroses showed weak correlations with magnetic resonance imaging (MRI) assessed outcomes. The data available in our study do not provide the opportunity to discriminate between these possibilities. The strongest associations were seen with SNPs in the NOD2 (nucleotide-binding oligomerisation domain containing 2) and TLR10 (toll-like receptor 10) genes. NOD2 genes have been reported to play a role in musculoskeletal manifestations in inflammatory bowel disease (36), and the protein encoded by the TLR10 family plays a fundamental role in pathogen recognition and activation of innate immunity. Studies of osteoarthritis in the general population have shown that genetic, epigenetic factors as well as life style may impact development of joint disease (37, 38). Haemophilic arthropathy is caused primarily by blood in the joint but the course of development is likely to be impacted by several other factors, as is the case for osteoarthritis, even though the relatively young age of our study group makes it unlikely that lifestyle factors have influenced the arthropathic development to any great degree.
Although validation by replication in a different cohort should be considered an integral part of the study design, the practicalities of doing so prevent that as our next step. Because of the low prevalence of haemophilia A in the general population, acquiring a sample size for an appropriately powered replication study is difficult and costly. The biologic mechanisms underlying the findings cannot be explained in a straightforward way. Development of haemophilic arthropathy requires intraarticular blood which may have direct toxic effects, as well as other long term processes in which chronic inflammation is of importance (39, 40). Therefore, prevention of bleeding events early in life is of utmost importance. It is long known, first demonstrated in a large cohort in the Orthopaedic Outcome Study (3), and subsequently in another large study (5) that the clinical bleeding phenotype and development of arthropathy varies substantially in severe haemophilia (3) and that regular prophylaxis, rather than high doses of clotting factor given episodically, provides the best joint outcome (18). Interestingly, the complex model showed that when a current or history of an inhibitor to FVIII was present the average odds ratio of the effect, across all SNPs tested, was 1.75, indicating that the less efficacious treatment for patients with haemophilia complicated by inhibitors has, in general, a greater negative impact than the SNPs examined. For the entire study group, the risk of having a higher ROM score increased from 1.07 to 2.39 during the follow-up period.
We conclude from this descriptive study that haemophilic arthropathy is associated with a group of genetic markers, chiefly immune response and immune modifier genes, selected from a literature review of inflammatory and immune genes and pathway public databases. Hypothetically, these genes are involved in the aetiology of synovial inflammation, cartilage and bone destruction which causes haemophilic arthropathy. The practical clinical potential of the findings could be their use in the construction of risk scores to predict the development and progression of joint disease in haemophilia. This is a complex and multifactorial problem and the calculation of any such risk score would consider not only genetic factors but also environmental parameters. In the young child though, such parameters are not readily obvious as many manifest later in life. Patients at high risk for arthropathy should consequently be started on prophylaxis very early in life (41) and be given more intensive clotting factor therapy than those at low risk, i.e. a greater degree of individualisation should be the goal (42). More studies are required, but our findings support a genetic role in the development of haemophilic arthropathy which needs to be better addressed in the context of future therapeutic interventions.
What is known about this topic?
Arthropathy is a major complication of severe haemophilia and is associated with joint haemorrhages. The degree of arthropathy is variable across individuals.
Severity and management of haemophilia do not explain all variation.
What does this paper add?
Genetic predisposition may be a contributing factor, as has been shown in other studies of arthritis.
Our work provides support for the involvement of genetic factors in the development of haemophilic arthropathy.
Study findings have the potential to contribute to the individualisation of therapy to limit or prevent this major complication.
Acknowledgements
We are grateful to the participants and parents who volunteered to take part in the Haemophilia Growth and Development Study.
Financial support:
This work is supported by the National Institutes of Health, National Institute of Child Health and Human Development, R01-HD-41224; by the Lund University, Centre for Thrombosis and Haemostasis, Skåne University Hospital Malmö/Lund, Malmö, Sweden; by an investigator-initiated grant from Baxter BioScience; and in part with federal funds from the NIH National Cancer Institute (NCI) under Contract No. HHSN261200800001E, and the Intramural Research Program of the NIH-NCI Center for Cancer Research.
Footnotes
Conflicts of interest
None declared.
References
- 1.Arnold WD, Hilgartner M. Hemophilic arthropathy. Current concepts of pathogenesis and management. J Bone Joint Surg Am 1977; 59: 287–305. [PubMed] [Google Scholar]
- 2.Madhok R, York J, Sturrock RD. Haemophilic arthritis. Ann Rheum Dis 1991; 50: 588–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aledort L, Haschmeyer RH, Pettersson H. A longitudinal study of orthopaedic outcomes for severe factor-VIII-deficient haemophiliacs. J Intern Med 1994; 236: 391–399. [DOI] [PubMed] [Google Scholar]
- 4.Pollmann H, Richter H, Ringkamp H, et al. When are children diagnosed as having severe haemophilia and when do they start to bleed? A 10-year single-centre PUP study. Eur J Pediatr 1999; 158: S166–S170. [DOI] [PubMed] [Google Scholar]
- 5.Soucie JM, Cianfrini C, Janco RL, et al. Joint range-of-motion limitations among young males with hemophilia: prevalence and risk factors. Blood 2004; 103: 2467–2473. [DOI] [PubMed] [Google Scholar]
- 6.Nijdam A, Altisent C, Carcao MD, et al. Bleeding before prophylaxis in severe hemophilia: paradigm shift over two decades. Haematologica 2015; 100: e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Miners A, Sabin C, Tolley K, et al. Assessing health-related quality-of-life in individuals with haemophilia. Haemophilia 1999; 5: 378–385. [DOI] [PubMed] [Google Scholar]
- 8.Mainardi CL, Levine PH, Werb Z, et al. Proliferative synovitis in hemophilia. Biochemical and morphologic observations. Arthritis Rheum 1978; 21: 137–144. [DOI] [PubMed] [Google Scholar]
- 9.Valentino L. Blood-induced joint disease: the pathophysiology of hemophilic arthropathy. J Thromb Heamost 2010; 8: 1895–1902. [DOI] [PubMed] [Google Scholar]
- 10.Hakobyan N, Enockson C, Cole A, et al. Experimental haemophilic arthropathy in a mouse model of a massive haemarthrosis: gross, radiological and histological changes. Haemophilia 2008; 14: 804–809. [DOI] [PubMed] [Google Scholar]
- 11.Forsyth A, Rivard GÉ, Valentino L, et al. Consequences of intra-articular bleeding in haemophilia: science to clinical practice and beyond. Haemophilia 2012; 18 (Suppl 4): 112–119. [DOI] [PubMed] [Google Scholar]
- 12.Roosendaal G, Vianen ME, Wenting MJ, et al. Iron deposits and catabolic properties of synovial tissue from patients with haemophilia. J Bone Joint Surg Br 1998; 80: 540–545. [DOI] [PubMed] [Google Scholar]
- 13.Roosendaal G, Van Rinsum A, Vianen M, et al. Haemophilic arthropathy resembles degenerative rather than inflammatory joint disease. Histopathology 1999; 34: 144–153. [DOI] [PubMed] [Google Scholar]
- 14.Decker J, Malone D, Haraoui B, et al. Rheumatoid arthritis: evolving concepts of pathogenesis and treatment. Ann Intern Med 1984; 101: 810–824. [DOI] [PubMed] [Google Scholar]
- 15.Hooiveld MJ, Roosendaal G, Van Den Berg H, et al. Haemoglobin-derived iron-dependent hydroxyl radical formation in blood-induced joint damage: an in vitro study. Rheumatology 2003; 42: 784–790. [DOI] [PubMed] [Google Scholar]
- 16.Wen F-Q, Jabbar AA, Chen Y-X, et al. C-myc proto-oncogene expression in hemophilic synovitis: in vitro studies of the effects of iron and ceramide. Blood 2002; 100: 912–916. [DOI] [PubMed] [Google Scholar]
- 17.Hooiveld M, Roosendaal G, Wenting M, et al. Short-term exposure of cartilage to blood results in chondrocyte apoptosis. Am J Pathol 2003; 162: 943–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manco-Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med 2007; 357: 535–544. [DOI] [PubMed] [Google Scholar]
- 19.Ragni M, Fogarty P, Josephson N, et al. Survey of current prophylaxis practices and bleeding characteristics of children with severe haemophilia A in US haemophilia treatment centres. Haemophilia 2012; 18: 63–68. [DOI] [PubMed] [Google Scholar]
- 20.Nilsson I, Berntorp E, Löfqvist T, et al. Twenty-five years’ experience of prophylactic treatment in severe haemophilia A and B. J Intern Med 1992; 232: 25–32. [DOI] [PubMed] [Google Scholar]
- 21.Carcao MD, van den Berg HM, Ljung R, et al. Correlation between phenotype and genotype in a large unselected cohort of children with severe hemophilia A. Blood 2013; 121: 3946–3952. [DOI] [PubMed] [Google Scholar]
- 22.Ahlberg A. Indicence, treatment and prophylaxis of arthropathy and other musculoskeletal manifestations of haemophilia A and B. Acta Orthop Scand 1965; 5: 20–39. [DOI] [PubMed] [Google Scholar]
- 23.Astermark J, Donfield S, Gomperts E, et al. The polygenic nature of inhibitors in hemophilia A: results from the Hemophilia Inhibitor Genetics Study (HIGS) Combined Cohort. Blood 2013; 121: 1446–1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilgartner MW, Donfield SM, Willoughby A, et al. Hemophilia Growth and Development Study: Design, Methods Entry Data. J Pediatr Hematol Oncol 1993; 15: 208–218. [DOI] [PubMed] [Google Scholar]
- 25.Lakich D, Kazazian H, Antonarakis S, et al. Inversions disrupting the factor VIII gene are a common cause of severe haemophilia A. Nat Genet 1993; 5: 236–241. [DOI] [PubMed] [Google Scholar]
- 26.Oldenburg J. Mutation profiling in haemophilia A. Thromb Haemost 2001; 85: 577–579. [PubMed] [Google Scholar]
- 27.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006; 38: 904–909. [DOI] [PubMed] [Google Scholar]
- 28.Eleftherohorinou H, Hoggart CJ, Wright VJ, et al. Pathway-driven gene stability selection of two rheumatoid arthritis GWAS identifies and validates new susceptibility genes in receptor mediated signalling pathways. Hum Mol Gen 2011; 20: 3494–3506. [DOI] [PubMed] [Google Scholar]
- 29.Hüffmeier U, Uebe S, Ekici AB, et al. Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nature Genet 2010; 42: 996–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng B, Sun L, Soltani-Arabshahi R, et al. Multiple loci within the major histocompatibility complex confer risk of psoriasis. PLoS Genet 2009; 5: e1000606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Y, Helms C, Liao W, et al. A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet 2008; 4: e1000041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gregersen P, Amos C, Lee A, et al. REL, encoding a member of the NF-kappaB family of transcription factors, is a newly defined risk locus for rheumatoid arthritis. Nat Genet 2009; 41: 820–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ellinghaus E, Stuart PE, Ellinghaus D, et al. Genome-wide meta-analysis of psoriatic arthritis identifies susceptibility locus at REL. J Invest Dermatol 2012; 132: 1133–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Bansal A, Martin M, et al. Genome-wide association analysis implicates the involvement of eight loci with response to tocilizumab for the treatment of rheumatoid arthritis. Pharmacogenomics 2013; 13: 235–241. [DOI] [PubMed] [Google Scholar]
- 35.Stahl E, Raychaudhuri S, Remmers E, et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nature Genet 2010; 42: 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sheth T, Pitchumoni C, Das K. Musculoskeletal Manifestations in Inflammatory Bowel Disease: A Revisit in Search of Immunopathophysiological Mechanisms. J Clin Gastroenterol 2014; 48: 308–317. [DOI] [PubMed] [Google Scholar]
- 37.Zhang M, Wang J. Epigenetics and Osteoarthritis. Genes Dis 2015; Epub ahead of print. [DOI] [PMC free article] [PubMed]
- 38.Fransen M, Simic M, Harmer A. Determinants of MSK health and disability: Lifestyle determinants of symptomatic osteoarthritis. Best Pract Res Clin Rheumatol 2014; 28: 435–460. [DOI] [PubMed] [Google Scholar]
- 39.Roosendaal G, Jansen NW, Schutgens R, et al. Haemophilic arthropathy: the importance of the earliest haemarthroses and consequences for treatment. Haemophilia 2008; 14 (Suppl 6): 4–10. [DOI] [PubMed] [Google Scholar]
- 40.Roosendaal G, Lafeber FP. Pathogenesis of haemophilic arthropathy. Haemophilia 2006; 12 (Suppl 3): 117–121. [DOI] [PubMed] [Google Scholar]
- 41.Astermark J, Petrini P, Tengborn L, et al. Primary prophylaxis in severe haemophilia should be started at an early age but can be individualized. Brit J Haematol 1999; 105: 1109–1113. [DOI] [PubMed] [Google Scholar]
- 42.Fischer K, Carlsson K, Petrini P, et al. Intermediate-dose versus high-dose prophylaxis for severe hemophilia: comparing outcome and costs since the 1970s. Blood 2013; 122: 1129–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]