

Key Points
Question
Can poloxamer 188, an agent that is reported to reduce blood viscosity and cell-cell interactions, effectively reduce the duration of vaso-occlusive episodes (painful crises) in hospitalized patients with sickle cell disease?
Findings
In this randomized clinical trial that included 388 children and adults with sickle cell disease, treatment with poloxamer 188 vs placebo resulted in mean time to last dose of parenteral opioids during vaso-occlusive episodes of 81.8 vs 77.8 hours, a difference that was not statistically significant.
Meaning
Among patients with sickle cell disease, poloxamer 188 did not significantly shorten the duration of painful vaso-occlusive episodes.
Abstract
Importance
Although effective agents are available to prevent painful vaso-occlusive episodes of sickle cell disease (SCD), there are no disease-modifying therapies for ongoing painful vaso-occlusive episodes; treatment remains supportive. A previous phase 3 trial of poloxamer 188 reported shortened duration of painful vaso-occlusive episodes in SCD, particularly in children and participants treated with hydroxyurea.
Objective
To reassess the efficacy of poloxamer 188 for vaso-occlusive episodes.
Design, Setting, and Participants
Phase 3, randomized, double-blind, placebo-controlled, multicenter, international trial conducted from May 2013 to February 2016 that included 66 hospitals in 12 countries and 60 cities; 388 individuals with SCD (hemoglobin SS, SC, S-β0 thalassemia, or S-β+ thalassemia disease) aged 4 to 65 years with acute moderate to severe pain typical of painful vaso-occlusive episodes requiring hospitalization were included.
Interventions
A 1-hour 100-mg/kg loading dose of poloxamer 188 intravenously followed by a 12-hour to 48-hour 30-mg/kg/h continuous infusion (n = 194) or placebo (n = 194).
Main Outcomes and Measures
Time in hours from randomization to the last dose of parenteral opioids among all participants and among those younger than 16 years as a separate subgroup.
Results
Of 437 participants assessed for eligibility, 388 were randomized (mean age, 15.2 years; 176 [45.4%] female), the primary outcome was available for 384 (99.0%), 15-day follow-up contacts were available for 357 (92.0%), and 30-day follow-up contacts were available for 368 (94.8%). There was no significant difference between the groups for the mean time to last dose of parenteral opioids (81.8 h for the poloxamer 188 group vs 77.8 h for the placebo group; difference, 4.0 h [95% CI, −7.8 to 15.7]; geometric mean ratio, 1.2 [95% CI, 1.0-1.5]; P = .09). Based on a significant interaction of age and treatment (P = .01), there was a treatment difference in time from randomization to last administration of parenteral opioids for participants younger than 16 years (88.7 h in the poloxamer 188 group vs 71.9 h in the placebo group; difference, 16.8 h [95% CI, 1.7-32.0]; geometric mean ratio, 1.4 [95% CI, 1.1-1.8]; P = .008). Adverse events that were more common in the poloxamer 188 group than the placebo group included hyperbilirubinemia (12.7% vs 5.2%); those more common in the placebo group included hypoxia (12.0% vs 5.3%).
Conclusions and Relevance
Among children and adults with SCD, poloxamer 188 did not significantly shorten time to last dose of parenteral opioids during vaso-occlusive episodes. These findings do not support the use of poloxamer 188 for vaso-occlusive episodes.
Trial Registration
ClinicalTrials.gov Identifier: NCT01737814
This phase 3 trial examines the effectiveness of poloxamer 188 in reducing the duration of painful vaso-occlusive episodes in children and adults with sickle cell disease compared with placebo.
Introduction
Sickle cell disease (SCD) is a group of inherited hemoglobinopathies for which the hallmark feature is the acute painful vaso-occlusive episode. There are 3 agents approved by the US Food and Drug Administration for the prevention of painful vaso-occlusive episodes (hydroxyurea, L-glutamine, and crizanlizumab-tmca). No available agent effectively manages vaso-occlusive episodes once they have begun.1 Current treatment remains supportive, with analgesia and hydration. Frequent vaso-occlusive episodes are also associated with higher mortality in individuals with SCD.2 Acute pain is estimated to account for 95% of hospital admissions for those with SCD, creating a burden for individuals with SCD, their families, and health care systems.3 The ability to reduce the severity and duration of vaso-occlusive episodes would be a significant advance.
The pathophysiology of vaso-occlusive episodes involves inflammation, hemolysis, hemostasis, cell adhesion, vaso-occlusion, and reperfusion injury. Vaso-occlusion results from complex interactions of sickle red blood cells, leukocytes, and platelets with the endothelium, mediated by adhesive molecules and receptors, including P-selectins, E-selectins, vascular cellular adhesion molecule 1, von Willebrand factor, glycoprotein Ib, thrombospondin, CD36 receptors, fibronectin, and vitronectin.4,5,6,7 Inhibition of these interactions remains an attractive therapeutic target. Poloxamer 188 (vepoloxamer, MST-188, RheothRx) is a nonionic block polymer surfactant for which numerous activities have been reported, including improved microvascular blood flow by blocking cell-to-cell interactions, reduced blood viscosity and adhesion of sickle cells to endothelium, and antithrombotic and anti-inflammatory properties.8
Poloxamer 188 has been evaluated in 3 clinical trials of SCD demonstrating safety and possible efficacy for painful vaso-occlusive episodes and acute chest syndrome, which involves intrapulmonary vascular occlusion and/or infection.9,10,11,12 These studies included a previous phase 3 trial that suggested efficacy for painful vaso-occlusive episodes, particularly in children and participants receiving hydroxyurea.10
Because intravenous poloxamer 188 is neither approved by the US Food and Drug Administration nor available for clinical use and because other drugs for managing ongoing vaso-occlusive episodes are absent, the present trial was designed to determine whether poloxamer 188 is efficacious for painful vaso-occlusive episodes in SCD.
Methods
Study Design
Overview
The Evaluation of Purified Poloxamer 188 in Vaso-Occlusive Crisis of Sickle Cell Disease (EPIC) study was a phase 3, randomized, double-blind, placebo-controlled, multicenter trial designed to assess the effectiveness of poloxamer 188 in reducing the duration of painful vaso-occlusive episodes in SCD. The protocol and statistical analysis plan are included in Supplement 1 and Supplement 2. Institutional review board or ethics committee approval was obtained at each site as well as written informed consent for each participant, including assent from children, following institutional guidelines. A data and safety monitoring board met at least annually and actively monitored study conduct and participant safety throughout the study.
Patients
Participants were enrolled between May 2013 and February 2016 at 46 sites in the US and 20 non-US sites in 11 countries (Belgium, Brazil, Dominican Republic, Jamaica, Jordan, Lebanon, Oman, Panama, Saudi Arabia, Spain, and Turkey) (eTable 1 in Supplement 3). The final date of follow-up was April 7, 2016. The study included patients hospitalized for acute pain typical of vaso-occlusive episodes requiring treatment with parenteral opioid analgesia. Moderate to severe pain lasting at least 4 hours before randomization was required. Outpatient screening for eligibility and baseline studies was allowed, but hospital admission was required for randomization. Full inclusion/exclusion criteria are included in eTable 2 in Supplement 3. Race and ethnicity were self-reported because SCD disproportionately affects some populations and disease severity can vary by ethnic group and race. Categories were presented by the investigator, with the option for participants to self-define under “other.” The initial study design included participants aged 8 to 18 years with hemoglobin SS or S-β0 thalassemia phenotype, but was later amended to allow inclusion of individuals aged 4 to 65 years as well as other SCD phenotypes (hemoglobin SC and S-β+ thalassemia).
Randomization
Participants who met all eligibility requirements were centrally registered and randomly assigned in a 1:1 ratio to receive poloxamer 188 or placebo using permuted blocks with sizes of 4 to 8. Randomization was stratified by age (<16 or ≥16 years), use of hydroxyurea (yes or no), and pain score (<8 or ≥8) using the Wong-Baker FACES Pain Rating Scale13,14 at randomization.
Intervention
After randomization (target of <2 hours and not >24 hours after presentation), poloxamer 188 or placebo was administered intravenously as a 1-hour loading dose of 100 mg/kg, followed by a continuous infusion at 30 mg/kg/h for at least 13 hours (1-hour loading; 12-48 hours of maintenance infusion). The maximum infusion time was 49 hours of actual infusion, which could be given over a period of 53 hours to allow for up to 4 cumulative hours of interrupted infusion. The bag or bottle of infusate, infusion lines, and drip chambers were covered with opaque sheaths and foil to maintain blinding. Concurrent use of hydroxyurea was allowed if a stable dose was expected throughout the study. Use of parenteral opioids was initially restricted to morphine, hydromorphone, nalbuphine, and tramadol. Transdermal opioid patches were prohibited. Allowable oral opioids included codeine, hydrocodone, hydromorphone, morphine, and oxycodone. Ketorolac was the only allowable parenteral, nonsteroidal anti-inflammatory agent. Allowable nonopioid analgesics included acetaminophen, aspirin, diclofenac sodium, ibuprofen, and naproxen. Long-acting variants of oral analgesics were allowed. The protocol was later amended to include parenteral preparations of allowed oral opioids. Sites were encouraged to adhere to standardized pain treatment guidelines. All opioid and nonopioid analgesic use was recorded. Systemic corticosteroids for painful vaso-occlusive episodes, L-glutamine after randomization, and other investigational treatments for painful vaso-occlusive episodes were prohibited. Participants were followed up for 30 days after discharge. Screening procedures included chest radiograph, platelet count, creatinine, alanine transaminase, and pregnancy testing, if applicable.
Outcomes
The primary outcome was hours from randomization until the last administration of parenteral opioid analgesic for painful vaso-occlusive episodes prior to hospital discharge.
Secondary outcomes included hospitalization for recurrence of painful vaso-occlusive episodes within 14 days of initial hospital discharge and occurrence of acute chest syndrome within 120 hours of randomization. Acute chest syndrome was defined per the National Acute Chest Syndrome Study Group.15
Prespecified subgroups included age at randomization (<16, ≥16 years); sex; use of hydroxyurea (yes, no), with “yes” defined as receiving hydroxyurea at least 14 days before randomization and continuing to receive hydroxyurea until at least the day before randomization; Wong-Baker FACES Pain Rating Scale score (range, 0 [no pain] to 10 [worst pain]) at randomization (<8, ≥8); duration of pain at presentation (≤12, >12 hours); region/country (US, non-US); hemoglobin phenotypes; and younger than 16 years with hydroxyurea use at a US study site. Because of a shorter duration of crisis for children younger than 16 years and those receiving hydroxyurea in the previous phase 3 study, age and hydroxyurea use were prespecified for comparison.
Other prespecified analyses included length of study drug infusion, total opioid use (morphine equivalents/kg) from time of randomization to discharge, time from randomization to discharge, re-hospitalization at 30 days, and pharmacokinetics and pharmacodynamics (not analyzed in the current study).
Statistical Analyses
Sample Size
Using data from the previous phase 3 study, the sample size was calculated based on a mean time to last dose of parenteral opioids of 96 hours for the placebo group vs 80 hours for the poloxamer 188 group (presumed 20% improvement) (Marty Emanuel, PhD, Visgenx, email, February 15, 2021), a 2-sided α of .05, and a coefficient of variation of 54% (accounting for log-normal distribution). Under these assumptions, a sample size of 376 (188 per group) achieved 90% power to detect a significant difference between treatment groups using a 2-sample t test. To account for dropout of 3%, the sample size was adjusted to 388 participants (194 per group). Sample size calculations were performed using SAS, version 9.3, PROC POWER for a 2-sample mean comparison. The same statistical software was used for all other statistical analyses.
Primary Outcome Analysis
Primary outcomes were analyzed by randomization group, including all randomized patients (primary analysis population; Figure 1). For participants who received no parenteral opioids after randomization, 1 hour (zero on log scale) was imputed as their time to last dose of parenteral opioids. For participants who were receiving parenteral opioids for reasons other than vaso-occlusive episodes, if the time of the last vaso-occlusive episode–related parenteral opioid dose was not documented, 3 blinded observers adjudicated the primary outcome. No other imputations were performed.
Figure 1. Participant Flow in a Study of the Effect of Poloxamer 188 vs Placebo on Painful Vaso-Occlusive Episodes in Children and Adults With Sickle Cell Disease.

aIncluding 1 participant with known or suspected bleeding disorder, 1 with pain crisis requiring hospitalization in the preceding 14 days, 1 for whom it was the investigator’s belief that the participant was suffering from chronic pain and not acute pain associated with an ongoing vaso-occlusive episode, and 1 who was not an appropriate study candidate (investigator decision).
bIncluding 3 participants who did not receive the investigational drug, 3 whose total duration of infusion exceeded 53 hours, 1 for whom infusion could not be started within 24 hours of presentation to site, and 1 who withdrew immediately after the start of the loading dose.
The primary outcome was compared between treatment groups using analysis of covariance (ANCOVA) modeling with natural logarithm time as the outcome,16 adjusting for the prespecified stratification groups.
Least squares means and differences from ANCOVA modeling were calculated to obtain geometric means and mean ratios due to the log-normally distributed primary outcome. The reported P values were from the 2-sided t test. For prespecified sensitivity analyses of primary outcomes and subgroups, nonparametric stratified van Elteren tests were used, as well as time-to-event Kaplan-Meier analyses with log-rank test statistics.
Interaction terms were added to the main ANCOVA model, in the form of treatment × covariate, to test for differential treatment effects in subgroups. Two-sided P values <.05 were considered significant, without multiplicity adjustment.
To detect heterogeneity of treatment effect across sites, an ANCOVA model for the primary outcome was fit with treatment as a fixed effect.17 Random effects were site and treatment × site interaction, using type III tests to determine overall significance of the random effects. To maintain stability of treatment effect within site, only sites with at least 5 participants were included in the analysis.
Secondary Outcome Analyses
The number of participants hospitalized for painful vaso-occlusive episode recurrences within 14 days of initial discharge was tabulated by treatment group. The denominator was the number of participants with at least 1 follow-up visit or contact at least 14 days after initial hospital discharge. A likelihood ratio test was used to compare the percentage of re-hospitalized participants in each treatment group. A logistic model was constructed to adjust for stratification factors. In a prespecified sensitivity analysis, a time-to-event analysis was conducted for time from hospital discharge to re-hospitalization. The statistical analysis was hierarchical; significance for the secondary outcomes was not declared unless the P value for the primary analysis was less than .05.
The number and percentage of participants with acute chest syndrome occurring within 120 hours of randomization were tabulated by treatment group. The primary and survival sensitivity analyses were performed in the same way as they were for re-hospitalization; for analyses of percentages by treatment group, adjustment was made for the same predictive factors.
Other Prespecified Analyses
For the length of study drug infusion, total opioid use, and time from randomization to discharge, the approach was analogous to the primary outcome. Thirty-day re-hospitalization was analyzed as a binary outcome, analogous to 14-day re-hospitalization. Predictors were modeled as binary variables with a chosen reference level.
Significance of each variable was based on the ANCOVA model parameter Z test. Geographic region (based on similarities and differences in standard treatment protocols for patients with SCD) was tested as a center-pooled variable, with smaller sites combined into a single prespecified region (eg, the 3 Saudi Arabian sites were included as “Saudi Arabia”). Because non-US regions were not significantly different among themselves, region was condensed to a dichotomous variable (US, non-US).
Adverse Event Analyses
The safety population included participants who received a study infusion, with groups defined by treatment received. Summaries of treatment-emergent adverse events (TEAEs), coded using the Medical Dictionary for Regulatory Activities, were screened to identify TEAEs for which 95% CIs of the difference between treatment groups did not include 0. Multiple adverse event records for an individual were counted as a single adverse event if preferred terms were the same and the dates were contiguous. TEAEs were analyzed by incidence in each treatment group, as well as severity, seriousness, and potential relationship to study medication.
Results
Patient Population
A total of 388 participants were enrolled in the trial (194 randomized to receive poloxamer 188 and 194 randomized to receive placebo). Key demographic variables and baseline characteristics are shown in Table 1. A total of 227 participants (58.5%) were younger than 16 years, with an overall mean age of participants of 15.2 years. The groups were well-balanced for other demographic characteristics, laboratory parameters, and baseline study characteristics, including duration of pain at presentation and randomization, baseline pain scores, and time from randomization to study drug administration.
Table 1. Demographic and Baseline Characteristics of Participants in a Study of the Effect of Poloxamer 188 vs Placebo on Painful Vaso-Occlusive Episodes in Children and Adults With Sickle Cell Disease.
| Characteristic | Mean (SD) | Normal valuesa | |
|---|---|---|---|
| Poloxamer 188 (n = 194) | Placebo (n = 194) | ||
| Demographic characteristics | |||
| Age at randomization, y | 15.2 (6.2) | 15.1 (7.3) | |
| Age <16 y, No. (%)b | 115 (59.3) | 112 (57.7) | |
| Sex, No. (%) | |||
| Male | 105 (54.1) | 107 (55.2) | |
| Female | 89 (45.9) | 87 (44.8) | |
| Ethnicity, No. (%) | |||
| Hispanic or Latino | 31 (16.0) | 20 (10.3) | |
| Race, No. (%)c | |||
| Black/African descent | 109 (57.4) | 118 (61.5) | |
| White | 63 (33.2) | 62 (32.3) | |
| Other | 18 (9.5) | 12 (6.3) | |
| Weight at screening, kg | 47.8 (18.9) | 48.7 (22.3) | |
| Hemoglobin phenotype, No. (%)d | |||
| SS disease | 134 (69.1) | 129 (66.5) | |
| S-β+ thalassemia disease | 21 (10.8) | 28 (14.4) | |
| S-β0 thalassemia disease | 20 (10.3) | 19 (9.8) | |
| SC disease | 19 (9.8) | 18 (9.3) | |
| Participants receiving hydroxyurea, No. (%) | 117 (60.3) | 119 (61.3) | |
| US hospital, No. (%) | 107 (55.2) | 121 (62.4) | |
| Baseline laboratory characteristicse | |||
| White blood cell count, ×10−3/μL | 15.2 (5.9) [n = 188] | 13.7 (5.6) [n = 190] | 4.5-15.5 |
| Hemoglobin, g/dL | 9.1 (1.5) [n = 188] | 9.2 (1.6) [n = 190] | 11.7-16.3 |
| Mean corpuscular volume, fL | 85.9 (14.4) [n = 188] | 84.0 (13.2) [n = 190] | 75.0-100.0 |
| Mean corpuscular hemoglobin concentration, g/dL | 34.4 (1.8) [n = 186] | 34.1 (1.8) [n = 190] | 31.0-37.0 |
| Platelet count, ×10−3/μL | 373.7 (171.2) [n = 188] | 351.0 (162.3) [n = 190] | 150.0-350.0 |
| Reticulocyte count, % | 8.8 (5.8) [n = 167] | 8.2 (5.6) [n = 163] | 0.5-1.8 |
| Creatinine, mg/dL | 0.5 (0.2) [n = 189] | 0.5 (0.2) [n = 188] | 0.3-1.3 |
| Lactate dehydrogenase, median (IQR), U/Le | 594.0 (482.0) [n = 171] | 532.0 (464.0) [n = 169] | 110.0-295.0 |
| Bilirubin, mg/dL | |||
| Total | 2.8 (2.0) [n = 185] | 2.5 (1.7) [n = 182] | 0.0-1.2 |
| Direct | 0.5 (0.4) [n = 175] | 0.4 (0.3) [n = 165] | 0.0-0.4 |
| Aspartate aminotransferase, U/L | 52.5 (30.4) [n = 185] | 47.9 (23.5) [n = 182] | ≤37.0 |
| Alanine transaminase, U/L | 28.4 (22.4) [n = 188] | 25.0 (17.5) [n = 185] | ≤40.0 |
| Key baseline study characteristics | |||
| Duration of pain at presentation, hf | 6.5 (5.4) [n = 193] | 7.2 (6.0) [n = 192] | |
| Time from presentation to randomization, h | 13.7 (6.6) | 13.6 (6.4) | |
| Duration of pain prior to randomization, h | 20.2 (7.8) | 20.6 (8.4) | |
| Wong-Baker FACES Pain Rating Scale score at randomization, No. (%)g |
|||
| <8 | 94 (48.5) | 95 (49.0) | |
| ≥8 | 100 (51.5) | 99 (51.0) | |
| Time from randomization to study drug start, h | 2.2 (1.7) [n = 188] | 2.0 (1.5) [n = 192] | |
Abbreviation: IQR, interquartile range.
SI conversion factors: To convert white blood cell count and platelet count to 109/L, multiply values by 0.001; hemoglobin and mean corpuscular hemoglobin concentration to g/L, multiply by 10.0; creatinine to μmol/L, multiply by 88.4; bilirubin to μmol/L, multiply by 17.104; and lactate dehydrogenase, aspartate aminotransferase, and alanine transaminase to μkat/L, multiply by 0.0167.
For laboratory values for which normal values vary significantly for children and adults, the values represented are the lowest and highest values for either children or adults.18
The age of 16 years was chosen as the cutoff to mirror that used in the previous phase 3 trial so that comparisons of results could be made between the trials.
Race was not reported for 2 participants in the poloxamer 188 group and 2 participants in the placebo group. Race was reported as unknown for 2 participants in the poloxamer 188 group. “Other” race was designated on the demographic form and further detail was not available.
Hemoglobin SS (HbSS) is homozygous HbS (sickle cell anemia). HbS-β+ thalassemia is double heterozygosity for HbS and β plus thalassemia trait. Some normal adult hemoglobin (HbA) is present. HbS-β0 thalassemia is double heterozygosity for HbS and beta null thalassemia. No HbA is present. HbSC is double heterozygosity for HbS and HbC.
Mean (SD) is shown when normally distributed and median (IQR) is shown otherwise.
Duration of pain at presentation was self-reported and measured as time of onset of moderate/severe vaso-occlusive episode to time of hospitalization.
The Wong-Baker FACES Pain Rating Scale (range, 0-10) is a 6-item ordinal scale depicting painful and nonpainful faces that are each assigned an ordinal value, with 0 representing no pain and 10 representing the worst pain.13
Eight participants received no study treatment after randomization (6 in the poloxamer 188 group and 2 in the placebo group; Figure 1). Five participants received their last dose of parenteral opioids before randomization (1 in the poloxamer 188 group and 4 in the placebo group) and were assigned a time of 1 hour (natural logarithm time to last dose of parenteral opioids = 0). Time and date of the last dose of parenteral opioids were unavailable for 4 participants (1 in the poloxamer 188 group and 3 in the placebo group). They were assigned a time to last dose of parenteral opioids of 1 hour; thus, the primary outcome was available for 384 of 388 (99.0%) participants and imputed for 4 (1.0%). Fifteen-day follow-up contacts occurred for 357 participants (92.0%) and 30-day follow-up contacts occurred for 368 (94.8%). Fourteen-day re-hospitalization data were available for 192 participants (99.0%) in the poloxamer 188 group and 190 (97.9%) in the placebo group and 30-day re-hospitalization data were available for 132 participants (68.0%) in the poloxamer 188 group and 142 (73.2%) in the placebo group.
Primary Outcome
For the primary analysis population, there was no significant difference between treatment groups in the mean time from randomization to last administration of parenteral opioids by ANCOVA (81.8 hours in the poloxamer 188 group vs 77.8 hours in the placebo group; difference, 4.0 hours [95% CI, −7.8 to 15.7]; geometric mean ratio, 1.2 [95% CI, 1.0-1.5]; P = .09; Table 2). Box and Kaplan-Meier plots are shown in Figure 2.
Table 2. Outcomes in a Study of the Effect of Poloxamer 188 vs Placebo on Painful Vaso-Occlusive Episodes in Children and Adults With Sickle Cell Diseasea,b.
| Outcome | Poloxamer 188 | Placebo | Difference (95% CI) | Least squares geometric mean ratio (95% CI)c | P valued |
|---|---|---|---|---|---|
| Primary outcome | |||||
| Time from randomization to last administration of parenteral opioids for treatment of vaso-occlusive episodes, mean, h | |||||
| Primary analysis population | 81.8 (n = 194) | 77.8 (n = 194) | 4.0 (−7.8 to 15.7) | 1.2 (1.0 to 1.5) | .09 |
| Participants aged <16 y | 88.7 (n = 115) | 71.9 (n = 112) | 16.8 (1.7 to 32.0) | 1.4 (1.1 to 1.8) | .008 |
| Participants aged ≥16 y | 71.7 (n = 79) | 86.0 (n = 82) | −14.3 (−32.8 to 4.3) | 0.9 (0.7 to 1.4) | .76 |
| Secondary outcomes, No./total No. (%) | |||||
| Acute chest syndrome | 32/194 (16.5) | 22/194 (11.3) | 5.2 (−1.7 to 12.0) | ||
| Re-hospitalization for recurrence of vaso-occlusive episodes within 14 d of initial hospital discharge | 16/192 (8.3) | 13/190 (6.8) | 1.5 (−3.8 to 6.8) | ||
All primary analysis population results are shown. Only prespecified subgroups for which interaction testing was significant are shown. The interaction between treatment and age was significant (P = .01). Interactions between treatment and other subgroups of interest, including hydroxyurea usage and region, were tested and found to be not significant (P = .51 and P = .87). All other nonsignificant prespecified analyses are shown in eTables 3, 4, and 5 in Supplement 3.
Comparisons between treatment groups in the primary efficacy analysis were adjusted for age (<16, ≥16), hydroxyurea use (yes, no), Wong-Baker FACES Pain Rating Scale score at randomization (<8, ≥8), and region (US, non-US) in an analysis of covariance model. Secondary outcome comparisons between treatment arms were adjusted for the same variables, using logistic regression.
For the primary outcome, the natural log transformation was applied to individual values prior to conducting each analysis. The least squares means and least squares mean differences were calculated on a log scale and transformed back to the original scale using the exponential function to obtain geometric means and geometric mean ratios. The least squares geometric mean ratio is an indication of the difference between the log mean time for poloxamer 188 and placebo adjusted for other factors in the model. A CI that does not include 1.0 indicates a significant difference.
Computed using a geometric analysis of covariance test.
Figure 2. Time From Randomization to Last Administration of Parenteral Opioids in a Study of the Effect of Poloxamer 188 vs Placebo on Painful Vaso-Occlusive Episodes in Children and Adults With Sickle Cell Disease.
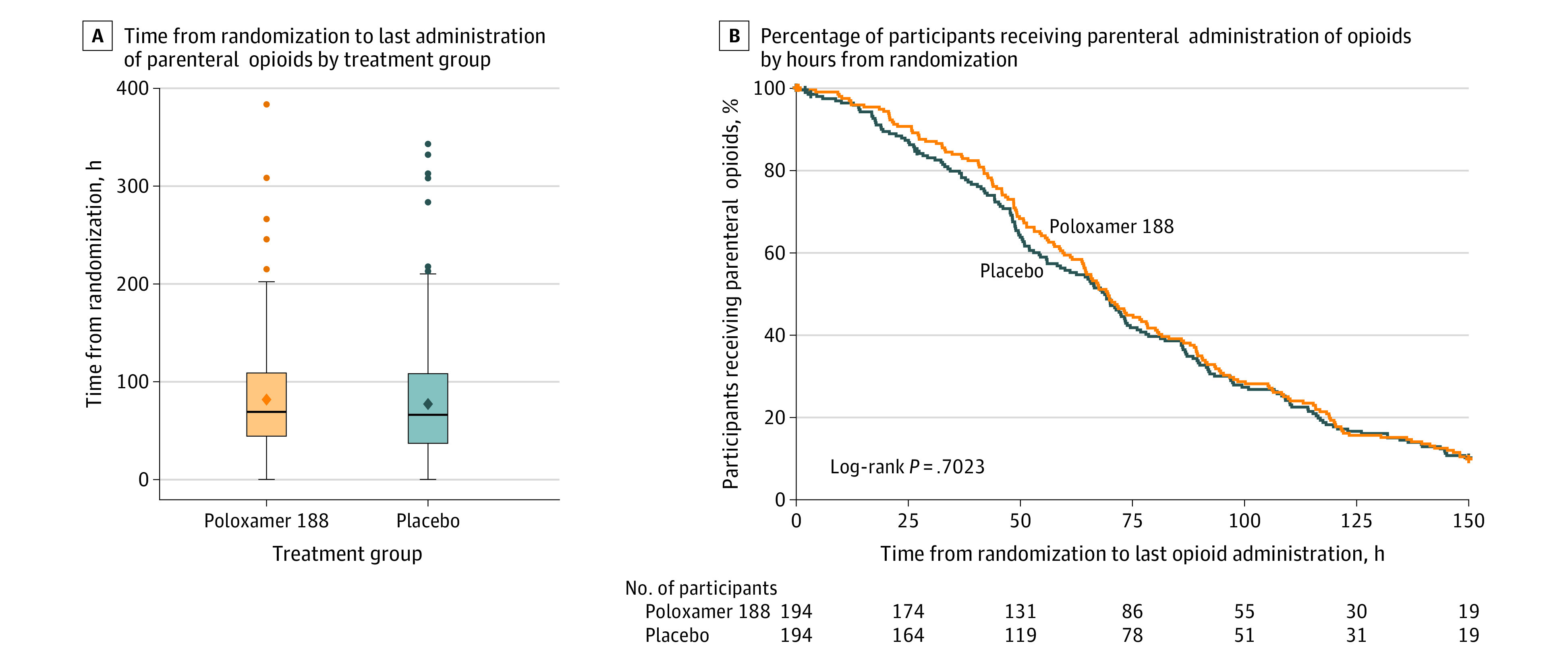
A, The mean values are represented as diamonds. Outliers are represented as circles beyond the whiskers. Error bars indicate the highest and lowest values within 1.5 × the interquartile range. The median (interquartile range) time from randomization to last administration of parenteral opioids was 69.2 (44.7-108.7) hours in the poloxamer 188 group and 66.4 (37.8-107.3) hours in the placebo group. B, No significant difference in time from randomization to last administration of parenteral opioids was observed between treatment groups in the primary analysis population.
Secondary Outcomes
For the primary analysis population, there were 32 participants with acute chest syndrome in the poloxamer 188 group (16.5%) and 22 (11.3%) in the placebo group (difference, 5.2% [95% CI, −1.7% to 12.0%]). Sixteen of 192 participants (8.3%) in the poloxamer group and 13 of 190 (6.8%) in the placebo group with 15-day follow-up visits were re-hospitalized for recurrence of painful vaso-occlusive episodes within 14 days (difference, 1.5% [95% CI, −3.8% to 6.8%]) (Table 2). Because of the hierarchical statistical testing, significance was not declared for the secondary outcomes.
Prespecified Subgroup Analyses
Based on a significant interaction of age and treatment (P = .01), there was a treatment difference in the mean time from randomization to last administration of parenteral opioids by ANCOVA for participants younger than 16 years, favoring the placebo group (88.7 hours in the poloxamer 188 group vs 71.9 hours in the placebo group; difference, 16.8 hours [95% CI, 1.7-32.0]; geometric mean ratio, 1.4 [95% CI, 1.1-1.8]; P = .008; Van Elteren test P < .05). For participants aged 16 years and older, the mean time from randomization to last administration of parenteral opioids was 71.7 hours in the poloxamer 188 group vs 86.0 hours in the placebo group (difference −14.3 hours [95% CI, −32.8 to 4.3]; geometric mean ratio, 0.9 [95% CI, 0.7-1.4]; P = .76; Van Elteren test P = .82) (Table 2). For participants who received hydroxyurea (n = 236), the results of the interaction test between hydroxyurea (yes, no) and treatment were not significant. Among the subgroup of participants younger than 16 years, there were 24 acute chest syndrome events (20.9%) in the poloxamer 188 group vs 12 (10.7%) in the placebo group (difference, 10.2% [95% CI, 0.8%-19.5%]); among participants aged 16 years or older, 8 acute chest syndrome events (10.1%) occurred in the poloxamer 188 group and 10 (12.2%) occurred in the placebo group (difference, −2.1% [95% CI, −11.8% to 7.7%]). Because acute chest syndrome was a secondary outcome, significance was not declared for either subgroup. Results of all other subgroup analyses, including duration of pain at presentation, pain score at randomization, and region, were nonsignificant (eTable 4 in Supplement 3).
Site Effects
A post hoc test for site × treatment interaction indicated no significant heterogeneity of treatment effect across the 25 sites with at least 5 participants (F23 = 0.91; P = .58).
Other Prespecified Analyses
No significant differences were seen for length of study drug infusion, total opioid use in morphine equivalents, time from randomization to discharge, or re-hospitalization for painful vaso-occlusive episode within 30 days (eTable 5 in Supplement 3).
Adverse Events
The 189 participants in the poloxamer 188 group and 191 participants in the placebo group safety population were compared for adverse events across all system organ classes and specific adverse events. Adverse events that were more common in the poloxamer 188 group than the placebo group included abdominal distension, hepatobiliary disorders, hyperbilirubinemia, and upper respiratory infections; hypoxia and infusion site swelling and pain were more common in the placebo group than in the poloxamer 188 group (Table 3). The majority of hepatobiliary disorders (77.3%) were elevated direct or total bilirubin, known adverse effects of poloxamer 188,10 which resolved in all participants in the poloxamer 188 group who were tested at the 30-day follow-up visit. Serious adverse events were similar between the poloxamer 188 and placebo groups, but anemia was more common in the placebo group (4 vs 0 participants) (Table 3).
Table 3. Adverse Events in a Study of the Effect of Poloxamer 188 vs Placebo on Painful Vaso-Occlusive Episodes in Children and Adults With Sickle Cell Disease.
| Adverse event | No. (%) | |
|---|---|---|
| Poloxamer 188 (n=189) | Placebo (n=191) | |
| Treatment-emergent adverse eventsa | ||
| ≥1 Adverse event | 184 (97.4) | 186 (97.4) |
| Abdominal distension | 4 (2.1) | 0 |
| Hepatobiliary disorders | 30 (15.9) | 14 (7.3) |
| Hyperbilirubinemia | 24 (12.7) | 10 (5.2) |
| Hypoxia | 10 (5.3) | 23 (12.0) |
| Infusion site pain | 0 | 4 (2.1) |
| Infusion site swelling | 0 | 4 (2.1) |
| Upper respiratory tract infection | 15 (7.9) | 5 (2.6) |
| Treatment-emergent serious adverse eventsb | ||
| ≥1 Serious adverse event | 64 (33.9) | 57 (29.8) |
| Anemia | 0 | 4 (2.1) |
Treatment-emergent adverse events (TEAEs) were defined as AEs occurring after the start of infusion; existing AEs that worsened during the study; or AEs that began prior to the start of infusion but were reported as possibly, probably, or definitely related to treatment. The safety group and all subgroups were screened by the Medical Dictionary for Regulatory Activities system organ class and preferred terms for TEAEs for which the 95% CIs of the difference between treatment groups did not include 0. Multiple AE records for an individual participant were counted as a single AE occurrence if preferred terms were the same and the stop date of the first AE matched the start date of the subsequent AE. TEAEs were analyzed with respect to incidence in each randomized treatment group, as well as by severity, seriousness, and potential relationship of the AE to the study medication.
TEAEs that were fatal, were life-threatening, required hospitalization, or prolonged hospitalization.
Discussion
In the current study, there was no evidence of favorable effects of poloxamer 188 on time to last dose of parental opioids. In contrast to the apparent benefit to participants younger than 16 years in the previous phase 3 study, apparent harm with poloxamer 188 was found in this age group in the current study.
Prior to the current study, more than 300 participants with SCD and painful vaso-occlusive episodes or acute chest syndrome were treated with poloxamer 188 in phase 1 to phase 3 studies. A placebo-controlled, randomized, phase 2 study of 28 participants treated with poloxamer 188 and 22 who were given placebo showed statistically significant differences in use of parenteral opioids, with non–statistically significant reductions in pain intensity and durations of crisis and hospitalization, all favoring the poloxamer 188 group.9 These results led to a multicenter, double-blind, randomized, placebo-controlled, phase 3 study of 255 children and adults with painful vaso-occlusive episodes that reported a reduction in mean duration of crisis (the primary outcome) of 8.8 hours (P = .04).10 The reduction was greater (approximately 21 hours) in participants younger than 16 years (127.1 hours in the poloxamer 188 group and 148.6 hours in the placebo group; P = .01) and in those receiving hydroxyurea (approximately 16 hours) (141.4 hours in the poloxamer 188 group vs 157.2 hours in the placebo group; P = .02). The study did not reach its enrollment goal (350 participants), raising the question of whether a larger study might have shown more significant results. The authors concluded that these results needed to be confirmed in subsequent clinical trials.
In evaluating such discrepant results in 2 phase 3 studies, several factors were considered. The findings may have occurred by chance; however, assuming the null hypothesis is correct (no difference between poloxamer 188 and placebo), the odds that 2 studies would produce such differing results in participants younger than 16 years by chance would be less than 1 in 10 000 (0.008 × 0.01). Thus, it seems likely that additional factors affected the results. The current study enrolled a younger population (mean age of 15.2 years vs 21.1 years), but was otherwise similar in design. The most significant difference was the primary outcome.
The choice of outcome measure has been critical to studies of SCD, especially for painful vaso-occlusive episodes. Assessment of pain is subjective and difficult to quantify. There are no precise biomarkers for painful vaso-occlusive episodes. In the first phase 3 study, stringent crisis termination criteria were used as an outcome. It proved difficult for study personnel and participants to adhere to these criteria, with a high rate of incomplete documentation of crisis resolution criteria (61 of 255 participants [23.9%]). In addition, these occurrences were unevenly distributed; more participants in the placebo group (38 of 128 [29.7%]) fell into this category than participants in the poloxamer 188 group (23 of 127 [18.1%]). This resulted in imputation for more participants in the placebo group than in the poloxamer 188 group, with assignment of the worst possible outcome of 168 hours, the imbalance favoring the poloxamer 188 group. Participants whose crises did not resolve in 168 hours were also assigned the worst possible outcome. Overall, 145 participants (56.9%) did not meet crisis resolution criteria within 168 hours, including 62 of 127 (48.8%) in the poloxamer 188 group and 83 of 128 (64.8%) in the placebo group, and were assigned the worst possible outcome. This included 6 randomized participants who did not receive the study drug (5 in the placebo group and 1 in the poloxamer 188 group) who were assigned the worst possible outcome, contributing to the imbalance in results for duration of crisis. Without these 6 participants, the P value increased from .04 to .07.10
In the current study, the study team attempted to develop an easily verified, quantifiable outcome measure more likely to be attained for each participant. Thus, time to last dose of parenteral opioids was selected as the primary outcome measure. This primary outcome was available for 99% of study participants, limiting imputations to 1%, which avoided imbalances in participants reaching verifiable outcomes seen in the prior study.
In the first phase 3 study, a small, nonsignificant difference was seen in the incidence of acute chest syndrome for children (3 of 37 in the poloxamer 188 group vs 6 of 36 in the placebo group). No beneficial effect on acute chest syndrome was seen in the current study. Rather, although not statistically significant, there were more participants younger than 16 years who developed acute chest syndrome in the poloxamer 188 group than in the placebo group, paralleling the direction of effects on the primary outcome for participants younger than 16 years.
In the current study, there were no apparent effects on acute chest syndrome or readmission for painful vaso-occlusive episodes in participants receiving hydroxyurea, despite the known effect of hydroxyurea in reducing rates of painful vaso-occlusive episodes and acute chest syndrome. Also, significant reductions in duration of crisis for participants receiving hydroxyurea in either treatment group were not seen. Cautious interpretation is warranted because the study was not designed to address these questions, more severely affected patients may be prescribed hydroxyurea, and hydroxyurea adherence for participants in the trial is unknown.
Limitations
This study has several limitations. First, the primary outcome measure, although it is quantitative and relatively easy to determine, still has subjective aspects. A variety of factors contribute to decisions to discontinue parenteral opioids, including patient, family, and clinician preferences and timing of assessments, which can vary widely among sites and individuals. Second, poloxamer 188 is difficult to blind effectively. As a detergent, agitation can cause foaming that might unblind participants or staff to treatment assignment. Although appropriate blinding procedures were in place, assessment of their success was not performed in this study or previous studies of poloxamer 188.
Conclusions
Among children and adults with SCD, poloxamer 188 did not significantly shorten time to last dose of parenteral opioids during vaso-occlusive episodes. These findings do not support the use of poloxamer 188 for vaso-occlusive episodes.
Trial protocol
Statistical analysis plan
eTables and eFigure
Data sharing statement
References
- 1.Cooper TE, Hambleton IR, Ballas SK, Johnston BA, Wiffen PJ. Pharmacological interventions for painful sickle cell vaso-occlusive crises in adults. Cochrane Database Syst Rev. 2019;2019(11):CD012187. doi: 10.1002/14651858.CD012187.pub2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Platt OS, Brambilla DJ, Rosse WF, et al. Mortality in sickle cell disease: life expectancy and risk factors for early death. N Engl J Med. 1994;330(23):1639-1644. doi: 10.1056/NEJM199406093302303 [DOI] [PubMed] [Google Scholar]
- 3.Ballas SK, Lusardi M. Hospital readmission for adult acute sickle cell painful episodes: frequency, etiology, and prognostic significance. Am J Hematol. 2005;79(1):17-25. doi: 10.1002/ajh.20336 [DOI] [PubMed] [Google Scholar]
- 4.Sundd P, Gladwin MT, Novelli EM. Pathophysiology of sickle cell disease. Annu Rev Pathol. 2019;14(1):263-292. doi: 10.1146/annurev-pathmechdis-012418-012838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017;376(16):1561-1573. doi: 10.1056/NEJMra1510865 [DOI] [PubMed] [Google Scholar]
- 6.Hebbel RP, Belcher JD, Vercellotti GM. The multifaceted role of ischemia/reperfusion in sickle cell anemia. J Clin Invest. 2020;130(3):1062-1072. doi: 10.1172/JCI133639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato GJ, Piel FB, Reid CD, et al. Sickle cell disease. Nat Rev Dis Primers. 2018;4:18010. doi: 10.1038/nrdp.2018.10 [DOI] [PubMed] [Google Scholar]
- 8.Hunter RL, Luo AZ, Zhang R, Kozar RA, Moore FA. Poloxamer 188 inhibition of ischemia/reperfusion injury: evidence for a novel anti-adhesive mechanism. Ann Clin Lab Sci. 2010;40(2):115-125. [PubMed] [Google Scholar]
- 9.Adams-Graves P, Kedar A, Koshy M, et al. RheothRx (poloxamer 188) injection for the acute painful episode of sickle cell disease: a pilot study. Blood. 1997;90(5):2041-2046. doi: 10.1182/blood.V90.5.2041 [DOI] [PubMed] [Google Scholar]
- 10.Orringer EP, Casella JF, Ataga KI, et al. Purified poloxamer 188 for treatment of acute vaso-occlusive crisis of sickle cell disease: a randomized controlled trial. JAMA. 2001;286(17):2099-2106. doi: 10.1001/jama.286.17.2099 [DOI] [PubMed] [Google Scholar]
- 11.Gibbs WJ, Hagemann TM. Purified poloxamer 188 for sickle cell vaso-occlusive crisis. Ann Pharmacother. 2004;38(2):320-324. doi: 10.1345/aph.1D223 [DOI] [PubMed] [Google Scholar]
- 12.Ballas SK, Files B, Luchtman-Jones L, et al. Safety of purified poloxamer 188 in sickle cell disease: phase I study of a non-ionic surfactant in the management of acute chest syndrome. Hemoglobin. 2004;28(2):85-102. doi: 10.1081/HEM-120035919 [DOI] [PubMed] [Google Scholar]
- 13.Garra G, Singer AJ, Taira BR, et al. Validation of the Wong-Baker FACES Pain Rating Scale in pediatric emergency department patients. Acad Emerg Med. 2010;17(1):50-54. doi: 10.1111/j.1553-2712.2009.00620.x [DOI] [PubMed] [Google Scholar]
- 14.Ware LJ, Epps CD, Herr K, Packard A. Evaluation of the Revised Faces Pain Scale, Verbal Descriptor Scale, Numeric Rating Scale, and Iowa Pain Thermometer in older minority adults. Pain Manag Nurs. 2006;7(3):117-125. doi: 10.1016/j.pmn.2006.06.005 [DOI] [PubMed] [Google Scholar]
- 15.Vichinsky EP, Neumayr LD, Earles AN, et al. ; National Acute Chest Syndrome Study Group . Causes and outcomes of the acute chest syndrome in sickle cell disease. N Engl J Med. 2000;342(25):1855-1865. doi: 10.1056/NEJM200006223422502 [DOI] [PubMed] [Google Scholar]
- 16.Wagenmakers E-J, Brown S. On the linear relation between the mean and the standard deviation of a response time distribution. Psychol Rev. 2007;114(3):830-841. doi: 10.1037/0033-295X.114.3.830 [DOI] [PubMed] [Google Scholar]
- 17.Feaster DJ, Mikulich-Gilbertson S, Brincks AM. Modeling site effects in the design and analysis of multi-site trials. Am J Drug Alcohol Abuse. 2011;37(5):383-391. doi: 10.3109/00952990.2011.600386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Test directory for The Johns Hopkins Hospital . Johns Hopkins Medical Laboratories Services. Accessed January 12, 2021. http://pathology.jhu.edu/jhml-services/test-directory/
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Trial protocol
Statistical analysis plan
eTables and eFigure
Data sharing statement