

Abstract
Background:
Staphylococcus aureus and Staphylococcus epidermidis are the most abundant bacteria found on the skin of patients with atopic dermatitis (AD). S aureus is known to exacerbate AD, whereas S epidermidis has been considered a beneficial commensal organism.
Objective:
In this study, we hypothesized that S epidermidis could promote skin damage in AD by the production of a protease that damages the epidermal barrier.
Methods:
The protease activity of S epidermidis isolates was compared with that of other staphylococcal species. The capacity of S epidermidis to degrade the barrier and induce inflammation was examined by using human keratinocyte tissue culture and mouse models. Skin swabs from atopic and healthy adult subjects were analyzed for the presence of S epidermidis genomic DNA and mRNA.
Results:
S epidermidis strains were observed to produce strong cysteine protease activity when grown at high density. The enzyme responsible for this activity was identified as EcpA, a cysteine protease under quorum sensing control. EcpA was shown to degrade desmoglein-1 and LL-37 in vitro, disrupt the physical barrier, and induce skin inflammation in mice. The abundance of S epidermidis and expression of ecpA mRNA were increased on the skin of some patients with AD, and this correlated with disease severity. Another commensal skin bacterial species, Staphylococcus hominis, can inhibit EcpA production by S epidermidis.
Conclusion:
S epidermidis has commonly been regarded as a beneficial skin microbe, whereas S aureus has been considered deleterious. This study suggests that the overabundance of S epidermidis found on some atopic patients can act similarly to S aureus and damage the skin by expression of a cysteine protease.
Keywords: Atopic dermatitis, microbiome, dysbiosis, Staphylococcus epidermidis, protease, skin, epidermal barrier, inflammation, cytokine
Graphical Abstract
Atopic dermatitis (AD) is a common inflammatory skin disease that is often associated with other allergic conditions and can affect up to 15% to 20% of children and 1% to 3% of adults worldwide.1–4 The prevalence of AD is highest in industrialized countries and has grown over the past decades. Meta-analyses have shown that exposures to some environmental conditions, such as having 3 or more siblings, daycare, pet ownership, and farm residence during childhood, are associated with a lower risk of AD.5,6 Although a clear explanation for these associations remains unknown, early-life exposure to a healthy microbial community on the skin and gut have been hypothesized to protect against AD.7,8
Increasingly detailed analyses have been performed to describe the composition of microbes that inhabit AD skin with the hope of better understanding how they may influence the development and progression of disease. AD skin is often characterized by an overabundance of staphylococcal bacterial species and a decrease in diversity of microbes. In particular, depending on the methods used, 30% to 100% of subjects with AD have been observed to be colonized on their skin by the common infectious pathogen Staphylococcus aureus.9,10 As a consequence of the close association between the presence of S aureus and severity of skin inflammation, this bacterial species has been a main focus for study of how the skin microbiome can promote AD. Multiple experimental models have clearly shown that S aureus can disrupt the epidermal barrier and alter both adaptive and innate immune responses.9–17 Several gene products of S aureus, including adhesions, superantigens, toxins, phenol-soluble modulins (PSMs), TLR ligands, and proteases, have been identified to promote these negative effects.
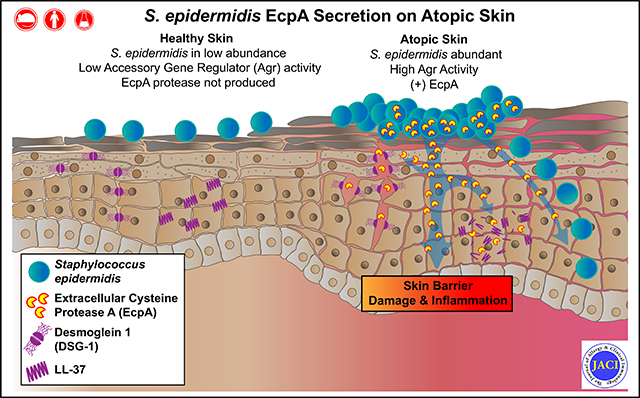
Although an overgrowth of Staphylococcus epidermidis on AD lesional skin has been reported in several studies, 10,18–20 this common commensal bacterium is generally regarded as a key member of the healthy skin microbiota with multiple benefits for the host when present on the skin.21–23 For example, S epidermidis has been reported to protect the skin against S aureus colonization through the production of various antimicrobial and anti-inflammatory molecules.24–26 However, S epidermidis is also an opportunistic pathogen, especially after contamination of implanted medical devices.27–30 In these conditions, the overgrowth and invasion of S epidermidis result in serious life-threatening infections.
In this study, we hypothesized that the overabundance of S epidermidis colonizing some patients with AD may alter the usual commensal host-microbe relationship and result in skin disease. Because S epidermidis is the coagulase-negative Staphylococcus (CoNS) that is most closely related to S aureus, we investigated whether protease activity from S epidermidis could induce skin damage in AD by enzymatic mechanisms similar to those used by S aureus. We have demonstrated that some S epidermidis strains can cause skin barrier damage and inflammation through secretion of the cysteine protease EcpA and that this correlated with human disease severity. These findings suggest that the permissive growth conditions of AD skin enable S epidermidis to shift from a beneficial commensal to a deleterious pathogen similar to S aureus.
METHODS
Human subjects and skin swab collection
Experiments involving human subjects were done according to protocols approved by the University of California, San Diego, institutional review board (project no. 140144). Written informed consent was obtained from all subjects. Table E1 (available in this article’s Online Repository at www.jacionline.org ) summarizes age, sex, ethnicity, and disease severity (local Eczema Area and Severity score and Scoring Atopic Dermatitis score) of all subjects. Swabs of surface microbiota from a 5-cm2 area of the antecubital fossa skin of both the left and right arms were collected from 14 healthy subjects and 13 patients with AD as previously described.26 For subjects with AD, swabs were collected from both lesional and nonlesional skin.
TABLE E1.
Human subjects involved in this study
| Group | Subject no. | Date of collection | Sex | Age (y) | Ethnicity | Local EASI score |
oSCORAD disease score | |
|---|---|---|---|---|---|---|---|---|
| Right arm | Left arm | |||||||
| Healthy subjects | H01 | 15/07/2019 | F | 51 | Hispanic or Latino/White | N/A | N/A | N/A |
| H02 | 15/07/2019 | F | 22 | Hispanic or Latino/White | N/A | N/A | N/A | |
| H03 | 15/07/2019 | F | 40 | Hispanic or Latino/White | N/A | N/A | N/A | |
| H04 | 15/07/2019 | F | 45 | White | N/A | N/A | N/A | |
| H05 | 15/07/2019 | M | 27 | White | N/A | N/A | N/A | |
| H06 | 17/07/2019 | F | 23 | Asian | N/A | N/A | N/A | |
| H07 | 19/07/2019 | F | 23 | Hispanic or Latino/White | N/A | N/A | N/A | |
| H08 | 22/07/2019 | F | 31 | Black/White | N/A | N/A | N/A | |
| H09 | 30/07/2019 | F | 28 | White | N/A | N/A | N/A | |
| H10 | 14/08/2019 | M | 40 | Asian | N/A | N/A | N/A | |
| H11 | 16/09/2019 | F | 55 | Asian | N/A | N/A | N/A | |
| H12 | 16/09/2019 | M | 58 | Asian | N/A | N/A | N/A | |
| H13 | 18/09/2019 | M | 21 | White | N/A | N/A | N/A | |
| H14 | 01/10/2019 | M | 27 | Hispanic or Latino/White | N/A | N/A | N/A | |
| Subjects with atopic dermatitis | AD01 | 16/07/2019 | M | 44 | Black | 0.095 | 0.095 | 66 |
| AD02 | 18/07/2019 | F | 26 | Asian | 0.075 | 0.055 | 54.3 | |
| AD03 | 25/07/2019 | M | 53 | White | 0.14 | 0.14 | 55.9 | |
| AD04 | 25/07/2019 | M | 23 | Asian | 0.12 | 0.12 | 34.2 | |
| AD05 | 25/07/2019 | F | 21 | Asian | 0.11 | 0.12 | 34.9 | |
| AD06 | 27/08/2019 | M | 40 | White | 0.055 | 0.075 | 67.2 | |
| AD07 | 19/09/2019 | F | 20 | Asian | 0.07 | 0.07 | 64.3 | |
| AD08 | 20/09/2019 | M | 20 | White | 0.055 | 0.055 | 24.6 | |
| AD09 | 30/09/2019 | F | 36 | Hispanic or Latino/White | 0.015 | 0.0125 | 22.68 | |
| AD10 | 04/10/2019 | F | 51 | White | 0.095 | 0.095 | 55.5 | |
| AD11 | 04/10/2019 | F | 21 | White | 0.04 | 0.04 | 33.4 | |
| AD12 | 08/10/2019 | M | 18 | Asian | 0.18 | 0.18 | 53.2 | |
| AD13 | 08/10/2019 | F | 18 | Asian | 0.15 | 0.15 | 44.8 | |
EASI, Eczema Area and Severity Index; F, female; M, male; N/A, nonapplicable; oSCORAD, Objective Scoring Atopic Dermatitis.
All samples were collected on the antecubital fossa skin. None of the subjects were undergoing treatment at the time of collection.
Mouse experiments
Age-matched 8- to 10-week-old female C57BL/6J mice were used in all experiments (n ≥ 4 per condition). The mice were cohoused at a rate of 3 to 5 mice per cage. All animal experiments were approved by the University of California, San Diego, Institutional Animal Care and Use Committee (protocol no. S09074). For both live bacteria or EcpA topical treatment, the dorsal skin of anesthetized mice (2% isoflurane) was shaved and depilated by using Nair cream followed by removal with alcohol wipes. The skin was allowed to recover from hair removal for at least 48 hours before application of bacteria. For topical bacterial exposure, S epidermidis or S aureus on agar disks (3% tryptic soy broth [TSB], 2% agar; diameter 8 mm) at a final density of 106 colony-forming units (CFUs)/cm2 was applied to the skin for 48 hours as previously described.13 An agar disk without bacteria was used as a vehicle control. For the S epidermidis agr inhibition experiments, live Staphylococcus hominis (106 CFUs/cm2), S hominis supernatant (5 μL), or synthetic autoinducing peptide (AIP) from S hominis C5 (10 μg)31 was loaded onto the agar disk at the same time as the S epidermidis. For topical application of EcpA on mouse skin, 2.5 μg of the purified enzyme (for details about purification, see the Methods section of this article’s Online Repository at www.jacionline.org ) in PBS containing 5 mM L-cysteine (EcpA activator) was applied (final volume 35 μL) for 24 hours on a 1 × 1-cm2 sterile gauze. PBS containing 5 mM L-cysteine was used as a vehicle control. The dorsal skin was then covered with wound dressing film (Tegaderm [3M]) and a bandage to hold the agar disk or gauze in place for the duration of the treatment. For the AD mouse model, age- and sex-matched BALB/c Flg−/− (flaky tail) mouse dorsal skin was tape-stripped and treated 3 times at 2-week intervals (for8 days with replacement every second day) with ovalbumin patches (6 weeks total) before removal of hair and application of live bacteria for 24 hours as previously published.13
Primary keratinocyte culture
Normal neonatal human primary epidermal keratinocytes (NHEKs) (Thermo Fisher Scientific, Waltham, Mass) were cultured in Epilife complete medium containing 60 mM CaCl2 (Thermo Fisher Scientific) supplemented with 1× human keratinocyte growth supplement (Thermo Fisher Scientific) and a 1× antibiotic-antimycotic (100 U/mL of penicillin, 100 U/mL of streptomycin, and 250 ng/mL of amphotericin B [Thermo Fisher Scientific]) at 37°C and 5% CO2. NHEKs were used only for experiments between passages 3 to 5. NHEKs were grown to about 90% to 100% confluency followed by differentiation in EpiLife complete medium with 2 mM CaCl2 for 72 hours. For bacterial supernatant treatments, differentiated NHEKs were treated with sterile-filtered bacterial supernatant at 5% (vol) in Epilife medium for 8 hours and then harvested for RNA extraction. For treatment with EcpA, the purified enzyme (final concentration 2.5 μg/mL) was added to the culture medium along with 5 mM of L-cysteine over the course of either 8 hours for RNA extraction or 20 hours for protein extraction. Some NHEKs were treated with cycloheximide (final concentration 20 μg/mL) (Sigma-Aldrich, St Louis, Mo) 2 hours before treatment with EcpA and for the entire duration of the treatment.
Protease activity assays
The protease activity assays were performed by using the EnzChek Elastase Assay Kit and EnzChek Gelatinase/Collagenase Assay Kit (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, 20 μL of bacteria supernatant from various CoNS strains (see Table E2 in this article’s Online Repository at www.jacionline.org ) was incubated with 1 μg of DQ gelatin, DQ elastin, DQ collagen I, or DQ collagen IV (Thermo Fisher Scientific) in the supplied digestion buffer in 96-well black plates (Corning, NY) for 20 hours, in the presence or absence of various protease inhibitors: E64 (Sigma-Aldrich) at 5 μM; EDTA (Thermo Fisher Scientific) at 2 mM; 1,10-phenanthroline (Sigma-Aldrich) at 2 mM; or aprotinin (Sigma-Aldrich) at 0.5 mg/mL. Relative fluorescent intensity was analyzed with a SpectraMAX Gemini EM fluorometer (Thermo Fisher Scientific; excitation wavelength, 485 nm; emission excitation wavelength, 538 nm).
TABLE E2.
Bacteria strains used in this study
| Name | Source | Reference |
|---|---|---|
| Staphylococcus epidermidis 1457 (AH2490) | Horswill (UC Denver) | Olson et al, 2014E2 |
| Staphylococcus epidermidis 1457 ΔecpA (AH2924) | Horswill (UC Denver) | Olson et al, 2014E2 |
| Staphylococcus epidermidis 12228 agr type I P3-GFP Ermr (AH3408) | Horswill (UC Denver) | Olson et al, 2014E2 |
| Staphylococcus epidermidis 1457 agr type II P3-GFP Ermr (AH273) | Horswill (UC Denver) | Olson et al, 2014E2 |
| Staphylococcus epidermidis 8247 agr type III P3-GFP Ermr (AH3409) | Horswill (UC Denver) | Olson et al, 2014E2 |
| Staphylococcus epidermidis (SE) ATCC12228 | Gallo (UCSD) | Zhang et al, 2003E5 |
| Staphylococcus epidermidis (SE) A9 | Gallo (UCSD) | This study |
| Staphylococcus epidermidis (SE) A11 | Gallo (UCSD) | This study |
| Staphylococcus epidermidis (SE) 09-G7 | Gallo (UCSD) | This study |
| Staphylococcus epidermidis (SE) 18-F3 | Gallo (UCSD) | This study |
| Staphylococcus epidermidis (SE) 05-A5 | Gallo (UCSD) | This study |
| Staphylococcus epidermidis (SE) 28-H4 | Gallo (UCSD) | This study |
| Staphylococcus hominis (SH) A9 | Gallo (UCSD) | Nakatsuji et al, 2017E6 |
| Staphylococcus hominis (SH) C5 | Gallo (UCSD) | Williams et al, 2019E7 |
| Staphylococcus capitis (SC) H8 | Gallo (UCSD) | This study |
| Staphylococcus warneri (SW)G2 | Gallo (UCSD) | This study |
| Staphylococcus lugdunensis (SL) E7 | Gallo (UCSD) | This study |
| Staphylococcus aureus USA300 (AH1263) | Horswill (UC Denver) | Mootz et al, 2015E8 |
UC, University of Colorado; UCSD, University of California, San Diego.
S epidermidis agr activity assay
S epidermidis 12228 agr type I P3-GFPErmr (AH3408), S epidermidis 1457 agr type II P3-GFPErmr (AH273), and S epidermidis 8247 agr type IIIP3-GFP Ermr (AH3409) reporter strains were used to detect S epidermidis agr activity as previously described.32 Strains were first cultured overnight (~16 hours) in the presence of 10 μg/mL of erythromycin (plasmid antibiotic selection). The bacteria were then inoculated at 107 CFUs/mL in 3% TSB (final volume 500 μL) along with CoNS sterile-filtered supernatant (≤10% vol/vol) or S hominis C5 synthetic AIP (≤100 nM) and shaken at 300 rpm in an incubator at 37°C. RNA was isolated after 12 hours of incubation, and the agr activity was measured after 24 hours. Bacteria were diluted 1:20 in PBS (final volume 200 μL) in 96-well black bottom plates (Corning), and GFP fluorescence was detected by using a SpectraMax Gemini EM fluorometer (Thermo Fisher Scientific; excitation wavelength, 485 nm; emission wavelength, 538 nm).
Quantification and statistical analysis
One-way ANOVA, 2-way ANOVA, the Student t test, nonparametric unpaired Kruskal-Wallis analysis, and Pearson and Spearman correlations were used throughout this study for statistical analysis, as indicated in the figure legends. All statistical analyses were performed by using GraphPad Prism, version 8.0 (GraphPad, La Jolla, Calif). All data are presented as means plus or minus SEMs, and a P value of .05 or lower was considered significant.
Details about the other methods used in this study are available in the Methods section of the Online Repository. For details about the specific primers and antibodies used in this study, see Tables E3 and E4 (available in this article’s Online Repository at www.jacionline.org ).
TABLE E3.
qPCR primers used in this study
| Gene target | Forward primer | Reverse primer | Reference |
|---|---|---|---|
| SE ecpA | TGTGCTTAAAACGCCACGTA | GTATAGCCGGCACACCAACT | Olson et al, 2014E2 |
| SE sepA | TGGTGTAACACAGCAAACTGC | ACTTCTTAAGGCATCTCCGCC | This study |
| SE esp | ACGTTGTTAATGGAGCTAAGGGT | TGCAGGTTTAACTACTTGACCA | This study |
| SE sodA | TCAGCAGTTGAAGGGACAGAT | CCAGAACAATGAATGGTTAAG | Nakatsuji et al, 2013E4 |
| SA femA | AACTGTTGGCCACTATGAGT | CCAGCATTACCTGTAATCTCG | Nakatsuji et al, 2013E4 |
| hHPRT | CCTGGCGTCGTGATTAGTGAT | AGACGTTCAGTCCTGTCCATAA | Zhang et al, 2016E9 |
| hIL-6 | AATTCGGTACATCCTCGACGG | TTGGAAGGTTCAGGTTGTTTTCT | Zhang et al, 2016E9 |
| hIL-8 | TCCAAGCTGGCCGTGGCTCT | CTGTGTTGGCGCAGTGTGGTCC | Zhang et al, 2016E9 |
| hTSLP | ATGTTCGCCATGAAAACTAAGGC | GCGACGCCACAATCCTTGTA | This study |
| hTNFα | GCTGCACTTTGGAGTGATCG | GGGTTTGCTACAACATGGGC | Zhang et al, 2016E9 |
| hIL-1α | TGGTAGTAGCAACCAACGGGA | ACTTTGATTGAGGGCGTCATTC | This study |
| hIL-1β | ATGATGGCTTATTACAGTGGCAA | GTCGGAGATTCGTAGCTGGA | This study |
| hDSG-1 | AACCCAATCGCCAAAATTCACT | ACCTCTCGATCAACTATGGATGT | This study |
| hCDSN | ACTGCTGCTGGCTGGTCT | AGAGCTTCTGGCACTGGAAA | Cau et al, 2017E10 |
| hIVL | GCCAGGTCCAAGACATTCAAC | GGGTGGTTATTTATGTTTGGGTGG | Cau et al, 2017E10 |
| hFLG | CCCTCGGTTTCCACTGTCTC | CCCTCGGTTTCCACTGTCTC | Cau et al, 2017E10 |
| hCAMP | AGGTCCTCAGCTACAAGGAAG | TCTTGAAGTCACAATCCTCTGGT | Liggins et al, 2019E11 |
| mHprt | GTTAAGCAGTACAGCCCCAAA | AGGGCATATCCAACAACAAACTT | Zhang et al, 2016E9 |
| mIl6 | ACAAAGCCAGAGTCCTTCAGAGAGA | AGCCACTCCTTCTGTGACTCCAG | Liggins et al, 2019E11 |
| mIl4 | GAGCCATATCCACGGATGCGAC | ATGCGAAGCACCTTGGAAGCCC | This study |
| mIl13 | TGCTTGCCTTGGTGGTCTCGC | GCGGCCAGGTCCACACTCCA | This study |
| mIl17α | ACGCGCAAACATGAGTCCAGGG | TGAGGGATGATCGCTGCTGCCT | This study |
| mIl22 | CCTACATGCAGGAGGTGGTG | AAACAGCAGGTCCAGTTCCC | This study |
h, Human; m, mouse; SA, S aureus: SE, S epidermidis.
TABLE E4.
Antibodies used in this study
| Antigen | Antibody description | Source | Dilution used for WB |
|---|---|---|---|
| Human DSG-1 | Mouse monoclonal IgG2b B-11 (sc-137164) | Santa Cruz Biotechnology, Santa Cruz, Calif | 1:100 |
| Human corneodesmosin (CDSN) | Rabbit polyclonal IgG H-80 (sc-134451) | 1:100 | |
| Human involucrin (IVL) | Goat polyclonal IgG N-17 (sc-15223) | 1:100 | |
| Human β-actin | Goat polyclonal IgG I-19 (sc-1616) | 1:500 | |
WB, Western blot.
RESULTS
S epidermidis produces proteolytic activity through expression of EcpA
The production of proteases by S aureus promotes damage to the epidermis in subjects with AD,13,33 but whether other bacterial species that reside on skin may also display proteolytic activity that could damage the skin is not well understood. To test this hypothesis, CoNS was collected from healthy human skin and lesional skin of subjects with AD and then assayed in vitro for proteolytic activity against a gelatin substrate. Among the initial CoNS strains tested, some S epidermidis isolates showed gelatinase activity and the level of enzymatic activity varied greatly between strains of the species (Fig 1, A and see Table E5 in this article’s Online Repository at www.jacionline.org ). The activity produced by S epidermidis 05-A5 was identified as likely to be a cysteine protease, as it was inhibited only by the cysteine protease inhibitor E64, whereas other classes of protease inhibitors had no effect (Fig 1, B). S epidermidis was known to only produce 1 cysteine protease (termed extracellular cysteine protease A [EcpA]); the expression of EcpA is under the control of the accessory gene regulator (agr) quorum sensing system.32,34 We therefore next sought to test whether EcpA was responsible for the proteolytic activity that we observed in S epidermidis.
FIG 1.

The cysteine protease EcpA secreted by S epidermidis (SE) presents a unique proteolytic activity. A, Protease activity against gelatin measured in the supernatant of CoNS strains cultured for 24 hours: SE, S hominis (SH), S capitis (SC), S warneri (SW), and S lugdunensis (SL) (n = 3). B, Protease activity against gelatin measured in the supernatant of SE 05-A5 cultured for 24 hours in the presence of various protease inhibitors: E64 (a cysteine protease inhibitor), EDTA, 1,10-phenanthroline (a metalloprotease inhibitor), and aprotinin (a serine protease inhibitor) (n = 3). C and D, ecpA mRNA levels across SE strains and Pearson correlation to gelatinase activity. E, Assessment of EcpA specific activity (FRET substrate) in supernatant of SE isolates from both healthy (n = 12) and atopic (n = 33) individuals after the isolates had been cultured for 24 hours (n = 3). F, Protease activity against gelatin, elastin, collagen I, and collagen IV substrates measured in the supernatant of SE 1457 WT or ΔecpA strains cultured for 24 hours. All data are representative of at least 2 independent experiments, and the results are means ± SEMs. F, Student t tests were used to determine statistical significance: *P < .05; **P < .01; ***P < .001; **** P < .0001. RFU, Relative fluorescence unit.
TABLE E5.
Growth of the various CoNS cultures (24-hour) used to prepare supernatants for protease assays
| Strain | OD 600 nm | Equivalent CFU/mL |
|---|---|---|
| Staphylococcus epidermidis (SE) 1457 | 12.91 | 3.87E+09 |
| Staphylococcus epidermidis (SE) ATCC12228 | 10.68 | 3.21E+09 |
| Staphylococcus epidermidis (SE) A9 | 11.03 | 3.31E+09 |
| Staphylococcus epidermidis (SE) A11 | 6.34 | 1.90E+09 |
| Staphylococcus epidermidis (SE) 09-G7 | 7.19 | 2.16E+09 |
| Staphylococcus epidermidis (SE) 18-F3 | 10.17 | 3.05E+09 |
| Staphylococcus epidermidis (SE) 05-A5 | 9.22 | 2.77E+09 |
| Staphylococcus epidermidis (SE) 28-H4 | 4.28 | 1.29E+09 |
| Staphylococcus hominis (SH) A9 | 6.48 | 1.94E+09 |
| Staphylococcus hominis (SH) C5 | 10.13 | 3.04E+09 |
| Staphylococcus capitis (SC) H8 | 14.26 | 4.28E+09 |
| Staphylococcus warneri (SW) G2 | 13.02 | 3.90E+09 |
| Staphylococcus lugdunensis (SL) E7 | 9.29 | 2.79E+09 |
To determine whether EcpA was responsible for the enzymatic activity observed in the S epidermidis strains, we next measured ecpA mRNA levels. S epidermidis strains such as the clinical isolate 05-A5 that we observed with maximal activity in Fig 1, A also had increased ecpA transcript levels (Fig 1, C and D). Use of a specific EcpA substrate on S epidermidis isolates from healthy and AD skin revealed that multiple strains from both populations had the capacity to produce active enzyme (Fig 1, E). Furthermore, targeted deletion of the ecpA gene (ΔecpA)32 in the strain S epidermidis 1457 eliminated specific EcpA substrate activity as well as gelatinase activity (Fig 1, F and see also Fig E1, A and B and Table E6 in this article’s Online Repository at www.jacionline.org ). The expression of other known S epidermidis-secreted proteases, sepA and esp, was not affected by ecpA deletion (Fig E1, C and D), and in addition to gelatin, EcpA also degraded elastin, collagen I, and collagen IV in vitro (Fig 1, F and see Fig E2 in this article’s Online Repository at www.jacionline.org ). These results therefore showed that the gene responsible for proteolytic activity from S epidermidis was EcpA and that this protease was actively secreted by multiple clinical isolates. Notably, EcpA was also recently shown to be associated with skin damage in patients with Netherton syndrome, who possess a mutation in the human protease inhibitor SPINK5.35
FIG E1.

Characterization of S epidermidis (SE) 1457 ΔecpA. A, Analysis of the mRNA levels of ecpA in both the SE 1457 WT or ΔecpA strain after 12 hours of culturing. B, EcpA activity with use of a specific FRET substrate measured in the sterile-filtered supernatant of SE 1457 WT or ΔecpA after 24 hours of culturing (n = 3). C and D, Analysis of mRNA levels for 2 other SE-secreted protease genes, sepA and esp, in both SE 1457 WT or ΔecpA after 12 hours of culturing. Data are representative of at least 2 independent experiments, and the results are represented as means ± SEMs. RFU, Relative fluorescence unit.
TABLE E6.
Growth of S epidermidis 1457 WT and ΔecpA cultures (24-hour) used to prepare supernatants for protease assays
| Strain | OD 600 nm | Equivalent in CFU/mL |
|---|---|---|
| Staphylococcus epidermidis (SE) 1457 WT | 14.05 | 4.21E+09 |
| Staphylococcus epidermidis (SE) 1457 ΔecpA | 13.99 | 4.20E+09 |
FIG E2.

Characterization of S epidermidis (SE) protease activity. A-C, Measurement of protease activity against the substrates elastin (A), collagen I (B), and collagen IV (C) with CoNS sterile-filtered supernatants that had been cultured for 24 hours (n = 3). D, Protease activity against the substrates elastin, collagen I, and collagen IV measured in the sterile-filtered supernatant of SE 05-A5 (after 24 hours of culturing) in the presence of various protease inhibitors: E64 (a cysteine protease inhibitor), EDTA, and 1,10-phenanthroline (a metalloprotease inhibitor), and aprotinin (a serine protease inhibitor) (n = 3). Data are representative of at least 2 independent experiments, and results are represented as means ± SEMs. SH, S hominis; SC, Staphylococcus capitis; SW, Staphylococcus warneri; SL, Staphylococcus lugdunensis.
S epidermidis cysteine protease EcpA disrupts the skin barrier
The potential of the S epidermidis protease to influence the skin barrier in AD was next evaluated by comparing S epidermidis strains that express EcpA with S aureus, S epidermidis 1457, and the clinical isolate S epidermidis 05-A5 from AD skin, which have a high EcpA expression (Fig 1).Both S epidermidis 1457 and 05-A5 induced disruption of the epidermal barrier as shown by an increase in transepidermal water loss (TEWL). S epidermidis 1457 ΔecpA and the AD skin isolate 18-F3 (which has low EcpA expression) did not (Fig 2, A and B). The difference in activity between the strains was not due to different amounts of live bacteria colonizing the skin (see Fig E3 in this article’s Online Repository at www.jacionline.org ). The severity of skin damage and epidermal barrier disruption correlated with the amount of S epidermidis 1457 wild-type (WT) when applied on the skin at a physiologically relevant bacterial density between 103 and 106 CFUs/cm2 (Fig 2, C and D). Furthermore, using a mouse model of AD, we also observed that S epidermidis 1457 increased TEWL and that this effect was dependent on EcpA (Fig 2, E). Additionally, EcpA expression was necessary for S epidermidis to penetrate the skin (Fig 2, F), and purified EcpA was able to increase TEWL even without application of live bacteria (Fig 2, G and H). These observations support the potential of S epidermidis EcpA to damage the epidermal barrier and exacerbate AD through a mechanism similar to that of S aureus.
FIG 2.

S epidermidis (SE) cysteine protease EcpA disrupts the skin barrier and degrades DSG-1 and LL-37. A and B, Representative pictures of the murine back skin treated with 106 CFUs/cm2 SA or SE isolates for 48 hours and TEWL measurements (n = 5). C and D, Representative pictures of the back skin after colonization with 103to 106 CFUs/cm2 SE 1457 WT for 48 hours and TEWL measurements (n = 4). E, TEWL measurements of AD mouse model back skin (Balb/c Flg−/− + ovalbumin [OVA]) colonized for 24 hours with live 106 CFUs/cm2 SE 1457 WT or SE 1457 ΔecpA (n = 5 or 6). F, Gram-positive bacteria staining (in purple) of skin sections from mice treated with either 106 CFUs/cm2 of SE 1457 WT or SE 1457 ΔecpA (n = 5). G and H, Assessment of C57BL/6 murine back skin and TEWL measurements after treatment with 2.5 μg/cm2 of EcpA or vehicle for 24 hours (n = 3). I and J, Differentiated NHEKs were treated for 20 hours with 2.5 μg/mL of EcpA ± 20 μg/mL of cycloheximide (protein synthesis inhibitor). Immunoblotting for DSG-1, involucrin (IVL), corneodesmosin (CDSN), and β-actin. Quantification of DSG-1 was normalized on β-actin (n = 3) (see Table E4). K and L, Coomassie blue staining of in vitro proteolysis of LL-37 by EcpA and S aureus (strain 113) growth inhibition assay (n = 3). All data are representative of at least 2 independent experiments, and results are means ± SEMs. One-way ANOVAs (B, D, and E), Student t tests (H and J), and 2-way ANOVA (L) were used to determine statistical significance: *P < .05; **P < .01; ***P < .001, ****P < .0001. Ctl, Control.
FIG E3.

Quantification of live staphylococci on 8- to 10-week-old female C57BL/6 mice skin after topical treatment with S aureus (SA) or S epidermidis (SE) live bacteria for 48 hours.
EcpA degrades DSG-1 and the antimicrobial peptide LL-37
To better understand how EcpA may damage the epidermis, human keratinocytes in culture were exposed to purified EcpA. This treatment was not cytolytic to keratinocytes and did not result in lactate dehydrogenase release (see Fig E4, A in this article’s Online Repository at www.jacionline.org ). However, Western blot analysis showed that desmoglein-1 (DSG-1), which is a key component of the corneodesmosome, was cleaved by EcpA. In contrast, other important epidermal barrier components, including corneodesmosin (CDSN) and involucrin (IVL), were not degraded and showed increased expression by treatment with EcpA (Fig 2, I and J). Treatment of keratinocytes with cycloheximide to inhibit new protein synthesis inhibited the ability of EcpA to induce CDSN and IVL (Fig 2, I) and increased the reduction in full-length DSG-1 (Fig 2, I and J). Interestingly, transcripts for DSG-1 (Dsg1), CDSN (Cdsn), IVL (Ivl), and filaggrin (Flg) were all increased by EcpA exposure (see Fig E4, B). Overall, these data suggest that EcpA can degrade an essential barrier protein such as DSG-1 and that human keratinocytes attempt to compensate for this by increased synthesis of several barrier proteins. The loss of full-length DSG-1, and potentially other substrates not measured here, can explain the loss of barrier function of the stratum corneum after exposure to S epidermidis EcpA.
FIG E4.

Effect of EcpA on cultured keratinocytes. A, Evaluation of the cytotoxicity of EcpA on differentiated NHEKs by measuring lactate dehydrogenase (LDH) release in the culture medium after 20 hours of treatment alone or in the presence of 20 μg/mL of cycloheximide. B, Differentiated NHEKs were treated for 8 hours with 2.5 μg/ mL of EcpA followed by analysis of the mRNA level of several epidermal differentiation markers and CAMP by qPCR (n = 3). Data are representative of at least 2 independent experiments, and results are represented as means ± SEMs. B, Student t tests were used to determine statistical significance: *P < .05; **P < .01; ***P < .001; ****P < .0001.
To identify additional potential substrates for EcpA, we next evaluated its capacity to degrade an antimicrobial peptide (AMP) that is partly responsible for controlling the composition of the skin microbiome.36–40 SspB is a cysteine protease produced by S aureus; it has been previously shown to degrade the cathelicidin AMP LL-37 and share strong homology with EcpA.35,41 Incubation of synthetic LL-37 with purified EcpA showed that this enzyme degraded LL-37 and decreased the capacity of this AMP to inhibit S aureus bacterial growth (Fig 2, K and L). This illustrates an additional potential substrate for EcpA that would result in a deleterious effect on cutaneous defense function.
EcpA induces skin inflammation
To further assess the potential for EcpA to influence AD, the ability of S epidermidis strains that express EcpA to promote an inflammatory response was directly tested on human keratinocytes and on mouse back skin. The WT S epidermidis 1457 with intact EcpA activity, but not the S epidermidis ΔecpA mutant strain, induced skin inflammation characterized by epidermal thickening;, immune cell infiltration; and increased expression of cytokines, including IL-6, IL-4, and IL-17α (Fig 3, A and B). EcpA-induced inflammation was also observed by using an AD mouse model colonized with either WT S epidermidis 1457 or the S epidermidis ΔecpA mutant strain. Notably, the expression of EcpA resulted in a significant increase in TH2 cytokines (Fig 3, C) in addition to the disruption of barrier function previously shown in Fig 2, E.
FIG 3.

EcpA induces skin inflammation in combination with other S epidermidis toxins. A, Representative hematoxylin and eosin staining of mouse skin sections across treatments. B and C, Analysis of the mRNA level of various cytokines after treatment of murine back skin with 106 CFUs/cm2 of S epidermidis (SE) 1457 WT or SE 1457 ΔecpA for 48 hours in the C57BL/6 mouse model (B) or AD mouse model (BALB/c Fig−/− 1 ovalbumin [OVA]) (C), respectively (n = 4–6). mRNA levels normalized by using the housekeeping gene Hprt. D, Analysis of the mRNA level of various cytokines after treatment of C57BL/6 murine back skin with vehicle or 2.5 μg/cm2 of EcpA for 24 hours. E, Differentiated NHEK treated for 8 hours with 2.5 μg/mL of EcpA or with 5% bacterial supernatant followed by analysis of the mRNA level of various cytokines normalized to the housekeeping gene HPRT (n = 3). All data are representative of at least 2 independent experiments, and results are means ± SEMs. One-way ANOVAs (B, C, and E) and Student t tests (D) were used to determine statistical significance: *P < .05; **P < .01; ***P < .001; **** P < .0001.
Treatment of mouse skin with purified EcpA resulted in a response different from that to treatment with the live bacteria. Purified EcpA induced skin inflammation but only Il-8 appeared to be significantly increased with application of the enzyme alone (Fig 3, A and D). Similarly, cultured human keratinocytes treated with purified EcpA showed an increase in levels of IL-6, IL-8, TLSP, IL-1α, and IL-1β (Fig 3, E). Interestingly, exposure to crude supernatant from S epidermidis 1457 without EcpA (ΔecpA) also induced a strong cytokine response in cultured human keratinocytes—even more than the S epidermidis 1457 WT supernatant did (Fig 3, E). These results suggest that the activity of EcpA is important for S epidermidis to induce skin inflammation on live skin, where an intact stratum corneum protects underlying live cells. However, without an established physical barrier, other bacterial components can also contribute to stimulation of the inflammatory response.
EcpA expression is increased in AD
Having established that some S epidermidis strains can cause damage and inflammation to mice in an EcpA-dependent manner, we next conducted an observational study to evaluate S epidermidis abundance and ecpA mRNA on subjects with AD. A cohort of 13 subjects with AD and 14 healthy subjects (see Table E1) was sampled by skin surface swabs from the antecubital crease to evaluate S aureus and S epidermidis genomic DNA (gDNA) absolute abundance by real-time quantitative PCR (qPCR) with species-specific primers.42 S epidermidis gDNA abundance was significantly increased on AD lesional skin compared with on healthy control skin, with no difference based on sex of the subjects assessed (Fig 4, A and see also Fig E5 in this article’s Online Repository at www.jacionline.org ). Two populations were identified within the AD cohort: 1 group with S epidermidis colonization similar to that on healthy skin (around 103 CFUs/cm2) and a second group with high to very high S epidermidis abundance (between 104 and 106 CFUs/cm2). This result is consistent with that in prior reports and demonstrates that S epidermidis overgrowth, similar to S aureus overgrowth, occurs in only some patients with AD.10,18–20 The abundance of gDNA from S epidermidis correlated with S aureus abundance (Fig 4, B and see also Fig E6, A in this article’s Online Repository at www.jacionline.org ).
FIG 4.

S epidermidis colonization and ecpA expression are increased on some subjects with AD. A, Measurement of gDNA absolute abundance of S epidermidis CFUs/cm2 from swabs of healthy control and AD nonlesional and lesional skin normalized to skin area. B, Spearman correlation between the gDNA absolute abundance of S epidermidis and S aureus from skin swabs. C, Relative abundance of S epidermidis ecpA mRNA isolated from swabs of healthy control and AD nonlesional and lesional skin normalized to skin area. D, Spearman correlation between S epidermidis EcpA mRNA relative abundance and S epidermidis CFUs/cm2 from swabs of AD skin. E, Spearman correlation between the local Eczema Area and Severity Index (EASI) score and S epidermidis CFUs/cm2 from swabs of AD lesional skin. F, Spearman correlation between the local EASI score and ecpA mRNA relative abundance from swabs of AD lesional skin. Each dot represents a single swab, and the bars represent means ± SEMs. A and C, A nonparametric unpaired Kruskal-Wallis analysis was used to determine statistical significance. *P < .05; **P < .01; ***P < .001; ****P < .0001.
FIG E5.

Sex differences in S epidermidis colonization and EcpA expression on AD skin. A and B, Measurement of gDNA absolute abundance of S epidermidis CFUs/cm2 or ecpA mRNA levels from swabs of healthy control and AD nonlesional and lesional skin normalized to skin area. Data are represented as means ± SEMs. A and B, Nonparametric unpaired Kruskal-Wallis analysis and Spearman correlation were used to determine statistical significance.
FIG E6.

Colonization of AD skin by S aureus and correlation to disease severity. A, Measurement of gDNA absolute abundance of S aureus CFUs/cm2 from swabs of healthy control and AD nonlesional and lesional skin normalized to skin area. B, Spearman correlation between the local Eczema Area and Severity Index (EASI) score and S aureus CFUs/cm2 from swabs of AD lesional skin. Each dot represents a single swab and the bars represent means ± SEMs. A, Nonparametric unpaired Kruskal-Wallis analysis was used to determine statistical significance: *P < .05; **P < .01; ***P < .001; ****P < .0001.
Next, the expression of ecpA mRNA by S epidermidis on AD skin was measured by qPCR analysis of ecpA mRNA extracted from skin swabs. Expression of mRNA for ecpA demonstrates the presence of live bacteria. Furthermore, because this gene is under control of the agr quorum sensing system, the expression of ecpA indicates that the density of S epidermidis is sufficiently high to permit expression. Analysis of RNA from AD skin swabs showed a high abundance of ecpA mRNA on the lesional skin of some subjects with AD (Fig 4, C). The abundance of ecpA mRNA correlated with the absolute abundance of S epidermidis gDNA (Fig 4, D). Subjects with the highest S epidermidis and S aureus absolute abundance, as well as ecpA mRNA relative expression, had the most severe disease as assessed by local Eczema Area and Severity Index score (Fig 4, E and F and see Fig E6, B).
EcpA activity is pH dependent, and its expression by S epidermidis is downregulated by CoNS AIPs
We next examined factors that could influence the capacity of S epidermidis to contribute to disease in AD. Inflamed skin in subjects with AD has increased surface pH compared with that in healthy subjects.43 Analysis of EcpA activity at a range of physiologic pH values showed that EcpA enzymatic activity is low at the pH of normal skin (pH 4.5–6) but becomes active at higher pH values (6.5–8) consistent with the pH of lesional AD skin (see Fig E7 in this article’s Online Repository at www.jacionline.org ). Therefore, the environmental conditions of AD skin are supportive of EcpA enzymatic activity.44
FIG E7.

pH-dependent activity of S epidermidis EcpA. EcpA (100 ng) was incubated with EcpA FRET substrate (250 nM) in sweat-like buffer (40 mM NaCl, 10 mM KCl, 1 mM sodium dihydrogen phosphate, 5 mM l-cysteine) at different pH ranges (4–8) for 1 hour at 37°C (n = 3). Data are representative of at least 2 independent experiments, and results are represented as means ± SEMs.
Finally, we sought to better understand how other members of the surface microbial community could influence S epidermidis EcpA expression. As discussed previously, S epidermidis protease activity is dependent on cell density, an observation that is consistent with regulation of EcpA by the agr quorum sensing system.32 We have previously shown that AIPs from certain S hominis strains can inhibit the S aureus agr quorum sensing system and thus inhibit the production of virulence factors under agr control.31,45,46 To investigate whether S hominis could also inhibit S epidermidis EcpA production, an S epidermidis 1457 agr type II reporter strain that contains an agr activity fluorescent reporter was cultured in the presence of either S hominis A9 supernatant or the S hominis C5 synthetic AIP peptide. Both S hominis A9 and C5 were able to inhibit S epidermidis 1457 agr reporter activity. Furthermore, S hominis A9 supernatant and the S hominis C5 synthetic AIP peptide inhibited ecpA mRNA expression and EcpA protease activity (Fig 5, B and D and see Fig E8, C in this article’s Online Repository at www.jacionline.org ). S hominis A9 supernatant and the S hominis C5 synthetic AIP were also able to inhibit both the agr activity and EcpA activity of the 2 other known S epidermidis agr types (see Fig E8, A and B). Importantly all of these agr inhibition and reduced EcpA expression phenotypes were independent of a reduction in S epidermidis growth rate as assessed through OD measurements at 600 nm, indicating that this is specifically due to quorum sensing inhibition and not due to inhibition of growth. Finally, we demonstrated that this quorum sensing interaction between S hominis and S epidermidis can occur when in vivo murine skin models are used. Application of live S hominis A9 or C5, S hominis A9 supernatant, or S hominis C5 synthetic AIP alongside S epidermidis 1457 WT resulted in decreases in S epidermidis–induced skin barrier damage without a decrease in S epidermidis abundance (Fig 5, E and F and see Fig E8, D). Taken together, these data show how the local skin microenvironment and microbial community will influence the activity of S epidermidis.
FIG 5.

EcpA production is downregulated by CoNS AIPs. A and C, Modulation of the S epidermidis (SE) 1457 agr type II P3-GFP reporter strain activity by S hominis (SH) A9 supernatant (A) or synthetic SH C5 AIP (C) after 24 hours of culturing (n = 3). OD values at 600 nm used to assess bacterial growth at the hour 24 time point. B and D, ecpA mRNA levels after 12 hours of culturing of SE 1457 agr reporter strain with SH A9 supernatant (10%) (B) or SH C5 AIP (100 nM) (n = 3) (D). E and F, C57BL/6 murine back skin was topically treated with 106 CFUs/cm2 of SE 1457 for 48 hours either alone (control) or with 106 CFUs/cm2 of S hominis (SH) A9,106 CFUs/cm2 of SH C5, SH A9 supernatant (5 μL), or SH C5 synthetic AIP (10 μg). E, Representative pictures of the treated back skin and measurement of TEWL (F) (n = 4). All data are representative of at least 2 independent experiments, and the results are means ± SEMs. One-way ANOVAs (A, C, and F) and Student t tests (B and D) were used to determine statistical significance: *P < .05; **P < .01; ***P < .001; ****P < .0001.
FIG E8.

Modulation of S epidermidis (SE) agr activity by some S hominis (SH) strains. A, Modulation of the agr activity and gelatinase activity of SH 12228 agr type I P3-GFP reporter strain by either SH A9 supernatant or SH C5 synthetic AIP (n = 3). B, Modulation of the agr activity and gelatinase activity of SE 8247 agr type III P3-GFP reporter strain by either SH A9 supernatant or SH C5 synthetic AIP (n = 3). c, Modulation of the gelatinase activity of SE 1457 agr type II P3-GFP reporter strain by either SH A9 supernatant or SH C5 synthetic AIP (n = 3). A-C, OD values recorded at 600 nm to approximate bacterial growth overtime. D, Quantification of live staphylococci on 8-to 10-week-oldfemaleC57BL/6mice skin after topical treatment with SE 1457 live bacteria for 48 hours either alone (vehicle) or along with SH A9, C5 live bacteria, SH A9 supernatant (5 μL), or SH C5AIP (n = 4). Data are representative of at least 2 independent experiments, and results are represented as means ± SEMs. A-C, One-way ANOVAs were used to determine statistical significance: *P < .05; **P < .01; *** P <.001; ****P <.0001.
DISCUSSION
Current understanding of the pathophysiology of AD suggests that the skin microbiome can strongly influence this disease.47–50 The presence of S aureus in ADand the negative consequences of colonization by this major human pathogen have long been known and extensively described.9–13 One mechanism by which S aureus has been shown to damage the skin and exacerbate AD is through its capacity to increase proteolysis of the stratum corneum. 31,33,51,52 This increased proteolytic activity can combine with a wide range of inherited mutations in human genes of the epidermal differentiation complex53–56 to result in disruption of the epidermal barrier and worsening of disease symptoms.13 However, despite the presence of many other species of Staphylococcus in AD besides S aureus and the large fraction of patients with AD who are not culture positive for S aureus, the potential harmful effect of other species of Staphylococcus has been poorly explored. In this study we have shown that S epidermidis, a common species of commensal bacteria found on healthy skin, can also drive skin injury through secretion of the cysteine protease EcpA.
S epidermidis has often been regarded as a key member of the healthy skin microbiota with multiple benefits for the host.21–23 For example, various S epidermidis strains have been shown to limit pathogen infections, tune skin immune development, promote wound repair, and protect against skin cancer.26,57–61 However, S epidermidis is also known to behave as an opportunistic pathogen, especially following contamination of implanted medical devices.27–30 Despite the infectious potential of S epidermidis, its frequency on healthy human skin has led to the conclusion that it is harmless when colonizing the skin surface. Indeed, even though S epidermidis overgrowth on lesional skin has been reported in several studies,10,18–20 its capacity to damage skin and contribute to AD has not been clearly addressed. A previous study reported that the abundance of S epidermidis on AD lesions correlated with disease severity,19 but a mechanism that could suggest causation was not clear. In the present work, we have confirmed that S epidermidis overgrowth occurs on the lesional skin of some patients with AD and that S epidermidis abundance is correlated with local disease severity. Interestingly, in this cohort, we observed a correlation between S aureus and S epidermidis abundance on AD lesional skin, suggesting that the 2 species may both contribute in some patients. Furthermore, we showed that the capacity of S epidermidis to promote skin barrier damage is associated with its abundance. This finding is in agreement with the findings of 2 recent studies suggesting that S epidermidis overgrowth could contribute to lowered skin barrier function in facial seborrheic dermatitis62 and in Netherton syndrome.35
We have now identified the proteolytic activity of the cysteine protease EcpA as a mechanism through which S epidermidis can induce skin damage. Several observations support this conclusion, as the deletion of ecpA in S epidermidis 1457 eliminated the in vitro proteolytic effect and prevented the capacity of the bacteria to disrupt the barrier and increase TEWL. We further showed that purified EcpA can disrupt both physical and antimicrobial barriers of the skin through the proteolysis of DSG-1 and the cathelicidin AMP LL-37. Furthermore, EcpA appeared essential to the enabling of S epidermidis to induce inflammation in vivo, where an intact stratum corneum protects underlying skin. In vitro however, EcpA did not seem to be the major factor responsible for S epidermidis–induced keratinocyte immune response. Other factors such as TLR ligands or toxins produced by S epidermidis may promote inflammation after EcpA has enhanced penetration past the stratum corneum. This would be similar to that shown for S aureus, in which case toxins such as PSMα or δ-toxin can induce a strong proinflammatory immune response if the barrier is first disrupted.14–17 S epidermidis also produces PSMs, and some of them have been shown to induce release of proinflammatory cytokines and cytolysis in neutrophils.63–65 We speculate that S epidermidis may use a similar 2-step process in which a protease first disrupts the barrier and other factors then trigger inflammation. Further investigations of the contribution of S epidermidis PSMs to skin inflammation are needed.
It is unlikely that the capacity of S epidermidis to induce skin injury is specific to the number of strains isolated from subjects with AD, as we have observed that isolates from both healthy and AD skin are able to produce EcpA. Indeed, both the population with AD and the healthy population may have a similar frequency of strains within the CoNS community that produce EcpA. This does not argue against the importance of EcpA in promoting the pathogenesis of some patients with AD. Rather than the presence or absence of the strain, our results suggest that it is the absolute abundance (in CFUs/cm2) that is important. Skin damage by EcpA is dependent on bacterial density on the skin, pH, and lack of other CoNS strains that could inhibit the S epidermidis agr system. On healthy skin, the activity of S epidermidis EcpA would be low owing to a low abundance of the bacteria and a high relative abundance of inhibitory CoNS strains. Furthermore, if EcpA were expressed, the enzymatic activity of the protein would be minimal as a result of the low pH of healthy skin. This could explain why, on the lesional skin of some patients with AD, S epidermidis may outcompete the other CoNS strains and increase production and activity of EcpA. This in turn would increase skin barrier damage and further exacerbate disease.
Why some S epidermidis strains appear to have lost the capacity to express EcpA is not clear. Studying the regulatory mechanisms of EcpA expression in S epidermidis will provide a more complete understanding of the factors predicting the ability of S epidermidis to become pathogenic on skin. In addition, the overgrowth of S epidermidis on the skin of some patients with AD remains unexplained, and the abundance of S epidermidis and S aureus appears to evolve in parallel on the skin of such patients. It is therefore difficult to determine whether 1 of the bacteria has a greater deleterious effect than the other. Also unclear is how endogenous cysteine protease inhibitors or even expression of the well-characterized staphostatin A (ecpB) secreted by S epidermidis itself can inhibit EcpA activity in AD.66 Thus, further work is needed to ultimately understand how S epidermidis and S aureus are coregulated and to explore the interaction between the 2 bacteria on AD skin.
We show that some unique CoNS strains, and S hominis A9 and C5 in particular, are able to inhibit the S epidermidis agr system and thus prevent S epidermidis–induced skin damage. These 2 strains are of interest as S hominis A9 was previously shown to have the capacity to kill S aureus though the production of a lantibiotic, whereas S hominis C5 AIP was shown to inhibit the S aureus agr system.16,31 Therefore, such commensal CoNS strains could constitute potential therapeutic tools to fight S aureus colonization and both S aureus and S epidermidis toxin production in AD. Clinical trials to evaluate the benefit of S hominis in AD are currently under way.
In conclusion, this study adds an extra level of complexity to our understanding of the interspecies and interkingdom communication that occurs between bacteria and host on the skin in AD. Our data suggest that S epidermidis can promote disease and identify a specific mechanism that is consistent with current understanding of the pathogenesis of AD.1,67,68 It is possible that other microbial species, including fungi such as Malassezia furfur, could have a similar detrimental effect.49,69–72 Such observations emphasize that AD should be considered a complex multifactorial disease that is influenced not only by multiple host genes but also by multiple microbial genes from multiple species. This highlights the need for multitargeted and more personalized therapeutic strategies in AD.
METHODS
Bacterial preparation
All bacteria strains used in this study are listed in Table E2 (in this article’s Online Repository at www.jacionline.org ). For preparation of CoNS supernatant, all strains were grown for 24 hours in 3% tryptic soy broth (TSB) at 300 rpm in an incubator at 37°C. The OD at 600 nm was read to evaluate bacterial concentration, and cultured bacteria were pelleted (for 15 minutes at 4,000 rpm and room temperature) followed by filter sterilization of the supernatant (0.22 μm). For RNA extraction, bacteria were cultured for 12 hours in the conditions described earlier. After the OD measurements, the bacteria suspension was mixed with 2 volumes of RNAprotect Reagent (Qiagen, Hilden, Germany) for 10 minutes before centrifugation (for 10 minutes at 13,000 rpm and room temperature) and storage of pellet at −80°C for further analysis. For mouse experiments with live bacteria colonization, bacteria were grown overnight (~16 hours), and the number of CFUs was approximated by measuring OD at 600 nm before application to mouse back skin followed by confirmation of the actual number of CFUs the next day.
Purification of the protease EcpA from S epidermidis supernatant
EcpA was purified from S epidermidis supernatant by cation exchange chromatography using a protocol based on the purification published by Dubin et al.E1 Specifically, 3% TSB(300 mL) was inoculated with a colony of S epidermidis strain 05-A5 and incubated at 37°C with shaking (220 rpm) for 24 hours. Bacteria were removed by centrifugation (2,900 g for 45 minutes at 4°C) followed by filtration (0.45 mm). Ammonium sulfate was added to 85% saturation, and the filtered supernatant was stirred gently at 4°C for 1 hour before the precipitated protein was pelleted by centrifugation (20,000 g for 45 minutes at 4°C). The resulting protein pellet was dissolved in 20 mM sodium phosphate (pH 6.0) containing 5 mM NaCl and then dialyzed extensively (4 × 4.5 L) at 4°C against this same buffer using Spectra/Por dialysis tubing (3.5-kDa cutoff). The protein solution was concentrated to 1.5 mL by using Amicon Ultra-15 concentrators (3-kDa cutoff) and then loaded on a HiScreen CaptoS column (3 columns in series) that had been equilibrated with 20 mM sodium phosphate (pH 6.0) containing 5 mM NaCl. The column was washed with 7 column volumes of equilibration buffer before the protein was eluted with use of a linear gradient of 5 to 500 mM NaCl (in 20 mM sodium phosphate [pH 6.0]) over 27 column volumes. Elution fractions containing active EcpA were identified by using a FRET-based, EcpA-specific protease assay (described later)E2 and analyzed for purity by SDS-PAGE. Those fractions estimated to contain EcpA (approximately 90% purity) were pooled and dialyzed against PBS. The resulting protein was confirmed to be EcpA by mass spectrophotometric analysis of tryptic peptides conducted at the University of Colorado School of Medicine Biological Mass Spectrometry Facility. The final recombinant EcpA used in this study was more than 95% pure with a molecular weight slightly greater than 20 kDa, as indicated by SDS-PAGE. This molecular weight is a little higher than previously reported as 19.8 kDa for a mature protease.E1 Because EcpA activates by autoprocessing, there can sometimes be slight differences in the final mature protease depending on the cleavage site. The remaining processed fragments of the preprotease are removed during the purification steps. Aliquots containing either 10 or 20 mg of purified EcpA, as determined by Bradford protein concentration assay, were lyophilized and stored at −80°C until they were used in various assays.
EcpA-specific protease assay
The measurement of EcpA activity was performed by using a specific FRET substrate with the sequence (5-FAM)-Lys-Leu-Leu-Asp-Ala-Ala-Pro-Lys-(QXL520)-OH (AnaSpec, Fremont, Calif) in 96-well black bottom plates (Corning) as previously described.E2 For measurement of EcpA activity in S epidermidis supernatant, 20 μL of supernatant was incubated with 250 nM of EcpA FRET substrate in Tris-HCl (pH 7.8) in a final concentration of 10 mM (total volume 100 μL). To test the effect of pH on EcpA activity, 2× sweat-like buffer (80 mM NaCl, 20 mM KCl, 2 mM sodium dihydrogen phosphate, and 10 mM L-cysteine) was prepared at different pH values (4 to 8). Then, 100 ng of EcpA was incubated with 250 nM EcpA FRET substrate with a 1× final concentration of sweat-like buffer in a total volume of 100 μL. Relative fluorescent intensity was measured with a SpectraMAX Gemini EM fluorometer (Thermo Fisher Scientific) (excitation wavelength, 485 nm; emission wavelength, 538 nm) after incubation at 37°C for 1 hour.
Keratinocyte protein extraction and Western blot analysis
For cell lysis, 200 μL of cold 1× RIPA buffer (Sigma-Aldrich) containing 1X protease inhibitor cocktail (cOmplete [Roche, Basel, Switzerland]) was applied to NHEKs (in a 12-well plate) followed by scraping. The cell lysates were incubated on ice and vortexed for 30 seconds every 15 minutes during a total of 1 hour and then centrifuged (at 14000 g for 20 minutes at 4°C) to remove debris. Protein concentration was determined by using a Pierce BCA protein assay kit (Thermo Fisher Scientific). Equal amounts of protein (12 μg) were run on 4% to 20% Tris-Glycine precast TGX gels (Bio-Rad, Hercules, Calif) and electrotransferred onto polyvinylidene difluoride membranes (Bio-Rad). Membranes were blocked in Odyssey Blocking Buffer (LI-COR Biosciences, Lincoln, Neb), incubated overnight at 4°C with primary antibodies, and finally incubated for 2 hours at room temperature with Odyssey (LI-COR Biosciences) fluorescent secondary antibodies (for details about the antibodies used in this study, see Table E4 in this article’s Online Repository at www.jacionline.org ). Images were acquired with an Odyssey infrared imaging system (LI-COR Biosciences). ImageJ software was used to quantify immunoreactive bands. Signals were normalized to actin immunodetection.
Lactate dehydrogenase cytotoxicity assay
The Pierce Lactate Dehydrogenase Cytotoxicity Assay Kit (Thermo Fisher Scientific) was used according to the manufacturer’s instructions to test whether EcpA was inducing cytotoxicity in NHEKs.
Quantification of live staphylococci from mouse skin swabs
After live bacteria treatment of mouse skin, the dressing film and agar disk were removed and surface bacteria were collected by using a swab soaked in TSB-glycerol solution. The swab head was then placed in 1 mL of TSB-glycerol solution, vortexed (for ~1 minute), serial-diluted, and plated onto mannitol salt agar plates supplemented with 3% egg yolk. After overnight incubation at 37°C, the number of CFUs was determined.
TEWL measurement
To determine damage to the epidermal skin barrier, TEWL of murine skin treated for 48 hours with S aureus or S epidermidis was measured by using a TEWAMETER TM300 (Courage + Khazaka Electronic, Köln, Germany).
Histologic staining
Full-thickness murine skin samples were collected, fixed in paraformaldehyde (4%), and embedded in paraffin. Sections (5-μm) were mounted onto Superfrost Plus glass slides (Thermo Fisher Scientific). Hematoxylin and eosin staining was performed by the University of California, San Diego, Dermatopathology Core. Gram staining was performed by a using Richard-Allan Scientific Chromaview Gram Stain Tissue kit (Thermo Fisher Scientific), which includes Crystal Violet to stain gram-positive bacteria and tartrazine (yellow dye) to counterstain the tissue elements. Pictures were taken on an Olympus BX51 fluorescent microscope.
LL-37 in vitro proteolysis and killing assay
A quantity of 20 μM of LL-37 dissolved in H2O (Genomemed; LLGDFFRKSKEKIGKEFKRIVQRIKDFLRN LVPRTES) was incubated with 10 μg/mL of EcpA (or with PBS at equal volume) in the presence of 5 mM of L-cysteine for 18 hours at 37°C. Then, 10 μL of the reaction mix was mixed with Laemmli buffer (Bio-Rad; 1× final concentration), incubated at 95°C for 5 minutes, run on 4% to 20% Tris-glycine precast TGX gels (BioRad), and stained with Simply Blue Safe Stain (Thermo Fisher Scientific) following the manufacturer’s instructions. For the S aureus killing assay, S aureus 113 was inoculated at a concentration of 105 CFUs/mL in 0.6% TSB in Dulbecco PBS (Life Technologies, Carlsbad, Calif) withthe LL-37 solution pretreated with either EcpA or PBS vehicle (with a final concentration of LL-37 of 16, 8, 4, 2, or 0 μM) and incubated at 30°C for 20 hours. OD was measured at 600 nm to evaluate S aureus growth.
Skin swab collection from human subjects
For collection of skin microbiome DNA, a swab head was soaked in molecular biology–grade TE buffer (Invitrogen, Carlsbad, Calif) containing 0.1% TritonX100 and 0.05% Tween-20 before rubbing the skin, after which it was placed in a clean microcentrifuge tube. For collection of skin microbiome RNA, the swab head was soaked in a 3% TSB and 16.67% glycerol solution (TSB-glycerol solution) before rubbing the skin; it was then placed in 1 mL of the same solution. The skin of the subjects was finally cleaned with an alcohol swab, and the samples were stored at −80°C for further analysis.
RNA isolation and real-time qPCR
All RNA was isolated by using the Purelink RNA isolation kit according to manufacturer’s instructions (Thermo Fisher Scientific). For isolation of human skin microbiome RNA, the swab head in TSB-glycerol solution was first vortexed for 1 minute. Then, 500 μL of bacteria suspension was incubated with 2 volumes of RNAprotect reagent (Qiagen) for 10 minutes before centrifugation (10 minutes at 13,000 rpm and room temperature), resuspension in 700 μL of RNA lysis buffer, bead beating (two 1-minute cycles with 5 minutes on ice after each) using lysing matrix B tubes, and centrifugation again. For isolation of RNA from the bacterial suspension, bacteria pellets were resuspended in 700 μL of RNA lysis buffer, bead-beated (two 1-minute cycles with 5 minutes on ice after each sequence) by using lysing matrix B tubes, and centrifuged (10 minutes at 13,000 rpm and room temperature). For cultured keratinocytes, 350 μL of RNA lysis buffer was directly added to the well (of a 24-well plate) after the cells had been washed with 1 Dulbecco PBS and incubated for 2 minutes at room temperature. For mouse tissue, full-thickness skin was preserved in RNAlater (Qiagen) at 4°C before bead beating in 700 μL of RNA lysis buffer (two 30-second cycles with 5 minutes on ice after each; 2.0-mm zirconia bead). The tissue was then centrifuged (for 10 minutes at 13,000 rpm and 4°C). For all samples, 350 μL of clear lysate was then added to 350 μL of 70% EtOH and column-based isolation of RNA. After RNA isolation, an equal volume of human skin swab microbiome RNA elution (11 μL) or equal quantity of bacterial, mouse, or keratinocyte RNA (500 ng) was reverse-transcribed by using the Verso cDNA Synthesis Kit (Thermo Fisher Scientific). Oligodeoxythymidines were not added to the reverse transcription mix for skin swab microbiome RNA samples to limit amplification of contaminant human DNA. qPCR assays were run on a CFX96 Real-Time Detection System (Bio-Rad) by using SYBR Green qPCR Master Mix (Biotool, Stratech, Ely, United Kingdom) along with specific primers (for details about primers, see Table E3 in this article’s Online Repository at www.jacionline.org ). The housekeeping genes used were Hprt/HPRT for both mouse and keratinocyte samples and gyrB for S epidermidis cultures. ecpA mRNA abundance from the skin swabs was normalized on skin area.
Microbial DNA extraction from human skin swabs and quantification of staphylococcal gDNA
Microbial DNA was extracted from skin swabs by using the PureLink Microbiome DNA Purification Kit according to the manufacturer’s instructions (Thermo Fisher Scientific). The absolute abundance of S aureus and S epidermidis gDNA in the microbial DNA elution was determined by qPCR as previously described.E3,E4 Briefly, qPCR was performed with iTaq Universal SYBR Green Supermix (Bio-Rad) by using S epidermidis– and S aureus–specific primers, targeting the S epidermidis sodA gene and S aureus femA genes, respectively. To determine the relative CFUs of S aureus- or S epidermidis–specific DNA, a standard curve was generated with gDNA extracted from known CFUs of S aureus (ATCC113) or S epidermidis (ATCC12228), respectively. The specificity of all primer pairs was confirmed by melting curve analysis and comparison with standard curves.
Key messages.
S epidermidis produces the protease EcpA under quorum sensing control.
EcpA can damage the skin barrier and promote inflammation in mouse skin.
The abundance of S epidermidis ecpA correlates with disease severity in some subjects with atopic dermatitis.
Acknowledgments
This work was funded by National Institutes of Health grants R01 AR076082, R37 AI052453, and U19 AI117673. Postdoctoral fellowship support for L. Cau was provided by SILAB. A. R. Horswill was supported by a Merit Award (BX002711) from the US Department of Veteran Affairs.
We thank Dr Brigitte Closs, Mrs Sylvie Bordes, and Dr Carine Mainzer (Silab) for providing advice and ideas for the project. We also thank the University of California, San Diego, Dermatopathology Core, and Julie Albright in particular, for performing histologic staining.
Abbreviations used
- AD
Atopic dermatitis
- Agr
Accessory gene regulator
- AIP
Autoinducing peptide
- AMP
Antimicrobial peptide
- CFU
Colony-forming unit
- CoNS
Coagulase-negative Staphylococcus
- DSG-1
Desmoglein-1
- EcpA
Extracellular cysteine protease A
- IVL
Involucrin
- HEK
Neonatal human primary epidermal keratinocyte
- PSM
Phenol-soluble modulin
- qPCR
Quantitative PCR
- TEWL
Transepidermal water loss
- TSB
Tryptic soy broth
- WT
Wild-type
Footnotes
Disclosure of potential conflict of interest: R. L. Gallo is a cofounder, scientific advisor, and consultant of MatriSys Biosciences and has equity in the company; in addition, he receives income from and has equity in Sente. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Bieber T Atopic dermatitis. N Engl J Med 2008;358:1483–94. [DOI] [PubMed] [Google Scholar]
- 2.Kapoor R, Menon C, Hoffstad O, Bilker W, Leclerc P, Margolis DJ. The prevalence of atopic triad in children with physician-confirmed atopic dermatitis. J Am Acad Dermatol 2008;58:68–73. [DOI] [PubMed] [Google Scholar]
- 3.Carroll CL, Balkrishnan R, Feldman SR, Fleischer AB Jr, Manuel JC. The burden of atopic dermatitis: impact on the patient, family, and society. Pediatr Dermatol 2005;22:192–9. [DOI] [PubMed] [Google Scholar]
- 4.Nutten S Atopic dermatitis: global epidemiology and risk factors. Ann Nutr Metab 2015;66(Suppl 1):8–16. [DOI] [PubMed] [Google Scholar]
- 5.Benn CS, Melbye M, Wohlfahrt J, Bjorksten B, Aaby P. Cohort study of sibling effect, infectious diseases, and risk of atopic dermatitis during first 18 months of life. BMJ 2004;328:1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flohr C, Yeo L. Atopic dermatitis and the hygiene hypothesis revisited. Curr Probl Dermatol 2011;41:1–34. [DOI] [PubMed] [Google Scholar]
- 7.Ring J, Kramer U, Schafer T, Behrendt H. Why are allergies increasing? Curr Opin Immunol 2001;13:701–8. [DOI] [PubMed] [Google Scholar]
- 8.Williams H, Stewart A, von Mutius E, Cookson W, Anderson HR. International Study of Asthma and Allergies in Childhood (ISAAC) Phase One and Three Study Groups. Is eczema really on the increase worldwide? J Allergy Clin Immunol 2008;121:947–54.e15. [DOI] [PubMed] [Google Scholar]
- 9.Leyden JJ, Marples RR, Kligman AM. Staphylococcus aureus in the lesions of atopic dermatitis. Br J Dermatol 1974;90:525–30. [DOI] [PubMed] [Google Scholar]
- 10.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, et al. Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 2012;22:850–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Williams MR, Nakatsuji T, Gallo RL. Staphylococcus aureus: master manipulator of the skin. Cell Host Microbe 2017;22:579–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geoghegan JA, Irvine AD, Foster TJ. Staphylococcus aureus and atopic dermatitis: a complex and evolving relationship. Trends Microbiol 2018;26:484–97. [DOI] [PubMed] [Google Scholar]
- 13.Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, et al. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol 2016;136:2192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Syed AK, Reed TJ, Clark KL, Boles BR, Kahlenberg JM. Erratum for Syed et al. , Staphylococcus aureus phenol-soluble modulins stimulate the release of proinflammatory cytokines from keratinocytes and are required for induction of skin inflammation. Infect Immun 2015;83:4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu H, Archer NK, Dillen CA, Wang Y, Ashbaugh AG, Ortines RV, et al. Staphylococcus aureus epicutaneous exposure drives skin inflammation via IL-36-mediated T cell responses. Cell Host Microbe 2017;22:653–66.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nakagawa S, Matsumoto M, Katayama Y, Oguma R, Wakabayashi S, Nygaard T, et al. Staphylococcus aureus virulent psmalpha peptides induce keratinocyte alarmin release to orchestrate IL-17-dependent skin inflammation. Cell Host Microbe 2017;22:667–77.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nakamura Y, Oscherwitz J, Cease KB, Chan SM, Munoz-Planillo R, Hasegawa M, et al. Staphylococcus delta-toxin induces allergic skin disease by activating mast cells. Nature 2013;503:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Soares J, Lopes C, Tavaria F, Delgado L, Pintado M. A diversity profile from the staphylococcal community on atopic dermatitis skin: a molecular approach. J Appl Microbiol 2013;115:1411–9. [DOI] [PubMed] [Google Scholar]
- 19.Hon KL, Tsang YC, Pong NH, Leung TF, Ip M. Exploring Staphylococcus epidermidis in atopic eczema: friend or foe? Clin Exp Dermatol 2016;41:659–63. [DOI] [PubMed] [Google Scholar]
- 20.Byrd AL, Deming C, Cassidy SKB, Harrison OJ, Ng WI, Conlan S, et al. Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol 2018;16:143–55. [DOI] [PubMed] [Google Scholar]
- 22.Nakatsuji T, Gallo RL. The role of the skin microbiome in atopic dermatitis. Ann Allergy Asthma Immunol 2019;122:263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stacy A, Belkaid Y. Microbial guardians of skin health. Science 2019;363:227–8. [DOI] [PubMed] [Google Scholar]
- 24.Otto M Staphylococcus colonization of the skin and antimicrobial peptides. Expert Rev Dermatol 2010;5:183–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT, et al. Selective antimicrobial action is provided by phenol-soluble modulins derived from Staphylococcus epidermidis, a normal resident of the skin. J Invest Dermatol 2010;130:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dong Y, Speer CP, Glaser K. Beyond sepsis: Staphylococcus epidermidis is an underestimated but significant contributor to neonatal morbidity. Virulence 2018; 9:621–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Le KY, Park MD, Otto M. Immune evasion mechanisms of Staphylococcus epidermidis biofilm infection. Front Microbiol 2018;9:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Otto M Staphylococcus epidermidis-the ‘accidental’ pathogen. Nat Rev Microbiol 2009;7:555–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Uckay I, Pittet D, Vaudaux P, Sax H, Lew D, Waldvogel F. Foreign body infections due to Staphylococcus epidermidis. Ann Med 2009;41:109–19. [DOI] [PubMed] [Google Scholar]
- 31.Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, et al. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J Bacteriol 2014;196: 3482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams MR, Nakatsuji T, Sanford JA, Vrbanac AF, Gallo RL. Staphylococcus aureus induces increased serine protease activity in keratinocytes. J Invest Dermatol 2017;137:377–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dubin G, Chmiel D, Mak P, Rakwalska M, Rzychon M, Dubin A. Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol Chem 2001;382:1575–82. [DOI] [PubMed] [Google Scholar]
- 35.Williams MR, Cau L, Wang Y, Kaul D, Sanford JA, Zaramela LS, et al. Interplay of staphylococcal and host proteases promotes skin barrier disruption in Netherton syndrome. Cell Rep 2020;30:2923–33.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Larrick JW, Hirata M, Zhong J, Wright SC. Anti-microbial activity of human CAP18 peptides. Immunotechnology 1995;1:65–72. [DOI] [PubMed] [Google Scholar]
- 37.Braff MH, Zaiou M, Fierer J, Nizet V, Gallo RL. Keratinocyte production of cathelicidin provides direct activity against bacterial skin pathogens. Infect Immun 2005;73:6771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nizet V, Ohtake T, Lauth X, Trowbridge J, Rudisill J, Dorschner RA, et al. Innate antimicrobial peptide protects the skin from invasive bacterial infection. Nature 2001;414:454–7. [DOI] [PubMed] [Google Scholar]
- 39.Murakami M, Ohtake T, Dorschner RA, Schittek B, Garbe C, Gallo RL. Cathelicidin anti-microbial peptide expression in sweat, an innate defense system for the skin. J Invest Dermatol 2002;119:1090–5. [DOI] [PubMed] [Google Scholar]
- 40.Zhang LJ, Guerrero-Juarez CF, Hata T, Bapat SP, Ramos R, Plikus MV, et al. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015;347:67–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sonesson A, Przybyszewska K, Eriksson S, Morgelin M, Kjellstrom S, Davies J, et al. Identification of bacterial biofilmandthe Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci Rep 2017;7:8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun 2013; 4:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Danby SG, Cork MJ. pH in atopic dermatitis. Curr Probl Dermatol 2018;54: 95–107. [DOI] [PubMed] [Google Scholar]
- 44.Oleksy A, Golonka E, Banbula A, Szmyd G, Moon J, Kubica M, et al. Growth phase-dependent production of a cell wall-associated elastinolytic cysteine proteinase by Staphylococcus epidermidis. Biol Chem 2004;385:525–35. [DOI] [PubMed] [Google Scholar]
- 45.Otto M, Echner H, Voelter W, Gotz F. Pheromone cross-inhibition between Staphylococcus aureus and Staphylococcus epidermidis. Infect Immun 2001;69: 1957–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Paharik AE, Parlet CP, Chung N, Todd DA, Rodriguez EI, Van Dyke MJ, et al. Coagulase-negative staphylococcal strain prevents Staphylococcus aureus colonization and skin infection by blocking quorum sensing. Cell Host Microbe 2017; 22:746–56.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Paller AS, Kong HH, Seed P, Naik S, Scharschmidt TC, Gallo RL, et al. The microbiome in patients with atopic dermatitis. J Allergy Clin Immunol 2019;143:26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Williams MR, Gallo RL. Evidence that human skin microbiome dysbiosis promotes atopic dermatitis. J Invest Dermatol 2017;137:2460–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Glatz M, Bosshard PP, Hoetzenecker W, Schmid-Grendelmeier P. The role of Malassezia spp. in atopic dermatitis. J Clin Med 2015;4:1217–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ong PY, Leung DY. Bacterial and viral infections in atopic dermatitis: a comprehensive review. Clin Rev Allergy Immunol 2016;51:329–37. [DOI] [PubMed] [Google Scholar]
- 51.Kanangat S, Postlethwaite A, Hasty K, Kang A, Smeltzer M, Appling W, et al. Induction of multiple matrix metalloproteinases in human dermal and synovial fibroblasts by Staphylococcus aureus: implications in the pathogenesis of septic arthritis and other soft tissue infections. Arthritis Res Ther 2006;8:R176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shaw L, Golonka E, Potempa J, Foster SJ. The role and regulation of the extracellular proteases of Staphylococcus aureus. Microbiology 2004;150:217–28. [DOI] [PubMed] [Google Scholar]
- 53.Bieber T Atopic dermatitis 2.0: from the clinical phenotype to the molecular taxonomy and stratified medicine. Allergy 2012;67:1475–82. [DOI] [PubMed] [Google Scholar]
- 54.Kato A, Fukai K, Oiso N, Hosomi N, Murakami T, Ishii M. Association of SPINK5 gene polymorphisms with atopic dermatitis in the Japanese population. Br J Dermatol 2003;148:665–9. [DOI] [PubMed] [Google Scholar]
- 55.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet 2006;38:441–6. [DOI] [PubMed] [Google Scholar]
- 56.De Benedetto A, Rafaels NM, McGirt LY, Ivanov AI, Georas SN, Cheadle C, et al. Tight junction defects in patients with atopic dermatitis. J Allergy Clin Immunol 2011;127:773–86.e1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, et al. Activation of TLR2 by a small molecule produced by Staphylococcus epidermidis increases antimicrobial defense against bacterial skin infections. J Invest Dermatol 2010;130: 2211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, et al. Commensal bacteria regulate Toll-like receptor 3-dependent inflammation after skin injury. Nat Med 2009;15:1377–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linehan JL, Harrison OJ, Han SJ, Byrd AL, Vujkovic-Cvijin I, Villarino AV, et al. Non-classical immunity controls microbiota impact on skin immunity and tissue repair. Cell 2018;172:784–-96.e18.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Naik S, Bouladoux N, Linehan JL, Han SJ, Harrison OJ, Wilhelm C, et al. Commensal-dendritic-cell interaction specifies a unique protective skin immune signature. Nature 2015;520:104–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakatsuji T, Chen TH, Butcher AM, Trzoss LL, Nam SJ, Shirakawa KT, et al. A commensal strain of Staphylococcus epidermidis protects against skin neoplasia. Sci Adv 2018;4:eaao4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.An Q, Sun M, Qi RQ, Zhang L, Zhai JL, Hong YX, et al. High Staphylococcus epidermidis colonization and impaired permeability barrier in facial seborrheic dermatitis. Chin Med J (Engl) 2017;130:1662–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mehlin C, Headley CM, Klebanoff SJ. An inflammatory polypeptide complex from Staphylococcus epidermidis: isolation and characterization. J Exp Med 1999;189: 907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liles WC, Thomsen AR, O’Mahony DS, Klebanoff SJ. Stimulation of human neutrophils and monocytes by staphylococcal phenol-soluble modulin. J Leukoc Biol 2001;70:96–102. [PubMed] [Google Scholar]
- 65.Cheung GY, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR, et al. Staphylococcus epidermidis strategies to avoid killing by human neutrophils. PLoS Pathog 2010;6:e1001133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dubin G, Stec-Niemczyk J, Dylag T, Silberring J, Dubin A, Potempa J. Characterisation of a highly specific, endogenous inhibitor of cysteine protease from Staphylococcus epidermidis, a new member of the staphostatin family. Biol Chem 2004;385:543–6. [DOI] [PubMed] [Google Scholar]
- 67.Dainichi T, Kitoh A, Otsuka A, Nakajima S, Nomura T, Kaplan DH, et al. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat Immunol 2018;19:1286–98. [DOI] [PubMed] [Google Scholar]
- 68.Czamowicki T, Krueger JG, Guttman-Yassky E. Novel concepts of prevention and treatment of atopic dermatitis through barrier and immune manipulations with implications for the atopic march. J Allergy Clin Immunol 2017; 139:1723–34. [DOI] [PubMed] [Google Scholar]
- 69.Casagrande BF, Fluckiger S, Linder MT, Johansson C, Scheynius A, Crameri R, et al. Sensitization to the yeast Malassezia sympodialis is specific for extrinsic and intrinsic atopic eczema. J Invest Dermatol 2006;126:2414–21. [DOI] [PubMed] [Google Scholar]
- 70.Johansson C, Eshaghi H, Linder MT, Jakobson E, Scheynius A. Positive atopy patch test reaction to Malassezia furfur in atopic dermatitis correlates with a T helper 2-like peripheral blood mononuclear cells response. J Invest Dermatol 2002;118:1044–51. [DOI] [PubMed] [Google Scholar]
- 71.Prohic A, Jovovic Sadikovic T, Kuskunovic-Vlahovljak S, Baljic R. Distribution of Malassezia species in patients with different dermatological disorders and healthy individuals. Acta Dermatovenerol Croat 2016;24:274–81. [PubMed] [Google Scholar]
- 72.Sparber F, De Gregorio C, Steckholzer S, Ferreira FM, Dolowschiak T, Ruchti F, et al. The skin commensal yeast Malassezia triggers a type 17 response that coordinates anti-fungal immunity and exacerbates skin inflammation. Cell Host Microbe 2019;25:389–403.e6. [DOI] [PubMed] [Google Scholar]
REFERENCES
- E1.Dubin G, Chmiel D, Mak P, Rakwalska M, Rzychon M, Dubin A. Molecular cloning and biochemical characterisation of proteases from Staphylococcus epidermidis. Biol Chem 2001;382:1575–82. [DOI] [PubMed] [Google Scholar]
- E2.Olson ME, Todd DA, Schaeffer CR, Paharik AE, Van Dyke MJ, Buttner H, et al. Staphylococcus epidermidis agr quorum-sensing system: signal identification, cross talk, and importance in colonization. J Bacteriol 2014;196:3482–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E3.Nakatsuji T, Chen TH, Two AM, Chun KA, Narala S, Geha RS, et al. Staphylococcus aureus exploits epidermal barrier defects in atopic dermatitis to trigger cytokine expression. J Invest Dermatol 2016;136:2192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E4.Nakatsuji T, Chiang HI, Jiang SB, Nagarajan H, Zengler K, Gallo RL. The microbiome extends to subepidermal compartments of normal skin. Nat Commun 2013;4:1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E5.Zhang YQ, Ren SX, Li HL, Wang YX, Fu G, Yang J, et al. Genome-based analysis of virulence genes in a non-biofilm-forming Staphylococcus epidermidis strain (ATCC 12228). Mol Microbiol 2003;49:1577–93. [DOI] [PubMed] [Google Scholar]
- E6.Nakatsuji T, Chen TH, Narala S, Chun KA, Two AM, Yun T, et al. Antimicrobials from human skin commensal bacteria protect against Staphylococcus aureus and are deficient in atopic dermatitis. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E7.Williams MR, Costa SK, Zaramela LS, Khalil S, Todd DA, Winter HL, et al. Quorum sensing between bacterial species on the skin protects against epidermal injury in atopic dermatitis. Sci Transl Med 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E8.Mootz JM, Benson MA, Heim CE, Crosby HA, Kavanaugh JS, Dunman PM, et al. Rot is a key regulator of Staphylococcus aureus biofilm formation. Mol Microbiol 2015;96:388–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E9.Zhang LJ, Sen GL, Ward NL, Johnston A, Chun K, Chen Y, et al. Antimicrobial peptide LL37 and MAVS signaling drive interferon-beta production by epidermal keratinocytes during skin injury. Immunity 2016;45:119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E10.Cau L, Pendaries V, Lhuillier E, Thompson PR, Serre G, Takahara H, et al. Lowering relative humidity level increases epidermal protein deimination and drives human filaggrin breakdown. J Dermatol Sci 2017;86:106–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- E11.Liggins MC,Li F, Zhang LJ, Dokoshi T, Gallo RL. Retinoids enhance the expression of cathelicidin antimicrobial peptide during reactive dermal adipogenesis. J Immunol 2019;203:1589–97. [DOI] [PMC free article] [PubMed] [Google Scholar]