

Abstract
Background
The UK Biobank contains data with varying degrees of reliability and completeness for assessing depression. A third of participants completed a Mental Health Questionnaire (MHQ) containing the gold-standard Composite International Diagnostic Interview (CIDI) criteria for assessing mental health disorders.
Aims
To investigate whether multiple observations of depression from sources other than the MHQ can enhance the validity of major depressive disorder (MDD).
Method
In participants who did not complete the MHQ, we calculated the number of other depression measures endorsed, for example from hospital episode statistics and interview data. We compared cases defined this way with CIDI-defined cases for several estimates: the variance explained by polygenic risk scores (PRS), area under the curve attributable to PRS, single nucleotide polymorphisms (SNPs)-based heritability and genetic correlations with summary statistics from the Psychiatric Genomics Consortium MDD genome-wide association study.
Results
The strength of the genetic contribution increased with the number of measures endorsed. For example, SNP-based heritability increased from 7% in participants who endorsed only one measure of depression, to 21% in those who endorsed four or five measures of depression. The strength of the genetic contribution to cases defined by at least two measures approximated that for CIDI-defined cases. Most genetic correlations between UK Biobank and the Psychiatric Genomics Consortium MDD study exceeded 0.7, but there was variability between pairwise comparisons.
Conclusions
Multiple measures of depression can serve as a reliable approximation for case status where the CIDI measure is not available, indicating sample size can be optimised using the entire suite of UK Biobank data.
Keywords: Depressive disorders, genetics, UK Biobank, classification, prospective health study
Background
The emergence of large-scale biobank resources has enabled genetic association studies of complex human traits to be performed with unprecedented sample sizes, and led to novel implication of common genetic variants with psychiatric disorders, including major depressive disorder (MDD).1 One of the analytical challenges in using national biobank resources is deciding on an approach to define disorder case and control status using the multiple sources of information available, each having varying degrees of reliability and completeness. The UK Biobank contains extensive data items that are relevant to psychiatric phenotyping, ranging from electronic health records to self-reported health data, and questionnaires that rely on retrospective recall of symptoms.2 The extent to which each source of information accurately classifies cases and controls for a given trait influences any study that is performed, by affecting power and interpretation of effect sizes.3 In genetic studies of polygenic traits, large sample sizes are a prerequisite for performing a genome-wide association study (GWAS), but investigators must balance phenotypic rigour against sample size, and missing data, where individuals do not meet criteria for either ‘cases’ or ‘controls’. These issues are particularly salient in disorders such as MDD, which encompass a spectrum of symptom severity and within-disorder phenotypic heterogeneity.4
Findings from existing GWASs
The impact of sampling decisions, as they relate to the balance between sample size and misclassification bias, has been demonstrated in the MDD GWAS literature. The CONVERGE study5 adopted a strategy to reduce phenotypic heterogeneity by recruiting only patients with recurrent MDD, diagnosed by a health professional, from a population of Han Chinese females. This was the first GWAS to identify and replicate genome-wide significant loci, despite having fewer participants (5303 cases and 5337 controls) than the largest MDD GWAS at the time (9240 cases and 9519 controls6), indicating the advantage of a comparatively homogeneous sample.
Other authors have leveraged minimal phenotyping to increase sample size in MDD GWASs. Using data collected by 23andMe, Inc., Hyde et al7 identified 75 607 individuals who reported receiving a clinical diagnosis of depression and 231 747 without a history of depression, and performed a GWAS in which 15 genome-wide significant loci were identified. Leveraging data from the UK Biobank, Howard, et al8 defined ‘broad depression’ as participants who endorsed ever having seen a general practitioner or psychiatrist for ‘nerves, anxiety, tension or depression’. This help-seeking phenotype generated a sample of 113 769 cases and 208 811 controls in which 14 genome-wide significant loci were identified.
The Psychiatric Genomics Consortium (PGC) leveraged minimal phenotyping by combining samples from 23andMe and a subset of the UK Biobank, with clinically ascertained cases to generate a sample of 116 404 cases and 314 990 controls, yielding 44 genome-wide significant loci.1 A meta-analysis of the latter three GWAS1,7,8 produced a sample size of 246 363 cases and 561 190 controls, revealing 102 genome-wide significant loci.9
Although increasing sample sizes have ostensibly increased genetic discovery, some authors have argued that the genetic architecture differs between minimally defined and strictly defined depression phenotypes, and that the former definition may yield associations with variants that are not specific to MDD. Cai et al10 compared the genetic architecture of depression phenotypes derived from different sources of information in the UK Biobank. The highest single nucleotide polymorphisms (SNP)-based heritability ( = ~26%) was observed in participants who met criteria for lifetime depression according to the Composite International Diagnostic Criteria Short Form11 (CIDI-SF) that comprised part of an online Mental Health Questionnaire (MHQ). The observed
= ~26%) was observed in participants who met criteria for lifetime depression according to the Composite International Diagnostic Criteria Short Form11 (CIDI-SF) that comprised part of an online Mental Health Questionnaire (MHQ). The observed  was comparatively lower in depression phenotypes derived from other sources of information; touchscreen questionnaires used to define symptom-based depression12 (
was comparatively lower in depression phenotypes derived from other sources of information; touchscreen questionnaires used to define symptom-based depression12 (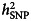 = 19%) and ‘broad depression’ (
= 19%) and ‘broad depression’ (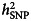 = 14%), hospital episode statistics coded as ICD-1013 diagnoses (
= 14%), hospital episode statistics coded as ICD-1013 diagnoses (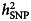 = 12%), and nurse interviews used to define self-reported depression (
= 12%), and nurse interviews used to define self-reported depression ( = 11%). Although a high degree of shared genetic liability was observed between these depression phenotypes, pairwise genetic correlations (rG) differed significantly from 1, suggesting phenotype-specific genetic effects.10
= 11%). Although a high degree of shared genetic liability was observed between these depression phenotypes, pairwise genetic correlations (rG) differed significantly from 1, suggesting phenotype-specific genetic effects.10
One interpretation of these findings is that the MHQ derivation of lifetime depression is the gold-standard for depression phenotyping in the UK Biobank, compared with the other sources of phenotypic data available. However, the MHQ was only completed by a subset of 157 366 UK Biobank participants. It is unclear whether repeated endorsement of depression, from sources other than the MHQ, can be used to reduce misclassification in participants who did not complete the MHQ, and thereby increase the sample size of credible depression ‘cases’.
Aims
Here, we establish five depression measures available in all UK Biobank participants and create case groups determined by the number of depression measures endorsed by individuals who did not complete the MHQ. We observe the strength of the genetic contribution to each case group by estimating the variance in depression liability explained by polygenic risk scores (PRS), area under the curve (AUC) attributable to PRS, and SNP-based heritability. We compare the strength of the genetic contribution in cases determined by number of endorsements with MHQ-derived lifetime depression cases to assess whether sample size can be optimised using all available phenotyping sources, without substantially increasing misclassification bias.
The choice of a control group also influences effect size estimates in genetic studies,14 and we additionally explore the use of partially screened or screened controls. We anticipate that our approach will encourage researchers to consider the benefit of using multiple phenotype sources to aid classifying cases and controls, not just for depression, but for the extensive range of complex human disorders available in the UK Biobank.
Method
Participants and phenotyping
The UK Biobank is a prospective health study of over 500 000 individuals located across the UK. Participants were aged between 40 and 69 at recruitment (2006–2010) and attended a baseline assessment where information on health was collected with a touchscreen questionnaire and verbal interview.2 Subsets of participants completed repeat assessments:
instance (1) n = 20 335 between 2012–2013;
instance (2) n = 42 961 (interview) and n = 48 340 (touchscreen) in 2014; and
instance (3) n = 2843 (interview) and n = 3081 (touchscreen) in 2019.
Participants with valid email addresses (n = 339 092) were invited to complete the online MHQ in 2017.15
The UK Biobank received ethical approval from the North West - Haydock Research Ethics Committee (reference 16/NW/0274). This study was conducted under application number 18177. Participants provided electronic signed consent at recruitment.2
We identified six measures for depression phenotyping (summarised below) and tabulated the number of individuals who met the criteria for each. Full definitions and UK Biobank field codes are given in Supplementary materials, Section 1, available at https://10.1192/bjo.2020.145.
Help-seeking
‘Help-seeking’ cases endorsed either of the following questions at baseline or instance 1 or 2: ‘Have you ever seen a general practitioner for nerves, anxiety, tension or depression?’, and ‘Have you ever seen a psychiatrist for nerves, anxiety, tension or depression?’.
Self-reported depression
‘Self-reported depression’ cases endorsed having experienced depression (past or present) during the verbal interview at baseline or instance 1 or 2.
Antidepressant usage
‘Antidepressant usage’ cases endorsed currently taking antidepressant medications during the verbal interview at baseline or instance 1 or 2.
Depression (Smith)
At baseline, 172 751 participants completed an extended touchscreen questionnaire that was enriched for psychosocial questions in addition to the help-seeking question. From these data, Smith et al12 defined three depression phenotypes, all of which required endorsement for ‘help-seeking’: (a) single episode of probable major depression, (b) probable recurrent major depression (moderate), and (c) probable recurrent major depression (severe). We refer to these individuals who endorsed ‘help-seeking’ and also met the additional criteria defined by Smith et al12 as ‘depression (Smith)’.
Hospital (ICD-10)
Hospital episode statistics contain diagnoses recorded with the ICD-10.13 We accessed the UK Biobank Data Portal Record Repository to identify ICD-10 diagnoses recorded between April 1997 to October 2016. ‘Hospital (ICD-10)’ cases were individuals assigned a primary or secondary diagnosis for depressive episode (F32–F32.9) or recurrent depressive disorder (F33–F33.9).
Lifetime depression (MHQ)
A total of 157 366 participants completed the MHQ. We identified individuals with a lifetime history of depression from responses to the CIDI depression module.11 We adopted scoring criteria previously defined,15 which is equivalent to the DSM criteria for MDD.16 We classified ‘lifetime depression (MHQ)’ cases as individuals meeting those criteria.
Screening
We defined five potential psychosis phenotypes: ‘self-reported psychosis’, ‘antipsychotic usage’, ‘bipolar (Smith)’, ‘hospital (ICD-10) psychosis’, and ‘psychosis (MHQ screen)’. Individuals meeting the criteria for any psychosis phenotype were excluded from analysis (n = 5482). The derivation of the psychosis phenotypes is provided in the Supplementary materials, Section 2.
Depression phenotypes determined by number of observed depression measures
We split the UK Biobank cohort by MHQ participation. In individuals who did not participate in the MHQ, we calculated endorsement for five depression phenotypes (‘help-seeking’, ‘self-reported depression’, ‘antidepressant usage’, ‘depression (Smith)’, or ‘hospital (ICD-10)’) to derive five independent depression case groups. These groups are referred to as: ‘one measure’, ‘two measures’, ‘three measures’, ‘four measures’ and ‘five measures’. We performed the same exercise in individuals who completed the MHQ to observe the phenotypic correlation between depression measures (excluding the MHQ) in those that met the criteria for lifetime depression (MHQ) and those that did not.
Controls
Two control groups were defined. Controls comprised all UK Biobank participants who did not meet the criteria for any of the depression or psychosis phenotypes. MHQ controls were restricted to those who participated in the MHQ and showed no psychiatric pathology in the MHQ responses. The criteria for controls and MHQ controls is provided in Supplementary materials, Section 3.
Genetic quality control
The UK Biobank performed preliminary quality control on genotype data.2 Using genetic principal components provided by the UK Biobank, we performed 4-means clustering on the first two principal components to identify and retain individuals of European ancestry. Quality control was performed using PLINK v1.917 to remove: variants with missingness >0.02 (before individual quality control), individuals with missingness >0.02, gender-discordant observations, variants with missingness >0.02 (after individual quality control), variants departing from Hardy–Weinberg equilibrium (P < 1 × 10−8), and variants with minor allele frequency <0.01. Relatedness kinship estimates provided by the UK Biobank were used to identify related pairs (KING r2 > 0.044)18 and the GreedyRelated19 algorithm was used to remove one individual from each pair. FlashPCA220 was used to generate principal components for the European-ancestry subset. The UK Biobank imputed genotype data to the Haplotype Reference Consortium21 and the UK10K Consortium22 using the IMPUTE4 software.2 We removed imputed variants with INFO score <0.4 and/or minor allele frequency <0.01.
Statistical analyses
We summarised sociodemographic data taken at baseline assessment: age, gender, socioeconomic status (SES), body mass index (BMI), smoking status and self-reported overall health rating, where participants were asked to rate their overall health on a scale of 1 (excellent) to 4 (poor). We tested for significant differences in sociodemographic variables between cases and controls using Welch Two Sample t-tests in R v3.6.2.23 To investigate the impact of control sampling, all statistical analyses were performed using controls and MHQ controls.
PRS analyses
The PRSice-2 software24 was used to perform PRS analyses. PRS were calculated using summary statistics from the latest PGC MDD GWAS.1 The PGC MDD GWAS was performed on multiple cohorts with varying phenotyping strategies including self-report (UK Biobank and 23andMe), electronic medical records and clinical ascertainment. We compared the predictive utility of PRS calculated using summary statistics from (a) the full PGC MDD sample (excluding UK Biobank), and (b) a subset of the PGC MDD sample with self-reported cases removed (additionally excluding 23andMe). Quality control was performed on summary statistics to remove variants within the major histocompatibility complex, and variants in linkage disequilibrium (r2 > 0.1) with the lead variant within a 250 kb region.
We tested for association between PRS calculated at eight P-value thresholds (PT; 0.001, 0.05, 0.1, 0.2, 0.3, 0.4, 0.5 and 1.0) and case–control status in each UK Biobank depression phenotype using logistic regressions adjusted for six principal components, genotyping batch and assessment centre (n = 128 variables). To control for multiple testing across PT, ten thousand permutations were performed for each model using linear regression for computational efficiency. We report observed and empirical P-values at the optimal PT and the corresponding R2 estimates, transformed to the liability scale using lifetime risk of 15%.1 To increase sample size, ‘four measures’ and ‘five measures’ cases were combined in subsequent analyses. The predictive ability of PRS was assessed using AUC with the R pROC package.25 We compared AUC for the null model (six principal components, genotyping batch and assessment centre on depression phenotypes) with the full model with PRS at the optimal PT, using DeLong's test for two correlated receiver operating characteristic (ROC) curves.
SNP-based heritability and genetic correlation analyses
To overcome computational limitations when performing GWASs with a large number of covariates (n = 128), we regressed six principal components, genotyping batch and assessment centre on depression case–control status using logistic regression with the glm function in R v3.6.2.23 GWASs were performed on residuals for the five depression groups (one; two; three; four and five measures combined; and lifetime depression (MHQ)) using both controls sets. GWASs were performed in BGENIE v1.22 and summary statistics were uploaded to FUMA26 to create Manhattan and QQ plots.
SNP-based heritabilities were calculated with linkage disequilibrium score regression (LDSC v1.0.027,28) using summary statistics excluding variants with INFO scores <0.9 and pre-computed linkage disequilibrium scores (1000 Genomes European data). SNP-based heritabilities were transformed to the liability scale using lifetime risk of 15%1 and, for comparison across a range of population prevalences (1% to 60%), using the transformation proposed by Lee et al29 (equation 8).
Genetic correlations (rG) were estimated using LDSC v1.0.0.27,28 The rG between each UK Biobank depression phenotype and PGC depression phenotype was calculated using summary statistics from both the full PGC MDD sample (excluding UK Biobank, 116 404 cases and 314 990 controls), and the subset of the PGC MDD sample with self-reported cases removed (excluding UK Biobank and 23andMe, 45 591 cases and 97 674 controls).1
The study design is summarised in Fig. 1.
Fig. 1.

Study design.
Nine-item Patient Health Questionnaire (PHQ-9) included in the Mental Health Questionnaire (MHQ); Section A, MHQ: participants indicated prior diagnosis for any of 16 mental health disorders. Refer to Supplementary section 3.2 for PHQ-9 and MHQ Section A details. a. Controls, UK Biobank (UKB) participants screened for any of the five psychosis and six depression phenotypes: help-seeking, self-reported depression, antidepressant usage, depression (Smith), hospital (ICD-10) or lifetime depression (MHQ); 162 130 controls are non-MHQ participants; 70 422 controls are MHQ participants (57 805 MHQ controls + 12 617 who did not meet Composite International Diagnostic Criteria Short Form (CIDI-SF) criteria for lifetime depression (MHQ) but were excluded from MHQ controls because of psychopathology indicated in MHQ Section A, or above threshold on PHQ-9). QC, quality control; AUC, area under the curve; GWAS, genome-wide association study; SNP, single nucleotide polymorphism; MDD, major depressive disorder; PRS, polygenic risk score; PGC, Psychiatric Genomics Consortium.
Results
Of individuals who did not participate in the MHQ, 93 414 met the criteria for at least one other depression phenotype (Table 1). These cases had poorer sociodemographic characteristics than lifetime depression (MHQ) cases (n = 28 982) and controls (n = 232 552), including lower SES, higher current smoking prevalence, higher BMI and poorer self-reported health (all P-values <1 × 10−109 in pairwise comparisons). The magnitude of difference increased when compared with MHQ controls (n = 57 805), who on average had more favourable sociodemographic outcomes than the larger set of controls. Lifetime depression (MHQ) cases also had poorer sociodemographic characteristics compared with both control groups (excluding current smoking status and BMI compared with the larger set of controls), although the magnitude of case–control differences was attenuated (all P-values <6 × 10−25 in pairwise comparisons) from that observed with the 93 414 cases derived from sources other than the MHQ.
Table 1.
Sociodemographic information for depression cases and controls
| n | Age, mean (s.d.) | Female, % | Townsend Deprivation Index, mean (s.d.)a | Current smoker, % | Mean BMI (s.d.) | Health rating,b mean (s.d.) | |
|---|---|---|---|---|---|---|---|
| Phenotyping in individuals who did not participate in the MHQ | |||||||
| By source of information | |||||||
| Hospital (ICD-10) | 10 198 | 56.7 (8.09) | 62 | −0.29 (3.46) | 22 | 29.0 (5.94) | 2.7 (0.83) |
| Self-reported depression | 15 091 | 55.8 (7.88) | 65 | −0.65 (3.35) | 18 | 28.5 (5.62) | 2.6 (0.82) |
| Depression (Smith) | 15 037 | 56.1 (8.12) | 63 | −1.00 (2.98) | 15 | 28.0 (5.19) | 2.4 (0.77) |
| Antidepressant usage | 20 057 | 57.0 (7.83) | 68 | −0.70 (3.35) | 18 | 28.9 (5.68) | 2.7 (0.82) |
| Help-seeking | 89 278 | 56.6 (7.97) | 64 | −1.02 (3.20) | 14 | 27.9 (5.16) | 2.3 (0.77) |
| By degree of endorsement | |||||||
| One measure | 57 321 | 56.9 (7.96) | 63 | −1.15 (3.18) | 13 | 27.6 (4.95) | 2.3 (0.74) |
| Two measures | 21 468 | 56.5 (8.08) | 64 | −0.92 (3.17) | 15 | 28.0 (5.24) | 2.4 (0.78) |
| Three measures | 9 738 | 56.1 (7.90) | 66 | −0.67 (3.35) | 19 | 28.6 (5.60) | 2.6 (0.81) |
| Four measures | 4 245 | 56.0 (7.88) | 67 | −0.37 (3.34) | 21 | 29.3 (6.14) | 2.8 (0.84) |
| Five measures | 642 | 56.4 (7.63) | 67 | −0.47 (3.23) | 22 | 29.4 (5.64) | 2.8 (0.83) |
| Total | 93 414 | 56.7 (7.98) | 64 | −1.01 (3.21) | 14 | 27.9 (5.17) | 2.4 (0.78) |
| Phenotyping in individuals who participated in the MHQ | |||||||
| Lifetime depression (MHQ) | 28 982 | 54.3 (7.53) | 69 | −1.46 (2.95) | 9 | 27.2 (5.04) | 2.1 (0.73) |
| MHQ controls | 57 805 | 56.8 (7.65) | 48 | −2.02 (2.65) | 6 | 26.5 (4.14) | 1.8 (0.62) |
| Phenotyping in all UK Biobank participants (screening for any indication of depression or psychosis) | |||||||
| Controls | 232 552 | 57.1 (8.10) | 47 | −1.65 (2.89) | 9 | 27.2 (4.53) | 2.0 (0.68) |
MHQ, Mental Health Questionnaire.
a. Negative scores indicate less deprivation.
b. Health rating was self-reported on a scale of 1 (excellent) to 4 (poor).
Comparing groups within and outside the MHQ sample, those who participated in the MHQ (n = 126 261) had more favourable sociodemographic characteristics than those who did not participate in the MHQ (n = 259 443), including higher SES, fewer current smokers, lower BMI and higher self-reported health (all P-values <3 × 10−89 in pairwise comparisons).
Supplementary Tables 1–11 provide the number of participants within subcategories (for example by ICD-10 code) for depression and psychosis in the entire UK Biobank sample.
Figure 2 shows the 93 414 individuals who did not participate in the MHQ but met the criteria for at least one other depression phenotype, stratified into independent groups according to the number of depression measures endorsed. For each stratum, the number of cases and prevalence as a proportion of controls (n = 232 552) was: one measure n = 57 321 (19.8%); two measures n = 21 468 (8.5%); three measures n = 9738 (4.0%); four measures n= 4245 (1.8%); and five measures n = 642 (0.3%).
Fig. 2.

Number of depression measures observed in participants who did not complete the Mental Health Questionnaire (MHQ).
To the left of the main graph the horizontal green bars indicate the number of individuals who met the criteria for any of the corresponding depression phenotypes. Vertical bars indicate the number of individuals endorsing combinations of the five depression phenotypes. Vertical bars are coloured by the number of depression measures endorsed (see key).
Of the 28 982 individuals who met CIDI-SF criteria for lifetime depression, 9304 (32%) did not endorse any of the five non-MHQ depression measures and 19 678 (68%) endorsed at least one. Of the 95 486 MHQ participants who did not meet CIDI-SF criteria for lifetime depression, 71 848 (75%) did not endorse any of the five non-MHQ depression measures and 23 638 (25%) endorsed at least one. Of individuals who did not meet CIDI-SF criteria, 37 681 (39%) were excluded from MHQ controls for psychopathology indicated within or outside the MHQ as follows: 19 165 excluded for recent depressive symptoms indicated on the nine-item Patient Health Questionnaire (PHQ-9)30 within the MHQ; 11 288 excluded for prior diagnosis of mental health disorders indicated in screening section A of the MHQ; 7228 had no indication of psychopathology according to the MHQ but met the criteria for at least one of the five non-MHQ depression measures. The remaining 57 805 participants that did not meet CIDI-SF criteria and endorsed no other measure of depression within or outside the MHQ were defined as MHQ controls. These data are summarised in Fig. 3. Supplementary Fig. 1 shows the phenotypic agreement between each of the five non-MHQ depression measures within MHQ participants.
Fig. 3.

Number of depression measures endorsed by Mental Health Questionnaire (MHQ) participants.
Categories on the x-axis represent the number of endorsements for the five non-MHQ depression phenotypes (help-seeking, self-reported depression, antidepressant usage, depression (Smith), or hospital (ICD-10)), with the total number of MHQ participants in each category shown above each bar. Bars are partitioned by MHQ outcomes. Excluded (MHQ responses), individuals that did not meet Composite International Diagnostic Criteria Short Form (CIDI-SF) criteria but had other indications for psychopathology within the MHQ (i.e. nine-item Patient Health Questionnaire (PHQ-9) or Screening Section A). Excluded (non-MHQ measures), individuals that did not meet CIDI-SF criteria and had no indication for psychopathology within the MHQ, but met the criteria for at least one of the five non-MHQ depression measures.
The associations between MDD PRS and case–control status of UK Biobank depression phenotypes were significant (all empirical P-values = 1 × 10−4) (Fig. 4). The variance in liability (R2) explained by the PRS ranged between 0.52% (one measure) and 3.54% (four measures). Across depression phenotypes, R2 increased when cases were compared with MHQ controls, and when PRS were calculated using summary statistics from the full PGC MDD sample (excluding UK Biobank), compared with the subset of the PGC MDD (excluding UK Biobank and 23andMe). Full results of each test of association are shown in Supplementary Table 12 and Supplementary Fig. 2. Four and five measures were combined in subsequent analyses to increase power.
Fig. 4.

Variances in depression liability explained by polygenic risk score (PRS).
Excluding (excl.) 23andMe, PRS calculated using summary statistics from the subset of the Psychiatric Genomics Consortium (PGC) major depressive disorder (MDD) sample (excluding UK Biobank and 23andMe). Including (incl.) 23andMe, PRS calculated using summary statistics from the full PGC MDD sample (excluding UK Biobank). Results are shown for the optimal PT for each test of association. R2 estimates were transformed to the liability scale using a population prevalence of 15% across all UK Biobank phenotypes. Observed P-values are shown above each bar. MHQ, Mental Health Questionnaire
The differences in AUC between null and full models were significant for each depression phenotype (maximum P-value = 2 × 10−25). The increase in AUC attributable to PRS for models including controls ranged between 1.41% (one measure) and 3.01% (three measures). For models including MHQ controls, the increase in AUC attributable to PRS ranged between 1.29% (one measure), and 3.60% (lifetime depression (MHQ)). AUC attributable to PRS generally increased with the number of depression measures endorsed, maximising in lifetime depression (MHQ) when compared with MHQ controls (Fig. 5). Supplementary Figure 3 shows ROC curves for null and full models across depression phenotypes.
Fig. 5.

Area under the curve (AUC) increases attributable to polygenic risk score (PRS), calculated using full Psychiatric Genomics Consortium (PGC) major depressive disorder (MDD) summary statistics (including 23andMe), at the PT corresponding to each case–control combination.
y-axis: AUC for full model minus AUC for null model. Null versus full model P-values estimated with DeLong's test for two correlated receiver operating characteristic curves are shown above each bar. MHQ, Mental Health Questionnaire.
Assuming a population prevalence of 15% across depression phenotypes, SNP-based heritability (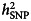 ) estimates ranged between 7% (s.e. = 0.005) in one measure and 21% (s.e. = 0.029) in four and five measures combined when GWAS were performed using controls (Fig. 6).
) estimates ranged between 7% (s.e. = 0.005) in one measure and 21% (s.e. = 0.029) in four and five measures combined when GWAS were performed using controls (Fig. 6). 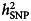 increased when GWAS were performed with MHQ controls, ranging between 17% (s.e. = 0.009) in one measure to 33.6% (s.e. = 0.034) in four and five measures combined. Supplementary Figs 4–8 show Manhattan and QQ plots, Supplementary Table 13 shows FUMA references for each GWAS performed, and Supplementary Tables 14 and 15 show the full results from BGENIE and LDSC.
increased when GWAS were performed with MHQ controls, ranging between 17% (s.e. = 0.009) in one measure to 33.6% (s.e. = 0.034) in four and five measures combined. Supplementary Figs 4–8 show Manhattan and QQ plots, Supplementary Table 13 shows FUMA references for each GWAS performed, and Supplementary Tables 14 and 15 show the full results from BGENIE and LDSC.
Fig. 6.

Single nucleotide polymorphisms (SNP)-based heritability ( ) transformed to the liability scale using a population prevalence of 15% across the UK Biobank depression phenotypes on the x-axis.
) transformed to the liability scale using a population prevalence of 15% across the UK Biobank depression phenotypes on the x-axis.
Error bars show 95% confidence intervals. MHQ, Mental Health Questionnaire.
Across a range of population prevalences between 1% and 60%, higher 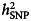 was observed for models including MHQ controls compared with controls (Fig. 7). In GWAS using controls, the lowest
was observed for models including MHQ controls compared with controls (Fig. 7). In GWAS using controls, the lowest  across the range of population prevalences was in one measure, followed by two measures, lifetime depression (MHQ), three measures, and four and five measures combined. We observed near complete overlap in
across the range of population prevalences was in one measure, followed by two measures, lifetime depression (MHQ), three measures, and four and five measures combined. We observed near complete overlap in  estimates between two measures and lifetime depression (MHQ), and between the three measures and the four and five measures combined. In GWAS using MHQ controls, the lowest
estimates between two measures and lifetime depression (MHQ), and between the three measures and the four and five measures combined. In GWAS using MHQ controls, the lowest  across the range of population prevalences was in lifetime depression (MHQ), followed by one measure, two measures, four and five measures combined, and three measures. Near complete overlap in
across the range of population prevalences was in lifetime depression (MHQ), followed by one measure, two measures, four and five measures combined, and three measures. Near complete overlap in  estimates was also observed between three measures and four and five measures combined.
estimates was also observed between three measures and four and five measures combined.
Fig. 7.

Single nucleotide polymorphisms (SNP)-based heritability ( ) transformed to the liability scale across population prevalence estimates between 1% to 60%.
) transformed to the liability scale across population prevalence estimates between 1% to 60%.
(a) genome-wide association study (GWAS) performed using controls; (b) GWAS performed using Mental Health Questionnaire (MHQ) controls. UKB, UK Biobank.
The genetic correlations (rG) between UK Biobank depression phenotypes and PGC depression phenotypes were between 0.62 and 0.90 (P-value <6 × 10−25 across all tests for the null hypothesis that rG = 0) (Fig. 8). The lowest estimate of rG was observed between three measures (compared with MHQ controls) and the PGC sample including 23andMe (rG = 0.62, 95% CI: 0.57–0.67). For the measures of depression, genetic correlations were highest for GWAS using controls, and with summary statistics excluding 23andMe. For Lifetime depression (MHQ), the highest genetic correlations were for GWAS using MHQ controls, and with the PGC sample excluding 23andMe (rG = 0.90, 95% CI: 0.80–1.00). Supplementary Table 16 and Supplementary Fig. 9 show estimates of rG between all UK Biobank depression phenotypes.
Fig. 8.

Genetic correlations between the UK Biobank depression phenotypes and Psychiatric Genomics Consortium (PGC) depression phenotypes.
Excluding (excl.) 23andMe, green points, summary statistics from the subset of the PGC major depressive disorder (MDD) sample (excluding UK Biobank and 23andMe). Including (incl.) 23andMe, black/grey points, summary statistics from the full PGC MDD sample (excluding UK Biobank). Summary statistics used to estimate genetic correlations (rG) were generated from genome-wide association study of UK Biobank depression phenotypes using controls and Mental Health Questionnaire (MHQ) controls. Error bars: 95% confidence intervals.
Discussion
Main findings
We examined whether multiple endorsements of depression can reduce misclassification and increase the sample of depression cases in the UK Biobank. Our investigation took an approach to classifying depression that aims to fully utilise the UK Biobank by incorporating all sources of information. We found that including at least two measures of depression can serve as a reliable approximation where the MHQ measure is not available and improve case–control classification. Further, increasing the number of measures provides an increasingly reliable approximation.
The results followed from defining independent groups of depression cases according to the number of depression measures endorsed in sources other than the MHQ. We compared cases defined using this approach with CIDI-defined cases for the following: variance explained by PRS, AUC attributable to PRS and SNP-based heritability. We further explored how these differ using partially screened controls compared with fully screened MHQ controls.
Our conclusion is based on three key observations.
We observed higher values of genetic estimates (variance explained by PRS, AUC attributable to PRS and SNP-based heritability) with increasing endorsement of depression measures.
When cases were defined by two or more measures of depression, these genetic estimates approximated or exceeded those observed in lifetime depression (MHQ).
Control sampling resulted in substantial differences between genetic estimates, which were higher when analyses were performed with MHQ controls.
PRS analyses showed the variance in depression liability increased with the number of measures endorsed, indicating increasing genetic similarity with the PGC MDD sample. The variance explained by PRS was comparable between one measure and lifetime depression (MHQ), although interpretation depends on population prevalence, which is difficult to estimate. By contrast, AUC allows comparisons that are independent of population prevalence. The highest AUC attributable to PRS was observed in lifetime depression (MHQ) and was more than double the estimate in one-measure cases. These results indicate that between-group differences in the variance explained by PRS on the liability scale may be masked by equivalent prevalence assumptions across the groups.
However, we found that SNP-based heritability estimates were approximately equivalent for lifetime depression (MHQ) and two measures across a range of population prevalences between 1% and 60%. Assuming lifetime risk of 15%,  for lifetime depression (MHQ) ranged between 11% and 13%, depending on the control group. This range is notably different to the
for lifetime depression (MHQ) ranged between 11% and 13%, depending on the control group. This range is notably different to the  estimate of 26% reported by Cai et al10 for the corresponding phenotype named ‘lifetimeMDD’. Much of the difference is accounted for by methodology and lifetime risk assumptions. Cai et al10 used phenotype correlation–genotype correlation (PCGC) software31 and the observed prevalence of ‘lifetimeMDD’ in the UK Biobank (24%) to determine liability scale
estimate of 26% reported by Cai et al10 for the corresponding phenotype named ‘lifetimeMDD’. Much of the difference is accounted for by methodology and lifetime risk assumptions. Cai et al10 used phenotype correlation–genotype correlation (PCGC) software31 and the observed prevalence of ‘lifetimeMDD’ in the UK Biobank (24%) to determine liability scale  . Using LDSC and lifetime risk of 15%, Cai et al10 report
. Using LDSC and lifetime risk of 15%, Cai et al10 report  of 16% for ‘lifetimeMDD’, which is modestly higher than our estimate, likely because of minor differences in the derivation of lifetime depression (MHQ). Notably, LDSC provides a lower bound of
of 16% for ‘lifetimeMDD’, which is modestly higher than our estimate, likely because of minor differences in the derivation of lifetime depression (MHQ). Notably, LDSC provides a lower bound of 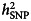 compared with other methods, thus our
compared with other methods, thus our  estimates would increase using other software packages.32 However, for computational efficiency and consistency with the published literature, we used LDSC and lifetime risk of 15% to calculate
estimates would increase using other software packages.32 However, for computational efficiency and consistency with the published literature, we used LDSC and lifetime risk of 15% to calculate 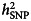 . Our estimate for lifetime depression (MHQ) broadly aligns to the aforementioned GWASs of depression that have adopted the same approach. Using LDSC and lifetime risk of 15%, Hyde et al,7 Howard et al,8 Wray et al1 and Howard et al9 reported liability
. Our estimate for lifetime depression (MHQ) broadly aligns to the aforementioned GWASs of depression that have adopted the same approach. Using LDSC and lifetime risk of 15%, Hyde et al,7 Howard et al,8 Wray et al1 and Howard et al9 reported liability  of 6%, 10%, 9% and 9% for their respective definitions of depression.
of 6%, 10%, 9% and 9% for their respective definitions of depression.
SNP-based heritability increased with the number of measures endorsed, and we posit this results from increasing phenotypic homogeneity within depression case groups. One measure was comprised mainly of help-seeking, but also included ostensibly stricter phenotypes including antidepressant usage, hospital (ICD-10) and self-reported depression (no participants had a single measure for depression (Smith) since it requires the endorsement of help-seeking). However, single observations may reflect indications other than depression. For example, help-seeking also captures indications for anxiety, and antidepressants can be prescribed for pain management. We therefore regard the number of endorsed measures as more important for phenotypic validity than the specific measure endorsed – those with only one measure are less likely to represent clinical populations than those with multiple measures.
The precision of  estimates declines in the smaller samples with three or more endorsements, however, the confidence internals in these groups showed little or no overlap with cases defined by fewer endorsements or with lifetime depression (MHQ), indicating significantly higher
estimates declines in the smaller samples with three or more endorsements, however, the confidence internals in these groups showed little or no overlap with cases defined by fewer endorsements or with lifetime depression (MHQ), indicating significantly higher 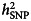 across a range of population prevalences. Multiple endorsements may also represent greater severity, but this is not easily demonstrable in the current study because we have not directly measured severity. Cai et al10 observed higher
across a range of population prevalences. Multiple endorsements may also represent greater severity, but this is not easily demonstrable in the current study because we have not directly measured severity. Cai et al10 observed higher 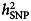 (32%) in the subset of ‘lifetimeMDD’ who met more stringent criteria for recurrent MDD. Further work is needed to explore disorder severity and SNP-based heritability, which may be possible in the UK Biobank using features such as length of episode and level of impairment.
(32%) in the subset of ‘lifetimeMDD’ who met more stringent criteria for recurrent MDD. Further work is needed to explore disorder severity and SNP-based heritability, which may be possible in the UK Biobank using features such as length of episode and level of impairment.
The pattern of pairwise correlations with the PGC MDD varied across UK Biobank depression phenotypes and was highest with lifetime depression (MHQ) (rG = 0.9). However, in cases determined by one, two, four or five measures of depression, genetic correlations with the PGC MDD were almost as high, ranging between 0.84 and 0.86. Across UK Biobank depression phenotypes, genetic correlations with the PGC MDD excluding 23andMe were higher than with the PGC MDD including 23andMe. This result indicates greater similarity with the clinically ascertained PGC sample, which may lend support to the validity of UK Biobank measures in general.
We observed lower genetic correlations with PGC MDD when GWAS of cases defined by number of endorsements were performed with MHQ controls. Recent work has demonstrated that estimates of genetic parameters increase when sampling controls from the left tail of an underlying liability distribution.14 We posit that MHQ controls represent the left tail of the liability distribution and this is supported by the observation that MHQ controls were healthier than controls for health indicators correlated with depression prevalence. That is, MHQ controls had higher SES, fewer smokers, lower BMI and better self-reported health ratings than controls. Our results also revealed larger effect sizes across PRS, AUC and SNP-based heritability analyses when using MHQ controls, compared with controls. MHQ control characteristics may make the UK Biobank dissimilar to the PGC, thus reducing the observed genetic correlation. However, we note that this is not universally supported in the analysis; with lifetime depression (MHQ) we observed higher genetic correlations with PGC phenotypes when models included MHQ controls.
Of participants who met CIDI-SF criteria, 32% would have otherwise gone undetected as cases of depression as they did not endorse any of the five non-MHQ measures of depression. Further, of participants who completed the MHQ and did not meet CIDI-SF criteria for lifetime depression, 39% were excluded from MHQ controls because they had some other indication for psychopathology within or outside the MHQ, for example roughly half were excluded because of recent depressive symptoms indicated on the PHQ-9, but did not fulfil CIDI-SF diagnostic criteria. This is consistent with the view that a percentage of cases would go undiagnosed in primary settings as they never sought help, and a percentage of those who sought help do not fulfil diagnostic criteria for MDD. This highlights the advantage of having both MHQ and non-MHQ sources of information to cross-validate depression phenotypes. Using both sources of information allowed us to define ‘super healthy’ MHQ controls, screened for subdiagnostic depressive symptoms. Although improving the definition of controls may increase power to detect genetic effects, the use of ‘super healthy’ controls omits the intermediate portion of the genetic liability distribution, which can increase SNP-based heritability estimates in the absence of a liability scale correction.14 We therefore regard the SNP-based heritabilities calculated using controls as the more accurate of the estimates reported here. Future studies with the main objective of genetic discovery may derive power benefits from strict control screening, such as used here to define MHQ controls.
Implications
Our results converge on the conclusion that repeated measures of depression may be used to reduce misclassification of depression cases and controls and increase the sample size of credible depression cases in addition to those defined using the MHQ. Cai et al10 compared depression phenotypes derived from different sources of information in the UK Biobank and showed that the strength of the genetic contribution was highest in CIDI-defined cases. We propose that our findings build upon this work by considering that the number of endorsed measures of depression can be used to decrease misclassification by identifying those participants who perhaps had a single mild episode of depression but would not meet the CIDI diagnostic criteria.
This study enhances the choices available for depression phenotyping in the UK Biobank. The appropriate balance between maximising sample size and minimising misclassification depends naturally on the study to be performed. For GWASs, two measures showed a high genetic correlation with PGC MDD summary statistics, and individuals with two or more measures would contribute 36 093 cases that could be combined with 28 982 lifetime depression (MHQ) cases. Amid increasing use of biobank resources for highly powered psychiatric studies, our study presents a framework that can be adopted for assessing mental health disorders in any biobank that contains multiple sources of information with varying degrees of validity and completeness.
Limitations
Representativeness is a noted limitation of UK Biobank phenotyping. A healthy volunteer bias has been observed in the UK Biobank,33 although it has been proposed that this bias does not invalidate exposure–outcome relationships, but may result in attenuated association.34 However, this selection bias extends to MHQ participation, where we observed more favourable sociodemographic characteristics in MHQ participants compared with non-participants. The differences that we observed in the genetic architecture of depression defined within and outside of the MHQ sample may be influenced by the polygenic basis of MHQ participation, which has been shown to correlate negatively with psychiatric phenotypes.35 A further limitation of the ability to extrapolate our results is the lack of representation in individuals of diverse ancestries. The literature has demonstrated attenuation in prediction between training and target samples of different ancestry,36 highlighting the need to build training data in varied ancestral populations.
A further relevant limitation relates to the completeness of the data, and to the opportunity individuals have to endorse specific measures. For example, the extended touchscreen questionnaire used to define ‘depression (Smith)’ was only available to approximately a third of the UK Biobank cohort. Regional, procedural or other criteria may have influenced the ability of all measures to be generically applied to the UK Biobank data-set. For instance, recording of data within Scotland excludes linkage to psychiatric hospital episode data. As a result, the reported number of measures may be lower than identified.
In conclusion, using a simple phenotyping approach, we created independent groups of depression cases determined by the number of depression measures endorsed in the UK Biobank. Our results indicate that two or more endorsements of depression can be used to reduce misclassification between cases and controls, often yielding genetic estimates that approximate, or exceed, the gold-standard CIDI criteria included in the MHQ. Although this study has not considered the relative benefit of considering one specific measure over another, the findings of the study highlight that any combination provides a good approximation of depression where the MHQ is not available. With the recent addition of primary care data for approximately half of UK Biobank participants, there is an opportunity to integrate this additional source of information to identify more credible depression cases. We anticipate that this phenotyping approach can be used across other complex traits, to fully utilise the UK Biobank resource.
Acknowledgements
We thank participants and scientists involved in making the UK Biobank resource available (http://www.ukbiobank.ac.uk/). This study was conducted under UK Biobank application number 18177. We thank the research participants and employees of 23andMe for making this work possible. Statistical analyses were carried out on the King's Health Partners High Performance Compute Cluster funded with capital equipment grants from the GSTT Charity (TR130505) and Maudsley Charity (980).
Data availability
Available from UK Biobank subject to standard procedures (www.ukbiobank.ac.uk). The full GWAS summary statistics for the 23andMe discovery data-set will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit https://research.23andme.com/collaborate/#publication for more information and to apply to access the data.
Author contributions
Conceptualisation and study design: K.P.G., C.M.L., P.F.O. Analysis and manuscript: K.P.G. Analytical consultation and interpretation: C.M.L., J.R.I.C., O.P., D.M.H., P.F.O., B.J. UK Biobank data curation and management: J.R.I.C., G.B., R.A., K.B.H., B.J. Genetic data preparation: J.R.I.C. Contributed to data preparation and analysis: C.H., O.P. Project supervisors: C.M.L., P.F.O. All authors critically edited the paper.
Funding
This work was supported by the UK Medical Research Council (PhD studentship to K.P.G.; grant MR/N015746/1). This paper represents independent research part-funded by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King's College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. D.M.H. is supported by a Sir Henry Wellcome Postdoctoral Fellowship (Reference 213674/Z/18/Z) and a 2018 NARSAD Young Investigator Grant from the Brain & Behavior Research Foundation (Ref: 27404).
Supplementary material
For supplementary material accompanying this paper visit http://dx.doi.org/10.1192/bjo.2020.145.
click here to view supplementary material
Declaration of interest
C.M.L. is a member of the scientific advisory board for Myriad Neuroscience. The remaining authors declare no competing interests.
ICMJE forms are in the supplementary material, available online at https://doi.org/10.1192/bjo.2020.145
References
- 1.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet 2018; 50: 668–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature 2018; 562: 203–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Visscher PM, Wray NR, Zhang Q, Sklar P, McCarthy MI, Brown MA, et al. 10 years of GWAS discovery: biology, function, and translation. Am J Hum Genet 2017; 101: 5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howard DM, Folkersen L, Coleman JRI, Adams MJ, Glanville K, Werge T, et al. Genetic stratification of depression in UK Biobank. Transl Psychiatry 2020; 10: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.CONVERGE Consortium. Sparse whole genome sequencing identifies two loci for major depressive disorder. Nature 2015; 523: 588–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Major Depressive Disorder Working Group of the Psychiatric GWAS Consortium. A mega-analysis of genome-wide association studies for major depressive disorder. Mol Psychiatry 2013; 18: 497–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hyde CL, Nagle MW, Tian C, Chen X, Paciga SA, Wendland JR, et al. Identification of 15 genetic loci associated with risk of major depression in individuals of European descent. Nat Genet 2016; 48: 1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun 2018; 9: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Howard DM, Adams MJ, Clarke T-K, Hafferty JD, Gibson J, Shirali M, et al. Genome-wide meta-analysis of depression identifies 102 independent variants and highlights the importance of the prefrontal brain regions. Nat Neurosci 2019; 22: 343–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cai N, Revez JA, Adams MJ, Andlauer TFM, Breen G, Byrne EM, et al. Minimal phenotyping yields genome-wide association signals of low specificity for major depression. Nat Genet 2020; 45: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kessler RC, Andrews G, Mroczek D, Ustun B, Wittchen HU. The world health organization composite international diagnostic interview short-form (CIDI-SF). Int J Methods Psychiatr Res 2006; 7: 171–85. [Google Scholar]
- 12.Smith DJ, Nicholl BI, Cullen B, Martin D, Ul-Haq Z, Evans J, et al. Prevalence and characteristics of probable major depression and bipolar disorder within UK biobank: cross-sectional study of 172,751 participants. PLoS One 2013; 8: e75362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Health Organization. International Classification of Diseases (10th edn). World Health Organization, 1992. [Google Scholar]
- 14.Schork A, Hougaard D, Nordentoft M, Mors O, Boerglum A, Mortensen PB, et al. Exploring contributors to variability in estimates of SNP-heritability and genetic correlations from the iPSYCH case-cohort and published meta-studies of major psychiatric disorders. bioRxiv [Preprint] 2019. Available from: 10.1101/487116 . [DOI] [Google Scholar]
- 15.Davis KAS, Coleman JRI, Adams M, Allen N, Breen G, Cullen B, et al. Mental health in UK Biobank – development, implementation and results from an online questionnaire completed by 157 366 participants: a reanalysis. BJPsych Open 2020; 6: e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (5th edn) (DSM-5). APA, 2013. [Google Scholar]
- 17.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. GigaScience 2015; 4: 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010; 26: 2867–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi SW. GreedyRelated Project. GitHub Repository, 2020. (https://gitlab.com/choishingwan/GreedyRelated).
- 20.Abraham G, Qiu Y, Inouye M. FlashPCA2: principal component analysis of Biobank-scale genotype datasets. Bioinformatics 2017; 33: 2776–8. [DOI] [PubMed] [Google Scholar]
- 21.The Haplotype Reference Consortium. A reference panel of 64,976 haplotypes for genotype imputation. Nat Genet 2016; 48: 1279–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.UK10 K Consortium, Walter K, Min JL, Huang J, Crooks L, Memari Y, et al. The UK10 K project identifies rare variants in health and disease. Nature 2015; 526: 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, 2008. (http://www.R-project.org). [Google Scholar]
- 24.Choi SW, O'Reilly PF. PRSice-2: polygenic risk score software for biobank-scale data. - PubMed - NCBI. GigaScience 2019; 8: 2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Robin X, Turck N, Hainard A, Tiberti N, Lisacek F, Sanchez J-C, et al. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics 2011; 12: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe K, Taskesen E, van Bochoven A, Posthuma D. Functional mapping and annotation of genetic associations with FUMA. Nat Commun 2017; 8: D1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bulik-Sullivan BK, Loh P-R, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics Consortium, et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47: 291–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh P-R, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015; 47: 1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Goddard ME, Wray NR, Visscher PM. A better coefficient of determination for genetic profile analysis. Genet Epidemiol 2012; 36: 214–24. [DOI] [PubMed] [Google Scholar]
- 30.Kroenke K, Spitzer RL. The PHQ-9: a new depression and diagnostic severity measure. Psychiatr Ann 2002; 32: 509–21. [Google Scholar]
- 31.Weissbrod O, Flint J, Rosset S. Estimating SNP-based heritability and genetic correlation in case-control studies directly and with summary statistics. Am J Hum Genet 2018; 103: 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Evans LM, Tahmasbi R, Vrieze SI, Abecasis GR, Das S, Gazal S, et al. Comparison of methods that use whole genome data to estimate the heritability and genetic architecture of complex traits. Nat Genet 2018; 50: 737–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of UK Biobank participants with those of the general population. Am J Epidemiol 2017; 186: 1026–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Batty GD, Gale C, Kivimaki M, Deary I, Bell S. Generalisability of results from UK biobank: comparison with a pooling of 18 cohort studies. medRxiv [Preprint] 2019. Available from: 10.1101/19004705. [DOI] [Google Scholar]
- 35.Adams MJ, Hill WD, Howard DM, Dashti HS, Davis KAS, Campbell A, et al. Factors associated with sharing e-mail information and mental health survey participation in large population cohorts. Int J Epidemiol 2019; 49: 410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duncan L, Shen H, Gelaye B, Meijsen J, Ressler K, Feldman M, et al. Analysis of polygenic risk score usage and performance in diverse human populations. Nat Commun 2019; 10: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
For supplementary material accompanying this paper visit http://dx.doi.org/10.1192/bjo.2020.145.
click here to view supplementary material
Data Availability Statement
Available from UK Biobank subject to standard procedures (www.ukbiobank.ac.uk). The full GWAS summary statistics for the 23andMe discovery data-set will be made available through 23andMe to qualified researchers under an agreement with 23andMe that protects the privacy of the 23andMe participants. Please visit https://research.23andme.com/collaborate/#publication for more information and to apply to access the data.