

Abstract
In aqueous solution, biological decarboxylation reactions proceed irreversibly to completion, whereas the reverse carboxylation processes are typically powered by the hydrolysis of ATP. The exchange of the carboxylate of ring-substituted arylacetates with isotope-labeled CO2 in polar aprotic solvents reported recently suggests a dramatic change in the partition of reaction pathways. Yet, there is little experimental data pertinent to the kinetic barriers for protonation, and thermodynamic data on CO2 capture by the carbanions of decarboxylation reactions. Employing a combined quantum mechanical and molecular mechanical simulation approach, we investigated the decarboxylation reactions of a series of organic carboxylate compounds in aqueous and in dimethylformamide solutions, revealing that the reverse carboxylation barriers in solution are fully induced by solvent effects. A linear Bell-Evans-Polanyi relationship was found between the rates of decarboxylation and the Gibbs energies of reaction, indicating diminishing recombination barriers in DMF. In contrast, protonation of the carbanions by the DMF solvent has large free-energy barriers, rendering the competing exchange of isotope-labeled CO2 reversible in DMF. The finding of an intricate interplay of carbanion stability and solute-solvent interaction in decarboxylation and carboxylation could be useful to designing novel materials for CO2 capture.
Graphical Abstract
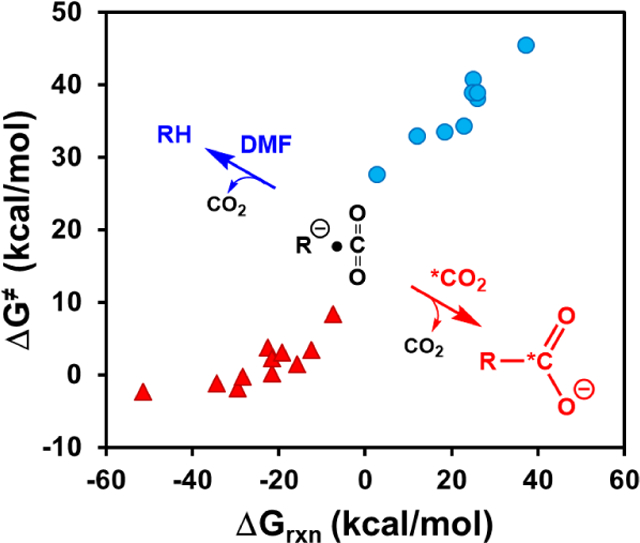
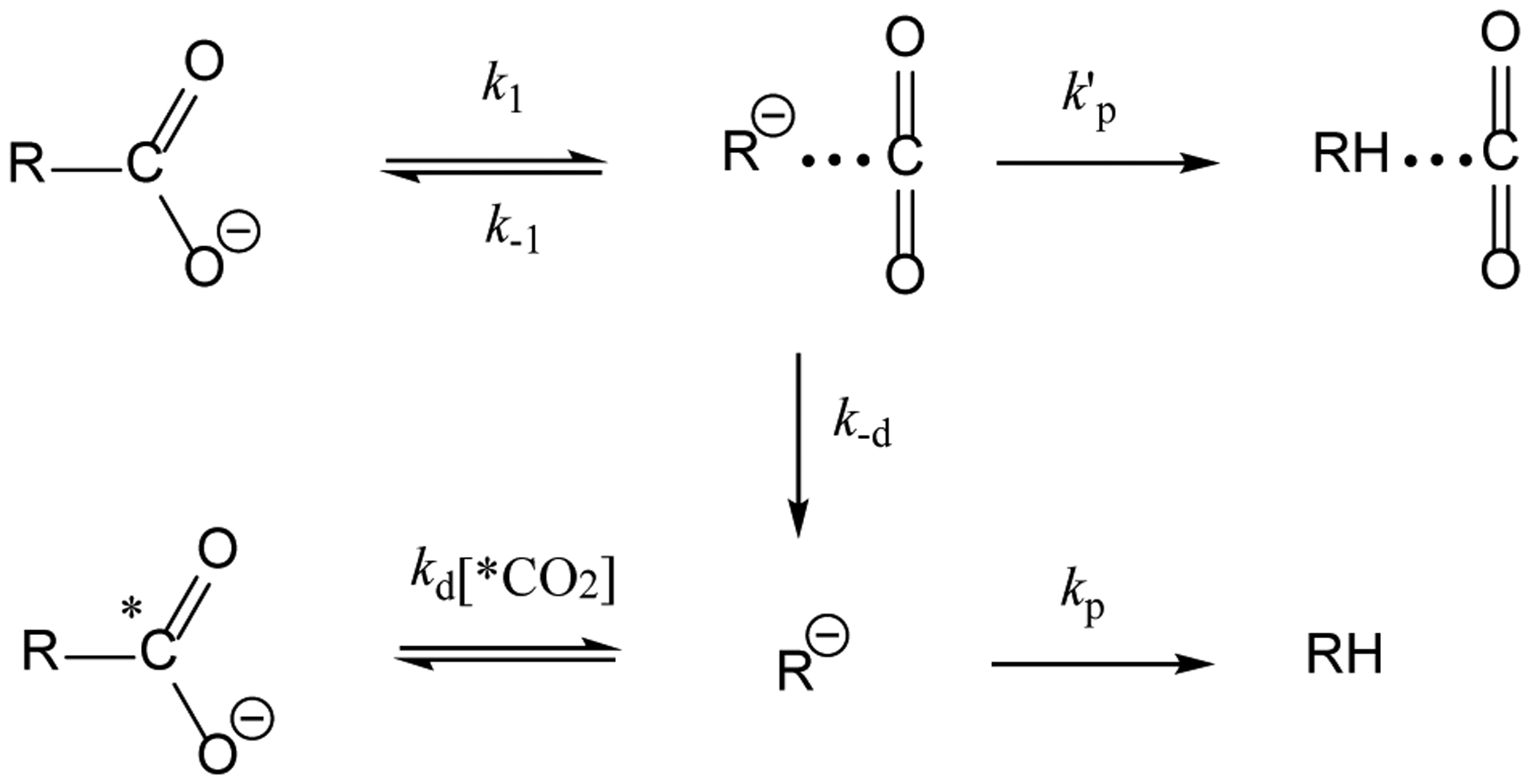
In aqueous solution, enzymatic decarboxylations, such as that of orotidine monophosphate (OMP) catalyzed by OMP decarboxylase, proceed effectively irreversibly to 100% completion,1, 2 while biosynthetic carboxylation reactions are coupled to thermodynamically favorable processes, particularly the hydrolysis of ATP. In Nature, the equilibrium of decarboxylation and carboxylation is established through CO2 release by respiring organisms and CO2 fixation in photosynthesis on a mammoth scale of billions of tons per year.3 Although the change from water to aprotic solvents can accelerate decarboxylation reactions,4 we were intrigued by reports5, 6 that exchange between the carboxylate of ring-substituted arylacetates (R-CO2−, Scheme 1) and labeled 13CO2 or 14CO2 in polar aprotic solvents dimethylformamide (DMF) or dimethylsulfoxide (DMSO) is substantially faster than their decarboxylation to form substituted toluenes (RH).5–11 These reaction conditions dramatically change the course of decarboxylation. Scheme 1 shows partitioning of the carbanion intermediate R−…CO2 of decarboxylation of R−…CO2 among recombination to form the reactant (k−1), protonation to form a ring-substituted toluene (k′p), and diffusional separation (k−d) to form a free carbanion, which then partitions between protonation (kp) and addition of isotope-labelled *CO2. Other competing patyways include a nucleophilic addition to the solvent DMF and a disproportionation process of arylacetates;5, 6 they have high reaction barriers (SI) and are not further discussed in this study. The observation of exchange of *CO2 requires a large kinetic barrier to protonation of ring substituted benzyl carbanions, compared to the barrier for addition of CO2: kd[*CO2]>> k′p, kp. However, there is little experimental data pertinent to the kinetic barriers for protonation of ring-substituted benzyl carbanions by DMF or DMSO,12–14 and no data for addition of these carbanions to CO2. We report here the results of a combined quantum mechanical and molecular mechanical (QM/MM) simulation study to determine the relative barriers to partitioning a series of carbanions between addition to CO2 and protonation by DMF.
Scheme 1.
We carried out molecular dynamics simulations to determine the potentials of mean force (PMF) along the CO2 dissociation coordinate for a set of 11 organic acids in water and in DMF, employing a dual-level QM/MM potential15, 16 to yield results at a quality of density functional theory with the M06-2X/aug-cc-pVTZ functional.17 The PMF for six decarboxylation reactions in DMF and in aqueous solution are depicted in Figure 1, along with the free energy profiles in the gas phase for comparison. The remaining data and details are given in the SI. The experimental free-energy barriers are obtained from rate constants mostly extrapolated to ambient temperature,18 whereas free energies of reaction are taken from the analysis by Guthrie.18,19 For the nine reactions where experimental data are available, the root-mean-square-errors (RMSE) between experiment and computation are 4.4 and 6.1 kcal/mol, respectively, for the barrier height and reaction free energy (Table S1), and the mean-signed-errors (MSE) are smaller (0.3 and 1.2 kcal/mol) than unsigned errors and RMSE. The agreement is good in view of the large span of rates and free energies. Quantitative rate constants for decarboxylation reactions in DMF are scarce,20 but in view of the good accord for the aqueous data and linear free energy relationships discussed below, the results demonstrate that the present simulations are adequate to provide insights on the origin of solvent effects on these decarboxylation reactions.
Figure 1.
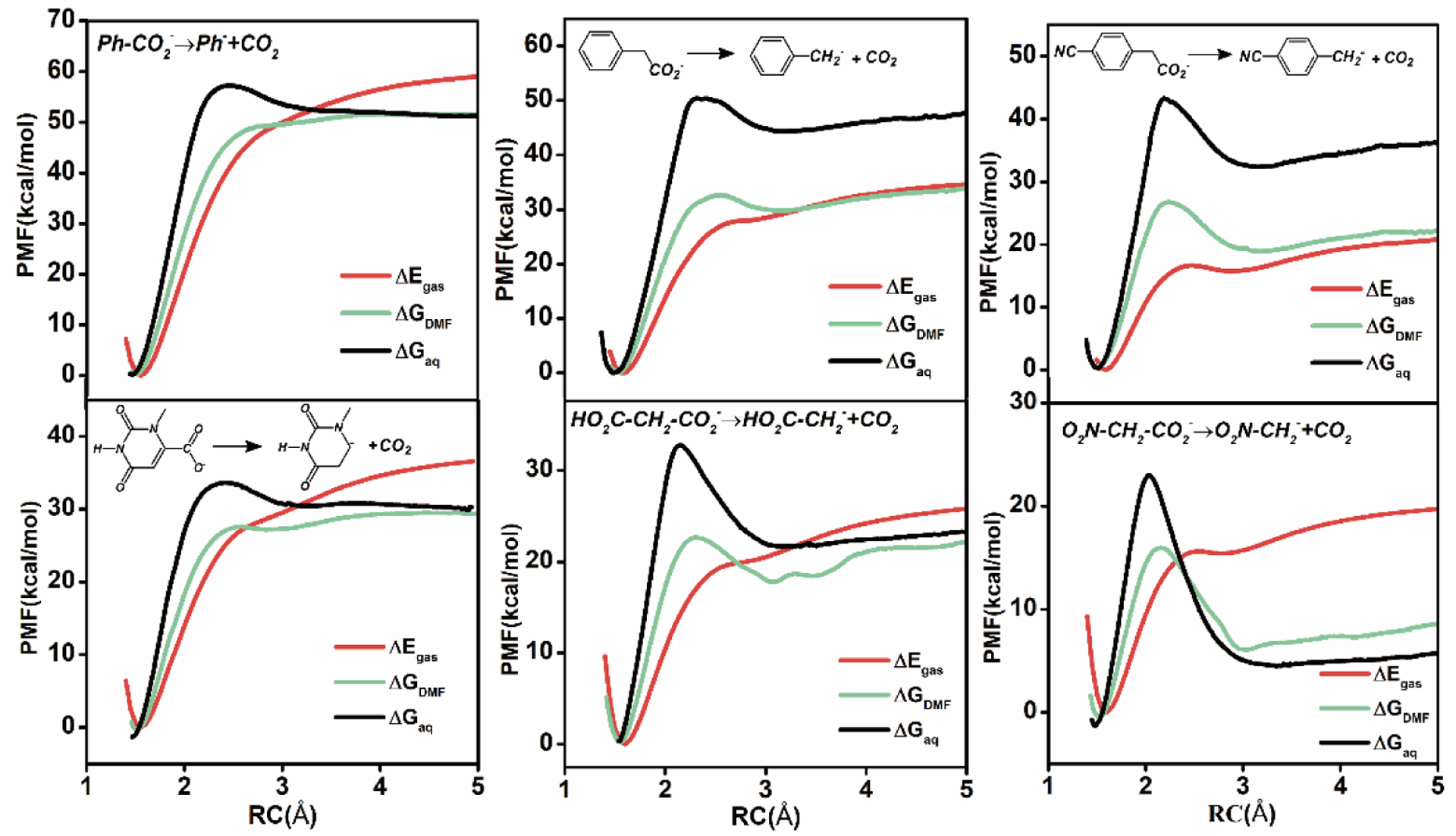
Computed potential of mean force for decarboxylation reactions in the gas phase (red), aqueous solution (black), and dimethylformamide (green) along the distance coordinate between the carbon atom of the carboxylate and the carbanion site.
First, decarboxylation reactions in water typically have high free energy barriers accompanied by strong solvation effects,4, 20–23 occurring at elevated temperatures. This is due to greater reactant stabilization by aqueous solvation relative to that at the decarboxylation transition state.22, 23 This effect is especially dramatic in the reverse, carboxylation process, and clearly depicted in Figures 1 and S2. In the gas phase, there is no or little recombination barrier for CO2 capture, but in contrast, aqueous solvation induces substantial free-energy barriers to CO2 recombination in all decarboxylation reactions. For nitroacetate ion, the recombination barrier is as large as 17 kcal/mol in water, reflecting an unusual Brønsted relationship between the rate and equilibrium constants for deprotonation, called nitroalkane anomaly.24, 25 Both deprotonation and decarboxylation reactions yield the same carbanion intermediate, with slow re-hybridization of the charge localized carbanion.26 It is remarkable that this phenomenon is carried over to the decarboxylation reaction. In protic solvents, protonation of the transient carbanion intermediate R−…CO2 to form RH (Scheme 1) renders decarboxylation reactions effectively irreversible. Consequently, in aqueous solution, the partition of R−…CO2 in Scheme 1 favors formation of RH because of the weak carbon acidity of the decarboxylation product. In DMF, the free energy barriers to CO2 recombination are much smaller than those in water (Figure 1). For several reactions, the overall free energy barriers of the recombination process are below the product state of decarboxylation or without a barrier at all. Overall, Figure 1 reveals that solvent reorganization, created by desolvation as the reacting species approach each other to form a chemical bond, induces CO2 recombination barriers, and the solvation-induced barriers are substantially smaller in DMF than in aqueous solution.
Secondly, we have examined the partition between CO2 recombination and protonation of the carbanions of decarboxylation by proton abstraction from DMF. For the latter calculations, we employed M06-2X/aug-cc-pVTZ along with the Minnesota solvation model.27 The method was validated by comparison between the computed gas-phase basicities and the experimental data,28 with an RMSE of 2.0 kcal/mol. The thermodynamic driving forces and free energy barriers for the proton transfer and CO2 recombination reactions are listed in Table 1. Except phenyl anion, all carbanions have smaller gas-phase basicity than DMF (385.9 kcal/mol) and exhibit positive free energies of reaction. On the other hand, the carboxylation reactions are substantially favorable both in the gas phase and in solution. Furthermore, proton abstraction from DMF by the carbanions of decarboxylation have large free-energy barriers, typically greater than 30 kcal/mol, in sharp contrast to the small barriers for CO2 recapture in DMF (Table 1). Therefore, the partition (Scheme 1) is strongly favored to the carboxylation process over protonation to form toluene derivatives in DMF.
Table 1.
Calculated and experimental gas-phase basicities, and calculated free energies of reaction and barriers for the proton abstraction by the carbanion of decarboxylation from dimethylformamide, and for CO2 recombination reactions in dimethyformamide. Geometries are optimized using M06-2X/aug-cc-pVTZ with which continuum solvation model is used for the proton transfer. All energies are given in kcal/mol.
| Carbanion | Gas-phase-basicity | Proton-transfer | CO2-recombination | |||
|---|---|---|---|---|---|---|
| ΔGcal | ΔGexp | |||||
| C6H5− | 391.8 | 392.4 | 2.6 | 27.7 | −51.5 | −2.3 |
| C6H5CH2− | 372.6 | 373.8 | 11.9 | 33.0 | −34.4 | −1.2 |
| p-cyanobenzyl ion | 349.6 | 353.3 | 24.9 | 40.8 | −22.6 | 3.7 |
| MUAa | 356.8 | - | 18.4 | 33.6 | −29.5 | −1.9 |
| F3C− | 369.0 | 370.2 | 22.7 | 34.4 | −28.4 | −0.3 |
| NCCH2− | 362.7 | 365.2 | 25.8 | 38.1 | −21.5 | 0.2 |
| HO2CCH2− | 361.3 | 361.1 | 24.7 | 39.0 | −21.6 | 2.2 |
| CH3COCH2− | 360.6 | 361.9 | 25.8 | 38.9 | −19.3 | 3.1 |
| Cl3C− | 348.9 | 349.9 | 30.9 | 34.2c | −12.5 | 3.4 |
| OIDb | 354.4 | - | 30.9 | 34.9c | −15.7 | 1.5 |
| O2NCH2− | 346.3 | 349.7 | 37.1 | 50.4c | −7.4 | 8.3 |
N-methyl-6-uracyl anion;
1-Oxoimidazolidinyl anion;
transition structures were not fully optimized.
The rate of decarboxylation reactions in water are known to correlate with the pKa values of the conjugate acids of the anions of decarboxylation through Brønsted plots at 25 °C.29 Interestingly, Shock and coworkers showed that a Brønsted plot of decarboxylation at 300 °C is also correlated with the pKa at 25 °C,30 suggesting that the use of rate extrapolation to ambient temperature is reasonable.29 Here, we found that the decarboxylation rates both in water and in DMF fall in a linear Bell-Evans-Polanyi plot. Figure 2 shows that the agreement between experiments and computation for the decarboxylation reactions in water is excellent, yielding a slope of 0.71 and 0.76, respectively. The intercept in the experimental correlation (16.7 kcal/mol) is 1.6 kcal/mol greater than that of computational data (15.0 kcal/mol). The Bell-Evans-Polanyi relationship for the decarboxylation reactions in DMF solution gives a slightly increased slope (0.79) and markedly reduced intercept (6.6 kcal/mol). In physical organic chemistry, the slope of a Bell-Evans-Polanyi plot correlates with the transition state along the reaction pathway.31 As there is no free energy barrier for the carboxylation process in the gas phase, this limiting case implies a slope of unity and zero intercept. The small increase in slope in DMF relative to that in water is significant, indicating further advancement of the transition state in an aprotic solvent. The smaller intercept value in DMF solution than aqueous solution corresponds to weak solvent reorganization and, thus, smaller solvation-induced free-energy barrier. Indeed, the average solvent-induced recombination barrier height is only 1.5 kcal/mol above the decomposition products, whereas it is 8.8 (9.6) kcal/mol in water from computation (experiments).
Figure 2.
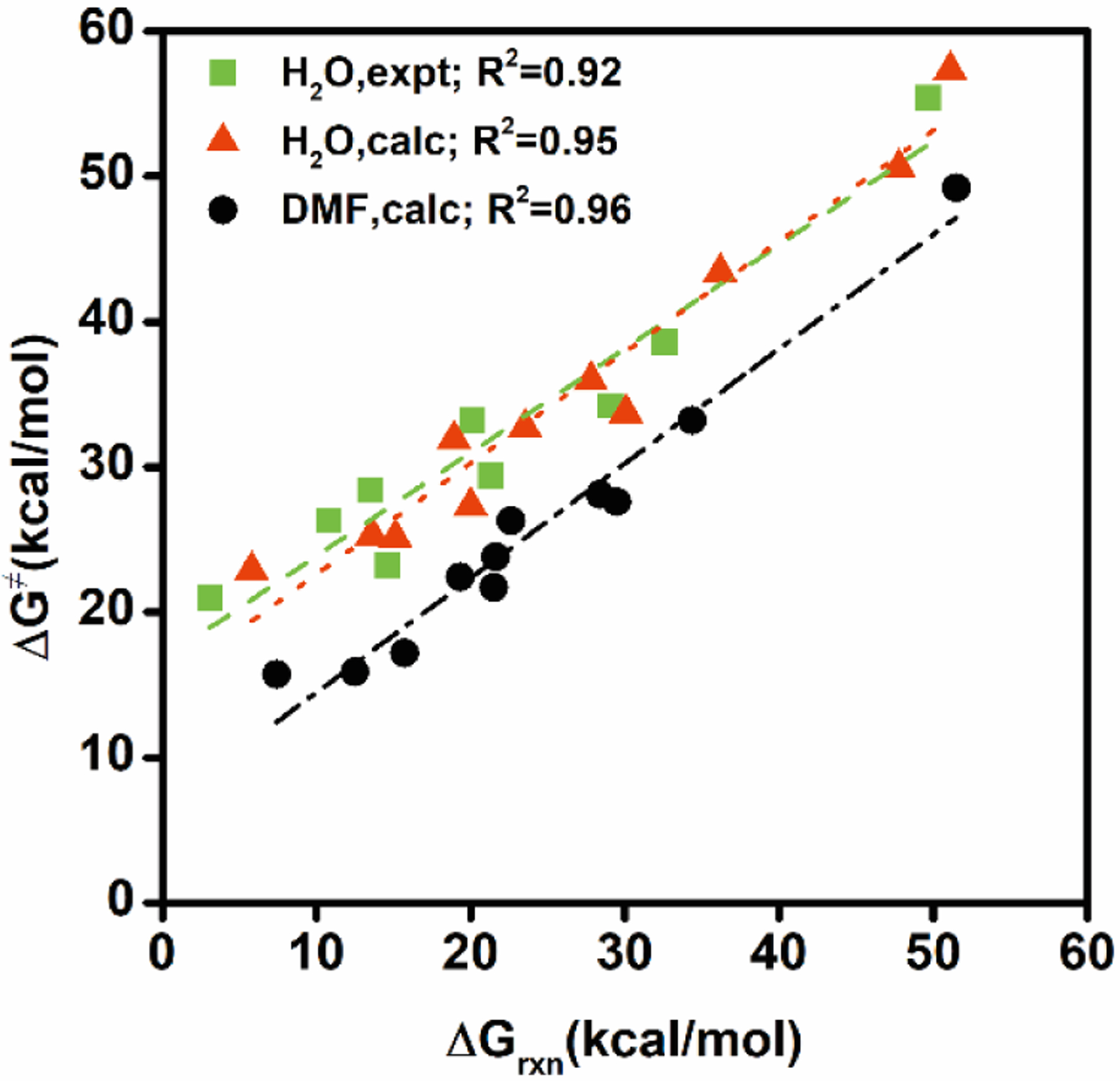
Bell-Evans-Polanyi plots of computed and experimental Gibbs energies of activation for decarboxylation reactions in water and in dimethylformamide solutions versus the corresponding free energies of reactions.
Finally, the average recombination barriers in aqueous and DMF solutions are not without trends. Figure 3 depicts the computed barriers for CO2 capture by the anionic nucleophiles of decarboxylation against the free energies of reaction. Interestingly, greater barriers are found to accompany smaller overall endergonicity in both solvents with similar slopes (−0.21 and −0.24 in DMF and in water). In dry DMF, a solvent favoring CO2 capture, recombination barriers are absent when decarboxylation reactions become highly endergonic (Figure 3). In other words, the free energy cost for a spontaneous decarboxylation reaction is largely dictated by the intrinsic, i.e., gas-phase, free energy of decarboxylation. Nevertheless, the Bell-Evans-Polanyi plot does not show direct correlation of the rates of decarboxylation or CO2 recombination with the Gibbs energy of reaction in the gas phase. It is the combined effects of the intrinsic stability of the carbanions and intermolecular interactions that lead to a highly correlated linear regression (R2 ≥ 0.95).
Figure 3.
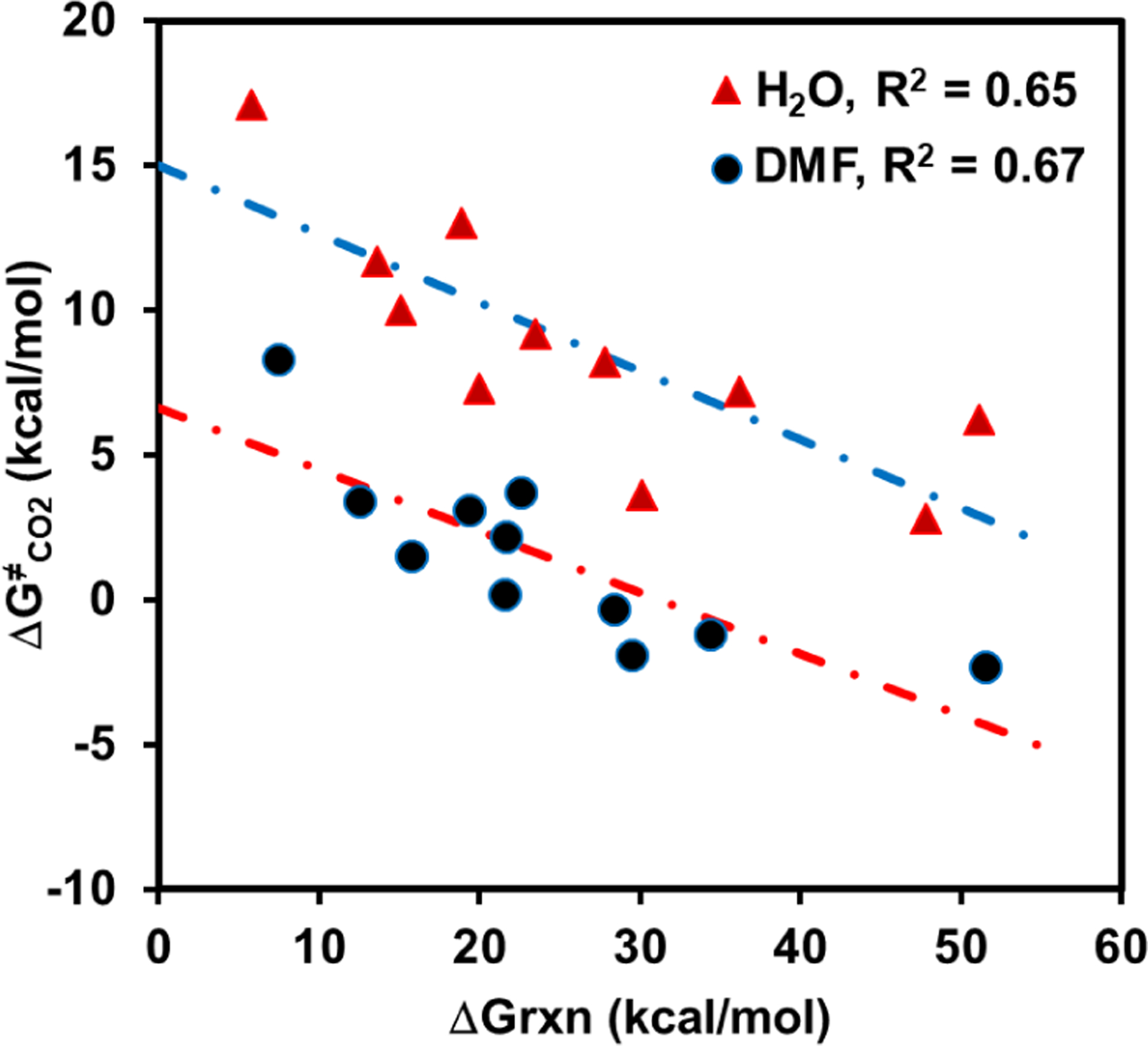
Computed free energy barriers for the reverse processes of decarboxylation reactions in aqueous and in dimethylformamide solutions plotted against the corresponding free energies of reactions.
The reversibility of decarboxylation and carboxylation was reported in the synthesis of α-nitro acids in methanol under basic and saturating CO2 conditions.32 Falvey and coworkers,8, 9 and others,33, 34 showed that decarboxylation of zwitterionic imidazolium-2-carboxylate is reversible by 13CO2 exchange in aprotic solvents, but protonation of the decarboxylation product in solvent mixtures containing a proton source inhibits the recovery of the carboxylate starting material. These N-heterocyclic carbene materials have been used for a variety of applications, including organic synthesis and CO2-transfer agents.35 Wolfenden and coworkers found a modest rate enhancement by transferring N-methyl orotate from water to DMF,20 in accord with the present results. Mayr and coworkers investigated a range of nucleophilic addition of carbanions to heteroallenes.36 Recently, a number of groups reported reversible exchange reactions of spontaneous decarboxylation and carboxylation of organic acids.5–11 Decarboxylation reactions are inherently slow, typically carried out at high temperatures.30 The Bell-Evans-Polanyi relationship in Figure 2 reveals that a balanced act of both carbanion stabilization to increase the overall decarboxylation rate and solvation effects to facilitate deprotonation of the conjugate acid of the decarboxylation anion may be considered in designing materials for CO2 capture, storage and release.34, 37 An organic solvent favors CO2 capture, but it would be of interest to balance carbanion stability and solvation effects to carry out reversible decarboxylation and CO2 capture in the presence of water.
In summary, the present study reveals that the barrier for CO2 capture corresponding to the reverse decarboxylation reaction is fully induced by solvent effects, especially in aqueous solution. The rates of decarboxylation in water and in DMF fall linearly to the Bell-Evans-Polanyi relationship, revealing diminishing recombination free-energy barriers in DMF relative to aqueous solution. For CO2 capture, the recombination reaction has an average solvent-induced free energy barrier of nearly 10 kcal/mol in water, and less than 2 kcal/mol in DMF. In contrast, protonation of the carbanions by the DMF solvent has large free-energy barriers, typically greater than 30 kcal/mol, rendering the competing diffusion and exchange of isotope-labeled CO2 reversible in DMF. Therefore, in DMF, the partition of the carbanions in Scheme 1 favors recombination with a CO2 molecule.
Supplementary Material
ACKNOWLEDGMENT
The computations were performed at the Supercomputing Center of Shenzhen Bay Laboratory, and completed at the Minnesota Supercomputing Institute.
Funding Sources
This work was partially supported by Shenzhen Municipal Science and Technology Innovation Commission (KQTD2017-0330155106581) and part of the study was completed at Minnesota, which is supported by the National Institutes of Health (GM046736). The work at Buffalo has been supported by the NIH (GM134881).
ABBREVIATIONS
- DL
dual level
- DMF
dimethylformamide
- DMSO
dimethyl sulfoxide
- MSE
mean signed errors
- OMP
orotidine monophosphate
- PMF
potential of mean force
- QM/MM
quantum mechanical and molecular mechanical
- RMSE
root-mean-square errors
- SI
supporting information
Footnotes
Supporting Information
A summary of computational methods, reaction schemes, computed potentials of mean force and solvation free energies for all reactions and optimized stationary and transition state structures (24 pages, PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Richard JP; Amyes TL; Reyes AC, Orotidine 5 ’-Monophosphate Decarboxylase: Probing the Limits of the & IT; Possible & IT; for Enzyme Catalysis. Acc. Chem. Res 2018, 51, 960–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Radzicka A; Wolfenden R, A proficient enzyme. Science 1995, 267, 90–3. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CT, Biologically generated carbon dioxide: nature’s versatile chemical strategies for carboxy lyases. Nat. Prod. Rep 2020, 37, 100–135. [DOI] [PubMed] [Google Scholar]
- 4.Kemp DS; Paul K, Decarboxylation of Benzisoxazole-3-Carboxylic Acids - Catalysis by Extraction of Possible Relevance to Problem of Enzymatic Mechanism. J. Am. Chem. Soc 1970, 92, 2553–4. [Google Scholar]
- 5.Kong DY; Moon PJ; Lui EKJ; Bsharat O; Lundgren RJ, Direct reversible decarboxylation from stable organic acids in dimethylformamide solution. Science 2020, 369, 557. [DOI] [PubMed] [Google Scholar]
- 6.Destro G; Horkka K; Loreau O; Buisson DA; Kingston L; Del Vecchio A; Schou M; Elmore CS; Taran F; Cantat T; Audisio D, Transition-Metal-Free Carbon Isotope Exchange of Phenyl Acetic Acids. Angew. Chem. Int. Ed 2020, 59, 13490–13495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kluger R, Decarboxylation, CO2 and the Reversion Problem. Acc. Chem. Res 2015, 48, 2843–2849. [DOI] [PubMed] [Google Scholar]
- 8.Denning DM; Falvey DE, Solvent-Dependent Decarboxylation of 1,3-Dimethylimdazolium-2-Carboxylate. J. Org. Chem 2014, 79, 4293–4299. [DOI] [PubMed] [Google Scholar]
- 9.Denning DM; Falvey DE, Substituent and Solvent Effects on the Stability of N-Heterocyclic Carbene Complexes with CO2. J. Org. Chem 2017, 82, 1552–1557. [DOI] [PubMed] [Google Scholar]
- 10.Hinsinger K; Pieters G, The Emergence of Carbon Isotope Exchange. Angew Chem Int Edit 2019, 58, 9678–9680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Destro G; Loreau O; Marcon E; Taran F; Cantat T; Audisio D, Dynamic Carbon Isotope Exchange of Pharmaceuticals with Labeled CO2. J. Am. Chem. Soc 2019, 141, 780–784. [DOI] [PubMed] [Google Scholar]
- 12.Llauger L; Cosa G; Scaiano JC, First determination of absolute rate constants for the reaction of aroyl-substituted benzyl carbanions in water and DMSO. J. Am. Chem. Soc 2002, 124, 15308–15312. [DOI] [PubMed] [Google Scholar]
- 13.Llauger L; Miranda MA; Cosa G; Scaiano JC, Comparative study of the reactivities of substituted 3-(benzoyl)benzyl carbanions in water and in DMSO. J. Org. Chem 2004, 69, 7066–7071. [DOI] [PubMed] [Google Scholar]
- 14.Haussermann A; Rominger F; Straub BF, CO2 on a Tightrope: Stabilization, Room-Temperature Decarboxylation, and Sodium-Induced Carboxylate Migration. Chem-Eur J 2012, 18, 14174–14185. [DOI] [PubMed] [Google Scholar]
- 15.Gao J, Hybrid Quantum Mechanical/Molecular Mechanical Simulations: An Alternative Avenue to Solvent Effects in Organic Chemistry. Acc. Chem. Res 1996, 29, 298–305. [Google Scholar]
- 16.Gao J, Enzymatic Kinetic Isotope Effects from Path-Integral Free Energy Perturbation Theory. Methods Enzymol 2016, 577, 359–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao Y; Truhlar DG, M06 DFT functionals. Theor. Chem. Acc 2008, 120, 215. [Google Scholar]
- 18.Guthrie JP; Peiris S; Simkin M; Wang Y, Rate constants for decarboxylation reactions calculated using no barrier theory. Can. J. Chem 2010, 88, 79–98. [Google Scholar]
- 19.Gao D; Pan Y-K, A QM/MM Monte Carlo Simulation Study of Solvent Effects on the Decarboxylation Reaction of N-Carboxy-2-imidazolidinone Anion in Aqueous Solution. J. Org. Chem 1999, 64, 1151–1159. [Google Scholar]
- 20.Lewis CA; Wolfenden R, Orotic Acid Decarboxylation in Water and Nonpolar Solvents: A Potential Role for Desolvation in the Action of OMP Decarboxylase. Biochemistry 2009, 48, 8738–8745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crosby J; Stone R; Lienhard GE, Mechanisms of Thiamine-Catalyzed Reactions - Decarboxylation of 2-(1-Carboxy-1-Hydroxyethyl)-3,4-Dimethylthiazolium Chloride. J. Am. Chem. Soc 1970, 92, 2891–2900. [DOI] [PubMed] [Google Scholar]
- 22.Gao J, An Automated Procedure for Simulating Chemical Reactions in Solution. Application to the Decarboxylation of 3-Carboxybenzisoxazole in Water. J. Am. Chem. Soc 1995, 117, 8600–7. [Google Scholar]
- 23.Lin Y.-l.; Gao J, Kinetic Isotope Effects of L-Dopa Decarboxylase. J. Am. Chem. Soc 2011, 133, 4398–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kresge AJ, Nitroalkane anomaly. Can. J. Chem 1974, 52, 1897–903. [Google Scholar]
- 25.Bernasconi CF, The principle of nonperfect synchronization: more than a qualitative concept? Acc. Chem. Res 1992, 25, 9–16. [Google Scholar]
- 26.Major DT; York DM; Gao J, Solvent Polarization and Kinetic Isotope Effects in Nitroethane Deprotonation and Implications to the Nitroalkane Oxidase Reaction. J. Am. Chem. Soc 2005, 127, 16374–16375. [DOI] [PubMed] [Google Scholar]
- 27.Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Petersson GA; Nakatsuji H; Li X; Caricato M; Marenich AV; Bloino J; Janesko BG; Gomperts R; Mennucci B; Hratchian HP; Ortiz JV; Izmaylov AF; Sonnenberg JL; Williams; Ding F; Lipparini F; Egidi F; Goings J; Peng B; Petrone A; Henderson T; Ranasinghe D; Zakrzewski VG; Gao J; Rega N; Zheng G; Liang W; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Throssell K; Montgomery JA Jr.; Peralta JE; Ogliaro F; Bearpark MJ; Heyd JJ; Brothers EN; Kudin KN; Staroverov VN; Keith TA; Kobayashi R; Normand J; Raghavachari K; Rendell AP; Burant JC; Iyengar SS; Tomasi J; Cossi M; Millam JM; Klene M; Adamo C; Cammi R; Ochterski JW; Martin RL; Morokuma K; Farkas O; Foresman JB; Fox DJ Gaussian 16, A.03; Wallingford, CT, 2016. [Google Scholar]
- 28.NIST NIST Chemistry WebBook. (accessed October 7).
- 29.Wolfenden R; Lewis CA; Yuan Y, Kinetic Challenges Facing Oxalate, Malonate, Acetoacetate, and Oxaloacetate Decarboxylases. J. Am. Chem. Soc 2011, 133, 5683–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Glein CR; Gould IR; Lorance ED; Hartnett HE; Shock EL, Mechanisms of decarboxylation of phenylacetic acids and their sodium salts in water at high temperature and pressure. Geochim. Cosmochim. Acta 2020, 269, 597–621. [Google Scholar]
- 31.Ritchie CD, Physical Organic Chemistry: The Fundmental Concepts. 2nd. ed.; CRC Press.: New York, 1989; p 376. [Google Scholar]
- 32.Finkbeiner HL; Stiles M, Chelation as a Driving Force in Organic Reactions .4 Synthesis of Alpha-Nitro Acids by Control of Carboxylation-Decarboxylation Equilibrium. J. Am. Chem. Soc 1963, 85, 616–22. [Google Scholar]
- 33.Voutchkova AM; Feliz M; Clot E; Eisenstein O; Crabtree RH, Imidazolium carboxylates as versatile and selective N-heterocyclic carbene transfer agents: Synthesis, mechanism, and applications. J. Am. Chem. Soc 2007, 129, 12834–12846. [DOI] [PubMed] [Google Scholar]
- 34.Fevre M; Pinaud J; Leteneur A; Gnanou Y; Vignolle J; Taton D; Miqueu K; Sotiropoulos JM, Imidazol(in)ium Hydrogen Carbonates as a Genuine Source of N-Heterocyclic Carbenes (NHCs): Applications to the Facile Preparation of NHC Metal Complexes and to NHC-Organocatalyzed Molecular and Macromolecular Syntheses. J. Am. Chem. Soc 2012, 134, 6776–6784. [DOI] [PubMed] [Google Scholar]
- 35.Kayaki Y; Yamamoto M; Ikariya T, N-Heterocyclic Carbenes as Efficient Organocatalysts for CO2 Fixation Reactions. Angew. Chem. Int. Edit 2009, 48, 4194–4197. [DOI] [PubMed] [Google Scholar]
- 36.Li Z; Mayer RJ; Ofial AR; Mayr H, From Carbodiimides to Carbon Dioxide: Quantification of the Electrophilic Reactivities of Heteroallenes. J. Am. Chem. Soc 2020, 142, 8383–8402. [DOI] [PubMed] [Google Scholar]
- 37.Long YD; Fang Z, Hydrothermal conversion of glycerol to chemicals and hydrogen: review and perspective. Biofuel Bioprod Bior 2012, 6, 686–702. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.