

Abstract
Background
Fenretinide is a synthetic retinoid that can induce cytotoxicity by several mechanisms. Achieving effective systemic exposure with oral formulations has been challenging. An intravenous lipid emulsion fenretinide formulation was developed to overcome this barrier. We conducted a study to establish the maximum tolerated dose (MTD), preliminary efficacy, and pharmacokinetics of intravenous lipid emulsion fenretinide in patients with advanced solid tumors.
Methods
Twenty-three patients with advanced solid tumors refractory to standard treatments received fenretinide as a continuous infusion for 5 consecutive days in 21-day cycles. Five different dose cohorts were evaluated between doses of 905mg/m2 and 1414mg/m2 per day using a 3+3 dose escalation design. A priming dose of 600 mg/m2 on day 1 was introduced in an attempt to address the asymptomatic serum triglyceride elevations related to the lipid emulsion.
Results
The treatment-related adverse events occurring in ≥ 20% of patients were anemia, hypertriglyceridemia, fatigue, aspartate aminotransferase (AST) / alanine aminotransferase (ALT) increase, thrombocytopenia, bilirubin increase, and dry skin. Five evaluable patients had stable disease as best response, and no patients had objective responses. Plasma steady state concentrations of the active metabolite were significantly higher than with previous capsule formulations.
Conclusion
Fenretinide emulsion intravenous infusion had a manageable safety profile and achieved higher plasma steady state concentrations of the active metabolite compared to previous capsule formulations. Single agent activity was minimal but combinatorial approaches are under evaluation.
Keywords: Retinoid, Ceramide, Phase I, Lipid emulsion
Background
The synthetic retinoid, N-(4-hydroxyphenyl) retinamide (fenretinide, 4-HPR), is cytotoxic to cell lines of multiple cancer types in vitro. Fenretinide is reported to induce cytotoxicity by multiple mechanisms, including p53- and caspase-independent apoptosis and/or non-apoptotic mechanisms independent of classic retinoid receptors.[1, 2] Induction of apoptosis by fenretinide coincides with induction of TGF-β and fenretinide cytotoxicity is associated with c-Jun N-terminal kinase (JNK) activation. Reactive oxygen species (ROS) contributing to fenretinide cytotoxicity were demonstrated in HL-60 myeloid leukemia and cervical squamous cell carcinoma cell lines in association with mitochondrial membrane permeability transition.[3–7] Fenretinide has also been shown to cause large increases of cytotoxic dihydroceramides in vitro in a time- and dose-dependent manner.[8–10]
Previously, an oral corn oil-containing capsule formulation intended for chemoprevention was tested at 200 – 900 mg/day resulting in 1 – 3 μmol/L plasma levels with minimal toxicity but also limited evidence of activity. [11, 12] Phase I and II high-dose trials of the oral capsule (500 – 4800 mg/m2/day) demonstrated a favorable safety profile and some suggestions of activity but resulted in 4-HPR steady state trough concentrations between 7.25 to 12.5 μmol/L. [13, 14] An improved oral formulation designed for use in pediatrics achieved greater bioavailability and achieved higher plasma levels in children with neuroblastoma and increased activity including four complete responses in the phase I trial.[15, 16] Additionally, there was significant interpatient variability with oral dosing. Thus, while fenretinide has potentially novel mechanisms of anticancer activity, its minimal aqueous solubility presented a significant challenge to achieving adequate systemic exposure which is in turn required to optimize anti-cancer activity. In order to overcome this barrier, an intravenous fenretinide formulation was developed using a lipid emulsion composed of a mixture of egg phospholipids, glycerin, alcohol, and soybean oil which resulted in significantly higher mean 4-HPR plasma concentrations in beagle dogs.[17]
We hypothesized that intravenous fenretinide delivery would be tolerable and result in higher and more consistent plasma drug level. The objectives of the present study were to determine the maximum tolerated dose in patients with solid tumors of an oil-in-water fenretinide emulsion administered as a continuous intravenous infusion, describe toxicities, evaluate fenretinide pharmacokinetics and, determine preliminary estimates of antitumor activity.
Patients and Methods
Drug Sources and Formulation
N-(4-hydroxyphenyl)retinamide (fenretinide, 4-HPR; NSC 374551) formulated as a 20% soy oil-in-water emulsion was provided by the Rapid Access to Intervention Development (RAID) Program, Developmental Therapeutics Program (DTP), National Cancer Institute (NCI). Fenretinide, N-(4-methoxyphenyl)retinamide (4-MPR), and N-(4-ethoxyphenyl)retinamide (4-EPR), for pharmacokinetic analyses were obtained from the NCI/DTP Open Chemicals Repository, Bethesda, MD.
Patient Selection
Patients aged ≥ 18 years were included if they had a histologically or cytologically confirmed solid tumor that was metastatic or unresectable, and for which standard therapies do not exist or were no longer effective. Other eligibility criteria included measurable disease by RECIST 1.0 criteria, Eastern Cooperative Oncology Group score of 0, 1, or 2, adequate organ function including hepatic (total bilirubin ≤ 1.5 × institutional upper limit of normal (ULN), AST/ALT ≤ 2.5 × ULN or ≤ 5 × ULN for patients with liver metastases), hematologic (absolute neutrophil count ≥ 1500/mcL, platelets ≥ 75,000/mcL) and renal (creatinine clearance ≥ 60mL/min/1.73m2). Exclusion criteria included patients with allergy to egg or history of allergic reactions to compounds of similar chemical or biologic composition to fenretinide, presence of untreated brain metastases, HIV infection, known hypertriglyceridemia requiring medication, poorly controlled diabetes mellitus with fasting serum glucose > 200 mg/dl or hemoglobin A1C over 7.5%. The study was approved by local investigational review boards in accordance with an assurance approved by the U.S. Department of Health and Human Services and registered at www.clinicaltrials.gov as NCT00387504. Written informed consents from each participant were obtained.
Study Treatment and Design
This was a phase I study designed to define the safety profile, maximum tolerated dose (MTD), pharmacokinetic (PK) parameters and initial anti-tumor activity of fenretinide in patients with advanced solid tumors. In parallel, there was an ongoing phase I study of fenretinide in patients with hematologic malignancies conducted by our group in the California Cancer Consortium.[18] Findings from the hematologic malignancies trial were used to guide dose schedule and treatment in the solid tumor trial as noted below. Patients were enrolled from the University of Southern California Norris Comprehensive Cancer Center, Los Angeles, California; University of California at Davis Comprehensive Cancer Center, Sacramento, California, and the UMC/Southwest Cancer Treatment Center/South Plains Oncology Consortium, Texas at Tech University Health Sciences Center, Lubbock, Texas.
Patients received fenretinide as a continuous intravenous (CIV) infusion for 5 consecutive days (D1 through D5) in 21-day cycles. Sequential dose-escalation in a standard 3 + 3 design was used.[19] The initial starting dose was chosen at 1280 mg/m2/day based on the expansion dose in the ongoing phase I trial in hematologic malignancies. However, after the first patient enrolled in the solid tumor trial experienced a dose-limiting toxicity, and based on evolving data in hematologic malignancies, the dose escalation schema was modified.[18] The initial starting dose level 1was decreased to 905 mg/m2/day, and a dose level 0 of 724 mg/m2/day was added in case 905 mg/m2/day was not tolerated. Escalation in 25% dose increments was planned (see Table 1). Another change that was introduced as detailed below was to fix the Day 1 infusion dose at 600 mg/m2/day and then continue the dosing on Days 2–5 according to the dose escalation table. Treatment was administered on an inpatient basis.
Table 1 –
Initial and Modified Dose Escalation Schedule
| Dose Level | Fenretinide (mg/m2) IV on Day 1 | Fenretinide (mg/m2/day) IV × Days 2–5 | Number of Patients Starting Treatment | Number of Patients Evaluable for DLT | Number of Patients with DLT |
|---|---|---|---|---|---|
| Level 0 | 724 mg/m2 | 724 mg/m2/day | 0 | - | - |
| Level 1i* | 1280 mg/m2 | 1280 mg/m2/day | 1 | 1 | 1 |
| Level 1 | 905 mg/m2 | 905 mg/m2/day | 3 | 3 | 0 |
| Level 2 | 1131 mg/m2 | 1131 mg/m2/day | 13 | 4 | 1 |
| Level 2a** | 600 mg/m2 | 1131 mg/m2/day | 5 | 4 | 0 |
| Level 3a** | 600 mg/m2 | 1414 mg/m2/day | 1 | 1 | 1 |
Level 1i of 1280 mg/m2/day was the initial planned dose, but after the first patient had a DLT and based on emerging data from the hematologic malignancy study Level 1 was modified to 905 mg/m2/day.
Levels 2a and 3a were added with Day 1 dose of 600 mg/m2/day. This was done in order to decrease the occurrence of minimally symptomatic lipid intolerance.
Patients were treated until evidence of tumor progression, or for 6 cycles. Patients could be treated for greater than 6 cycles after discussion with the Study Chair based upon considerations of response, toxicity, and available drug supply.
The MTD was defined as the highest dose tested with at least six patients treated, at which < 33% of patients experienced a dose limiting toxicity (DLT). For DLT assessment, patients had to receive ≥ 90% of the planned first cycle dose and be observed for ≥ 21 days from Day 1, or experience toxicity that met the definition of DLT at any time during the first cycle. Patients not evaluable for DLT assessment were removed from the study and replaced.
Treatment was held for grade 3 or 4 toxicity. In case of grade 4 toxicity, treatment with fenretinide was discontinued. Patients who experienced a DLT (other than grade 4) or grade 3 non-hematologic toxicity that resolved to ≤ grade 1 in 7 days were allowed to be retreated with a dose reduction in the next cycle. Patients were removed from treatment if a scheduled cycle was delayed > 3 weeks due to toxicity or for recurrence of the same DLT.
DLT was defined as (a) grade 4 thrombocytopenia, (b) grade 4 neutropenia that lasted more than 7 days or febrile neutropenia, (c) any toxicity related to study drug that resulted in treatment delays of > 3 weeks (one cycle), (d) any grade 3 or 4 non-hematological toxicity related to study drug excluding: nyctalopia; ≥ grade 3 headache with history of migraines; grade 3 nausea, vomiting, or diarrhea controllable with medical management; grade 3 alanine aminotransferase (ALT) or aspartate aminotransferase (AST) increase, or grade 3 or 4 alkaline phosphatase increase, that recovered to ≤ grade 2, or baseline, by Day 21; grade 3 fever or infection with ≤ grade 2 neutropenia; grade 4 fever or infection associated with a central venous catheter or other known cause; grade 3 fatigue that resolved by Day 21. Grade 3 or 4 asymptomatic triglyceride elevation that returned to baseline within 96 hours with ≤ grade 1 lipase elevation was not considered a DLT.
Safety and Efficacy Evaluations
A history and physical examination were done at baseline and on Days 1, 8, and 15 of each cycle. During the 5-day infusion, a complete blood count (CBC) was obtained daily, and a complete metabolic panel (CMP) and serum lipase were examined every other day. CBC and CMP were examined on Days 8 and 15 of each cycle. Triglycerides were checked every 12 hours for Days 1–5. Disease evaluation with CT or MRI was done at baseline and every 6 weeks while on treatment. Tumor response was evaluated based on the Response Evaluation Criteria in Solid Tumors (RECIST) version 1.0. Toxicities were graded using NCI Common Terminology Criteria for Adverse Events (version 3.0).
Definition of Minimally Symptomatic Lipid Intolerance
In the Phase I study of fenretinide in patients with hematologic malignancies, it was noted that four out of six DLTs at doses ranging between 640 mg/m2/day and 1810 mg/m2/day were related to asymptomatic ≥ grade 3 elevation of triglycerides. This was attributed to decreased plasma clearance of the lipid-based emulsion vehicle due to insufficient serum lipase capacity to metabolize the soy oil load and was not thought to reflect clinically significant drug related toxicity. As a result, asymptomatic grade 3 or 4 hypertriglyceridemia that returned to baseline within 96 hours after stopping infusion, with ≤ grade 1 lipase elevation that returned to baseline within 72 hours, and no signs/symptoms of pancreatitis was defined as “minimally symptomatic lipid intolerance” and was excluded from the DLT definition. Patients with grade 3 or 4 hypertriglyceridemia had their infusion stopped and were re-treated at a 50% dose reduction if the hypertriglyceridemia resolved within 48 hours. With the introduction of this definition, criteria were adopted to serve as flags to indicate whether the true probability of minimally symptomatic intralipid intolerance was greater than 0.30. Crossing this threshold would be used to determine whether a dose was practical and feasible.
Pharmacokinetics
Blood samples were collected at 0, 6, 12, 18, 24, 36, 48, 72, 96, and 120 hours during the infusion, and at 2 hours, 48 hours and 72 hours after completing infusion. The plasma concentrations of 4-HPR, 4-MPR, and 4-oxo-4-HPR were assayed using LC-MS/MS methodology as previously described.[20] The plasma concentrations of individual patients were fitted with a one-compartment model with weighted least squares regression in the ADAPT 5 software (Biomedical Simulation Resource, University of Southern California, Los Angeles, CA) to estimate model parameters. The maximum blood concentration (Cmax) was determined as the highest observed concentration during the infusion, and steady-state concentration (Css) and areas under the concentration–time curves from 0 to +192 h after the infusion (AUC0–192h) were determined from estimated data using one-compartmental analysis.
Statistical Methods
All analyses of data from this study were descriptive using standard summary statistics.
Results
Patient Demographics
Twenty-three patients were enrolled and started treatment between 11/27/06 and 4/15/2013. Sixteen (70%) were male, median age was 59 years (range 22–80); 9 (39%) had ECOG status of 0. Median number of prior chemotherapy regimens was 4 (range 1–10). The two most frequent malignancies recruited included colorectal and non-small cell lung cancer. (See Table 2)
Table 2 –
Patient Characteristics
| Characteristic | No. (%) |
|---|---|
| Number of Patients Treated | 23 |
| Age, years | |
| Median | 59 |
| Range | 22 – 80 |
| Gender | |
| Male | 16 (70%) |
| Female | 7 (30%) |
| Race/Ethnicity | |
| African American | 1 (4%) |
| Asian | 1 (4%) |
| White | 15 (65%) |
| Hispanic | 4 (17%) |
| Unknown | 2 (9%) |
| ECOG Performance Status | |
| 0 | 9 (39%) |
| 1 | 14 (61%) |
| Primary Cancer | |
| Colorectal | 6 (26%) |
| Non-Small Cell Lung | 5 (22%) |
| Liver | 2 (9%) |
| Esophagus | 2 (9%) |
| Connective soft tissue | 2 (9%) |
| Other GI (appendix, pancreas) | 2 (9%) |
| Other (larynx, ovary, prostate & unknown primary) | 4 (17%) |
| Prior treatment | |
| Chemotherapy | 23 (100%) |
| Radiation Therapy | 9 (39%) |
| Surgery | 16 (70%) |
| No. prior chemotherapy regimens | |
| Median | 4 (1–10) |
| 1 | 2 (9%) |
| 2 | 4 (17%) |
| 3–4 | 8(35%) |
| 5–10 | 9(39%) |
Dose Escalation and Determination of MTD and Recommended Phase II Dose
The first patient received a fenretinide dose of 1280 mg/m2/day and experienced a DLT with grade 3 thrombocytopenia and hemorrhage. The protocol was the amended to start at the lower dose of 905 mg/m2, with dose level 0 at 724 mg/m2 added in case the 905 mg/m2 dose was not tolerable. Three patients were treated at 905 mg/m2 with no DLT. At the next dose level, 1131 mg/m2/day; one of three evaluable patients had a DLT (grade 4 thrombocytopenia and grade 3 bilirubin) which resulted in treating two additional patients at the same dose level, one of whom was evaluable and did not experience any DLT. Five other patients treated at this dose level were not evaluable for DLT. One patient was deemed ineligible based on pre-treatment laboratory values, one experienced early death unrelated to study therapy, one had a treatment deviation, one had dose interruption for unrelated toxicity, and one had dose interruption after < 50% of total dose administration for grade 3 ALT elevation. The ALT elevation was deemed possibly related, but this did not meet DLT definition because it resolved within 8 days, and the patient was not evaluable because they received less than 90% of the planned dose.
Four other patients at this dose level experienced grade 3 or 4 triglyceride elevation without associated symptoms and were not evaluable for DLT; however, this resulted in the boundary of acceptable rate of minimally-symptomatic lipid intolerance being crossed. The protocol was subsequently revised to reduce the Cycle 1 Day 1 dose to 600 mg/m2/day for all dose levels as experience from the parallel hematologic Phase 1 study suggested that some patients experienced an apparent induction of serum lipase capacity if treated with a reduced dose on Day 1 which allowed them to tolerate full dosing on subsequent days.
Five patients were treated with the revised schedule on Dose Level 2a: 600 mg/m2 on Day 1, followed by 1131 mg/m2/day on Days 2–5. One patient experienced grade 3 hypertriglyceridemia and was inevaluable due to receiving < 50% of total dose. The other 4 patients did not experience a DLT. The dose was escalated to Dose Level 3a: 600 mg/m2 on Day 1 and 1414 mg/m2/day on all other days. One patient was enrolled who experienced DLT (grade 4 elevated bilirubin and grade 3 encephalopathy) and subsequently died of disease progression. Given that the recommended phase 2 dose in the fenretinide hematologic malignancies study was 1200 mg/m2/day, dose escalation was halted, and the study trial closed due to drug supply limitations.
A total of eight evaluable patients were treated at 1131 mg/m2/day between dose levels 2 and 2a and only one experienced a DLT; as a result, this dose was considered to be tolerable.
Toxicity and Tolerability
The median number of cycles completed was 1 (range 0 – 7). Grade 3 or 4 treatment-related adverse events were seen in twelve patients (52%), but most of these were asymptomatic laboratory findings. The most common treatment related grade 3 or 4 adverse events were hypertriglyceridemia, occurring in six patients (26%) followed by bilirubin increase, ALT increase and platelet count decrease in two patients each (9%) (Table 3). The most common toxicities of any grade were anemia (52%), hypertriglyceridemia (52%), fatigue (48%), AST/ALT increase (43%), thrombocytopenia (30%), bilirubin increased (30%), and dry skin (26%). There were no cases of pancreatitis. Five patients (22%) were taken off study for toxicity.
Table 3.
Grade 3 and 4 Treatment Related Adverse Events
| Toxicity | All Cycles | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Level 1 (N=1) | Level 1a (N=3) | Level 2 (N=13) | Level 2a (N=5) | Level 3a (N=1) | ||||||
| Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | Grade 3 | Grade 4 | |
| AST / ALT Increased | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| Hyperbilirubinemia | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 |
| Encephalopathy | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Fatigue | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 |
| Hemoglobin Reduced | 1 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| Neutrophils Reduced | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| Platelets Reduced | 1 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 |
| Hypertriglyceridemia | 0 | 0 | 0 | 0 | 3 | 3 | 0 | 0 | 0 | 0 |
Four patients died on treatment or within 30 days of last study treatment. Two of these were attributed to disease progression. One patient with esophageal cancer experienced perforation of the esophageal primary leading to sepsis and death. Autopsy demonstrated extensive tumor necrosis in liver metastases and chest lymph nodes with evidence of recent capillary bed collapse, suggesting that tumor necrosis was related to treatment response. This fourth patient with metastatic prostate adenocarcinoma experienced grade 3 encephalopathy on day 5 of cycle 1 related to dural metastases and leptomeningeal carcinomatosis but this was concurrent with grade 4 elevation of bilirubin and grade 2 elevation of AST/ALT suggestive of drug-induced liver injury. The patient died on cycle 1 day 15.
Efficacy
A variety of solid tumor types were enrolled, with a total of 23 subjects. Five subjects were inevaluable for response as they were taken off therapy in Cycle 1 and did not have follow-up imaging. The best response was stable disease in 5/18 subjects (28%) and progressive disease in 13 subjects (72%). The subjects that had stable disease were one each with hepatocellular carcinoma (22 weeks), soft tissue sarcoma (13 weeks), mucinous adenocarcinoma of the appendix (22 weeks), carcinosarcoma of unknown primary (22 weeks), and squamous cell carcinoma of the larynx (11 weeks).
Pharmacokinetics
End-of-infusion plasma samples were only available for six patients that completed an adequate number of infusions with no change in dose (Table 5). Analysis showed a dose-to-plasma level relationship with median steady-state drug levels of 9.9mcg/mL at 905mg/m2/day, and approximately 10.8mcg/mL at 1131mg/m2/day (Figure 1).
Table 5.
Pharmacokinetic Parameters of Intravenous Fenretinide (4-HPR)
| 905 mg/m2/d (n=3) | 1131 mg/m2/d (n=3) | |||
|---|---|---|---|---|
| Median | Range | Median | Range | |
| V (L) | 168.5 | 74.4–255.9 | 193.1 | 122.5–420.5 |
| CL (L/h) | 6.1 | 4.3–11.0 | 8.9 | 5.2–10.2 |
| t1/2 (h) | 16.2 | 11.9–19.0 | 16.3 | 15.0–28.5 |
| AUC0–192 (μg·h/mL) | 1279 | 897–1532 | 1297 | 809–1859 |
| Cmax (μg/mL) | 12.2 | 8.1–14.5 | 12.3 | 7.8–18.8 |
| Css (μg/mL) | 10.5 | 7.2–12.1 | 10.4 | 7.0–15.0 |
Figure 1 –
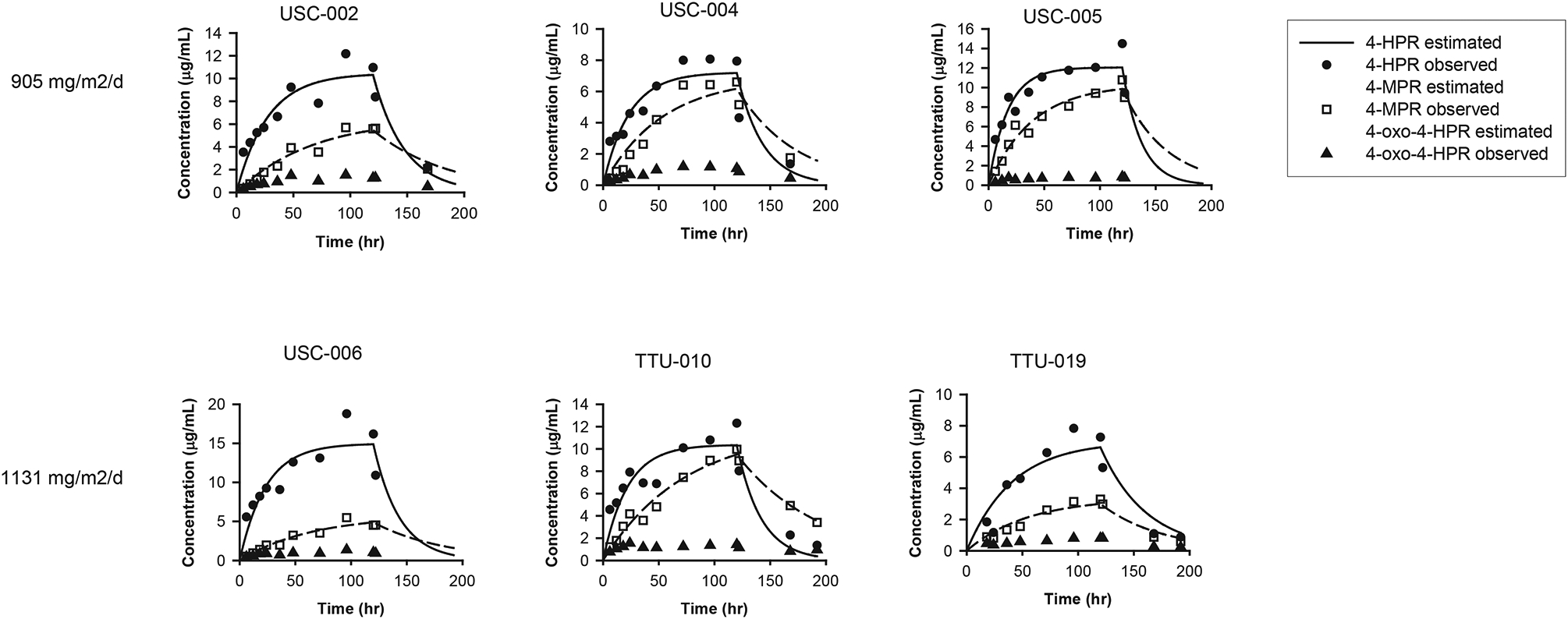
Drug and Metabolite Concentrations
Discussion
This phase I study evaluated the safety, tolerability and MTD of fenretinide monotherapy in a novel lipid formulation for IV administration. The dose escalation plan was significantly influenced by our parallel phase I trial of fenretinide in hematologic malignancies as detailed above. Furthermore, the evaluability of patients for DLT was influenced by the incidence of asymptomatic hypertriglyceridemia which we accounted for in the study design. Overall, the trial did establish the feasibility of using the fenretinide emulsion in adult subjects with solid tumors. Even though 52% of patients had grade 3 or 4 adverse events, these mostly consisted of asymptomatic laboratory abnormalities. There was one death on study with a possible attribution to study drug, but this was in the setting of advanced and heavily pretreated prostate adenocarcinoma with leptomeningeal carcinomatosis.
Fenretinide emulsion in patients with solid tumors obtained plasma steady state concentrations of 4-HPR that were significantly higher than previous capsule formulations and comparable to fenretinide emulsion in patients with hematologic malignancies.[13, 14, 18] It should be noted that the present study reports 4-HPR concentrations as μg/mL, while the capsule formulation studies referenced above report concentrations as μmol/L. The 4-HPR concentrations achieved with capsule formulations was 7.25 to 12.5 μmol/L, which is 2.8 to 4.9 μg/mL. Therefore, these data demonstrate that higher 4-HPR concentrations are achieved with the IV lipid formulation, with Css between 7.0 – 15.0 μg/mL (Table 5).
Fenretinide manifested minimal single agent activity in this diverse cohort of heavily pre-treated patients with a variety of solid tumors with no objective radiologic responses and with 28% of patients having stable disease ranging from 11 to 22 weeks. In contrast, fenretinide showed promising single-agent activity in hematological malignancies with a response rate of 75% in T-cell lymphomas.[18] A phase II trial of IV fenretinide in patients with T-cell lymphomas is ongoing (NCT02495415). Based on this, further development of fenretinide in solid tumors would benefit from either patient selection based on a predictive biomarker or from use of 4-HPR together with agents that synergistically enhance its activity in preclinical models of solid tumors, such as safingol, a microtubule inhibitor, or BCL-2 inhibitors.[15, 21–23]
The recommended Phase 2 dose (RP2D) of this formulation of fenretinide in hematological malignancies was 600 mg/m2/day on Day 1 and 1200 mg/m2/day on Days 2–5, every 21 days. In this study, we did not definitively determine a MTD but we were able to establish that schedules of 1131 mg/m2/day × 5 days, or 600 mg/m2/day on Day 1 and 1131 mg/m2/day on Days 2–5, every 21 days, appeared tolerable with one DLT out of 8 evaluable patients. Because of exhaustion of drug supply, it is unclear whether higher doses would be been tolerable in subjects with advanced solid tumors and would have resulted in higher plasma concentrations.
Several studies with this formulation of fenretinide are ongoing. A phase I study of the combination of IV fenretinide plus IV safingol, a stereochemical-variant dihydroceramide precursor is recruiting patients with all tumor types (NCT01553071).
Table 4.
All Treatment-Related Adverse Events Observed In > 10% Patients (N = 23)
| Toxicity Observed In >10% Of Patients | Any Grade N (%) | Grade 3 N (%) | Grade 4 N (%) |
|---|---|---|---|
| Hemoglobin Reduced | 12 (52%) | 2 (9%) | 0 |
| Hypertriglyceridemia | 12 (52%) | 3 (13%) | 3 (13%) |
| Fatigue | 11 (48%) | 1 (4%) | 0 |
| AST / ALT Increased | 10 (43%) | 2 (9%) | 0 |
| Hyperbilirubinemia | 7 (30%) | 1 (4%) | 1 (4%) |
| Platelets | 7 (30%) | 1 (4%) | 1 (4%) |
| Dry Skin | 6 (26%) | 0 | 0 |
| Alkaline Phosphatase | 5 (22%) | 0 | 0 |
| Burn | 5 (22%) | 0 | 0 |
| Dizziness | 5 (22%) | 0 | 0 |
| Dry Mouth | 5 (22%) | 0 | 0 |
| Flatulence | 5 (22%) | 0 | 0 |
| Nausea | 5 (22%) | 0 | 0 |
| Night Blindness | 5 (22%) | 0 | 0 |
| Ocular/Visual - Other | 5 (22%) | 0 | 0 |
| Pruritus/itching | 5 (22%) | 0 | 0 |
| Constipation | 4 (17%) | 0 | 0 |
| Creatinine | 4 (17%) | 0 | 0 |
| Pain | 4 (17%) | 0 | 0 |
| Vision-Blurred Vision | 4 (17%) | 0 | 0 |
| Vision-Flashing Lights/Floaters | 4 (17%) | 0 | 0 |
| Anorexia | 3 (13%) | 0 | 0 |
| Hypocalcemia | 3 (13%) | 0 | 0 |
| Dry Eye Syndrome | 3 (13%) | 0 | 0 |
| Dyspnea | 3 (13%) | 0 | 0 |
| Neutrophils Reduced | 3 (13%) | 1 (4%) | 0 |
Funding
The research reported was supported by the National Cancer Institute of the National Institutes of Health under Award Number UM1CA186717 and U01CA062505. Additional support was provided under National Institutes of Health awards P30CA33572, P30CA093373, and P30CA014089 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of Interest
C. P. Reynolds and B. J. Maurer reports being co-inventors on issued patents on intravenous formulations of fenretinide with financial interests through institutional intellectual property revenue sharing agreements, and are consultants to, and owns stock in, CerRx, Inc., that licenses this technology; M. H. Kang is an unpaid consultant to CerRx, Inc.
Ethics approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. The study was approved by local investigational review boards in accordance with an assurance approved by the U.S. Department of Health and Human Services and registered at www.clinicaltrials.gov as NCT00387504.
Consent to participate
Written informed consents from each participant were obtained.
Availability of data and material
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- 1.Clifford JL, et al. , Retinoid receptor-dependent and -independent effects of N-(4-hydroxyphenyl)retinamide in F9 embryonal carcinoma cells. Cancer Res, 1999. 59(1): p. 14–8. [PubMed] [Google Scholar]
- 2.Ponzoni M, et al. , Differential effects of N-(4-hydroxyphenyl)retinamide and retinoic acid on neuroblastoma cells: apoptosis versus differentiation. Cancer Res, 1995. 55(4): p. 853–61. [PubMed] [Google Scholar]
- 3.Oridate N, et al. , Involvement of reactive oxygen species in N-(4-hydroxyphenyl)retinamide-induced apoptosis in cervical carcinoma cells. J Natl Cancer Inst, 1997. 89(16): p. 1191–8. [DOI] [PubMed] [Google Scholar]
- 4.Kitareewan S, et al. , 4HPR triggers apoptosis but not differentiation in retinoid sensitive and resistant human embryonal carcinoma cells through an RARgamma independent pathway. Oncogene, 1999. 18(42): p. 5747–55. [DOI] [PubMed] [Google Scholar]
- 5.Suzuki S, et al. , Implication of mitochondria-derived reactive oxygen species, cytochrome C and caspase-3 in N-(4-hydroxyphenyl)retinamide-induced apoptosis in cervical carcinoma cells. Oncogene, 1999. 18(46): p. 6380–7. [DOI] [PubMed] [Google Scholar]
- 6.Hail N and Lotan R, Mitochondrial permeability transition is a central coordinating event in N-(4-hydroxyphenyl)retinamide-induced apoptosis. Cancer Epidemiol Biomarkers Prev, 2000. 9(12): p. 1293–301. [PubMed] [Google Scholar]
- 7.Hail N and Lotan R, Mitochondrial respiration is uniquely associated with the prooxidant and apoptotic effects of N-(4-hydroxyphenyl)retinamide. J Biol Chem, 2001. 276(49): p. 45614–21. [DOI] [PubMed] [Google Scholar]
- 8.O’Donnell PH, et al. , N-(4-hydroxyphenyl)retinamide increases ceramide and is cytotoxic to acute lymphoblastic leukemia cell lines, but not to non-malignant lymphocytes. Leukemia, 2002. 16(5): p. 902–10. [DOI] [PubMed] [Google Scholar]
- 9.Batra S, Reynolds CP, and Maurer BJ, Fenretinide cytotoxicity for Ewing’s sarcoma and primitive neuroectodermal tumor cell lines is decreased by hypoxia and synergistically enhanced by ceramide modulators. Cancer Res, 2004. 64(15): p. 5415–24. [DOI] [PubMed] [Google Scholar]
- 10.Wang H, et al. , N-(4-hydroxyphenyl)retinamide elevates ceramide in neuroblastoma cell lines by coordinate activation of serine palmitoyltransferase and ceramide synthase. Cancer Res, 2001. 61(13): p. 5102–5. [PubMed] [Google Scholar]
- 11.Costa A, et al. , Tolerability of the synthetic retinoid Fenretinide (HPR). Eur J Cancer Clin Oncol, 1989. 25(5): p. 805–8. [DOI] [PubMed] [Google Scholar]
- 12.Veronesi U, et al. , Randomized trial of fenretinide to prevent second breast malignancy in women with early breast cancer. J Natl Cancer Inst, 1999. 91(21): p. 1847–56. [DOI] [PubMed] [Google Scholar]
- 13.Jasti BR Phase I clinical trial of Fenretinide (NSC374551) in advanced solid tumors. in Proceedings of the American Society and Clinical Oncology. 2001. [Google Scholar]
- 14.Villablanca JG, et al. , Phase II study of oral capsular 4-hydroxyphenylretinamide (4-HPR/fenretinide) in pediatric patients with refractory or recurrent neuroblastoma: a report from the Children’s Oncology Group. Clin Cancer Res, 2011. 17(21): p. 6858–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Maurer BJ, et al. , Improved oral delivery of N-(4-hydroxyphenyl)retinamide with a novel LYM-X-SORB organized lipid complex. Clin Cancer Res, 2007. 13(10): p. 3079–86. [DOI] [PubMed] [Google Scholar]
- 16.Maurer BJ, et al. , Phase I trial of fenretinide delivered orally in a novel organized lipid complex in patients with relapsed/refractory neuroblastoma: a report from the New Approaches to Neuroblastoma Therapy (NANT) consortium. Pediatr Blood Cancer, 2013. 60(11): p. 1801–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X, Maurer B, and Reynolds C, Preclinical toxicology and pharmacokinetics of intravenous lipid emulsion fenretinide. 2007, AACR Meeting Abstracts. [Google Scholar]
- 18.Mohrbacher AM, et al. , Phase I Study of Fenretinide Delivered Intravenously in Patients with Relapsed or Refractory Hematologic Malignancies: A California Cancer Consortium Trial. Clin Cancer Res, 2017. 23(16): p. 4550–4555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Le Tourneau C, Lee JJ, and Siu LL, Dose escalation methods in phase I cancer clinical trials. J Natl Cancer Inst, 2009. 101(10): p. 708–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cho HE and Min HK, Analysis of fenretinide and its metabolites in human plasma by liquid chromatography-tandem mass spectrometry and its application to clinical pharmacokinetics. J Pharm Biomed Anal, 2017. 132: p. 117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maurer BJ, et al. , Synergistic cytotoxicity in solid tumor cell lines between N-(4-hydroxyphenyl)retinamide and modulators of ceramide metabolism. J Natl Cancer Inst, 2000. 92(23): p. 1897–909. [DOI] [PubMed] [Google Scholar]
- 22.Chen NE, et al. , Reactive Oxygen Species Mediates the Synergistic Activity of Fenretinide Combined with the Microtubule Inhibitor ABT-751 against Multidrug-Resistant Recurrent Neuroblastoma Xenografts. Mol Cancer Ther, 2016. 15(11): p. 2653–2664. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TH, et al. , Fenretinide via NOXA Induction, Enhanced Activity of the BCL-2 Inhibitor Venetoclax in High BCL-2-Expressing Neuroblastoma Preclinical Models. Mol Cancer Ther, 2019. 18(12): p. 2270–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]