

SUMMARY
Autophagy and phagocytosis are conserved cellular functions involved in innate immunity. However, the nature of their interactions remains unclear. We evaluated the role of autophagy in regulating phagocytosis in macrophages from myeloid-specific autophagy-related gene 7-deficient (Atg7−/−) mice. Atg7−/− macrophages exhibited higher bacterial uptake when infected with Mycobacterium tuberculosis (Mtb) or with M. tuberculosis var. bovis BCG (BCG). In addition, BCG-infected Atg7−/− mice showed increased bacterial loads and exacerbated lung inflammatory responses. Atg7−/− macrophages had increased expression of two class A scavenger receptors: macrophage receptor with collagenous structure (MARCO) and macrophage scavenger receptor 1 (MSR1). The increase in scavenger receptors was caused by increased activity of the nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) transcription factor resulting from accumulated sequestosome 1 (SQSTM1 or p62) in Atg7−/− macrophages. These insights increase our understanding of the host-pathogen relationship and suggest that therapeutic strategies should be designed to include modulation of both phagocytosis and autophagy.
INTRODUCTION
Autophagy and phagocytosis are related cellular functions. Autophagy is a process by which organelles and cellular proteins are encapsulated and then delivered to lysosomes for degradation or recycling (Mizushima and Komatsu, 2011). Furthermore, autophagy functions in diverse aspects of inflammation and immunity (Mizushima and Komatsu, 2011; Xu and Eissa, 2010). Phagocytosis is also involved in mammalian defense against infection (Stuart and Ezekowitz, 2005). In contrast to autophagy, which is involved in removal of intracellular sources, phagocytosis engulfs extracellular materials. Both pathways are stimulated by pattern recognition receptor ligands (Blander and Medzhitov, 2004; Xu et al., 2007) and have critical roles in pathogen capture and degradation, respectively (Xu and Eissa, 2010).Recent studies revealed interactions between phagosomes formed by phagocytosis and autophagosomes formed by autophagy (Gutierrez et al., 2004; Sanjuan et al., 2007; Xu et al., 2007), including a report that suggested that modulation of autophagy might affect phagocytosis (Lima et al., 2011). However, the nature of the interactions and the underlying mechanisms remain unknown.
Tuberculosis (TB) remains as one of the major threats to global public health (McMurray, 2001). Macrophages are the first line of defense against Mycobacterium tuberculosis (Mtb), being the main host cell and killing effector (Liu and Modlin, 2008). They phagocytose and sequester bacteria in phagosomes, which can then fuse with lysosomes for bacterial degradation. However, Mtb has the ability to survive and grow inside macrophages by preventing lysosomal destruction (Gutierrez et al., 2004). Autophagy stimulation can be harnessed toward enhancing macrophage defense against Mtb (Xu et al., 2007). However, there are several gaps in our knowledge, which prevent the translation of these promising studies into therapeutic strategies. It is not known whether there is interaction between the regulatory mechanisms governing the processes of phagocytosis and autophagy. In this study, we show that autophagy modulates phagocytosis by regulating the expression of class A scavenger receptors, revealing mechanisms by which accumulated SQSTM1 leads to enhanced expression of scavenger receptors. Finally, we confirm that autophagy is required for eradication of mycobacteria in vivo.
RESULTS
Atg7–/– Macrophages Are More Permissive to BCG Growth
Prior studies have shown that autophagy is critical for elimination of mycobacteria in macrophages (Castillo et al., 2012; Gutierrez et al., 2004; Jagannath et al., 2009; Watson et al., 2012; Xu et al., 2007). Execution of autophagy is mediated by a ubiquitination-like system that involves two conjugation pathways (Mizushima and Komatsu, 2011). In the first one, autophagy gene 12 (ATG12) is activated by linkage to ATG7 and then to ATG10 before becoming covalently linked to ATG5. ATG16 binding generates an ATG5-ATG12-ATG16 complex present in the autophagy isolation membrane. In the second pathway, microtubule-associated protein 1 light chain 3 alpha (LC3, also known as ATG8) is cleaved by ATG4 to leave an exposed conserved glycine residue required for autophagosome formation. Cleaved LC3 is then linked to ATG7, then to ATG3, and then to phosphatidylethanolamine. Lipidation of LC3 is one of the best markers for autophagy because modified LC3 remains associated with autophagosomes until destruction at the autolysosomal stage. Thus, both ATG5 and ATG7 are considered key autophagy proteins, although they both can play a role in other cellular functions besides degradative autophagy, such as LC3-associated phagocytosis (LAP) and restriction of viral replication (Hwang et al., 2012; Zhao et al., 2008). During LAP, LC3 is recruited to single-membrane phagosomes in response to infectious agents, requiring both Atg5 and Atg7 (Lerena and Colombo, 2011; Sanjuan et al., 2007).
We have generated myeloid-specific Atg7-deficient mice. We crossed mice bearing Atg7flox/flox alleles with LysM-Cre transgenic mice, which express Cre recombinase in a myeloid-specific manner. We then compared Atg7flox/flox LysM-Cre-positive mice, henceforth referred to as Atg7−/− mice, to Atg7flox/flox littermates, i.e., Atg7+/+. Consistent with this conditional deletion model, bone-marrow-derived macrophages (BMDMs) from Atg7−/− mice exhibited reduction of ATG7, inhibition of LC3II formation, and increase in SQSTM1, a known autophagy substrate (Figure 1A; Bjørkøy et al., 2005). We infected BMDMs from Atg7−/− and Atg7+/+ mice with BCG and estimated bacterial colony forming units (CFU). We found that ATG7 deficiency resulted in an impaired ability of the cell to control infection, as evidenced by greater numbers of viable BCG in Atg7−/− macrophages (Figure 1B). Surprisingly, we noticed that the increase of BCG in Atg7−/− cells was evident in cells lysed at the 0 time point, i.e., at the end of the 1 hr infection period. We reasoned that it would be highly unlikely that differences in bacterial killing by the autophagic-lysosomal system could account for the observed increase because of the short duration of infection. Instead, the data suggested that there might be an increased bacterial uptake by Atg7−/− macrophages.
Figure 1. Enhanced Mycobacterial Uptake and Growth in Atg7–/– Macrophages.
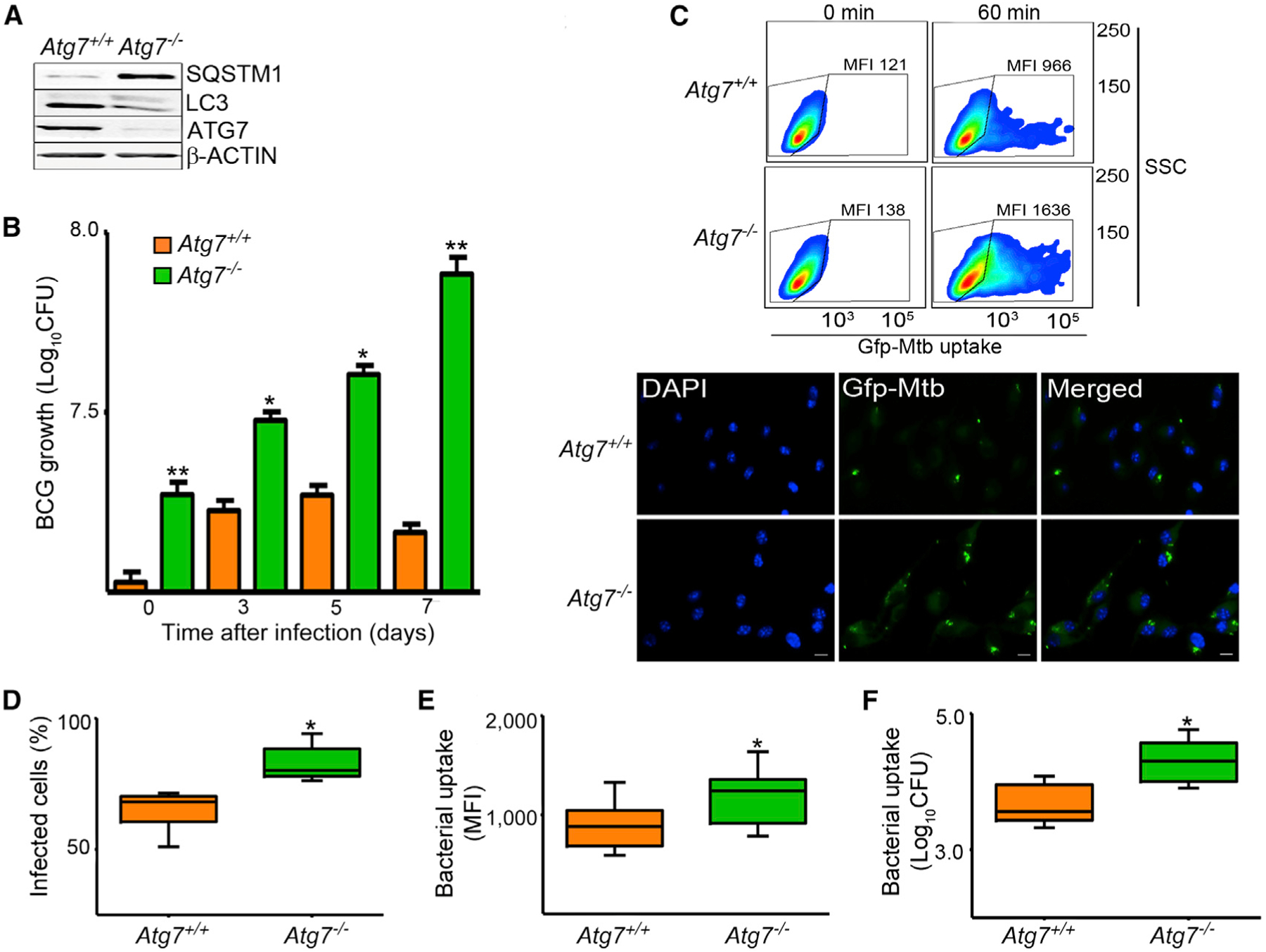
(A) Lysates of BMDMs from Atg7+/+ and Atg7−/− mice were evaluated by immunoblot analysis.(B) BMDMs were infected for 1 hr with BCG at an MOI of 10. Bacterial counts were estimated at 0, 3`, 5, and 7 days by the CFU assay.(C) BMDMs were infected with GFP-expressing H37Rv strain of Mtb (MOI 50) for 60 min. Phagocytic activity was measured by flow cytometry. Representative side scatter (SSC) versus GFP-Mtb dot plots with the corresponding mean fluorescence intensity (MFI) are shown. Mtb uptake was also measured by fluorescence microscopy at 1 hr after infection. DAPI was used to counterstain the nuclei. Scale bars represent 5 μm. Bacterial uptake was evaluated at the end of the 1 hr infection period by determining percent of infected cells (percentage of macrophages containing at least one bacterium). Representative images are shown.(D–F) BMDMs were infected with GFP-expressing Mtb (MOI 50). Bacterial uptake was quantified by fluorescence microscopy (D), flow cytometry (E), and CFU assay (F).
Data are mean ± SD, n = 3; *p < 0.05, **p < 0.001. See also Figure S1.
Atg7–/– Macrophages Showed Enhanced BCG and Mtb Uptake
To confirm that the higher bacterial CFU counts observed in Atg7−/− macrophages at the end of the 1 hr infection period were caused by changes in phagocytic activity, we infected BMDMs with Mtb or BCG and evaluated bacterial uptake. To minimize any potential role for autophagic degradation in these assays, we evaluated uptake at 30 and 60 min of infection. Uptake of Mtb (Figure 1) or BCG (Figures S1A–S1D available online) was substantially higher in Atg7−/− macrophages. The differences in BCG uptake between the Atg7+/+ and Atg7−/− macro phages were abolished when phagocytosis inhibitors (4°C incubation and 10 μM cytochalasin D treatment) were used, suggesting that the observed differences were due to increased BCG internalization (Figure S1E). Finally, we evaluated BCG phagocytosis in vivo. Atg7+/+ and Atg7−/− mice were infected intraperitoneally with 5 × 105 BCG for 2 hr and then macrophages were recovered by peritoneal lavage and evaluated for BCG internalization. Results showed that Atg7−/− mice had a higher percentage of BCG-infected macrophages compared to those from Atg7+/+ mice (Figure S1F). These results confirm that the increased phagocytosis in Atg7−/− mice is operative in vivo and is not an in-vitro-only phenomenon.
Atg7–/– Mice Exhibited Increased Susceptibilityto Mycobacterial Infection
It has been recently shown that autophagy reduces mycobacterial growth in macrophages (Gutierrez et al., 2004; Jagannath et al., 2009; Xu et al., 2007) and in Atg5 genetically deficient mice (Castillo et al., 2012; Watson et al., 2012). We studied the role of autophagy in mycobacterial elimination in vivo in our myeloid-specific Atg7−/− mice. Mice were inoculated with 1 × 106 BCG intranasally and bacteriological and histopathological changes were evaluated in lungs. Atg7−/− mice were more susceptible to the infection, as evidenced by higher bacterial counts and exacerbated inflammatory responses. We determined the extent and cellular composition of the inflammatory infiltrate in hematoxilin and eosin-stained lung sections. One month after infection, Atg7−/− mice had more pronounced inflammatory responses, with scattered accumulations of lymphocytes and macrophages and perivascular and peribronchiolar cellular infiltrates (Figures 2A and S2A). Quantification of inflammation (Figure 2B) and morphometric analysis (Figure 2C) showed that BCG-infected Atg7−/− mice had exacerbated inflammation. Similar findings were seen in mice infected for 2 or 4 months (Figures S2B–S2D). Consistent with enhanced inflammatory responses to BCG infection in Atg7−/− mice, lungs showed increased cellular counts. Analysis of T cells, helper T cells, cytotoxic T cells, macrophages, granulocytes, and B cells in lungs or spleens (Figures S2E and S2F) revealed proportional increase in all populations (Figures 2D and 2E). To evaluate bacterial dissemination in the lungs, the number of bacteria was determined by CFU assay and by acid fast staining. With CFU assays, we observed substantially increased bacterial counts in lungs from Atg7−/− mice at 1 month after infection (Figure 2G) and this difference continues to increase at 4 months after infection (Figure S2B). Ziehl-Nieelsen-stained lung sections revealed that infected Atg7−/− mice had increased numbers of bacilli per field as well (Figure 2F). Taken together, these results indicate that mice with ATG7 deficiency in myeloid cells exhibited increased inflammation and bacterial colonization when challenged with BCG.
Figure 2. Increased Susceptibility to Mycobacterial Infection in Atg7–/– Mice In Vivo.
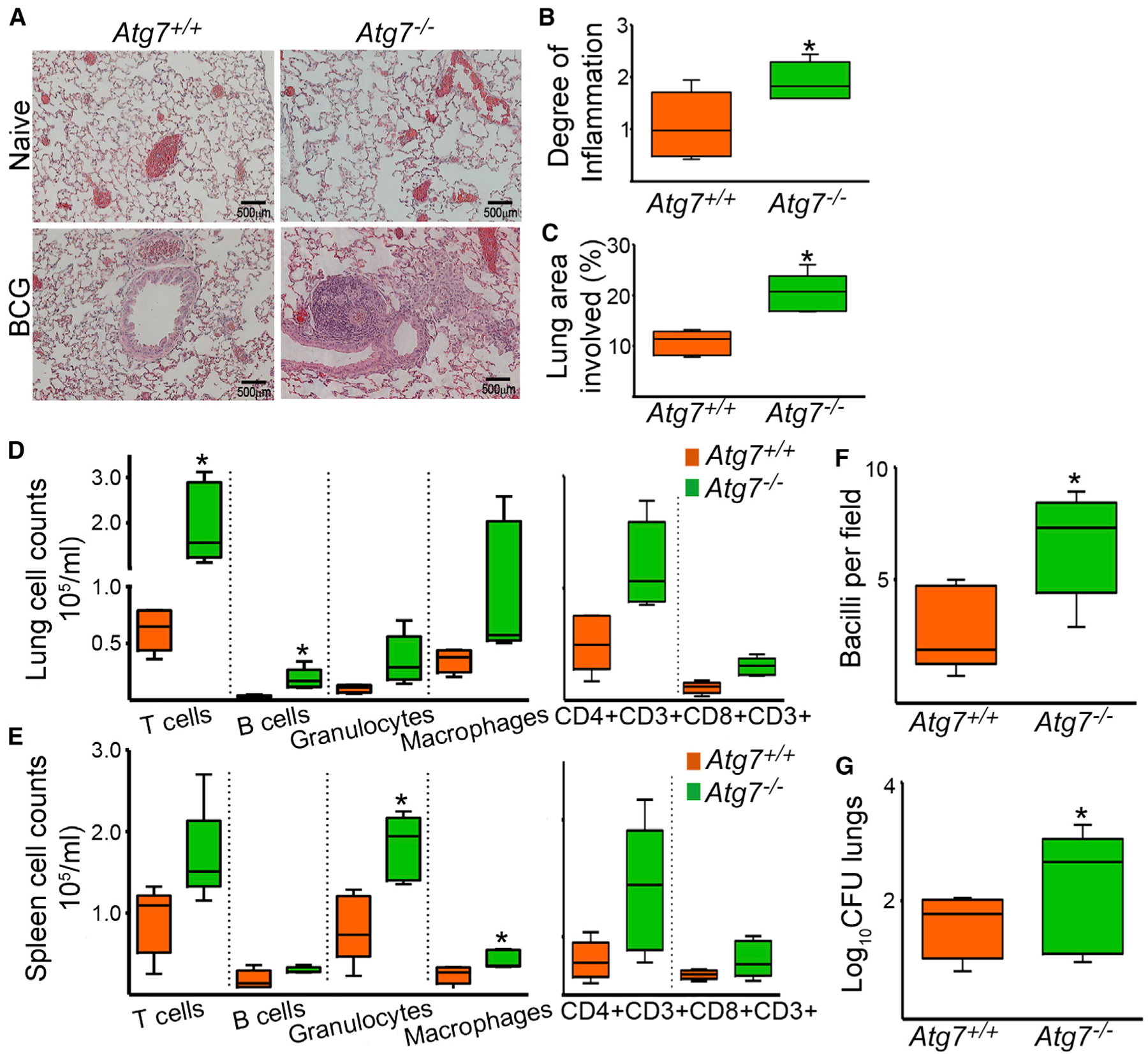
Atg7+/+ and Atg7−/− mice were infected with 1 × 106 BCG intranasally and bacteriological and histopathological changes were evaluated 1 month after infection.(A) Representative lung sections from naive and BCG-infected Atg7−/− and Atg7+/+ mice are shown.(B and C) Degree of lung inflammation was estimated (B), and the percentage of lung involved in the inflammatory response was determined by morphometric analysis (C).(D and E) The cellular composition of the inflammatory infiltrate in lungs (D) and spleens (E) was evaluated by flow cytometry, with conjugated antibodies against CD3+ (T cells), CD4+CD3+ (helper T cells), CD8+CD3+ (cytotoxic T cells), F4/80 (macrophages), Lys6c (granulocytes), or B220 (B cells).(F and G) Number of BCG bacilli in the lung was determined by counting Ziehl-Neelsen-stained bacilli per field of lung section (F) and the number of viable bacteria was estimated by the CFU assay (G). Data expressed as Log10 CFU.
Data are mean ± SD. *p < 0.05. Scale bars represent 500 μm. See also Figure S2.
Atg7–/– Macrophages Displayed Enhanced Surface Expression of the Class A Scavenger Receptors MARCO and MSR1
To elucidate the underlying mechanisms for enhanced bacterial uptake in Atg7−/− macrophages, we first examined whether ATG7 deficiency modulates phagocytic receptors involved in mycobacterial internalization. Mycobacteria are internalized through a broad array of phagocytic receptors (Ernst, 1998). We initially evaluated the surface expression of 22 different receptors in Atg7−/− and Atg7+/+ BMDMs at baseline (Table S1). Of these receptors, only MARCO was substantially increased in Atg7−/− macrophages (Figures 3A and 3B). These data prompted us to investigate other scavenger receptors. We found that Atg7−/− BMDMs had increase in surface expression of another class A scavenger receptor, namely MSR1 (Figures 3A–3D). No differences in other scavenger receptors were observed (Table S1).
Figure 3. Increased Surface Expression of Class A Scavenger Receptors MARCO and MSR1 in Atg7–/– Macrophages.
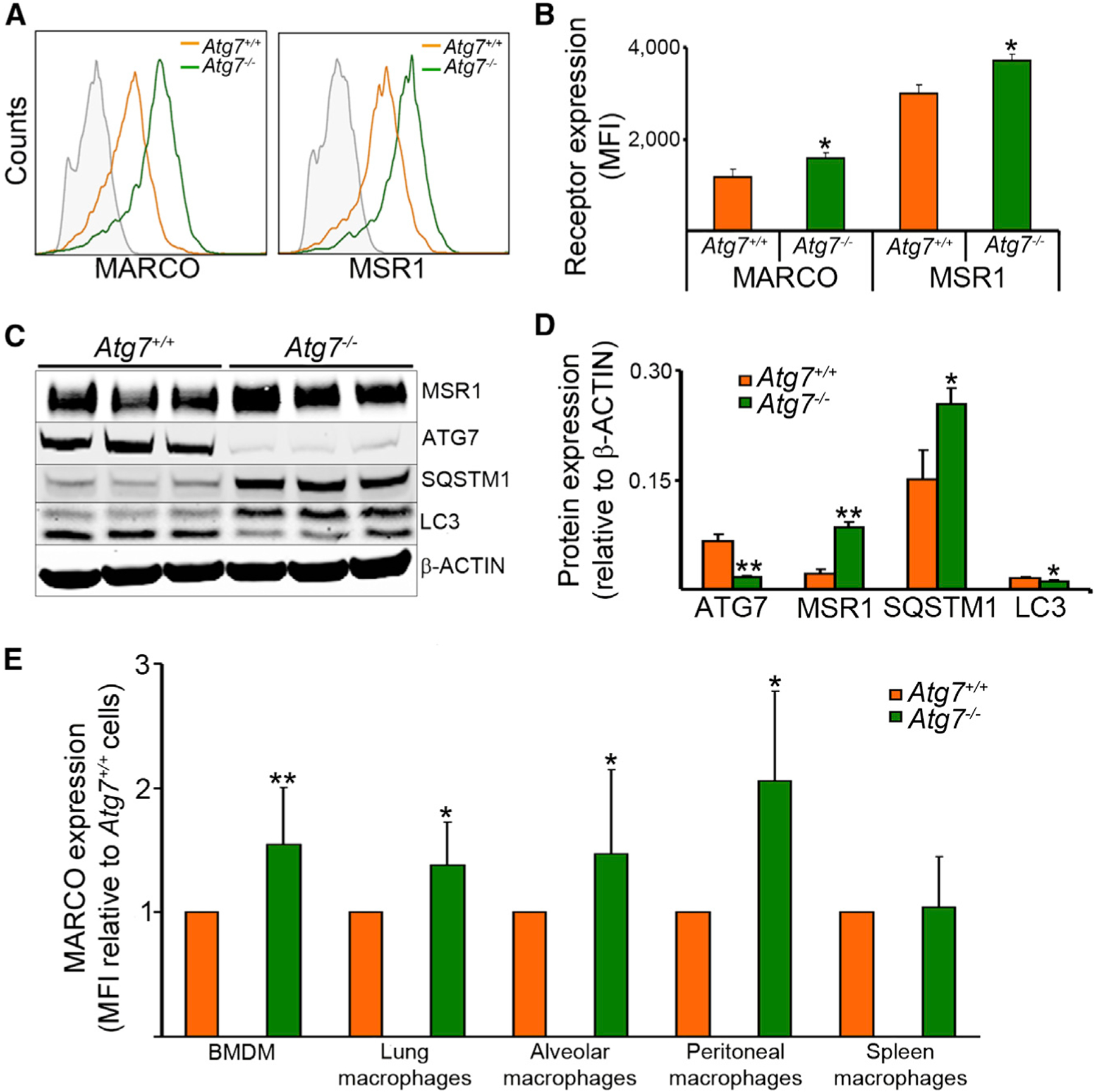
Receptor expression in BMDMs from Atg7+/+ and Atg7−/− mice was measured by flow cytometry. MSR1 expression was measured by immunoblot and flow cytometry with a goat polyclonal antibody from R&D Systems. An Alexa Fluor 594 donkey anti-goat antibody was used as secondary antibody for flow cytometry.(A and B) Representative histogram (A) and quantitative data (B) of surface expression of MARCO and SR-A are shown.(C and D) Representative blot (C) and quantitative analysis (D) of MSR1 expression. Antibodies to ATG7, SQSTM1, LC3, and ACTIN were used to confirm the phenotype. Immunoblot data are expressed relative to intensity of the ACTIN band and flow cytometry data are expressed as MFI.(E) MARCO surface expression was evaluated in different macrophage populations.
Data are expressed as fold increase in Atg7−/− relative to that of Atg7+/+ cells. Data are shown as mean ± SD (n = 6); *p < 0.05, **p < 0.001. See also Figure S3 and Table S1.
We then investigated whether MARCO surface expression was increased in Atg7−/− macrophages obtained from different body sites. In Atg7−/− mice, the surface expression of MARCO was enhanced in macrophages from other non-bone-marrow sources, including lung, brochoalveolar lavage, and peritoneal cavity, whereas substantial differences in spleen could not be detected (Figure 3E). Our data suggest that Cre expression is reduced in spleen macrophages and thus autophagy targeting is less efficient in these cells (Figure S3). The data above suggested that the receptor increase in Atg7−/− macrophages might be responsible for the increased BCG uptake. Prior studies have linked MARCO to mycobacterial uptake. MARCO interacts with the mycobacterial glicolipid trehalose dimycolate to modulate cell activation (Bowdish et al., 2009). The ability of MARCO to bind mycobacteria suggests an important role for MARCO in the macrophage response to this pathogen. We confirmed that MARCO and MSR1 were involved in BCG uptake. The phagocytic activity was partially reduced when MARCO or MSR1 were blocked by using a neutralizing antibody E31 against MARCO or one of two class A scavenger receptor antagonists: fucoidan or poly(I). (Figure S4A). These data support the assertion that MARCO and MSR1 are among the receptors involved in mycobacterial internalization by macrophages. It should be noted, however, that the receptor diversity for mycobacteria, accompanied by receptor cooperation, makes it difficult to dissect the role of these individual receptors during phagocytosis. Taken together, these results indicate that macrophages deficient in ATG7 displayed higher expression of MARCO and MSR1, which correlated with their increased phagocytic capacity.
Lack of Atg3, Atg5, or Atg7 Enhanced Scavenger Receptor Expression and Phagocytic Capacity
Mouse embryonic fibroblasts (MEFs) have been used as a cellular model to evaluate autophagy role in mycobacterial studies (Pilli et al., 2012). To confirm that the increased bacterial uptake in Atg7−/− cells was caused by autophagy deficiency, we evaluated bacterial internalization and scavenger receptor expression in MEFs obtained from Atg3−/−, Atg5−/−, or Atg7−/− mice, compared to their wild-type counterparts (Figure 4). Atg-deficient MEFs exhibited reduction of the corresponding ATG levels, inhibition of LC3II formation, and increase of SQSTM1 (Figure 4A). Furthermore, we found that Atg3−/−, Atg5−/−, and Atg7−/− MEFs had increase in total protein, surface expression and mRNA of MARCO and MSR1 (Figures 4B–4E), and increased ability to internalize BCG (Figure 4E). These data suggest that the observed changes in scavenger receptor expression and phagocytosis in Atg7−/− cells are autophagy dependent and not due to an autophagy-independent role of ATG7.
Figure 4. Increased Scavenger Receptor Expression and Bacterial Uptake in Autophagy-Deficient MEFs.
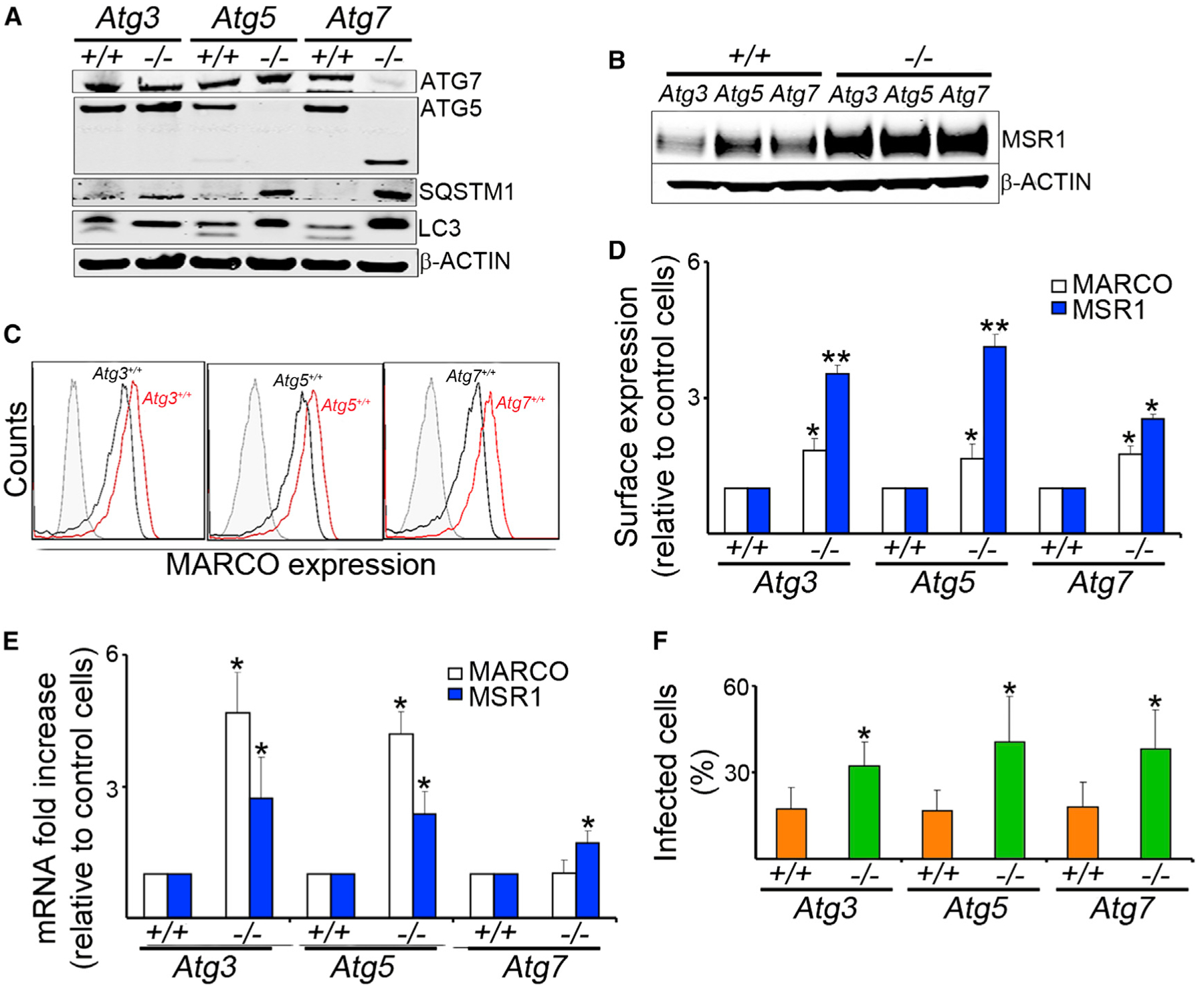
MEFs of Atg3−/−, Atg5−/−, Atg7−/−, and their wild-type controls were evaluated.(A) Antibodies to ATG7, ATG5, SQSTM1, LC3, and b-ACTIN were used to confirm the autophagy-deficient phenotypes.(B) Representative blot of MSR1 and β-ACTIN expression.(C and D) Receptor expression of MARCO and MSR1 was measured by flow cytometry. Representative histograms (C) and quantitative analyses (D) are shown.(E) Real-time PCR was done with mRNA of MEFs and specific primers for MARCO or MSR1.(F) MEFs were infected for 1 hr with GFP-BCG (MOI 10). Percentage of infected cells was determined by immunofluorescence.
Data are mean ± SD, n = 6, *p < 0.05, **p < 0.001.
Atg7–/– Macrophages Exhibited Increased Phagocytosis of Bacteria Known to Be Internalized through Class A Scavenger Receptors
Because scavenger receptors can mediate uptake of other bacteria, we sought to determine whether Atg7−/− macrophages would have increased uptake of such bacteria as well. We evaluated the uptake of S. aureus and E. coli because both are internalized through class A scavenger receptors. Phagocytosis of S. aureus and E. coli was measured by flow cytometry with pHrodo-labeled bacteria. Bacteria conjugated to pHrodo dye are nonfluorescent outside the cell or when surface bound but become bright red when they are intracellular. Atg7−/− macrophages exhibited increases E. coli and S. aureus internalization (Figure 5). In contrast, there were no differences in zymosan phagocytosis, which is known to be internalized via receptors other than scavenger receptors (Figure S4B). The uptake of latex beads remained unchanged (Figure S4B), even though these receptors have been implicated in bead internalization by alveolar macrophages (Palecanda et al., 1999; Zhou et al., 2008). These findings support the hypothesis that the observed increase in phagocytosis by Atg7−/− macrophages is due to enhanced surface expression of class A scavenger receptors.
Figure 5. Increased Phagocytosis of S. aureus and E. coli in Atg7–/– Macrophages.

BMDMs from Atg7−/− and Atg7+/+ mice were infected for 30 min or 1 hr with pHrodo-labeled S. aureus or E. coli (MOI 10) and bacterial uptake was measured by flow cytometry. Representative overlaid histograms (A) and quantitative analyses (B) are shown. Data show the MFI relative to uninfected cells (mean ± SD), n = 3; *p < 0.05, **p < 0.001. See also Figure S4.
Next, we investigated whether endocytosis was altered in Atg7−/− macrophages. We tested ligands for class A scavenger receptors (acetylated LDL, LPS, and titanium oxide) (Figure S4C). We also evaluated particles that are not ligands for scavenger receptors (transferrin, cholera toxin subunit B, and dextran) (Figure S4C). No differences in the endocytic capacity were observed. These results suggest that the differences in Atg7−/− cells apply only to the internalization by phagocytosis and not to that by endocytosis. Changes in other aspects, such as lipid membrane composition or formation of plasma membrane microdomains, could accompany the differential scavenger receptor expression to contribute together to the increased phagocytic capacity of Atg7−/− macrophages (Stuart and Ezekowitz, 2005). Taken together, these results indicate that MARCO and MSR1 are responsible for the increased uptake observed in ATG7-deficient macrophages.
Upregulation of MARCO and MSR1 mRNA in Atg7–/– Macrophages
We proceeded to identify the mechanism responsible for the upregulation of MARCO and MSR1 in Atg7−/− macrophages. We evaluated receptor mRNA expression in BMDMs from Atg7−/− and Atg7+/+ mice. We found that mRNAs of both MARCO and MSR1 were substantially elevated in Atg7−/− cells (Figures 6A and 6B). Because MARCO mRNA is known to be induced by LPS, we used LPS induction of MARCO in our assays as a positive control. BMDMs from Toll-like receptor 4 (TLR4)-, Toll-interleukin 1 receptor (TIR) domain containing adaptor protein (TIRAP)-, TIR-domain-containing adaptor-inducing interferon-β (TRIF)-, and p50-deficient mice were used as negative controls (Figures S5A–S5E).
Figure 6. Increased mRNA Expression of Class A Scavenger Receptors and NFE2L2 Activity in Atg7–/– Macrophages.
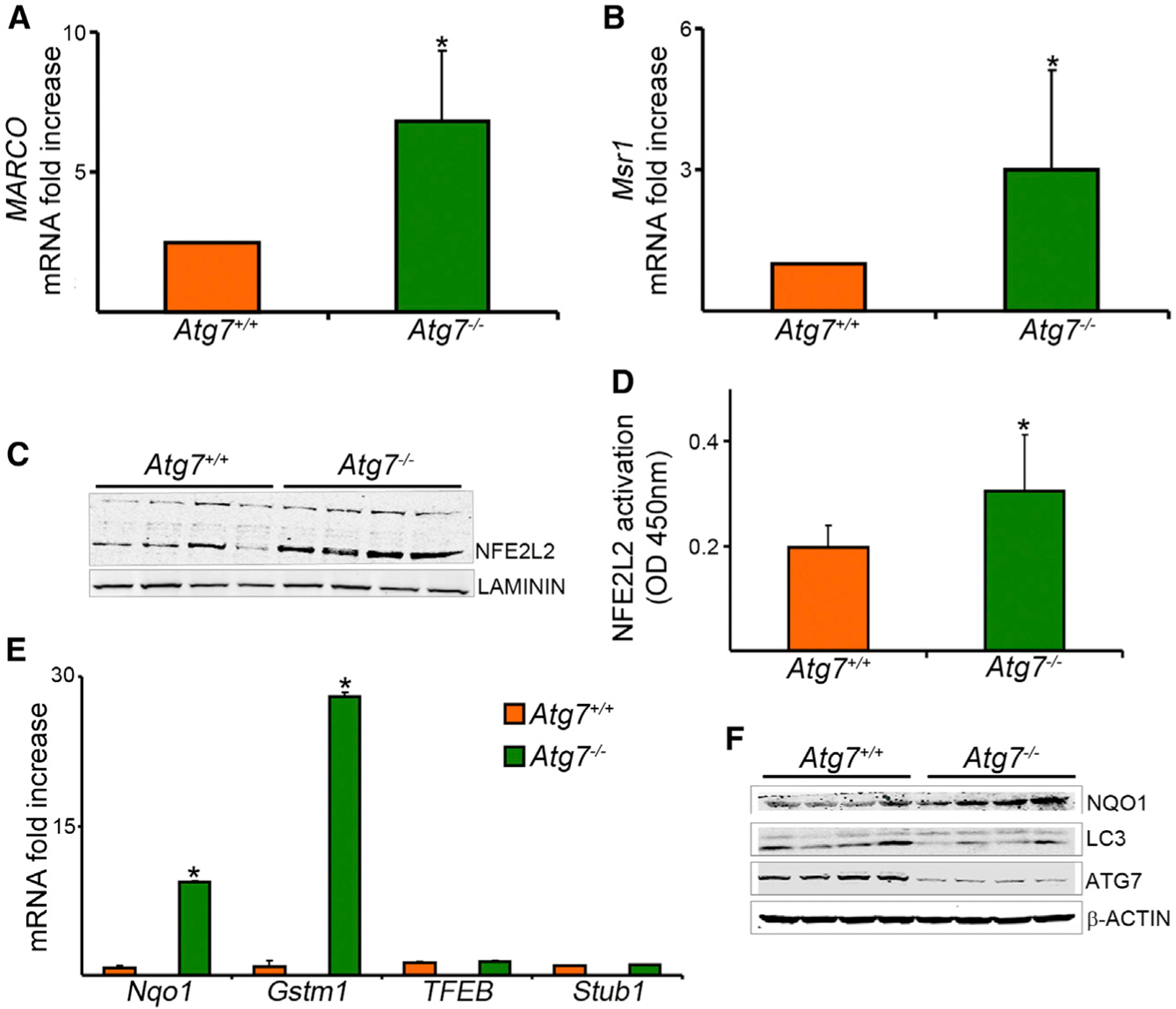
(A and B) Real-time PCR was done with mRNA of BMDMs from Atg7−/− and Atg7+/+ mice and specific primers for MARCO (A) or Msr1 (B). Data are expressed as fold increase, normalized to 18S mRNA.(C) Immunoblot analysis was done on nuclear fraction of the cell lysates with NFE2L2 antibody. Laminin was used as a loading control.(D) NFE2L2 activation in nuclear extracts was measured by quantification of the DNA binding activity of NFE2L2 by a colorimetric assay.(E) Expressions of Nqo1 and Gstm1, downstream gene targets of NFE2L2, and of TFEB and Stub1, as control genes, were analyzed by RT-PCR.(F) Cell lysates were analyzed by immunoblot with antibodies against NQO1, LC3, ATG7, or b-ACTIN.
Data are mean ± SD, n = 3, *p < 0.05. See also Figure S5.
A recent study revealed that MARCO is one of the target genes for the nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) (Harvey et al., 2011). The MARCO promoter was found to contain antioxidant response elements (ARE), which are known to mediate the effect of NFE2L2. We determined the presence of ARE sequences in the mouse and human genomic sequence in the 5′ untranslated region of Msr1, suggesting that Msr1 is also a potential transcriptional target for NFE2L2 (Figures S5F and S5G). NFE2L2, also known as Nrf2, is a key transcriptional regulator (Kensler et al., 2007). Under quiescent conditions, NFE2L2 interacts with Kelch-like ECH-associated protein 1 (KEAP1), which continuously targets NFE2L2 for proteasomal degradation, maintaining a low basal expression of NRF2-regulated genes. However, upon recognition of oxidative and electrophilic signals, NRF2 is released from KEAP1, escapes degradation, translocates to the nucleus, and transactivates expression of cytoprotective genes (Harvey et al., 2011). Additional recent evidence suggests that SQSTM1 interacts with the NFE2L2-binding site on KEAP1 and thus disrupts KEAP1 inhibitory effect on NFE2L2. SQSTM1, also known as p62, is one of the best known substrates for autophagy (Bjørkøy et al., 2005; Pankiv et al., 2007). Thus, autophagy deficiency is accompanied by accumulation of SQSTM1 (Figure 1A; Liu et al., 2012; Mizushima and Komatsu, 2011). Based on these recent findings, we hypothesized that in autophagy-deficient macrophages, accumulated SQSTM1 interacts with KEAP1, liberating it from NFE2L2, and then accumulation of NFE2L2 leads to upregulation of MARCO and MSR1. To test this hypothesis, we evaluated NFE2L2 expression and activity in Atg7−/− macrophages. We observed increased NFE2L2 amounts in nuclear fractions from Atg7−/− BMDMs (Figure 6C), as well as more NFE2L2 activity in Atg7−/− macrophages, as evidenced by the higher ability of the active form of NFE2L2 to bind to immobilized oligonucleotides containing the corresponding transcription factor binding site (Figure 6D). To confirm the functional consequences of increased NFE2L2 activity in Atg7−/− macrophages, we evaluated the expression of NAD(P)H dehydrogenase, quinone 1 (Nqo1) and glutathione S-transferase mu 1 (Gstm1), known downstream target genes for NFE2L2. NQO1 was upregulated in Atg7−/− macrophages at the mRNA and protein levels (Figures 6E and 6F), supporting the conclusion of increased NFE2L2 activity upon loss of ATG7. As a negative control, there were no substantial differences between Atg7−/− and Atg7+/+ macrophages in the expression of genes that are not under NFE2L2 regulation such as transcription factor EB (TFEB) and STIP1 homology and U-Box containing protein 1 (Stub1) (Figure 6E). Furthermore, the expression of MARCO and MSR1 was inhibited with SN-50, an inhibitor that prevents NRF2 nuclear translocation. (Figures S5H and S5I).
The data above suggested that the increase in MARCO and MSR1 in Atg7−/− macrophages is caused by accumulation of SQSTM1 leading to enhanced NFE2L2 activation. Although the baseline of NFE2L2 is kept low by its constant degradation, we tested whether SQSTM1-deficient macrophages might have even lower amounts of NFE2L2 and thus lower expression of MARCO and MSR1. To answer this question, we studied BMDMs from SQSTM1-deficient mice. Importantly, we found that nuclear NFE2L2 levels were reduced in Sqstm1−/− macrophages, and that was associated with reduction in both mRNA and surface expression of MARCO, compared to those in control cells (Figures S5J–S5L). Expression of mRNA of Nqo1, an NFE2L2 target gene, was also reduced. In contrast, the surface expression of transferrin receptor, CD14, and TLR2, which are not under the transcriptional regulation of NFE2L2, was not substantially different. These data confirm the critical role of SQSTM1 in modulating NFE2L2 levels and scavenger receptor expression. However, the ability to internalize BCG was not decreased in Sqstm1−/− macrophages (Figure S5M), consistent with a recent study (Ponpuak et al., 2010). This finding is probably due to compensatory increased by other receptors, as previously shown (Court et al., 2010). Taken together, these results indicate that accumulation of SQSTM1 leads to increased activation of NFE2L2 and the subsequent increase in MARCO and MSR1. A model for the mechanism of autophagy regulation of phagocytosis is shown in Figure S6.
DISCUSSION
This study shows that autophagy modulates phagocytosis. It shows that the underlying mechanism for increased phagocytosis in autophagy-deficient macrophages is through increased scavenger receptor expression, caused by higher activation of NFE2L2. Our study shows the physiological relevance of autophagy-mediated regulation of phagocytosis in the context of mycobacterial infection. Further, this study confirms that autophagy is important in the control of infection by mycobacteria in vivo in ATG7-deficient mice.
This study reveals a role for autophagy as a modulator of phagocytosis via scavenger receptors. The potential for cross talk between autophagy and phagocytosis has been recently suggested. The activation of both pathways requires hVPS34 PI3K kinase and can be promoted by Toll-like receptor agonists (Gutierrez et al., 2004; Sanjuan et al., 2007; Xu et al., 2007). Our study clearly shows that phagocytosis is enhanced in autophagy-deficient macrophages. Increased uptake of mycobacteria in autophagy-deficient macrophages was observed as early as 30 min after infection, making it unlikely to be explained by differences in lysosomal degradation. Supporting this notion, our previous ultrastructural analysis by electron microscopy showed that at an hour after infection, most of mycobacteria can be found inside single-membrane phagosomal compartments, not in autophagosomes (Xu et al., 2007). Because we focused our study on the role of ATG7 deficiency in macrophages, it remains to be elucidated whether other Atg genes can also modulate phagocytosis and receptor expression in these cells, as well as the contribution of differential LC3 recruitment to phagosomes in our experimental conditions.
It has been previously shown that autophagy is required for mycobacterial control in macrophages (Gutierrez et al., 2004; Xu et al., 2007). A recent study also discovered a link between the autophagy pathway, bacterial ESX-1 system, and cytosolic DNA-sensing pathway in the host in control of mycobacterial infection (Watson et al., 2012). We found that defects in autophagy lead to increased mycobacterial uptake in macrophages because of enhanced availability of phagocytic receptors at the cell surface. Thus, our study demonstrates another mechanism, at the level of receptor regulation, by which autophagy modulates the host immune response to mycobacteria as well as to other pathogens. Further, we show that autophagy is required for defense against mycobacteria in vivo. The increase in bacterial internalization in autophagy-deficient mice was shown both in cultured cells and in vivo. The increased uptake of bacteria in autophagy-deficient macrophages raises questions about the relative contribution of increased phagocytosis and of decreased lysosomal degradation to the enhanced survival of mycobacteria observed in these cells. The biological consequences of increased phagocytosis should vary depending on the type of pathogen and the integrity of other host defense machinery. In autophagy-competent cells, increased phagocytosis per se should be beneficial to the host to capture and kill extracellular pathogens. In contrast, in autophagy-deficient cells, increased phagocytosis of intracellular pathogens is likely to lead to exaggerated infection. The LysM-Cre system deletes ATG7 from all myeloid cells—not only macrophages but also monocytes and granulocytes. Our study focused on macrophages because they are the first cell encountered by mycobacteria within the respiratory tract, the primary host cell, and the main effector cell responsible for killing upon activation. However, the role(s) of granulocytes and monocytes in this model remain elusive. Although BCG lacks the ESX-1 secretion system important for autophagy induction (Gutierrez et al., 2004), in contrast to Mtb, we did not observe differences in the effect of autophagy on internalization between both species.
Mycobacteria are internalized through a broad array of phagocytic receptors (Ernst, 1998). We observed that the upregulation of phagocytosis was associated with increased surface expression of two phagocytic scavenger receptors, MSR1 and MARCO. Scavenger receptors are transmembrane glycoproteins that bind to different ligands and induce their uptake and clearance (Areschoug and Gordon, 2009). They play a role in innate immunity as pattern-recognition receptors mediating phagocytosis of different pathogens. MARCO and MSR1 are well-characterized class A scavenger receptors with a broad ligand specificity. They are expressed on macrophages, upregulated upon bacterial infection, and have been implicated in bacterial binding and removal (Arredouani et al., 2005; Rogers et al., 2009). In the lungs, these scavenger receptors mediate uptake and clearance of airborne pathogens and inhaled environmental particles (Józefowski et al., 2005). Expression of MARCO and MSR1 is increased in macrophages upon BCG (Ito et al., 1999) and Mtb (Court et al., 2010) infection, suggesting a role for these scavenger receptors in antimycobacterial response. However, redundancy of multiple receptors involved in phagocytosis of mycobacteria complicates interpretation of experiments in examining the effect of deficiency of a single cell-surface receptor on Mtb infection (Court et al., 2010). Supporting the hypothesis that these receptors play a role in host defense against infection, a recent study showed that genetic variants of MARCO are associated with susceptibility to tuberculosis in humans (Ma et al., 2011). It has been shown that mice lacking MARCO and MSR1 have defective bacterial clearance and higher proinflammatory responses when challenged with other bacteria. MARCO and MSR1 also protect against oxidant particles by downregulating lung inflammation after inhalation of titanium dioxide, ozone, silica, or environmental allergens (Arredouani et al., 2007).
We conclude that the increase in phagocytosis observed in ATG7-deficient macrophages was caused by scavenger receptor upregulation. The increased surface expression of these receptors observed in Atg7−/− macrophages was attributed to upregulation of NFE2L2 activity caused by SQSTM1 accumulation. It was recently shown that higher amounts of SQSTM1 lead to accumulation of NFE2L2. Indeed, accumulation of SQSTM1 in liver (Komatsu et al., 2007) or bronchiolar epithelial cells deficient in ATG7 (Inoue et al., 2011) has been shown to result in NFE2L2 activation and increased expression of detoxifying enzymes. Although we have demonstrated that in autophagy-deficient macrophages SQSTM1-mediated activation of NFE2L2 results in transcriptional increase in class A scavenger receptors, we cannot rule out the possibility that changes in other NFE2L2 target genes contribute to the observed phenotype. It has recently been shown that increased NFE2L2 can lead to increased phagocytosis by increased MARCO expression (Harvey et al., 2011). It is unlikely that the observed increase in scavenger receptor expression in Atg7−/− macrophages is caused by alternative activation of these macrophages, because no differences in mannose receptor or IL-10 and IL-12 were found.
Phagocytosis and endocytosis are related but quite distinct cellular processes. Although phagocytosis was increased in Atg7−/− macrophages, endocytosis of particles, known to use scavenger receptors for cell entry, was not substantially affected. The ingestion of particles by phagocytosis requires the formation of large endocytic vacuolar compartments by the organized movements of membranes and the actin cytoskeleton. In contrast to simple endocytosis, phagocytosis requires the integration of membrane receptors, cytoplasmic proteins that bind receptors and initiate and amplify signaling, plasma membrane lipids, and proteins that regulate actin polymerization and membrane fusion. We hypothesized that changes in other cellular components might synergize with the increased scavenger receptor expression to account for the observed increase in phagocytosis.
Collectively, this study reveals that defects in autophagy lead to increased susceptibility to mycobacterial infection, not only by impairing the macrophage-killing capacity but also by increasing the amount of bacteria internalized. Further, it reveals insights into the regulation of scavenger receptors by autophagy. These insights increase our understanding of the host-pathogen relationship and lay the groundwork for strategies aimed at combating infectious diseases. Understanding the interactions between phagocytosis and autophagy is fundamental to the identification of new therapeutic strategy against pathogens and modulation of both processes should be considered in any potential drug design.
EXPERIMENTAL PROCEDURES
Mice
We purchased C57BL/6, p50-deficient, and LysM-Cre transgenic mice from Jackson Laboratory. Mice genetically deficient for TLR4, TIRAP, or TRIF were kind gifts from R. Medzhitov and S. Akira, respectively. Atg7flox mice and SQSTM1-deficient mice were previously described (Komatsu et al., 2007). Mice were housed within a biosafety level 2 vivarium. Mice were used at 8–20 weeks of age. This study followed a protocol approved by the animal care committee of our institution.
Isolation and Culture of Mouse Macrophages
Mice were euthanized by asphyxia in a high CO2 environment. Bone marrow cells were cultured in DMEM+Glutamax (Gibco) with 10% FBS. Cells were differentiated into macrophages by using 10 ng/ml mouse M-CSF (Sigma) for 7 days in 5% CO2 at 37°C. Alveolar macrophages were collected by bronchoalveolar lavage (BAL) with complete DMEM containing 0.5 mM EDTA (Sigma). Based on F4/80 expression, more than 95% of BAL cells were macrophages. Peritoneal macrophages were collected by peritoneal lavage via complete DMEM. Cells were plated on coverslips and after 2 hr incubation at 37°C, and nonadherent cells were removed by gentle washing. Cell mono-layers were cultured overnight in complete medium. Viability of the collected cells was consistently >95% by trypan blue exclusion.
PCR and Immunoblot Analysis
Total RNA was purified with an RNEasy Mini Kit (QIAGEN), and cDNA synthesis was performed with cDNA Reverse Transcription Kit (Applied Biosystems). Target genes and 18S mRNA expression were measured in 96-well optical plates by using Taqman Universal PCR Master Mix and the StepOnePlus Real-Time PCR Systems (Applied Biosystems). For immunoblot analysis, cells were washed with cold PBS and lysed with RIPA buffer in the presence of a protease inhibitor cocktail (Beckton Dickinson) for 30 min on ice. Total protein concentrations were determined with the BCA protein assay kit (Pierce). Cell lysates (50 μg of protein) were heated at 100°C for 5 min in sample buffer, resolved on SDS-PAGE, and transferred to nitrocellulose membranes. Antibodies to β-ACTIN, ATG7, SQSTM1, LC3, ATG5, NQO1, NFE2L2, MSR1, and laminin were used. Immunoreactive bands were detected by infrared imaging system (Odyssey).
Flow Cytometry and Fluorescence Microscopy
Cells were washed in staining buffer (0.5% BSA-PBS) and incubated for 30 min at 4°C in the dark with conjugated mouse monoclonal antibodies diluted in staining buffer. Monoclonal antibodies against CD11a, CD11b, CD11c,CD14, CD206, CD36, CD16, CD71, TLR2, TLR4, CD40, CD86, CD69, CD195, CD184, CD119, CD120b, and CD212 were purchased from Becton Dickinson; MARCO antibody was from Serotec; CD209 antibody was from eBioscience; and polyclonal MSR1 antibody (AF1797) was from R&D Systems. Cells were pretreated with a rat anti-mouse CD16/CD32 (Becton Dickinson) to block nonspecific binding and isotypes were used as controls. After incubation, cells were washed twice in staining buffer, labeled with sytox blue for cell viability, and analyzed immediately. Flow cytometry was performed with LSR Fortessa (Becton Dickinson), FACSDiva software for data collection, and FlowJo software for analysis. Cells were first gated based on forward and scatter properties. Cell duplets were excluded and cells were gated on F4/80-positive (macrophages) and sytox-blue-negative (viable) events. In the gated events, both percentages of positivity and MFI for each marker were assessed and untreated and isotype controls were used to determine cutoffs. At least 10,000 cells were evaluated to determine surface receptor expression. Data are presented as mean ± SD of mean fluorescence intensity (MFI).
Mycobacterial Infection in Macrophages
BCG and the H37Rv strain of Mtb were cultured in Middlebrook 7H9 broth medium (Becton Dickinson) and stocks were prepared and stored at 80°C. At the time of infection, bacteria were thawed, vortexed, sonicated, and passed through a 28G needle 15 times to disrupt bacterial clumps. Cellular mono-layers were infected for 1 hr at an MOI of 10. Upon infection, extracellular and surface-bound bacteria were killed by the inclusion of 50 μg/ml gentamycin in culture media. Cells were washed three times with ice-cold PBS and incubated for 3, 5, and 7 days in fresh complete DMEM. After incubation, cells were lysed with 0.1% SDS and cell lysates were diluted, plated on 7H11 Middlebrook agar plates (Becton Dickinson), and incubated at 37°C in 5% CO2. Colony-forming units (CFU) were counted 3–4 weeks later and results were expressed as log10 CFU. The BCG and Mtb strains were kindly provided by D. McMurray from Texas A&M Health Science Center. Experiments involving Mtb handling were performed in BSL-3 facility.
In Vivo BCG Infection
Mice were intranasally infected with 1 × 106 CFU BCG (five mice per group, per time point). For bacterial counts, lungs were homogenized, diluted and plated in 7H11 Middlebrook agar plates (Becton Dickinson), and incubated at 37°C in 5% CO2. CFUs were counted 3–4 weeks later. For histological evaluation, lungs were inflated with 10% formalin, embedded in paraffin, sectioned, and stained with hematoxilin-eosin or acid fast staining for evaluation of the inflammatory infiltrate and the bacterial loads, respectively. The degree of inflammation was evaluated by scores from 0 to 3 for the following criteria: 0, no inflammation, occasional inflammatory cells; 1, mild inflammation, increased number of inflammatory cells; 2, moderate inflammation, confluent inflammatory cells; 3, extensive inflammation, extended infiltrate; and 4, severe inflammation, massive infiltration. The percentage of tissue involved in the inflammatory response was determined by morphometric analysis with the ImageJ 11.4 software from NIH.
Phagocytosis Assays
Macrophages were infected with GFP-expressing BCG or GFP-expressing Mtb at an MOI of 10 and then incubated at 37°C in a 5% CO2 environment. After infection, macrophages were washed, extracellular bacteria were removed by 50 μg/ml gentamycin treatment and PBS washing, and cells were fixed with 4% formaldehyde. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI) (Invitrogen). Coverslips were mounted onto slides with Slowfade Gold medium and analyzed on a Zeiss Axiovert 200M fluorescence microscope. Phagocytosis was evaluated by counting 300 macrophages per well at 633 magnification under oil immersion. Serial sections were taken with Z-stack spanning to confirm colocalization and images were deconvolved. Percent of infected macrophages was estimated by counting cells containing at least one ingested bacterium. Phagocytic index was generated by multiplying the percentage of phagocytosis by the mean number of internalized bacteria per cell. Cells incubated with 1.0 mm yellow-green polystyrene microbeads or zymosan were used as controls. Cytochalasin D treatment and 4°C incubation were used to prevent phagocytosis. To discriminate between intracellular and extracellular bacteria, 0.08 μg/ml trypan blue was added to quench extracellular fluorescent signal and serial images were taken across cells. For flow cytometry evaluation, a specific conjugated F4/80 antibody (eBioscience) was used to identify the cell type, sytox blue (Invitrogen) exclusion was used to determine cell viability, and RFP-expressing BCG was used to quantify bacterial uptake. Cells were infected at an MOI of 10 and then incubated at 37°C in a 5% CO2. After infection, macrophages were washed, extracellular and surface-bound bacteria were removed, and cells were fixed with 4% formaldehyde. Flow cytometry was performed as described above. In the gated events, phagocytosis percentage and MFI were assessed and uninfected controls were used to determine cutoffs for phagocytosis. Phagocytosis of S. aureus and E. coli was measured by flow cytometry with pHrodo-labeled bacteria (Invitrogen). To assess the macrophage ability to phagocytose bacteria in vivo, mice were challenged intraperitoneally with GFP-expressing BCG. After 2 hr, peritoneal lavage was collected and cells were fixed and analyzed by microscopy as described above. Scavenger receptors functions were inhibited by MARCO neutralizing antibody E31 (Hycult biotechnology) or the class A scavenger receptor antagonists Fucoidan (Sigma) or polyinosinic acid (Poly-I) (Sigma).
Endocytosis Assays
Cells were incubated in the presence of different ligands (titanium oxide [Sigma], Alexa Fluor 488-labeled acetylated LDL, dextran, transferrin, cholera toxin subunit B, or LPS [Invitrogen]) at 37°C for 30 or 60 min, washed with PBS, and assayed for fluorescence. Cells were then washed and treated with acid wash (50 mM glycine, 150 mM NaCl [pH 2.5]) to remove surface-bound particles (Lei and Martinez-Moczygemba, 2008).
Statistical Analysis
Parametric statistical calculations were performed with Microsoft Excel and GraphPad Prism software. Differences between two groups were determined by Student’s t test for samples with equal variance. Comparisons between more than two groups were done by ANOVA and the Tukey-Kramer test. p values <0.05 were considered significant. Data are expressed as means ± standard deviation of three independent experiments.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank scientists who contributed reagents, as outlined in the text, and members of the N.T.E. laboratory for useful discussions. This study is supported by Heart Lung and Blood Institute, National Institute of Allergy and Infectious Diseases, and the American Heart Association. This project was supported by the Cytometry and Cell Sorting Core at Baylor College of Medicine with funding from the National Institutes of Health (NIAID P30AI036211, NCI P30CA125123, and NCR S10RR024574) and the assistance of J.M. Sederstrom.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and one table and can be found with this article online at http://dx.doi.org/10.1016/j.immuni.2013.08.026.
REFERENCES
- Areschoug T, and Gordon S (2009). Scavenger receptors: role in innate immunity and microbial pathogenesis. Cell. Microbiol 11, 1160–1169. [DOI] [PubMed] [Google Scholar]
- Arredouani MS, Palecanda A, Koziel H, Huang YC, Imrich A, Sulahian TH, Ning YY, Yang Z, Pikkarainen T, Sankala M, et al. (2005). MARCO is the major binding receptor for unopsonized particles and bacteria on human alveolar macrophages. J. Immunol 175, 6058–6064. [DOI] [PubMed] [Google Scholar]
- Arredouani MS, Franco F, Imrich A, Fedulov A, Lu X, Perkins D, Soininen R, Tryggvason K, Shapiro SD, and Kobzik L (2007). Scavenger Receptors SR-AI/II and MARCO limit pulmonary dendritic cell migration and allergic airway inflammation. J. Immunol 178, 5912–5920. [DOI] [PubMed] [Google Scholar]
- Bjørkøy G, Lamark T, Brech A, Outzen H, Perander M, Overvatn A, Stenmark H, and Johansen T (2005). p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol 171, 603–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, and Medzhitov R (2004). Regulation of phagosome maturation by signals from toll-like receptors. Science 304, 1014–1018. [DOI] [PubMed] [Google Scholar]
- Bowdish DM, Sakamoto K, Kim MJ, Kroos M, Mukhopadhyay S, Leifer CA, Tryggvason K, Gordon S, and Russell DG (2009). MARCO, TLR2, and CD14 are required for macrophage cytokine responses to mycobacterial trehalose dimycolate and Mycobacterium tuberculosis. PLoS Pathog. 5, e1000474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo EF, Dekonenko A, Arko-Mensah J, Mandell MA, Dupont N, Jiang S, Delgado-Vargas M, Timmins GS, Bhattacharya D, Yang H, et al. (2012). Autophagy protects against active tuberculosis by suppressing bacterial burden and inflammation. Proc. Natl. Acad. Sci. USA 109, E3168–E3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Court N, Vasseur V, Vacher R, Frémond C, Shebzukhov Y, Yeremeev VV, Maillet I, Nedospasov SA, Gordon S, Fallon PG, et al. (2010). Partial redundancy of the pattern recognition receptors, scavenger receptors, and C-type lectins for the long-term control of Mycobacterium tuberculosis infection. J. Immunol 184, 7057–7070. [DOI] [PubMed] [Google Scholar]
- Ernst JD (1998). Macrophage receptors for Mycobacterium tuberculosis. Infect. Immun 66, 1277–1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, and Deretic V (2004). Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell 119, 753–766. [DOI] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Sethi S, Kong X, Yarmus L, Brown RH, Feller-Kopman D, Wise R, and Biswal S (2011). Targeting Nrf2 signaling improves bacterial clearance by alveolar macrophages in patients with COPD and in a mouse model. Sci. Transl. Med 3, 78ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Maloney NS, Bruinsma MW, Goel G, Duan E, Zhang L, Shrestha B, Diamond MS, Dani A, Sosnovtsev SV, et al. (2012). Nondegradative role of Atg5-Atg12/ Atg16L1 autophagy protein complex in antiviral activity of interferon gamma. Cell Host Microbe 11, 397–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Kubo H, Taguchi K, Suzuki T, Komatsu M, Motohashi H, and Yamamoto M (2011). Inducible disruption of autophagy in the lung causes airway hyper-responsiveness. Biochem. Biophys. Res. Commun 405, 13–18. [DOI] [PubMed] [Google Scholar]
- Ito S, Naito M, Kobayashi Y, Takatsuka H, Jiang S, Usuda H, Umezu H, Hasegawa G, Arakawa M, Shultz LD, et al. (1999). Roles of a macrophage receptor with collagenous structure (MARCO) in host defense and heterogeneity of splenic marginal zone macrophages. Arch. Histol. Cytol 62, 83–95. [DOI] [PubMed] [Google Scholar]
- Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL Jr., and Eissa NT (2009). Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat. Med 15, 267–276. [DOI] [PubMed] [Google Scholar]
- Józefowski S, Arredouani M, Sulahian T, and Kobzik L (2005). Disparate regulation and function of the class A scavenger receptors SR-AI/II and MARCO. J. Immunol 175, 8032–8041. [DOI] [PubMed] [Google Scholar]
- Kensler TW, Wakabayashi N, and Biswal S (2007). Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol 47, 89–116. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T, Mizushima N, Iwata J, Ezaki J, Murata S, et al. (2007). Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131, 1149–1163. [DOI] [PubMed] [Google Scholar]
- Lei JT, and Martinez-Moczygemba M (2008). Separate endocytic pathways regulate IL-5 receptor internalization and signaling. J. Leukoc. Biol 84, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerena MC, and Colombo MI (2011). Mycobacterium marinum induces a marked LC3 recruitment to its containing phagosome that depends on a functional ESX-1 secretion system. Cell. Microbiol 13, 814–835. [DOI] [PubMed] [Google Scholar]
- Lima JG, de Freitas Vinhas C, Gomes IN, Azevedo CM, dos Santos RR, Vannier-Santos MA, and Veras PS (2011). Phagocytosis is inhibited by autophagic induction in murine macrophages. Biochem. Biophys. Res. Commun 405, 604–609. [DOI] [PubMed] [Google Scholar]
- Liu PT, and Modlin RL (2008). Human macrophage host defense against Mycobacterium tuberculosis. Curr. Opin. Immunol 20, 371–376. [DOI] [PubMed] [Google Scholar]
- Liu XD, Ko S, Xu Y, Fattah EA, Xiang Q, Jagannath C, Ishii T, Komatsu M, and Eissa NT (2012). Transient aggregation of ubiquitinated proteins is a cytosolic unfolded protein response to inflammation and endoplasmic reticulum stress. J. Biol. Chem 287, 19687–19698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma MJ, Wang HB, Li H, Yang JH, Yan Y, Xie LP, Qi YC, Li JL, Chen MJ, Liu W, and Cao WC (2011). Genetic variants in MARCO are associated with the susceptibility to pulmonary tuberculosis in Chinese Han population. PLoS ONE 6, e24069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray DN (2001). Disease model: pulmonary tuberculosis. Trends Mol. Med 7, 135–137. [DOI] [PubMed] [Google Scholar]
- Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. [DOI] [PubMed] [Google Scholar]
- Palecanda A, Paulauskis J, Al-Mutairi E, Imrich A, Qin G, Suzuki H, Kodama T, Tryggvason K, Koziel H, and Kobzik L (1999). Role of the scavenger receptor MARCO in alveolar macrophage binding of unopsonized environmental particles. J. Exp. Med 189, 1497–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem 282, 24131–24145. [DOI] [PubMed] [Google Scholar]
- Pilli M, Arko-Mensah J, Ponpuak M, Roberts E, Master S, Mandell MA, Dupont N, Ornatowski W, Jiang S, Bradfute SB, et al. (2012). TBK-1 promotes autophagy-mediated antimicrobial defense by controlling autophagosome maturation. Immunity 37, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponpuak M, Davis AS, Roberts EA, Delgado MA, Dinkins C, Zhao Z, Virgin HW 4th, Kyei GB, Johansen T, Vergne I, and Deretic V (2010). Delivery of cytosolic components by autophagic adaptor protein p62 endows autophagosomes with unique antimicrobial properties. Immunity 32, 329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NJ, Lees MJ, Gabriel L, Maniati E, Rose SJ, Potter PK, and Morley BJ (2009). A defect in Marco expression contributes to systemic lupus erythematosus development via failure to clear apoptotic cells. J. Immunol 182, 1982–1990. [DOI] [PubMed] [Google Scholar]
- Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, and Green DR (2007). Toll-like receptor signalling in macrophages links the autophagy pathway to phagocytosis. Nature 450, 1253–1257. [DOI] [PubMed] [Google Scholar]
- Stuart LM, and Ezekowitz RA (2005). Phagocytosis: elegant complexity. Immunity 22, 539–550. [DOI] [PubMed] [Google Scholar]
- Watson RO, Manzanillo PS, and Cox JS (2012). ExtracellularM. tuberculosis DNA targets bacteria for autophagy by activating the host DNA-sensing pathway. Cell 150, 803–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, and Eissa NT (2010). Autophagy in innate and adaptive immunity. Proc. Am. Thorac. Soc 7, 22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Jagannath C, Liu XD, Sharafkhaneh A, Kolodziejska KE, and Eissa NT (2007). Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 27, 135–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, Cadwell K, Delgado MA, Ponpuak M, Green KG, et al. (2008). Autophagosomeindepende006Et essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe 4, 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Imrich A, and Kobzik L (2008). Characterization of immortalized MARCO and SR-AI/II-deficient murine alveolar macrophage cell lines. Part. Fibre Toxicol 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.