

Abstract
Background:
Factor XIII (FXIII) promotes fibrin crosslinking and red blood cell (RBC) retention in clots. The FXIII-A polymorphism, Val34Leu, is associated with protection against venous thrombosis. This effect is hypothesized to result from fibrinogen concentration-dependent changes in fibrin structure. Effects of the FXIII-A Val34Leu polymorphism in whole blood clots have not been investigated.
Aim:
Characterize effects of FXIII-A Val34Leu polymorphism and fibrinogen on whole blood clots.
Methods:
We isolated platelet-poor plasmas from human donors (FXIIIVal/Val, FXIIIVal/Leu, FXIIILeu/Leu), reconstituted plasmas with platelets and RBCs, and triggered clotting. We assessed contributions of gender, age, clotting times, thrombin generation, FXIII activity, FXIII-A Val34Leu polymorphism, and fibrinogen to clot mass. We also reconstituted FXIII-depleted plasma with platelets, RBCs, and purified FXIIIVal/Val or FXIIILeu/Leu, varied fibrinogen, and characterized effects on clot mass.
Results:
Clot mass was associated with age, fibrinogen, prothrombin time, and thrombin generation. Clots reconstituted with plasmas from individuals with FXIII-AVal/Val and FXIII-AVal/Leu did not differ in mass from clots with FXIII-ALeu/Leu. However, clots containing a 34Val allele demonstrated a fibrinogen concentration-dependent increase in mass, whereas clots with homozygous 34Leu did not. In plasmas with high fibrinogen, mass was higher for clots with 34Val alleles compared to clots with homozygous 34Leu. In clots reconstituted with purified FXIII, increasing fibrinogen enhanced clot mass in the presence of 34Val, but decreased mass in the presence of 34Leu.
Conclusions:
FXIII 34Leu mitigates the effect of elevated fibrinogen on whole blood clot mass. The Val34Leu polymorphism may protect against venous thrombosis by reducing clot mass.
Keywords: Thrombosis, factor XIII, transglutaminase, fibrinogen, erythrocyte
INTRODUCTION
Factor XIII (FXIII) is a protransglutaminase present in cells and plasma. Plasma FXIII is a non-covalent heterotetramer (FXIII-A2B2) that circulates bound to fibrinogen [1]. During coagulation, FXIII is activated by thrombin-mediated cleavage and release of 37-amino acid (4-kDa) activation peptides from the N-termini of the catalytic FXIII-A2 subunits, followed by calcium-mediated dissociation of the carrier FXIII-B2 subunits [2]. Activated FXIII (FXIIIa) catalyzes the formation of ε-N-(γ-glutamyl)-lysyl crosslinks between γ- and α-chains of fibrin [3] and between fibrin and other plasma proteins [4–6]. These crosslinks enhance clot mechanical and biochemical stability, respectively. FXIII deficiency is associated with poor wound healing, miscarriage, and intracranial hemorrhage [7].
The common gene variant in FXIII-A encoding a valine (Val) to leucine (Leu) substitution at codon 34 (Val34Leu) is present in ~25% of European Caucasians [8]. Findings on the role of this polymorphism in thrombosis risk have been inconsistent; however, both independent studies and meta-analyses suggest the 34Leu variant offers modest but significant protection against venous thrombosis [9–14]. Discovery that the effects of the Val34Leu polymorphism are mediated by gene-environment interactions between FXIII-A genotype and plasma fibrinogen concentration has resolved some of the discord [15]. For example, in the Leiden Thrombophilia Study, although Leu homozygosity showed weak protection against venous thrombosis in men [16], reanalysis adjusting for fibrinogen concentration strengthened the association for both men and women, especially in individuals older than 45 years [17].
The FXIII-A Val34Leu polymorphism is located 3 amino acids before the thrombin cleavage site and results in ~2.5-fold accelerated FXIII activation [18, 19] and accelerated fibrin crosslinking [20, 21]. Effects of the Val34Leu polymorphism on clot quality are hypothesized to manifest via fibrinogen-dependent changes in clot structure. Briefly, the normal range of fibrinogen in plasma is 1.5–4 g/L. Fibrinogen increases with age [22] and can exceed 7 g/L in individuals with congenital hyperfibrinogenemia or acquired hyperfibrinogenemia secondary to inflammatory disease or pregnancy. In plasmas with low/normal fibrinogen concentration, the 34Leu variant produces clots with thin fibrin fibers and low permeability. However, in plasmas with higher fibrinogen, it produces thicker fibers in clots that have increased permeability and susceptibility to fibrinolysis [15]. Since increased fibrinogen is associated with increased risk of cardiovascular disease [23], these observations suggest a mechanism whereby the FXIII-A 34Leu polymorphism mitigates effects of hyperfibrinogenemia on clot quality and reduces thrombotic risk.
We recently showed that FXIIIa activity promotes red blood cell (RBC) retention during clot contraction and is a major determinant of venous thrombus composition and mass [24–26]. Mice with heterozygous deficiency in FXIII-A (F13a1+/−) have delayed FXIII activation, decreased RBC retention in contracted whole blood clots, and reduced thrombus mass in vitro and in vivo [24, 25]. Mice with normal levels of FXIII but delayed FXIII activation secondary to decreased FXIII binding to fibrinogen (Fibγ390–396A) also show delayed FXIII activation and reduced thrombus mass [25]. In whole blood from persons with hemophilia, co-treatment with hemostatic agents and supraphysiologic concentrations of FXIII accelerate FXIII activation and increase clot mass ex vivo [27]. By extension, one might speculate that individuals with the 34Leu variant and accelerated FXIII activation would produce larger clots carrying increased venous thrombosis risk. However, the paradoxical association of the 34Leu variant with decreased venous thrombosis risk [11–14] warrants an explicit analysis of the interaction between this polymorphism and fibrinogen concentration during whole blood clot formation. Determining the impact of the 34Leu variant on clot characteristics is essential for understanding the clinical significance of the FXIII-A Val34Leu polymorphism in thrombosis risk.
Herein we used independent but complementary methods to determine the relationships between the FXIII-A Val34Leu polymorphism, fibrinogen concentration, and whole blood clot formation. Collectively, our data suggest the 34Leu variant mitigates the effect of hyperfibrinogenemia on whole blood clot mass.
MATERIALS and METHODS
Materials.
Sigmacote® was from Sigma-Aldrich (St. Louis, MO). Prostaglandin-I2 was from Cayman Chemical (Ann Arbor, MI). Lipidated tissue factor (TF, Innovin) was from Siemens (Munich, Germany). Non-reducing 6X sample buffer containing sodium dodecyl sulfate (SDS) was from Boston Bioproducts (Ashland, MA). β-mercaptoethanol was from Fisher Scientific (Hampton, NH). Tris-glycine polyacrylamide gels (10%) were from Bio-Rad (Hercules, CA). Odyssey Blocking Buffer was from LI-COR Biosciences (Lincoln, NE). Anti-human FXIII-A polyclonal antibody (SAF13A-AP) was from Enzyme Research Laboratories (South Bend, IN). FXIII-deficient plasma was from Affinity Biologicals (Ancaster, Ontario). Anti-human fibrinogen polyclonal antibody (A0080) was from Dako (Glostrup, Denmark). Alexa Fluor®−488 anti-rabbit and anti-sheep secondary antibodies were from Jackson Immunoresearch (West Grove, PA). Polyvinylidene difluoride membranes were from Invitrogen (Carlsbad, CA) and scanned on a GE Typhoon FLA-9000 Imager (GE Healthcare, Pittsburgh, PA). Calibrated automated thrombography reagents (fluorogenic thrombin substrate, TF/Lipid Reagents [PPP-Low], and thrombin calibrator) were from Diagnostica Stago (Parsippany, NJ).
Human blood draws and plasma preparation.
Approval for the use of human subjects was obtained from the University of Debrecen Ethics Committee (number: DE RKEB/IKEB: 3189-2010) and University of North Carolina Institutional Review Board. All participants provided signed informed consent in accordance with the Declaration of Helsinki. Donors had no known bleeding disorder, liver or kidney disease, cancer, were not pregnant, had no history of surgery or thrombotic event (including acute myocardial infarction or stroke) within the past 3 months, and were not on antiplatelet or anticoagulant therapy. Whole blood was obtained by venipuncture and drawn into tubes containing 0.105 M citrate (Becton Dickinson, Franklin Lakes, NJ, 10% v/v, final). Blood samples were processed immediately by centrifugation twice at 1220×g (15 minutes, room temperature) to obtain platelet-poor plasma. Hemostasis screening tests (prothrombin time and activated partial thromboplastin time) were measured immediately from the plasma samples. Remaining plasma was stored at −70°C until further analysis. DNA isolation was performed from buffy coats of blood samples by QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). The FXIII-A Val34Leu (c.103G>T; rs5985) polymorphism was identified by real-time PCR [28] using fluorescence resonance energy transfer detection and melting curve analysis on a LightCycler® 480 instrument (Roche Diagnostics GmbH, Mannheim, Germany; primers are available from the authors upon request). Normal, pooled plasma was prepared by sequential centrifugation as described [29] from a pool of 32 healthy donors with normal partial thromboplastin times; the fibrinogen concentration in the normal pool was 3.2 mg/mL (Clauss assay).
FXIII zymogen preparation.
Purified FXIII-A2B2 was isolated as described [30] from pooled plasma from individuals possessing only 34Val or only 34Leu FXIII-A alleles.
Western blotting.
FXIII antigen and activity were analyzed in Peak 1 (FXIII-free) fibrinogen preparations by immunoblot detection of FXIII-A subunit and fibrin crosslinking. Fibrinogen preparations were clotted with thrombin (1 U/mL) and EDTA (10 mM, final) or CaCl2 (10 mM, final) for 2 hours. Samples were then dissolved in 50 mM dithiothreitol, 12.5 mM EDTA, and 8 M urea at 60°C for 1 hour, diluted 120-fold in 6X reducing SDS sample buffer, boiled, separated on 10% Tris-Glycine gels, and transferred to polyvinylidene difluoride membranes. Membranes were blocked for 1 hour at room temperature with blocking buffer, incubated overnight at 4°C with primary antibodies against FXIII-A or fibrin(ogen), and then incubated with fluorescence-labeled secondary antibodies for 1 hour at room temperature.
Reconstituted whole blood assays.
Whole blood was centrifuged (150×g, 20 minutes). Platelet-rich plasma from a donor carrying the FXIIIVal/Val genotype was isolated and treated with prostaglandin-I2 (50 ng/mL, final) and then centrifuged to obtain platelet-poor plasma (700×g, 10 minutes) or platelets (400×g, 20 minutes). Platelet pellets were resuspended in Tyrode’s buffer (15 mM HEPES, 3.3 mM NaH2PO4, pH 7.4, 138 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 5.5 mM dextrose). RBCs were isolated from an O-negative donor; the RBC fraction was isolated, washed with citrated glycine saline buffer, and packed three times by centrifugation (400×g, 5 minutes). Platelets and RBCs were prepared and used on the day of the experiment.
In the first series of experiments, reconstituted whole blood was prepared by combining resuspended platelets (100 K/μL, final), washed RBCs (4.5 million/μL, final), and thawed platelet-poor plasma from donors with FXIIIVal/Val, FXIIIVal/Leu, or FXIIILeu/Leu. Clotting was triggered in siliconized wells with TF and CaCl2 (1 pM and 10 mM, final, respectively) at 37°C. After 2 hours, contracted clots were removed from the wells, dabbed on a Kimwipe to remove the serum, and weighed. These experiments were performed on three separate days, and clots from individual plasmas were normalized to the average mass of control clots made with normal pooled plasma reconstituted with platelets and RBCs on each day (36.8±2.5 mg [N=3 clots], 35.4±0.6 mg [N=2 clots], and 30.0±0.2 mg [N=2 clots], respectively).
In the second series of experiments, reconstituted whole blood was prepared by combining resuspended platelets (100 K/μL, final), washed RBCs (4.5 million/μL, final), and thawed FXIII-deficient plasma with purified FXIIIVal/Val or FXIIILeu/Leu zymogen (21 μg/mL, final) and FXIII-free fibrinogen (to achieve the final concentrations indicated). Clotting was triggered in siliconized wells with TF and CaCl2 (1 pM and 10 mM, final, respectively) at 37°C. After 2 hours, contracted clots were removed from the wells, dabbed on a Kimwipe to remove the serum, and weighed.
Hemostasis assays.
FXIII activity was measured according to Karpati et al [31]. Reference ranges were determined according to guidelines of the Clinical and Laboratory Standards Institute. Fibrinogen concentration was determined by the Clauss method [32] using reagents from Labexpert LTD (Debrecen, Hungary). Prothrombin time (PT) and activated partial thromboplastin time (APTT) analyses were performed on a BCS coagulometer (Siemens Healthcare Diagnostics, Marburg, Germany) using DADE Innovin PT and Actin FS reagents from Siemens (Munich, Germany). Normal control values are the result of mixed samples of 25 healthy individuals with no hemostasis disorders, measured with the same reagent and coagulometer.
Thrombin generation.
Thrombin generation was measured by calibrated automated thrombography, as described [29]. Briefly, thrombin generation was triggered in duplicate platelet-poor plasma samples with TF, phospholipids, and CaCl2 (1 pM, 4 μM, and 16.7 mM, final, respectively). Fluorescence was detected by a Fluoroskan Ascent® fluorometer (Thermo Fischer Scientific, Waltham, MA). Thrombograms were analysed with Thrombinoscope software v5.0 (Thrombinoscope BV, Maastricht, The Netherlands).
Statistical methods.
Normality of data was evaluated by Shapiro-Wilk test. Descriptive statistics (mean, standard deviation or median, interquartile range) were calculated for continuous variables. Differences between categorical variables were assessed by Fisher’s exact or χ2 test. In all two-group analyses, unpaired Student’s t test, or in case of non-parametric data Mann-Whitney U test, was used. ANOVA using Bonferroni post-hoc test or Kruskal-Wallis test using Dunn’s post-hoc test were applied for multiple comparisons. Pearson’s or Spearman’s correlation coefficient was used to determine the strength of correlation between parameters of normalized clot mass and other continuous variables. To test for differences between adjusted means, univariate analysis incorporating covariate testing (one-way ANCOVA) was performed. P<0.05 was considered significant. Statistical analysis was performed using the Statistical Package for Social Sciences 22.0 (SPSS, Chicago, IL), GraphPad Prism 5.0 (GraphPad Prism Inc., La Jolla, CA), and Stata 12.0 software (Stata Corp LP, TX).
RESULTS
Effects of the FXIII-A Val34Leu polymorphism on clinical presentation and clot properties has been challenging to characterize because of individual variation in other potential modifiers, including overall procoagulant activity, fibrinogen concentration, and co-existing polymorphisms [10]. We therefore approached this question using independent but complementary methods that combined the translational strengths of human samples with the experimental control offered by in vitro biochemical analyses.
Determinants of whole blood clot mass in a reconstituted model using individual plasmas.
In the first series of experiments, we isolated platelet-poor plasma from 86 healthy individuals with homozygous or heterozygous 34Val or 34Leu alleles: FXIIIVal/Val (N=40), FXIIIVal/Leu (N=28), and FXIIILeu/Leu (N=18). Demographic and clinical characteristics of all donors and donors separated by genotype are shown in Table 1. Subjects with the three genotypes did not differ in gender, age, FXIII activity, APTT, PT, or thrombin generation lag time or time to peak. Subjects with the FXIIIVal/Val genotype had lower fibrinogen than subjects with FXIIIVal/Leu or FXIIILeu/Leu genotypes. Subjects with the FXIIIVal/Leu genotype had increased thrombin peaks compared to individuals with FXIIIVal/Val or FXIIILeu/Leu genotypes. Endogenous thrombin potential (ETP) differed significantly between groups, although differences were small.
Table 1.
Demographic and clinical characteristics for all plasma donors and donors separated by genotype.
| Comparison of Genotypes (P): | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Reference | All (N=86) | FXIIIVal/Val (N=40) | FXIIIVal/Leu (N=28) | FXIIILeu/Leu (N=18) | P& | FXIIIVal/Val vs FXIIIVal/Leu | FXIIIVal/Leu vs FXIIILeu/Leu | FXIIIVal/Val vs FXIIILeu/Leu | |
| Clinical Characteristics | |||||||||
| Gender (female/male) | - | 71/15 | 33/7 | 24/4 | 14/4 | 0.7868 | - | - | - |
| Age (years) | - | 26 (23.0–38.8) | 25 (21.0–36.8) | 28.5 (23.8–38.3) | 28.0 (23.8–46.3) | 0.4053 | - | - | - |
| FXIII Activity (%) | 69 – 143 | 114 ± 20 | 114 ± 18 | 115 ± 26 | 110 ± 15 | 0.7127 | - | - | - |
| Fibrinogen (g/L) | 1.5 – 4 | 3.3 (2.9–4.2) | 3.0 (2.7–3.7) | 4.0 (2.9–4.5) | 3.2 (2.9–4.5) | 0.0432 | 0.0401 | - | 0.0443 |
| Clotting Tests | |||||||||
| APTT (seconds) | 28.6 – 37.6 | 29.7 ± 2.9 | 29.7 ± 3.2 | 29.6 ± 2.6 | 29.8 ± 2.7 | 0.9546 | - | - | - |
| PT (seconds) | 8.2 – 12.2 | 8.2 (7.8–8.6) | 8.3 (7.9–8.6) | 7.9 (7.6–8.8) | 8.2 (8.1–8.4) | 0.3436 | - | - | - |
| Thrombin Generation Parameters | |||||||||
| Lag time (minutes) | - | 2.3 (2.0–2.7) | 2.3 (2.0–2.7) | 2.3 (2.0–2.8) | 2.3 (2.0–2.3) | 0.5985 | - | - | - |
| Time to Peak (minutes) | - | 4.7 (4.0–5.5) | 4.7 (4.0–5.5) | 4.7 (4.1–5.4) | 4.8 (4.2–5.5) | 0.9720 | - | - | - |
| Peak Thrombin (nM) | - | 318 ± 93 | 317 ± 80 | 345 ± 107 | 283 ± 83 | 0.0470 | - | 0.0420 | - |
| ETP (nM*minute) | - | 1393 (1211–1581) | 1382 (1223–1581) | 1498 (1368–1797) | 1206 (1148–1363) | 0.0002 | 0.0179 | 0.0001 | 0.0085 |
APTT, activated partial thromboplastin time; PT, prothrombin time; ETP, endogenous thrombin potential. FXIII, factor XIII. Continuous data show means ± standard deviation or medians (interquartile range), as appropriate.
Differences between genotypes were analyzed by one-way ANOVA with Bonferroni post-hoc testing or Kruskal-Wallis test with Dunn’s post-hoc testing.
To identify relationships between general demographic and clinical characteristics and clot mass, we reconstituted the individual plasma samples with O-negative RBCs and washed platelets isolated from a FXIIIVal/Val donor, and triggered clotting by addition of TF and CaCl2 (Figure 1A). Following clot formation and contraction, we measured contracted clot mass. In aggregate, average normalized clot mass correlated significantly with age, fibrinogen, PT, and thrombin generation lag time, time to peak, and ETP, but did not correlate with total FXIII activity (Table 2).
Figure 1. In reconstituted whole blood with human donor plasma, the FXIII 34Leu variant mitigates the effect of high fibrinogen concentration on contracted clot mass.
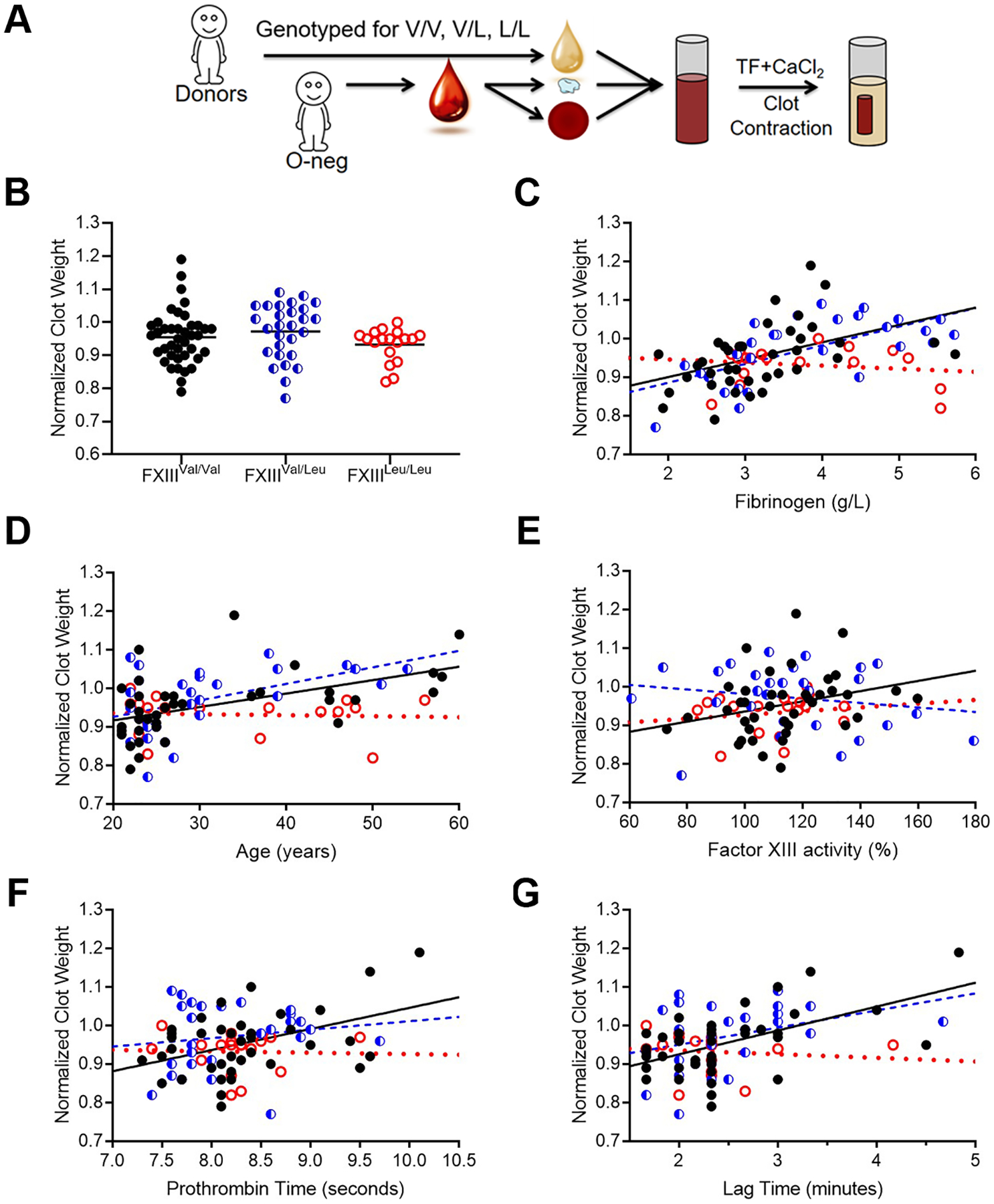
(A) Platelet-poor plasmas from 86 healthy donors (40 FXIIIVal/Val, 28 FXIIIVal/Leu, and 18 FXIIILeu/Leu) were reconstituted with washed platelets and O-negative red blood cells. Clotting was triggered with tissue factor and CaCl2, and contracted clots were weighed after 2 hours. Clot mass was normalized between assays performed on two different days. (B) Normalized clot mass. (C-F) Normalized clot mass were plotted as a function of (C) fibrinogen, (D) age, (E) FXIII activity, (F) prothrombin time, and (G) thrombin generation lag time. Lines indicate linear fits. Symbols are: FXIIIVal/Val (black closed circles), FXIIIVal/Leu (blue half-filled circles), and FXIIILeu/Leu (red open circles).
Table 2.
Characteristics associated with normalized clot mass.
| All (N=86) | FXIIIVal/Val (N=40) | FXIIIVal/Leu (N=28) | FXIIILeu/Leu (N=18) | |||||
|---|---|---|---|---|---|---|---|---|
| R | P | R | P | R | P | R | P | |
| Clinical Characteristics | ||||||||
| Age (years) | 0.3602 | 0.0007 | 0.5244 | 0.0005 | 0.4113 | 0.0297 | −0.1488 | 0.5557 |
| FXIII Activity (%) | 0.1126 | 0.3019 | 0.3506 | 0.0266 | −0.1700 | 0.3872 | 0.2193 | 0.3819 |
| Fibrinogen (g/L) | 0.5267 | <0.0001 | 0.5496 | 0.0002 | 0.6461 | 0.0002 | −0.0298 | 0.9066 |
| Clotting Tests | ||||||||
| APTT (seconds) | 0.0573 | 0.6004 | 0.0286 | 0.8610 | 0.1230 | 0.5330 | −0.1755 | 0.4862 |
| PT (seconds) | 0.2248 | 0.0374 | 0.3127 | 0.0495 | 0.1017 | 0.6065 | 0.0614 | 0.8088 |
| Thrombin Generation Parameters | ||||||||
| Lag time (minutes) | 0.3371 | 0.0015 | 0.4581 | 0.0030 | 0.2615 | 0.1790 | −0.3436 | 0.1627 |
| Time to Peak (minutes) | 0.3036 | 0.0045 | 0.3159 | 0.0471 | 0.3595 | 0.0603 | −0.1216 | 0.6306 |
| Peak Thrombin (nM) | −0.0531 | 0.6272 | −0.0297 | 0.8556 | −0.1061 | 0.5911 | 0.0606 | 0.8113 |
| ETP (nM*minute) | 0.2495 | 0.0205 | 0.2597 | 0.1056 | 0.0252 | 0.8986 | 0.2486 | 0.3199 |
R, Spearman coefficients; APTT, activated partial thromboplastin time; PT, prothrombin time; ETP, endogenous thrombin potential
Comparison of individual FXIII genotypes showed average normalized clot mass did not differ between groups (Figure 1B). Clot mass correlated strongly and significantly with fibrinogen in plasmas containing a 34Val allele (FXIIIVal/Val [R=0.5496] and FXIIIVal/Leu [R=0.6461], P=0.0002), whereas mass of clots with homozygous 34Leu did not change (R=−0.0298, P=0.9066) Table 2, Figure 1C). Clot mass also correlated with age, FXIII activity, PT, and thrombin generation lag time and time to peak in clots with FXIIIVal/Val, but not in clots with FXIIIVal/Leu (Table 2, Figure 1D–G). Clot mass did not correlate significantly with any of the measured demographic or clinical characteristic for clots with FXIIILeu/Leu.
Interaction between FXIII-A Val34Leu and fibrinogen in a reconstituted model using individual plasmas.
Given the modifying effects of fibrinogen concentration on FXIII-A genotype-dependent fibrin structure [15], we further interrogated specific relationships between fibrinogen and clot mass for each FXIII-A genotype. Clots formed from individual genotypes did not differ at low-to-normal fibrinogen concentrations. However, in plasmas with the highest fibrinogen (>3.5 g/L) the mass of clots formed from individuals carrying a 34Val allele was higher than those from individuals with homozygous 34Leu and these differences persisted even after adjusting for age, gender, and thrombin generation (Table 3).
Table 3.
Effects of fibrinogen and FXIII-A 34Val or 34Leu alleles on clot mass.
| FXIIIVal/Val | FXIIIVal/Leu | FXIIILeu/Leu | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| fibrinogen < 3.5 g/L (N=28) | fibrinogen > 3.5 g/L (N=12) | P | fibrinogen < 3.5 g/L (N=14) | fibrinogen > 3.5 g/L (N=14) | P | fibrinogen < 3.5 g/L (N=10) | fibrinogen > 3.5 g/L (N=8) | P | P% | |
| Normalized clot weight | 0.93±0.07 | 1.02±0.08 | 0.002* 0.002$ 0.010# |
0.92±0.08 | 1.02±0.05 | 0.001* <0.0001$ 0.001# |
0.93±0.04 | 0.93±0.06 | 1.000* 0.622$ 0.851# |
0.0001 <0.0001 <0.0001 |
Data show mean ± standard deviation. Groups were compared using
ANOVA with Bonferroni post-hoc test,
ANCOVA with Bonferroni post-hoc test adjusted for age and gender, or
ANCOVA with Bonferroni post-hoc test adjusted for age, gender, and endogenous thrombin potential. Findings were similar when adjusted for age, gender, and lag time or time to peak.
ANOVA comparing all groups
Interaction between FXIII-A Val34Leu and fibrinogen in a reconstituted model using purified FXIII zymogen.
To experimentally circumvent potential effects of age, thrombin generation, or other modifiers on clot mass, we then utilized a fully reconstituted system comprised of commercially-sourced FXIII-depleted plasma, washed O-negative RBCs, washed platelets (FXIIIVal/Val donor), and FXIII-free fibrinogen (Supplemental Figure 1), in which we added either purified FXIIIVal/Val or FXIIILeu/Leu zymogen (Figure 2A, Supplemental Figure 2). We then triggered clotting by addition of TF and CaCl2 and measured clot mass. Like that seen in experiments with individual plasmas, mass was similar for clots formed in the presence of lower fibrinogen (<4 g/L), and addition of fibrinogen induced a concentration-dependent increase in FXIIIVal/Val clot mass (R2=0.9536, P<0.005, Figure 2B). Interestingly, the biochemical control offered by this experimental design unveiled an inverse effect of fibrinogen on the mass of clots formed with FXIIILeu/Leu, in which clot mass decreased with increasing fibrinogen (R2=0.8416, P<0.03, Figure 2B). These surprising findings suggest an even higher specific impact of the FXIII-A Val34Leu polymorphism on clot structure than can be appreciated from analysis of heterogeneous plasma samples. Collectively, combined observations from these complimentary methods suggest the FXIII-A 34Leu allele mitigates prothrombotic effects of elevated fibrinogen on contracted whole blood clot mass.
Figure 2. In reconstituted whole blood with purified FXIII, the FXIII 34Leu variant alters contracted clot mass in a fibrinogen concentration-dependent manner.
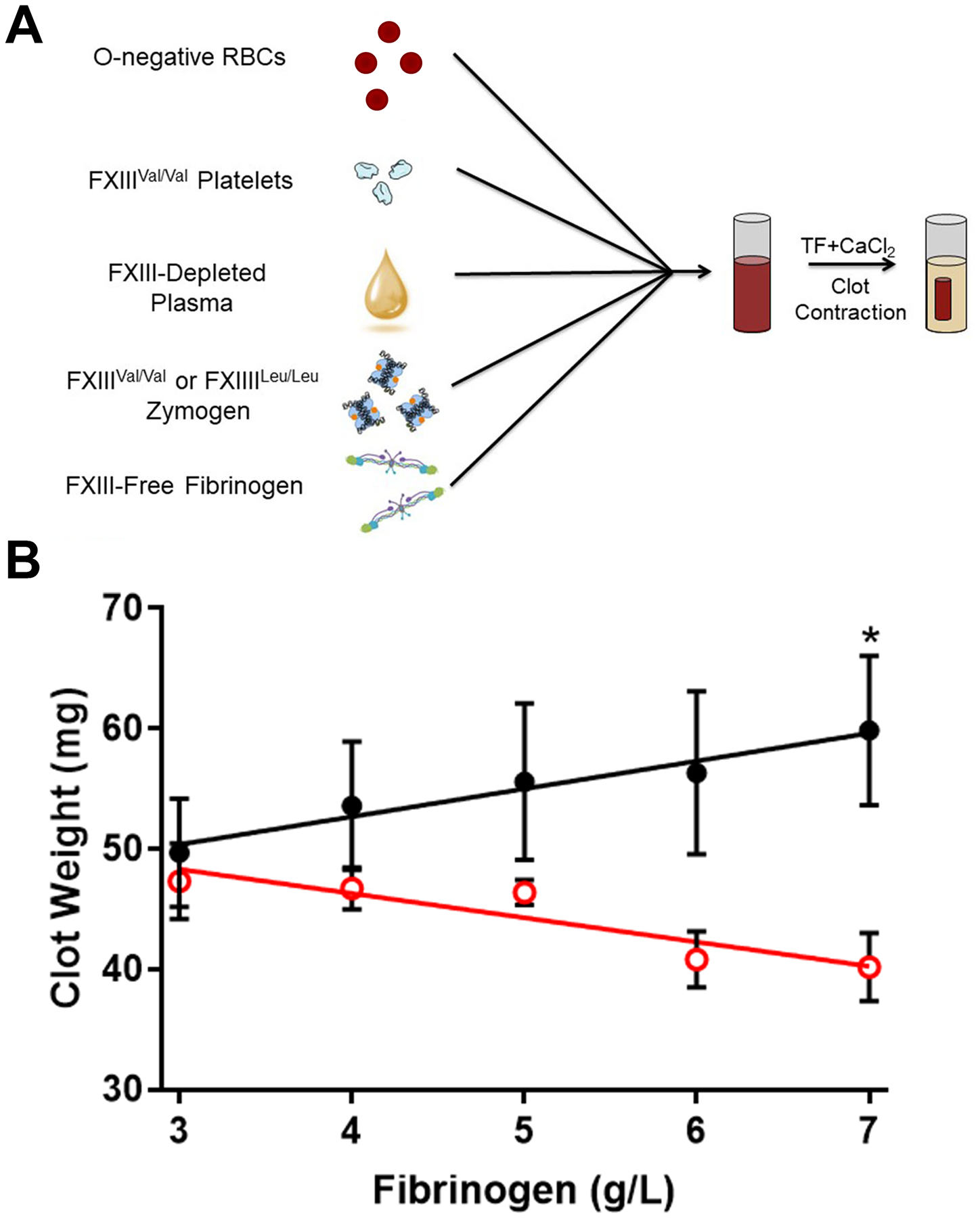
(A) O-negative red blood cells, FXIIIVal/Val platelets, and FXIII-depleted plasma reconstituted with either FXIIIVal/Val or FXIIILeu/Leu zymogen, were combined. FXIII-free fibrinogen (peak 1) was added to reach the final concentrations indicated in panel B. Clotting was triggered with tissue factor and CaCl2, and contracted clots were weighed at 2 hours. (B) Differences between FXIIIVal/Val (black closed circles) and FXIIILeu/Leu (red open circles) clot mass for each fibrinogen concentration were compared by t-test. Data show mean ± standard error of the mean (N=3). *P<0.05
DISCUSSION
Complex gene-environment interactions make univariable studies on humans or blood samples difficult to interpret. Early conflicting reports on the contribution of the FXIII-A Val34Leu polymorphism have been partly resolved by incorporation of co-modifiers into the analysis, revealing intriguing interactions with fibrinogen concentration as a determinant of fibrin structure. Although individuals with the 34Leu allele make clots with thinner fibrin fibers, effects of 34Leu are reversed in the presence of high fibrinogen [15]. These observations establish a rational, but complex, explanation for the protection of 34Leu against thrombosis by associating protective effects specifically with the high risk setting of elevated fibrinogen. Nonetheless, understanding the effects of the 34Leu allele on clot formation has remained challenging. A major advance of our study is the extension of observations from a reductionist setting in cell-free plasma to a more holistic model that includes blood cells, platelet-mediated contraction, and clot consolidation. Together with previous findings, our data suggest the 34Leu polymorphism affects not only fibrin network structure, but also the way in which fibrin modifies the composition and mass of whole blood clots.
Using a thrombosis model triggered by FeCl3 injury to the femoral vein, Duval et al previously observed that the FXIII Val34Leu polymorphism accelerates fibrin crosslinking, but does not alter clot size [20]. FeCl3 injures the endothelium in flowing blood and induces rapid formation (< 5 minutes) of a platelet-rich thrombus that remains unchanged in size over the 60-minute observation period [20]. In contrast, our in vitro clot contraction model recapitulates aspects of venous thrombosis in which stasis induces formation of RBC-rich thrombi that undergo subsequent clot contraction and consolidation. Indeed, we have observed differences in the contribution of FXIII to thrombi triggered by FeCl3 under flow [33] and thrombi triggered by venous stasis [24, 25], suggesting different effects of the FXIII V34L polymorphism on clot size in these two studies stem from differences in the role of FXIII in these experimental models.
Although mechanism(s) mediating the effects of the Val34Leu polymorphism on FXIII(a) activation are unclear, biochemical and structural analyses suggest the leucine side chain forms a more energetically favorable interaction with thrombin, enhancing substrate specificity and facilitating FXIII activation [34]. Fibrin crosslinking has only minimal effects on fibrin network density and pore size [26]. However, in plasmas with high fibrinogen, presence of the 34Leu allele produces thicker fibers in clots that have increased permeability [15], and we now show that these whole blood clots are smaller. Together with earlier studies [24–26], we speculate that changes in the timing of crosslinking affects the acquisition of viscoelastic properties of individual fibrin fibers within the clot, and disruption of this coordination compromises the events that stabilize fibrin, permitting loss of RBCs from the clot and the formation of smaller clots. It is interesting to note different effects of accelerated FXIII activation due to supraphysiologic FXIII [27] versus accelerated activation of physiologic levels of FXIII (present study); whereas supraphysiologic FXIII levels enhance crosslinking and increase clot size [27], accelerated activation of physiologic FXIII levels result in the production of smaller clots. These differences may reflect effects of altering the molecular ratio of FXIII:fibrin(ogen) and therefore the number of fibers that are crosslinked quickly in each setting. Studies that incorporate detailed analysis of crosslinking patterns induced during clot formation in concert with structural analysis of whole blood clots are needed to test this hypothesis.
Given the reproducible finding that 34Leu decreases the impact of elevated fibrinogen on clot mass, it is interesting to consider why this effect has been so difficult to detect in population studies. Other modifiers both within and external to the fibrinogen and FXIII loci likely alter the relative contribution of the Val34Leu polymorphism to clot formation, structure, and stability. For example, differential expression of the alternatively-spliced fibrinogen γ′ chain that binds and restricts thrombin activity also modifies the effect of elevated fibrinogen on clot formation [35–37]. In lieu of information on the relative expression of γA and γ′ fibrinogen in a given plasma, it would be difficult to predict the effect of increased fibrinogen a priori on clot formation. Fibrin(ogen) function may also be altered by post-translational modifications, including glycation and homocysteinylation in diabetes and other pathologies. Other polymorphisms in FXIII have also been implicated as modifiers of FXIII function, including the FXIII-B intron K c.1952+144 C>G polymorphism associated with lower FXIII activity and antigen, which appears to be protective against coronary artery disease only in the presence of both the FXIII-A 34Leu allele and elevated fibrinogen [38]. Thus, although our analysis resolved one aspect of these gene-environment interactions, complete understanding of how these mechanisms operate during thrombus formation in vivo will require further studies.
We previously showed that changes in fibrin crosslinking that reduce clot mass in vitro (observed in both human and mouse blood) decrease venous thrombus mass in mice [24–26]. We have also observed that elevated prothrombin, an established risk factor for venous thrombosis in humans, increases fibrin density and thrombus mass in mice [39]. Collectively, these observations suggest that mechanisms that enhance thrombus size promote thrombosis, whereas mechanisms that decrease thrombus size reduce venous thrombosis risk. It is unclear whether smaller thrombi would have reduced propensity to occlude blood flow, enhanced susceptibility to endogenous fibrinolytic mechanisms, or both. Further work is necessary to understand the implications of altered thrombus mass in venous thromboembolic risk in humans, as well how reduced thrombus RBC content caused by altered FXIII function affect thrombus formation and resolution.
In summary, we have shown that the FXIII-A 34Leu allele mitigates effects of elevated fibrinogen on the mass of contracted whole blood clots. The FXIII-A Val34Leu polymorphism may protect against venous thrombosis by decreasing clot RBC retention and consequently, reducing thrombus mass.
Supplementary Material
ESSENTIALS.
Factor XIII (FXIII) promotes fibrin crosslinking and red blood cell retention in clots
FXIII-A Val34Leu polymorphism offers modest but significant protection against venous thrombosis
Fibrinogen increases the mass of clots containing 34Val alleles, but not homozygous 34Leu
The FXIII-A Val34Leu polymorphism may protect against venous thrombosis by reducing clot mass
Acknowledgements:
The authors thank Drs. Lori Holle and Joan Beckman for excellent technical assistance.
Funding:
This study was supported by funding from the National Institutes of Health (Integrative Biology Training Program [T32HL69768 to UNC/S.K.], F31HL139100 to S.K., and R56HL094740 and R01HL126974 to A.S.W.), and the Hungarian National Research Development and Innovation Fund (FK128582, K120042) and GINOP-2.3.2-15-2016-00043 and GINOP-2.3.2.-15-2016-00050 co-financed by the European Union and the European Regional Development Fund (to Z.B. and L.M.).
Footnotes
Conflict-of-interest disclosure: The authors declare no competing financial interests.
REFERENCES
- 1.Byrnes JR, Wilson C, Boutelle AM, Brandner CB, Flick MJ, Philippou H, Wolberg AS. The interaction between fibrinogen and zymogen FXIII-A2B2 is mediated by fibrinogen residues gamma390–396 and the FXIII-B subunits. Blood. 2016; 128: 1969–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komaromi I, Bagoly Z, Muszbek L. Factor XIII: novel structural and functional aspects. J Thromb Haemost. 2011; 9: 9–20. [DOI] [PubMed] [Google Scholar]
- 3.Glover CJ, McIntire LV, Brown CH 3rd, Natelson EA. Rheological properties of fibrin clots. Effects of fibrinogen concentration, Factor XIII deficiency, and Factor XIII inhibition. J Lab Clin Med. 1975; 86: 644–56. [PubMed] [Google Scholar]
- 4.Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through alpha(2)-antiplasmin cross-linking. Blood. 2011; 117: 6371–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. 1998; 273: 27220–4. [DOI] [PubMed] [Google Scholar]
- 6.van Giezen JJ, Minkema J, Bouma BN, Jansen JW. Cross-linking of alpha 2-antiplasmin to fibrin is a key factor in regulating blood clot lysis: species differences. Blood Coagul Fibrinolysis. 1993; 4: 869–75. [PubMed] [Google Scholar]
- 7.Muszbek L, Katona E. Diagnosis and Management of Congenital and Acquired FXIII Deficiencies. Semin Thromb Hemost. 2016; 42: 429–39. [DOI] [PubMed] [Google Scholar]
- 8.Muszbek L Deficiency causing mutations and common polymorphisms in the factor XIII-A gene. Thromb Haemost. 2000; 84: 524–7. [PubMed] [Google Scholar]
- 9.Voko Z, Bereczky Z, Katona E, Adany R, Muszbek L. Factor XIII Val34Leu variant protects against coronary artery disease. A meta-analysis. Thromb Haemost. 2007; 97: 458–63. [PubMed] [Google Scholar]
- 10.Byrnes JR, Wolberg AS. Newly-recognized roles of factor XIII in thrombosis. Semin Thromb Hemost. 2016; 42: 445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Catto AJ, Kohler HP, Coore J, Mansfield MW, Stickland MH, Grant PJ. Association of a common polymorphism in the factor XIII gene with venous thrombosis. Blood. 1999; 93: 906–8. [PubMed] [Google Scholar]
- 12.Franco RF, Reitsma PH, Lourenco D, Maffei FH, Morelli V, Tavella MH, Araujo AG, Piccinato CE, Zago MA. Factor XIII Val34Leu is a genetic factor involved in the etiology of venous thrombosis. Thromb Haemost. 1999; 81: 676–9. [PubMed] [Google Scholar]
- 13.Wells PS, Anderson JL, Scarvelis DK, Doucette SP, Gagnon F. Factor XIII Val34Leu variant is protective against venous thromboembolism: a HuGE review and meta-analysis. Am J Epidemiol. 2006; 164: 101–9. [DOI] [PubMed] [Google Scholar]
- 14.Renner W, Koppel H, Hoffmann C, Schallmoser K, Stanger O, Toplak H, Wascher TC, Pilger E. Prothrombin G20210A, factor V Leiden, and factor XIII Val34Leu: common mutations of blood coagulation factors and deep vein thrombosis in Austria. Thromb Res. 2000; 99: 35–9. [DOI] [PubMed] [Google Scholar]
- 15.Lim BC, Ariens RA, Carter AM, Weisel JW, Grant PJ. Genetic regulation of fibrin structure and function: complex gene-environment interactions may modulate vascular risk. Lancet. 2003; 361: 1424–31. [DOI] [PubMed] [Google Scholar]
- 16.van Hylckama Vlieg A, Komanasin N, Ariens RA, Poort SR, Grant PJ, Bertina RM, Rosendaal FR. Factor XIII Val34Leu polymorphism, factor XIII antigen levels and activity and the risk of deep venous thrombosis. Br J Haematol. 2002; 119: 169–75. [DOI] [PubMed] [Google Scholar]
- 17.Vossen CY, Rosendaal FR. The protective effect of the factor XIII Val34Leu mutation on the risk of deep venous thrombosis is dependent on the fibrinogen level. J Thromb Haemost. 2005; 3: 1102–3. [DOI] [PubMed] [Google Scholar]
- 18.Balogh I, Szoke G, Karpati L, Wartiovaara U, Katona E, Komaromi I, Haramura G, Pfliegler G, Mikkola H, Muszbek L. Val34Leu polymorphism of plasma factor XIII: biochemistry and epidemiology in familial thrombophilia. Blood. 2000; 96: 2479–86. [PubMed] [Google Scholar]
- 19.Ariëns RA, Philippou H, Nagaswami C, Weisel JW, Lane DA, Grant PJ. The factor XIII V34L polymorphism accelerates thrombin activation of factor XIII and affects cross-linked fibrin structure. Blood. 2000; 96: 988–95. [PubMed] [Google Scholar]
- 20.Duval C, Ali M, Chaudhry WW, Ridger VC, Ariens RA, Philippou H. Factor XIII A-subunit V34L variant affects thrombus cross-Linking in a murine model of thrombosis. Arterioscler Thromb Vasc Biol. 2016; 36: 308–16. [DOI] [PubMed] [Google Scholar]
- 21.Undas A, Brzezinska-Kolarz B, Brummel-Ziedins K, Musial J, Szczeklik A, Mann KG. Factor XIII Val34Leu polymorphism and gamma-chain cross-linking at the site of microvascular injury in healthy and coumadin-treated subjects. J Thromb Haemost. 2005; 3: 2015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mari D, Mannucci PM, Coppola R, Bottasso B, Bauer KA, Rosenberg RD. Hypercoagulability in centenarians: the paradox of successful aging. Blood. 1995; 85: 3144–9. [PubMed] [Google Scholar]
- 23.Danesh J, Lewington S, Thompson SG, Lowe GD, Collins R, Kostis JB, Wilson AC, Folsom AR, Wu K, Benderly M, Goldbourt U, Willeit J, Kiechl S, Yarnell JW, Sweetnam PM, Elwood PC, Cushman M, Psaty BM, Tracy RP, Tybjaerg-Hansen A, Haverkate F, de Maat MP, Fowkes FG, Lee AJ, Smith FB, Salomaa V, Harald K, Rasi R, Vahtera E, Jousilahti P, Pekkanen J, D’Agostino R, Kannel WB, Wilson PW, Tofler G, Arocha-Pinango CL, Rodriguez-Larralde A, Nagy E, Mijares M, Espinosa R, Rodriquez-Roa E, Ryder E, Diez-Ewald MP, Campos G, Fernandez V, Torres E, Marchioli R, Valagussa F, Rosengren A, Wilhelmsen L, Lappas G, Eriksson H, Cremer P, Nagel D, Curb JD, Rodriguez B, Yano K, Salonen JT, Nyyssonen K, Tuomainen TP, Hedblad B, Lind P, Loewel H, Koenig W, Meade TW, Cooper JA, De Stavola B, Knottenbelt C, Miller GJ, Cooper JA, Bauer KA, Rosenberg RD, Sato S, Kitamura A, Naito Y, Palosuo T, Ducimetiere P, Amouyel P, Arveiler D, Evans AE, Ferrieres J, Juhan-Vague I, Bingham A, Schulte H, Assmann G, Cantin B, Lamarche B, Despres JP, Dagenais GR, Tunstall-Pedoe H, Woodward M, Ben-Shlomo Y, Davey Smith G, Palmieri V, Yeh JL, Rudnicka A, Ridker P, Rodeghiero F, Tosetto A, Shepherd J, Ford I, Robertson M, Brunner E, Shipley M, Feskens EJ, Kromhout D, Dickinson A, Ireland B, Juzwishin K, Kaptoge S, Lewington S, Memon A, Sarwar N, Walker M, Wheeler J, White I, Wood A. Plasma fibrinogen level and the risk of major cardiovascular diseases and nonvascular mortality: an individual participant meta-analysis. Jama. 2005; 294: 1799–809. [DOI] [PubMed] [Google Scholar]
- 24.Kattula S, Byrnes JR, Martin SM, Holle LA, Cooley BC, Flick MJ, Wolberg AS. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood advances. 2018; 2: 25–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Aleman MM, Byrnes JR, Wang JG, Tran R, Lam WA, Di Paola J, Mackman N, Degen JL, Flick MJ, Wolberg AS. Factor XIII activity mediates red blood cell retention in venous thrombi. The Journal of clinical investigation. 2014; 124: 3590–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Byrnes JR, Duval C, Wang Y, Hansen CE, Ahn B, Mooberry MJ, Clark MA, Johnsen JM, Lord ST, Lam WA, Meijers JC, Ni H, Ariens RA, Wolberg AS. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. 2015; 126: 1940–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beckman JD, Holle LA, Wolberg AS. Factor XIII cotreatment with hemostatic agents in hemophilia A increases fibrin alpha-chain crosslinking. Journal of thrombosis and haemostasis : JTH. 2018; 16: 131–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shemirani AH, Muszbek L. Rapid detection of the factor XIII Val34Leu (163 G-->T) polymorphism by real-time PCR using fluorescence resonance energy transfer detection and melting curve analysis. Clin Chem Lab Med. 2004; 42: 877–9. [DOI] [PubMed] [Google Scholar]
- 29.Machlus KR, Colby EA, Wu JR, Koch GG, Key NS, Wolberg AS. Effects of tissue factor, thrombomodulin and elevated clotting factor levels on thrombin generation in the calibrated automated thrombogram. Thromb Haemost. 2009; 102: 936–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lorand L, Credo RB, Janus TJ. Factor XIII (fibrin-stabilizing factor). Methods Enzymol. 1981; 80 Pt C: 333–41. [DOI] [PubMed] [Google Scholar]
- 31.Karpati L, Penke B, Katona E, Balogh I, Vamosi G, Muszbek L. A modified, optimized kinetic photometric assay for the determination of blood coagulation factor XIII activity in plasma. Clin Chem. 2000; 46: 1946–55. [PubMed] [Google Scholar]
- 32.Clauss A Rapid physiological coagulation method in determination of fibrinogen. Acta Haematol. 1957; 17: 237–46. [DOI] [PubMed] [Google Scholar]
- 33.Tang Z, Kattula S, Holle LA, Cooley BC, Lin F-C, Wolberg AS. Factor XIII deficiency does not prevent FeCl3-induced carotid artery thrombus formation in mice. Res Pract Thromb Haemost. 2020; in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trumbo TA, Maurer MC. Examining thrombin hydrolysis of the factor XIII activation peptide segment leads to a proposal for explaining the cardioprotective effects observed with the factor XIII V34L mutation. J Biol Chem. 2000; 275: 20627–31. [DOI] [PubMed] [Google Scholar]
- 35.Walton BL, Getz TM, Bergmeier W, Lin FC, Uitte de Willige S, Wolberg AS. The fibrinogen gammaA/gamma’ isoform does not promote acute arterial thrombosis in mice. J Thromb Haemost. 2014; 12: 680–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Allan P, Uitte de Willige S, Abou-Saleh RH, Connell SD, Ariens RA. Evidence that fibrinogen gamma’ directly interferes with protofibril growth: implications for fibrin structure and clot stiffness. J Thromb Haemost. 2012; 10: 1072–80. [DOI] [PubMed] [Google Scholar]
- 37.Cooper AV, Standeven KF, Ariens RA. Fibrinogen gamma-chain splice variant gamma’ alters fibrin formation and structure. Blood. 2003; 102: 535–40. [DOI] [PubMed] [Google Scholar]
- 38.Mezei ZA, Bereczky Z, Katona E, Gindele R, Balogh E, Fiatal S, Balogh L, Czuriga I, Adany R, Edes I, Muszbek L. Factor XIII B subunit polymorphisms and the risk of coronary artery disease. Int J Mol Sci. 2015; 16: 1143–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aleman MM, Walton BL, Byrnes JR, Wang JG, Heisler MJ, Machlus KR, Cooley BC, Wolberg AS. Elevated prothrombin promotes venous, but not arterial, thrombosis in mice. Arterioscler Thromb Vasc Biol. 2013; 33: 1829–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.