

Abstract
Background:
Parkinson’s disease is the second most common neurodegenerative disorder and affects people from all ethnic backgrounds, yet little is known about the genetics of Parkinson’s disease in non-European populations. In addition, the overall identification of copy number variants at a genome-wide level has been understudied in Parkinson’s patients. The objective of this study was to understand the genome-wide burden of copy number variants in Latinos and its association with Parkinson’s disease.
Methods:
We used genome-wide genotyping data from 747 Parkinson’s disease patients and 632 controls from the Latin American Research Consortium on the Genetics of Parkinson’s disease.
Results:
Genome-wide copy number burden analysis showed that patients were significantly enriched for copy number variants overlapping known Parkinson’s disease genes compared with controls (odds ratio, 3.97; 95%CI, 1.69–10.5; P = 0.018). PRKN showed the strongest copy number burden, with 20 copy number variant carriers. These patients presented an earlier age of disease onset compared with patients with other copy number variants (median age at onset, 31 vs 57 years, respectively; P = 7.46 × 10−7).
Conclusions:
We found that although overall genome-wide copy number variant burden was not significantly different, Parkinson’s disease patients were significantly enriched with copy number variants affecting known Parkinson’s disease genes. We also identified that of 250 patients with early-onset disease, 5.6% carried a copy number variant on PRKN in our cohort. Our study is the first to analyze genome-wide copy number variant association in Latino Parkinson’s disease patients and provides insights about this complex disease in this understudied population.
Keywords: Parkinson’s disease, genetics, copy number variants, Latin America
Parkinson’s disease (PD) is the second most common neurodegenerative disorder and the fastest-growing cause of disability because of a neurological disorder in the world.1,2 PD is a multifactorial syndrome that is thought to be caused by the complex interaction of genetics, environmental factors, and aging.3
Evidence for the genetic basis of PD has increased substantially over the past decades.4–6 The first causal gene for PD, SNCA, was discovered in 19977, and its protein product (α-synuclein) was further shown to be a major component of Lewy bodies, the pathological hallmark of PD. Dominant pathogenic single-nucleotide variants (SNVs) in SNCA,8–11 as well as copy number variants (CNVs) such as duplication or triplication of the entire gene with a clear dose effect, have been reported.12–14 The discovery of SNCA was followed by that of PRKN,15 where both pathogenic SNVs and CNVs are associated usually with an autosomal-recessive, early-onset form of the disease.6 Almost exclusively, genetic discoveries in PD have focused on SNVs, and studies on CNVs have been infrequent.17–19 CNVs in PRKN, SNCA, PINK1, PARK7, and ATP13A2 (from more to less frequent) have been reported using a candidate gene approach,20–22 whereas no CNVs have been shown for LRRK2. To date, only 2 studies have investigated the burden of CNVs in PD at a genome-wide level, including exclusively European and Ashkenazi Jewish individuals,17, 19 with a sample size of 1672 and 432, respectively.
PD is a global disease affecting all ethnicities. Unfortunately, the majority of studies do not include individuals of non-European ancestry, creating a large gap in knowledge. This is especially true for Hispanics/Latinos. It is worth mentioning here that these terms are not interchangeable. The term “Latino” refers to populations from Latin America (Mexico, most of Central and South America, and some islands in the Caribbean), whereas the term “Hispanic” refers only to Spanish-speaking populations and does not include those Latin American countries where Portuguese or French is spoken. Because our cohort includes individuals from Brazil (who speak Portuguese), we refer to individuals from our cohort as Latinos, wheres most studies in the United States only include Hispanics. Despite Hispanics/Latinos being the largest and fastest-growing ethnic minority in the United States,23 they are critically underrepresented in most genetic studies.24 This is probably because of their complex admixed ancestry with influences primarily from European, Amerindian, and African populations. In the United States, the incidence and prevalence rates of PD among Hispanics are at least as high, if not higher, than in non-Hispanic whites, whereas the rates are lower for Asians and Blacks.25,26 Yet little is known about the genetics of PD in Hispanics/Latinos, especially the frequency and characteristics of CNVs. No genome-wide studies in this population have been performed to date.
To address the lack of diversity in PD genetic studies and to understand the genetic architecture of PD in Latinos, we created the Latin American Research Consortium on the Genetics of PD (LARGE-PD).27 For this study, we used genome-wide genotypes of 1497 individuals from LARGE-PD. The aim of this study was to elucidate genomic structural changes as well as assess the CNV burden in this cohort of Latino PD patients and controls.
Methods
As part of our ongoing collaborative effort within LARGE-PD,27 we examined data from a total of 1497 individuals (807 PD patients and 690 controls) recruited from 9 different sites across the following 5 countries: Peru (n = 721), Colombia (n = 351), Brazil (n = 227), Uruguay (n = 191), and Chile (n = 13). All patients were evaluated by a movement disorder specialist at each of the sites and met the UK PD Society Brain Bank clinical diagnostic criteria.28 Controls were selected from individuals from the same countries who did not have symptoms compatible with neurodegenerative disorders. All PD patients and controls provided signed informed consent according to the local ethical requirements of each site. All individuals were genotyped on Illumina’s Multi-Ethnic Global Array (MEGA; Illumina, San Diego, California) at the Genomics Core at the University of Washington. A total of 1,779,819 markers were available before quality control (QC).
We performed an initial round of QC using PLINK 1.90,29 based on single-nucleotide polymorphism (SNP) genotype data for all samples and following established protocols described in Niestroj et al.30 In summary, we excluded samples if they had a call rate < 0.96 or a discordant sex status and SNPs that had a genotyping rate of <0.98, a minor allele frequency < 0.05, and deviation from Hardy–Weinberg equilibrium with P < 0.001. We did not perform a heterozygosity check in our QC to increase the sensitivity in detecting large CNVs. The presence of a true large deletion might make a sample look like an outlier in a heterozygosity check.
We then performed pruning of the SNPs for linkage disequilibrium (–indep-pairwise 200 100 0.2) using PLINK.29 The 1000 Genomes population31 was used as a reference for visual clustering of the principal components analysis (PCA) to assess for population stratification.
For CNV calling, we focused only on autosomal CNVs because of the higher quality of CNV calls from nonsex chromosomes. A custom population B-allele frequency (BAF) file was generated as a reference before calling CNVs. Then, we created guanine cytosine (GC) wave-adjusted log R ratio (LRR) intensity files for all samples and employed PennCNV32 software to detect CNVs in our data set.
We assessed cryptic relatedness using KING33 software and excluded individuals who were closely related (up to second degree) to another participant in our cohort by using the unrelated algorithm in KING. We performed an intensity-based QC to remove samples with low-quality data as previously described in Huang et al.34 Following this step, all samples had an LRR standard deviation of <0.27, absolute value of waviness factor < 0.03, and a BAF drift < 0.0014.
Called CNVs were removed from the data set if they spanned < 20 markers, were < 20 kilobases (kb) in length, and had a SNP density < 0.0001 (amount of markers/length of CNV). SNP density was not considered for CNVs spanning ≥ 20 markers and ≥1 Mb in length, as larger CNVs are not likely to be artifacts. To ensure that only high-quality CNVs passed our filters, we implemented a quality score calculation for each CNV following the methods of Macé et al,35 in which various CNV metrics are combined to estimate the probability of a called CNV to be a consensus call. Quality scores ranged from 0 (lowest) to 1 (highest) for duplications and similarly from 0 to −1 for deletions, and CNVs with quality scores between −0.5 and 0.5 were filtered out. A subset of final QC-passed CNVs were also inspected visually by 5 investigators with expertise in the interpretation of BAF and LRR plots. CNVs were annotated for gene content using Ensembl36 including gene name and the corresponding exonic coordinates in hg19 assembly using bedtools 2.27.0.37
We calculated CNV burden for PD using different categories to evaluate the relative contribution on PD risk: (1) the carrier status of overall CNV burden, including CNVs in nongenic regions, (2) the carrier status of CNVs intersecting “any gene” but none of the PD genes, (3) the carrier status of CNVs intersecting a list of “known PD genes,” and (4) the carrier status of large CNVs (≥1 Mb in length). P values were adjusted with the false discovery rate (FDR) method to correct for multiple testing. For the overall CNV burden category, deletions and duplications were also analyzed separately. We selected 19 genes for the “known PD genes” category that were grouped as follows: 6 are well-established causal genes for PD (LRRK2, PRKN, PARK7, PINK1, SNCA, and VPS35); 1 is a susceptibility factor for PD with a large effect size (GBA); and 12 either result in a parkinsonian syndrome that sometimes overlaps PD or are putative causal genes for PD that have not been adequately validated (ATP13A2, DNAJC6, DNAJC13, EIF4G1, FBXO7, GIGYF2, HTRA2, PLA2G6, RAB39B, SYNJ1, TMEM230, and VPS13C).6,38,39
In addition, for those individuals carrying a CNV in PRKN, we sequenced the entire coding region to identify those who may be compound heterozygous.
To assess for the difference in CNV burden between PD patients and controls, we fitted a logistic regression model using the “glm” function of the stats package40 in R 3.6.0.41 Cox proportional hazards regression analyses and Kaplan–Meier curves were calculated using the survival package.42 For all burden analyses, odds ratio (OR), 95% confidence interval (95%CI), and significance were calculated. ORs were calculated by the exponential of the logistic regression coefficient. For Cox proportional hazards regression, hazard ratios (HRs) were calculated to allow for censored observations. Potential confounding variables were used as covariates and included age, sex, and the first 5 ancestry principal components for all regression models.
Results
We had available data from a total of 1497 individuals in LARGE-PD.27 All the samples passed call rate and sex checks. We excluded 39 individuals because of relatedness, and 79 because of failing our intensity-based QC steps. Thus, after QC, our final cohort included 1379 individuals (747 PD patients and 632 controls) from Peru (n = 677), Colombia (n = 320), Brazil (n = 192), Uruguay (n = 177), and Chile (n = 13). There were more men in the PD patient group than in the control group (53.2% vs 33.1%, P < 0.001). Sample demographics are shown in Table 1. To visualize the ethnic composition of our cohort, we performed PCA using 1000 Genome populations31 as a reference. Our samples overlapped strongly with the projection of Admixed Amerindian samples (Fig. S1).
TABLE 1.
Sample demographics with number of PD patients and controls with their characteristics following QC steps
| PD patients | Controls | P | |
|---|---|---|---|
| Number of samples | 747 | 632 | ns |
| CNV carriers | 692 | 582 | NA |
| Age (mean) | 62 | 56.6 | a |
| Age at onset (mean) | 54.4 | NA | NA |
| Sex, male (%) | 395 (53.2) | 209 (33.1) | a |
PD, Parkinson’s disease; CNV, copy number variant; ns, nonsignificant; NA, not applicable.
P ≤ 0.001.
The initial number of CNV calls was 249,101 including 176,462 deletions and 72,639 duplications. After all QC steps, including filtering by consensus quality score as described in the Methods, the final number of high-quality CNVs was 8412, including 5155 CNVs in PD patients and 3257 CNVs in controls. CNV analysis showed 1274 of the samples (92.4%) carrying at least 1 QC-passed CNV. The length of the CNVs in the overall cohort ranged from 20 kilobases (kb) to 3.4 Megabases (Mb), with a median size of 52.4 kb. CNV characteristics are shown in Table S1.
We applied logistic regression with age, sex, and the first 5 ancestry principal components included as covariates to compare the CNV burden in PD patients and controls on all categories defined earlier, adjusting P values for multiple testing (see Methods section for details). We found no significant difference in overall CNV burden (OR, 1.19; 95%CI, 0.78–1.8; P = 0.64), CNVs in any gene (OR, 1.07; 95%CI, 0.81–1.4; P = 0.77), and large CNVs that were ≥1 Mb in length (OR, 1.46; 95%CI, 0.82–2.65; P = 0.4); see Figure 1. Interestingly, 9 PD patients and 6 controls carried a ≥1 Mb duplication on chromosome 11 (P = 0.8), which did not overlap with any known gene region. We also calculated overall CNV burden independently in duplications (OR, 1.36; 95%CI, 1.05–1.77; P = 0.06) and deletions (OR, 0.98; 95%CI, 0.74–1.29; P = 0.89), and neither was statistically significant.
FIG. 1.
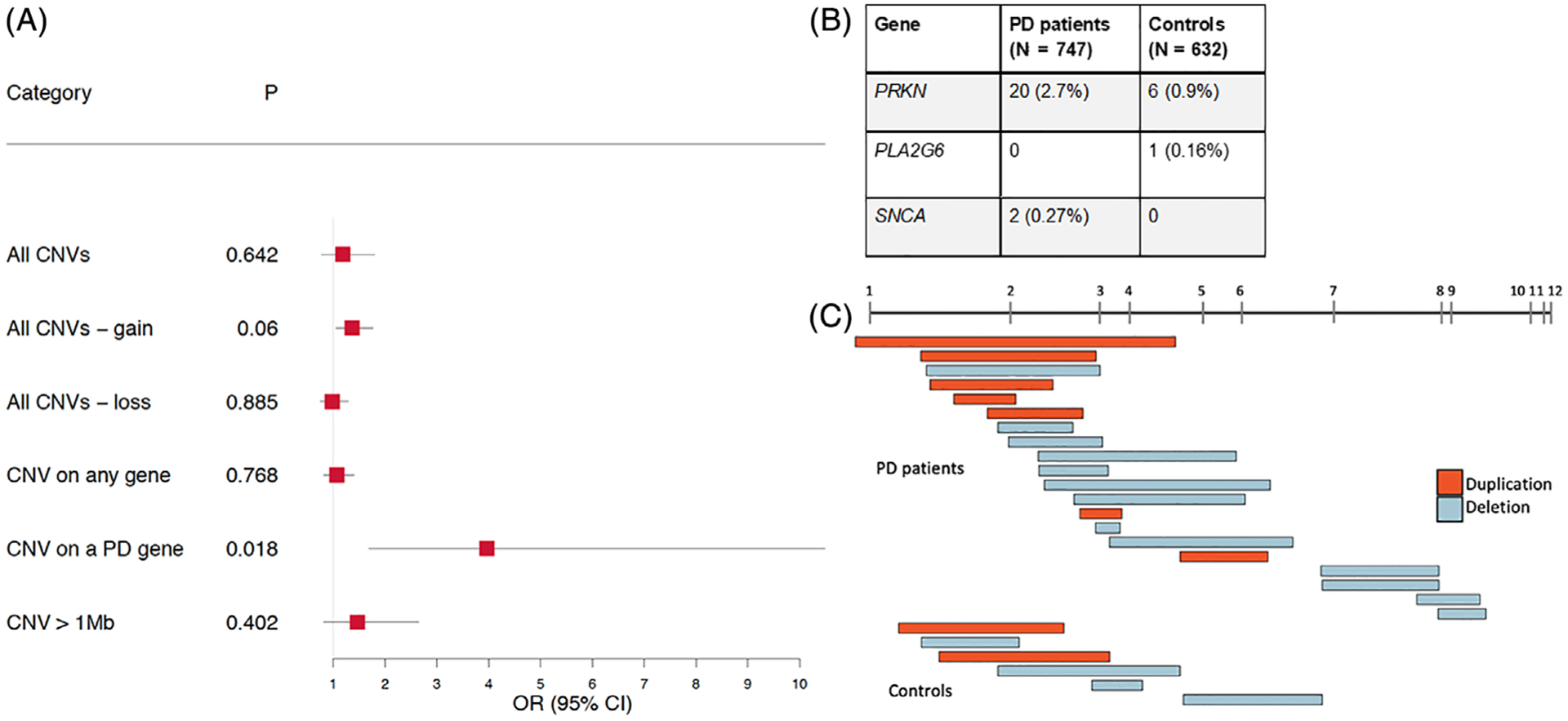
(A) Forest plot showing the CNV burden for PD patients compared with controls. Odds ratios (ORs) and P values were calculated using logistic regression for CNVs corrected for age, sex, and first 5 components of PCA. P values were adjusted with FDR for multiple testing. OR > 1 indicates an increased risk for PD per unit of CNV burden. (B) Table showing number of CNV carriers in any of the 19 known PD genes. (C) Visualization of CNVs in PRKN.
We then explored CNVs in genomic regions that were previously associated with typical PD and other parkinsonian phenotypes, and we found that PD patients were significantly enriched with CNVs overlapping these genes (OR, 3.97; 95%CI, 1.69–10.5; P = 0.018; Fig. 1). This finding was largely driven by CNVs in PRKN in 20 patients, followed by 2 patients with a CNV in SNCA, compared with 6 controls carrying a CNV in PRKN and none in SNCA. In addition, 1 control had a CNV in PLA2G6 (Fig. 1). Of the 20 PRKN CNV carriers, 2 were homozygous for a CNV and 5 were compound heterozygous for a CNV and an SNV (Table S2).
To assess whether PD patients carrying a PRKN CNV in our cohort had an earlier age at onset (AAO), we performed a Mann–Whitney test and compared patients who carry a CNV overlapping PRKN with those carrying a CNV overlapping any other gene. The median AAO for patients with a CNV overlapping PRKN was 31 years old, whereas that for patients with a CNV overlapping any other gene was 57 years old (P = 7.46 × 10−7).
To further investigate the CNV burden in early-onset PD (EOPD) patients, we performed a subset analysis in which cases with an AAO < 50 years old were compared with controls. Again, we observed a significant enrichment in CNVs in known PD genes in patients with EOPD (OR, 4.91; 95%CI, 1.92–13.68; P = 0.006). This result was also driven by CNVs in PRKN.
Kaplan–Meier estimates of AAO showed that individuals carrying a CNV in a known PD gene had significantly earlier onset of symptoms compared with individuals with other or no CNVs (log rank test, P < 0.001; Fig. 2). In this analysis, controls were also included but were censored observations because it is only known that they did not develop PD up to the age of their last visit. Using a Cox proportional hazards regression analysis with age, sex, and the first 5 ancestry principal components included as covariates, we found that the effect of carrying a CNV in a known PD gene on the hazard of AAO was highly significant (HR, 2.42; 95%CI, 1.57–3.71; P = 5.70 × 10−5).
FIG. 2.
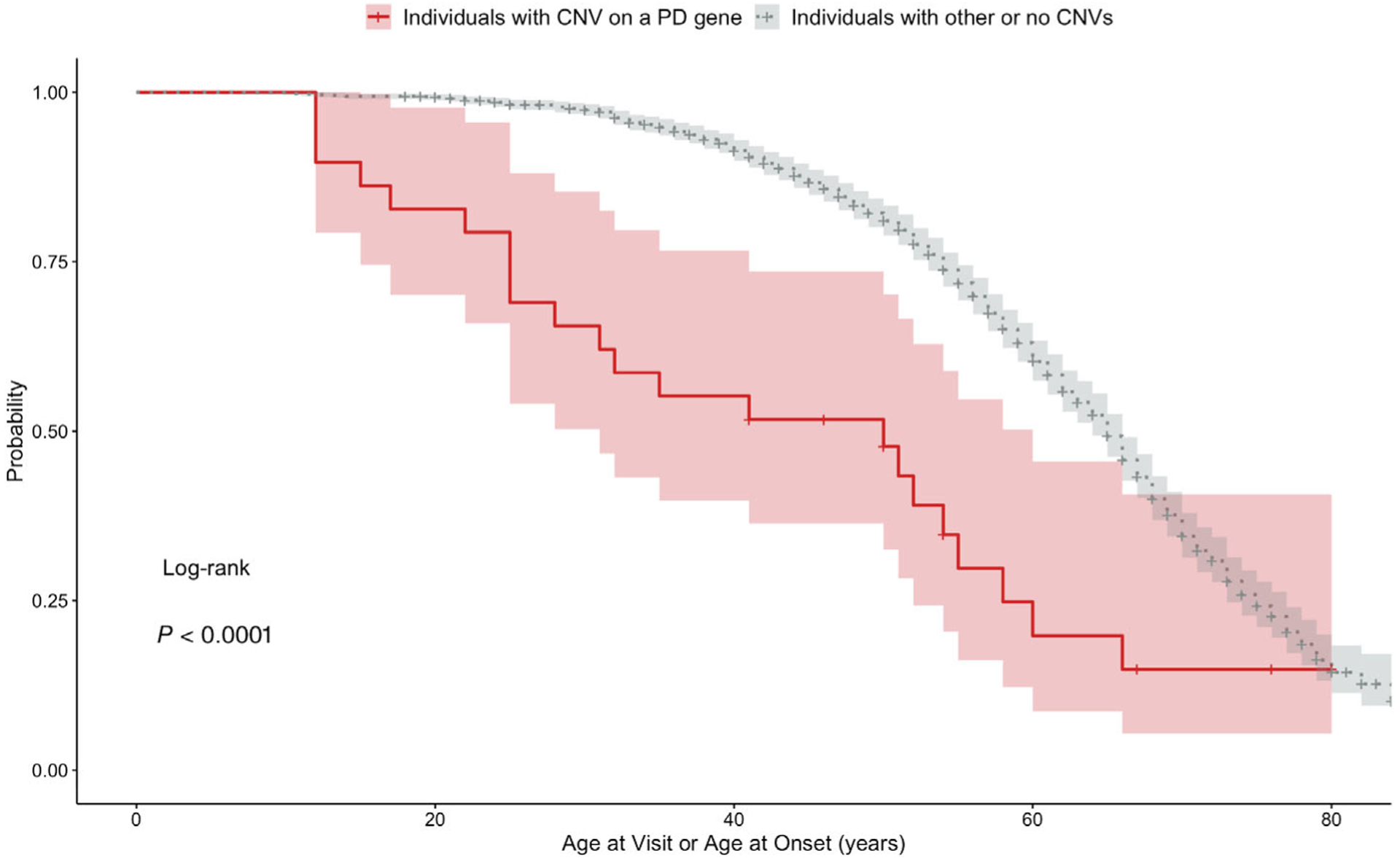
Kaplan–Meier estimates of individuals (PD patients and controls) carrying a CNV in a PD gene and individuals with other or no CNVs. Controls are censored observations because it is only known that they did not develop PD up to the age of their last visit. Probability is the probability of not having symptoms associated with PD. Age at visit or age at onset is time to onset of PD symptoms for cases and time to the last visit for controls. Highlight around the curves shows 95% confidence intervals.
We also assessed AAO in PD patients only, comparing CNV carriers on a known PD gene with PD patients with other or no CNVs and found that having at least 1 CNV results in earlier onset of symptoms (HR, 1.92; 95%CI, 1.22–3.02; P < 0.001; Fig. S2).
Discussion
Here, we present a genome-wide characterization of CNVs in a cohort of Latino PD patients and controls from LARGE-PD.27 We analyzed genotypes of 1497 individuals on the same platform and analyzed all samples with the same CNV calling and quality control pipeline. We used LARGE-PD controls from the same countries and controlled by ancestry using PCA. This is particularly important considering the large admixture present in these countries and that the data for Latino population frequency of CNVs is limited, especially in neurologically healthy adults.43,44 We assessed the CNV burden for different categories, and although we did not observe a difference in the overall genome-wide CNV burden, we did observe an increased burden of CNVs overlapping known PD genes in PD patients, largely driven by CNVs in PRKN. Our result agrees with what Pankratz and colleagues had previously shown in their genome-wide CNV analysis using only familial PD cases of European ancestry.17 Interestingly, no CNVs in PD genes were identified in the other genome-wide CNV burden analysis, which was performed in a cohort of Ashkenazi Jews.19 In our study, we identified 20 patients who carried CNVs overlapping PRKN (including 2 homozygote carriers) and 2 patients overlapping SNCA, 2 well-established PD genes, and found that 14 of these patients had a disease AAO < 50 years. The median AAO for patients with a CNV overlapping PRKN was almost 20 years earlier than that of other patients, in agreement with the literature.45–47
PRKN mutations are the most common genetic cause for EOPD (age of onset < 50 years),5,15,48 but an important caveat is that this information is mostly derived from studies in populations of European ancestry. The frequency of all PRKN pathogenic variants (CNVs and SNVs) in EOPD patients with European ancestry ranges from 49% in familial cases to 15%–18% in isolated patients, whereas the frequency of carrying a CNV in PRKN in isolated EOPD patients is approximately 10%.45,49,50 Some studies suggest that alterations in PRKN are more frequent in Hispanic populations. One study showed a 2.7-fold increase for carrying any PRKN alteration and a 2.8-fold increase for carrying a heterozygous mutation in Hispanic EOPD patients (n = 77) compared with white non-Hispanics, and the frequency of PRKN CNVs in Hispanic PD patients in this cohort was 6.4%.48 In another study examining Mexican-mestizo EOPD patients (n = 63), the frequency of PRKN CNVs was found to be 50%, and 18% of these patients were heterozygous.51 In our cohort, we had 250 patients with EOPD, and 5.6% of these patients (n = 14) carried a CNV in PRKN, with the majority being heterozygous for the CNV (12 of 14, 85.7%).
The role of homozygous and compound heterozygous variants, including CNVs in PRKN, is well known, especially in EOPD.5,15,45 However, there is also increasing evidence that PRKN heterozygosity is a risk factor for PD and is associated with a decreased AAO.45,46 In our cohort, most of the PRKN CNV carriers were heterozygous, and there was a significant association between the AAO and PRKN carrier status. Still, the role of heterozygous PRKN CNVs in altering PD susceptibility remains controversial.16,52 To correctly characterize PD patients, an integrated SNV-CNV analysis is needed, given the importance of both allele types for comprehensive genetic diagnosis in PD.53 Our sequencing of the entire PRKN coding region for all 26 PRKN CNV carriers identified that 5 of the 20 patients also carried a pathogenic SNV, whereas none of the 6 controls did. Two of these patients carried pathogenic variants that were not present in public databases — one with a frameshift variant in exon 6, and the other with an acceptor splice-site mutation (this family was reported by us elsewhere54). Details for these 5 compound heterozygous and the 2 CNV homozygous carriers are included in Table S2. This is very similar to the frequency reported in previous studies mentioned above.48,51
Pankratz et al showed that the frequency of carrying a single PRKN CNV was higher in PD patients compared with controls, whereas it was similar for carrying a single point mutation.55 Our results, together with previous studies,46,56 suggest that heterozygous PRKN CNV carrier status may still play a role in the development of PD despite its recessive inheritance, probably through a haploinsufficiency effect.57
One limitation of our study is that we did not validate all CNVs with a different method. However, from our overall cohort (n = 1379), 117 individuals (8.4%) were previously screened for CNVs in some PD genes (including PRKN and SNCA) using multiplex ligation-dependent probe amplification (MLPA; P052, MRC Holland) for other ongoing projects. This previous cohort included 77 of the 250 EOPD patients (30.8%) and 7 of the 22 patients that were found to be carrying a CNV in PRKN or SNCA in this current study. Results for all 77 samples matched, confirming negatives and all 7 positive carriers, showing that our CNV analysis pipeline was accurate and detected 100% of the known CNV carriers (with no false-positives) found with a different method.
The large genetic variation in Latinos because of admixture from several populations (mostly European, Amerindian, and African) creates a challenge when analyzing this population. For this reason, we established a workflow with rigorous quality control. We also constructed a Latino reference file from scratch for CNV calls, as publicly available reference files were all based on Europeans. In this study, we analyzed all samples together to boost statistical power. However, separate calling of CNVs in subpopulations based on admixture analysis is likely to yield more refined results. Thus, larger sample sizes will be needed to make discoveries specific to subpopulations of Latinos. Admixture mapping to examine the chromosomal location of the CNVs could also provide more insights about the relationship between PD genetics and ethnicity.58,59
To our knowledge, this is the first study that focuses on genome-wide CNVs in PD patients from Latin America. We believe that expanding the diversity of genetic studies for PD is necessary to understand the genetic profiles of these individuals and that our work will enrich current scientific knowledge about CNVs in this underrepresented population.
Supplementary Material
Acknowledgments:
We thank all the individuals who donated their samples as well as their time to participate in LARGE-PD, which made this and future projects possible. We would also thank all our collaborators at the different Latin American sites for their efforts and support for building this incredible resource.
Funding agencies:
Recruitment of LARGE-PD participants was funded by an International Research Program Grant from the Parkinson’s Disease Foundation (2010-2012), and this work was funded by a Stanley Fahn Junior Faculty Award from the Parkinson’s Foundation. This work was also supported by a research grant from the American Parkinson’s Disease Association, with resources and the use of facilities at the Veterans Affairs Puget Sound Health Care System. M. J. -D. -R. and C. V. -P. were supported by the Committee for Development and Research (Comite para el desarrollo y la investigación-CODI)-Universidad de Antioquia grant 2020-31455.
Appendix
Members of the Latin American Research Consortium on the Genetics of PD (LARGE-PD):
Argentina: Federico Micheli, Emilia Gatto.
Brazil: Vitor Tumas, Vanderci Borges, Henrique B. Ferraz, Carlos R.M. Rieder, Artur Shumacher-Schuh, Bruno L. Santos-Lobato.
Chile: Pedro Chaná.
Colombia: Carlos Velez-Pardo, Marlene Jimenez-Del-Rio, Francisco Lopera, Gonzalo Arboleda, Humberto Arboleda, Jorge Luis Orozco, Sonia Moreno, William Fernandez, Carlos E. Arboleda-Bustos.
Costa Rica: Jaime Fornaguera, Alvaro Hernández Guillén, Gabriel Torrealba Acosta.
Ecuador: Jorge Chang-Castello, Brennie Andreé Muñoz.
Honduras: Alex Medina, Anabelle Ferrera.
Mexico: Daniel Martinez-Ramirez, Mayela Rodriguez.
Peru: Mario Cornejo-Olivas, Pilar Mazzetti, Hugo Sarapura, Andrea Rivera, Luis Torres, Carlos Cosentino, Angel Medina.
Puerto Rico: Angel Viñuela.
Uruguay: Elena Dieguez, Victor Raggio, Andres Lescano, Ignacio Amorin.
Footnotes
Relevant conflicts of interest/financial disclosures: The authors report no financial disclosure/conflict of interest related to the current article.
Supporting Data
Additional Supporting Information may be found in the online version of this article at the publisher’s web-site.
References
- 1.de Lau LML, Breteler MMB. Epidemiology of Parkinson’s disease. Lancet Neurol 2006;5(6):525–535. [DOI] [PubMed] [Google Scholar]
- 2.GBD 2016 Parkinson’s disease collaborators. Global, regional, and national burden of Parkinson’s disease, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 2018;17(11):939–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noyce AJ, Bestwick JP, Silveira-Moriyama L, Hawkes CH, Giovannoni G, Lees AJ, Schrag A. Meta-analysis of early nonmotor features and risk factors for Parkinson disease. Ann Neurol 2012;72(6):893–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singleton AB, Farrer MJ, Bonifati V. The genetics of Parkinson’s disease: progress and therapeutic implications. Mov Disord 2013;28(1):14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Domingo A, Klein C. Chapter 14 — Genetics of Parkinson disease. In: Geschwind DH, Paulson HL, Klein C, editors. Handbook of Clinical Neurology. Amsterdam, Netherlands: Elsevier; 2018:211–227. [DOI] [PubMed] [Google Scholar]
- 6.Klein C, Westenberger A. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med 2012;2(1):a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Polymeropoulos MH, Lavedan C, Leroy E, et al. Mutation in the α-synuclein gene identified in families with Parkinson’s disease. Science 1997;276(5321):2045–2047. [DOI] [PubMed] [Google Scholar]
- 8.Krüger R, Kuhn W, Müller T, et al. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 1998;18(2):106–108. [DOI] [PubMed] [Google Scholar]
- 9.Zarranz JJ, Alegre J, Gómez-Esteban JC, et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 2004;55(2):164–173. [DOI] [PubMed] [Google Scholar]
- 10.Appel-Cresswell S, Vilarino-Guell C, Encarnacion M, et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 2013;28(6):811–813. [DOI] [PubMed] [Google Scholar]
- 11.Lesage S, Anheim M, Letournel F, et al. G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 2013;73(4):459–471. [DOI] [PubMed] [Google Scholar]
- 12.Singleton AB, Farrer M, Johnson J, et al. Alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003;302(5646):841. [DOI] [PubMed] [Google Scholar]
- 13.Ibáñez P, Bonnet AM, Débarges B, et al. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 2004;364(9440):1169–1171. [DOI] [PubMed] [Google Scholar]
- 14.Chartier-Harlin M-C, Kachergus J, Roumier C, et al. Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 2004;364(9440):1167–1169. [DOI] [PubMed] [Google Scholar]
- 15.Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998;392 (6676):605–608. [DOI] [PubMed] [Google Scholar]
- 16.Mata IF, Lockhart PJ, Farrer MJ. Parkin genetics: one model for Parkinson’s disease. Hum Mol Genet 2004;13(suppl_1): R127–R133. [DOI] [PubMed] [Google Scholar]
- 17.Pankratz N, Dumitriu A, Hetrick KN, et al. Copy number variation in familial Parkinson disease. PLoS One 2011;6(8):e20988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim J-S, Yoo J-Y, Lee K-S, et al. Comparative genome hybridization array analysis for sporadic Parkinson’s disease. Int J Neurosci 2008; 118(9):1331–1345. [DOI] [PubMed] [Google Scholar]
- 19.Liu X, Cheng R, Ye X, et al. Increased rate of sporadic and recurrent rare genic copy number variants in Parkinson’s disease among Ashkenazi Jews. Mol Genet Genomic Med 2013;1(3):142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Darvish H, Movafagh A, Omrani MD, et al. Detection of copy number changes in genes associated with Parkinson’s disease in Iranian patients. Neurosci Lett 2013;551:75–78. [DOI] [PubMed] [Google Scholar]
- 21.Toft M, Ross OA. Copy number variation in Parkinson’s disease. Genome Med 2010;2(9):62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ibáñez P, Lesage S, Janin S, et al. α-Synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol 2009;66(1):102–108. [DOI] [PubMed] [Google Scholar]
- 23.Colby SL, Ortman JM. Projections of the size and composition of the US population: 2014 to 2060, current population reports, Washington DC: Census Bureau; 2014: 25–1143. https://www.census.gov/content/dam/Census/library/publications/2015/demo/p25-1143.pdf [Google Scholar]
- 24.Popejoy AB, Fullerton SM. Genomics is failing on diversity. Nature 2016;538(7624):161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Van Den Eeden SK, Tanner CM, Bernstein AL, Fross RD, Leimpeter A, Bloch DA, Nelson LM. Incidence of Parkinson’s disease: variation by age, gender, and race/ethnicity. Am J Epidemiol 2003;157(11):1015–1022. [DOI] [PubMed] [Google Scholar]
- 26.Wright Willis A, Evanoff BA, Lian M, Criswell SR, Racette BA. Geographic and ethnic variation in Parkinson disease: a population-based study of US Medicare beneficiaries. Neuroepidemiology 2010; 34(3):143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zabetian CP, Mata IF. LARGE-PD: examining the genetics of Parkinson’s disease in Latin America. Mov Disord 2017;32(9): 1330–1331. [DOI] [PubMed] [Google Scholar]
- 28.Gibb WR, Lees AJ. The relevance of the Lewy body to the pathogenesis of idiopathic Parkinson’s disease. J Neurol Neurosurg Psychiatry 1988;51(6):745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chang CC, Chow CC, Tellier LCAM, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Niestroj L-M, Howrigan DP, Perez-Palma E, et al. Evaluation of copy number burden in specific epilepsy types from a genome-wide study of 18,564 subjects. preprint: Genetics; 2019; 2019/05/27/. [Google Scholar]
- 31.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature 2015;526(7571):68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang K, Li M, Hadley D, et al. PennCNV: an integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res 2007;17(11):1665–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, Chen W-M. Robust relationship inference in genome-wide association studies. Bioinformatics 2010;26(22):2867–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang AY, Yu D, Davis LK, et al. Rare copy number variants in NRXN1 and CNTN6 increase risk for Tourette syndrome. Neuron 2017;94(6):1101–1111.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Macé A, Tuke MA, Beckmann JS, et al. New quality measure for SNP array based CNV detection. Bioinformatics 2016;32(21):3298–3305. [DOI] [PubMed] [Google Scholar]
- 36.Ensembl. Nucleic acids research. Oxford, UK: Oxford Academic; 2019. [Google Scholar]
- 37.Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 2010;26(6):841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bandres-Ciga S, Diez-Fairen M, Kim JJ, Singleton AB. Genetics of Parkinson’s disease: an introspection of its journey towards precision medicine. Neurobiol Dis 2020;137:104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lunati A, Lesage S, Brice A. The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 2018;174(9):628–643. [DOI] [PubMed] [Google Scholar]
- 40.SurajGupta/r-source GitHub. https://github.com/SurajGupta/r-source
- 41.R: The R Project for Statistical Computing. https://www.r-project.org/
- 42.Therneau TM, Lumley T, Elizabeth A, et al. Survival: survival analysis. https://CRAN.R-project.org/package=survival. Accessed June 2020.
- 43.Redon R, Ishikawa S, Fitch KR, et al. Global variation in copy number in the human genome. Nature 2006;444(7118):444–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jakobsson M, Scholz SW, Scheet P, et al. Genotype, haplotype and copy-number variation in worldwide human populations. Nature 2008;451(7181):998–1003. [DOI] [PubMed] [Google Scholar]
- 45.Lücking CB, Dürr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000;342(21):1560–1567. [DOI] [PubMed] [Google Scholar]
- 46.Sun M, Latourelle JC, Wooten GF, et al. Influence of heterozygosity for Parkin mutation on onset age in familial Parkinson disease: the GenePD study. Arch Neurol 2006;63(6):826–832. [DOI] [PubMed] [Google Scholar]
- 47.Oliveira SA, Scott WK, Martin ER, et al. Parkin mutations and susceptibility alleles in late-onset Parkinson’s disease. Ann Neurol 2003;53(5):624–629. [DOI] [PubMed] [Google Scholar]
- 48.Marder KS, Tang MX, Mejia-Santana H, et al. Predictors of parkin mutations in early-onset Parkinson disease: the consortium on risk for early-onset Parkinson disease study. Arch Neurol 2010;67(6):731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedrich K, Marder K, Harris J, et al. Evaluation of 50 probands with early-onset Parkinson’s disease for parkin mutations. Neurology 2002;58(8):1239–1246. [DOI] [PubMed] [Google Scholar]
- 50.Periquet M, Latouche M, Lohmann E, et al. Parkin mutations are frequent in patients with isolated early-onset parkinsonism. Brain 2003;126(6):1271–1278. [DOI] [PubMed] [Google Scholar]
- 51.Camacho JLG, Jaramillo NM, Gómez PY, et al. High frequency of Parkin exon rearrangements in Mexican-mestizo patients with early-onset Parkinson’s disease. Mov Disord 2012;27(8): 1047–1051. [DOI] [PubMed] [Google Scholar]
- 52.Yu E, Rudakou U, Krohn L, et al. Analysis of heterozygous PRKN variants and copy number variations in Parkinson’s disease. Mov Disord 2020. 10.1002/mds.28299 [DOI] [PubMed] [Google Scholar]
- 53.Robak LA, Du R, Yuan B, et al. Integrated Sequencing & Array Comparative Genomic Hybridization in familial Parkinson’s disease. Neurol Genet 2020;6(5):e498 2019/11/11/. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cornejo-Olivas M, Torres L, Mata IF, et al. A Peruvian family with a novel PARK2 mutation: clinical and pathological characteristics. Parkinsonism Relat Disord 2015;21(5):444–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pankratz N, Kissell DK, Pauciulo MW, et al. Parkin dosage mutations have greater pathogenicity in familial PD than simple sequence mutations. Neurology 2009;73(4):279–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huttenlocher J, Stefansson H, Steinberg S, et al. Heterozygote carriers for CNVs in PARK2 are at increased risk of Parkinson’s disease. Hum Mol Genet 2015;24(19):5637–5643. [DOI] [PubMed] [Google Scholar]
- 57.Klein C, Lohmann-Hedrich K, Rogaeva E, Schlossmacher MG, Lang AE. Deciphering the role of heterozygous mutations in genes associated with parkinsonism. Lancet Neurol 2007;6(7):652–662. [DOI] [PubMed] [Google Scholar]
- 58.Genovese G, Handsaker RE, Li H, et al. Using population admixture to help complete maps of the human genome. Nat Genet 2013; 45(4):406–414.e1–e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lou H, Li S, Jin W, et al. Copy number variations and genetic admixtures in three Xinjiang ethnic minority groups. Eur J Hum Genet 2015;23(4):536–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.