

Summary:
Understanding resistance to BCL2 inhibition is a critical scientific and clinical challenge. In this issue of Cancer Discovery, two laboratories use unbiased approaches of large loss-of-function CRISPR/Cas 9 screens to discover targetable liabilities in cell signaling and metabolism to acute myeloid leukemia resistant to BCL2 inhibition.
Apoptosis is a carefully regulated cell death program initiated by noxious stimuli, inflammation, or cellular structural dysfunction orchestrated via the interactions of intra- and extra-mitochondrial proteins with competing roles in normal homeostasis. In apoptosis, the BH3-only protein BIM activates formation of mitochondrial membrane pores by interacting with BAX, cytochrome C is released through these mitochondrial pores, and caspase activation ensues (1). B-cell lymphoma 2 (BCL2) is a critical intramitochondrial protein that inhibits apoptosis by negatively regulating apoptosis by sequestering BIM. BIM binds to the hydrophobic groove of the BH3 binding site of BCL2, which depletes available BIM to activate BAX. This biological structural insight led to the development of BCL2 inhibitors, which were successfully modeled to competitively bind to the BH3 binding site for BIM; ergo, “BH3 mimetics.” Two studies in this issue provide new insight into the use and resistance to these drugs (refs. 2, 3).
The advent of BH3 mimetics and BCL2 inhibition has heralded transformational changes in treatment algorithms for hematologic malignancies, and led a slew of new approvals for acute myeloid leukemia (AML)—the first seen in decades (4, 5). Venetoclax (ABT-199; AbbVie) is a second-generation selective BCL2 inhibitor which has modest activity in relapsed/ refractory AML as a single agent (6), but in combination with the low-dose cytosine arabinoside (LDAC) or the DNA methyltransferase inhibitors (DNMTi) 5'azacitidine or decitabine led to remission in 55% to 67% of untreated elderly patients with AML deemed noncandidates for high-intensity chemotherapy in recent clinical trials (4, 5). These responses have provided hope to patients with AML and physicians hoping to provide efficacious, low-toxicity therapies. Still, most patients with AML treated with venetoclax-based regimens ultimately relapse, and a large number of patients, typically those previously treated with DNMTi or those with TP53 mutations, never respond at all (4, 5). This explains the urgency to understand venetoclax resistance and further liabilities in this “hallmark of cancer.”
Resistant BCL2 mutations have arisen in patients with venetoclax-treated chronic lymphocytic leukemia (CLL; ref. 7), although never noted in AML, and mechanisms of venetoclax resistance in wild-type BCL2 remain unclear. Just as BIM preferentially binds BCL2 (1), NOXA is an activator of apoptosis that preferentially binds to induced myeloid leukemia cell protein 1 (MCL1), another antiapoptotic factor in the BCL2 family of proteins. Whereas BCL2 upregulation is nearly universal in CLL (8), the heterogeneity of expression of antiapoptotic factors in AML is far more diverse. Some AMLs are BCL2-, MCL1-, or BCL-XL–dependent, and often multiple anti apoptotic proteins are upregulated in variable fashion with proportional sensitivity to selective inhibitors of antiapoptotic proteins (9, 10). Early observations revealed MCL1 was frequently upregulated in response to venetoclax treatment (9). This led to the hypothesis that sequential treatment with venetoclax followed by selective inhibitor of MCL1 or combination treatment with both agents may be an effective treatment strategy. Although this has yet to come to bear in the clinic, there are sufficient data to illustrate this approach is effective in synergistically removing AML cells in vitro and in xenografts (10). Though safe in early preclinical combination studies, safety concerns with potential dual inhibition of BCL2 and MCL1 have yet to be abated (10, 11), and despite a relatively bland safety signal with venetoclax in the clinic, neutropenia is not subtle, and can be significant (4, 5). Also, the predicted emergence of BCL-XL or other antiapoptotic protein–driven resistance remains a concern. So, although development of MCL1 inhibitors in the clinic is ongoing, a deeper understanding of venetoclax resistance from an unbiased perspective would be useful.
In this issue of Cancer Discovery, two laboratories harness the power of genome-wide CRISPR/Cas9 to conduct unbiased screens to best identify liabilities and synergies in venetoclax treatment in AML. In the study by Nechiporuk and colleagues (3), a CRISPR/Cas9 screen edified prior findings that reliance on alternative BCL2 family antiapoptotic family members (e.g., MCL1, BCL-XL, etc.) and absence of PMAIP1 (gene encoding for NOXA) both impart venetoclax resistance (see Fig. 1). Likewise, the absence of BAX led to resistance due to an inability to trigger intra-mitochrondrial BAX-dependent mitochondrial outer membrane permeabilization and apoptosis. TP53 deletion had similar effects, consistent with p53's central role as a transcriptional regulator of proapoptotic proteins under cellular stress (see Fig. 1). Although the authors did not illustrate correlation between specific loss/gain-of-function mutations and venetoclax resistance, the importance of functional p53 is clear, and the TP53-mutated (and underexpressed) patient samples from the BEAT AML study, although having variable loci and variant allele frequencies, tended to have greater resistance to venetoclax. Each of the identified genes effects apoptosis, edifying the understanding of apoptosis, control and highlighting the importance of disruption of the mitochondrial homeostasis in venetoclax resistance. Nechiporuk and colleagues went on to discover alternative signaling and dependence of venetoclax-resistant TP53 knockout AML cells to the NTRK signaling pathway. Neurotrophin receptors (NTRK1/2/3 or TRKA/B/C) are transmembrane receptors with tyrosine kinase activity, which are known to couple RAS/ MAPK, PI3K, or PLCγ signaling pathways in cancer (12). The importance of this pathway in the absence of functional p53 is unclear, but the activity in TP53 knockout venetoclax resistance is noteworthy.
Figure 1.
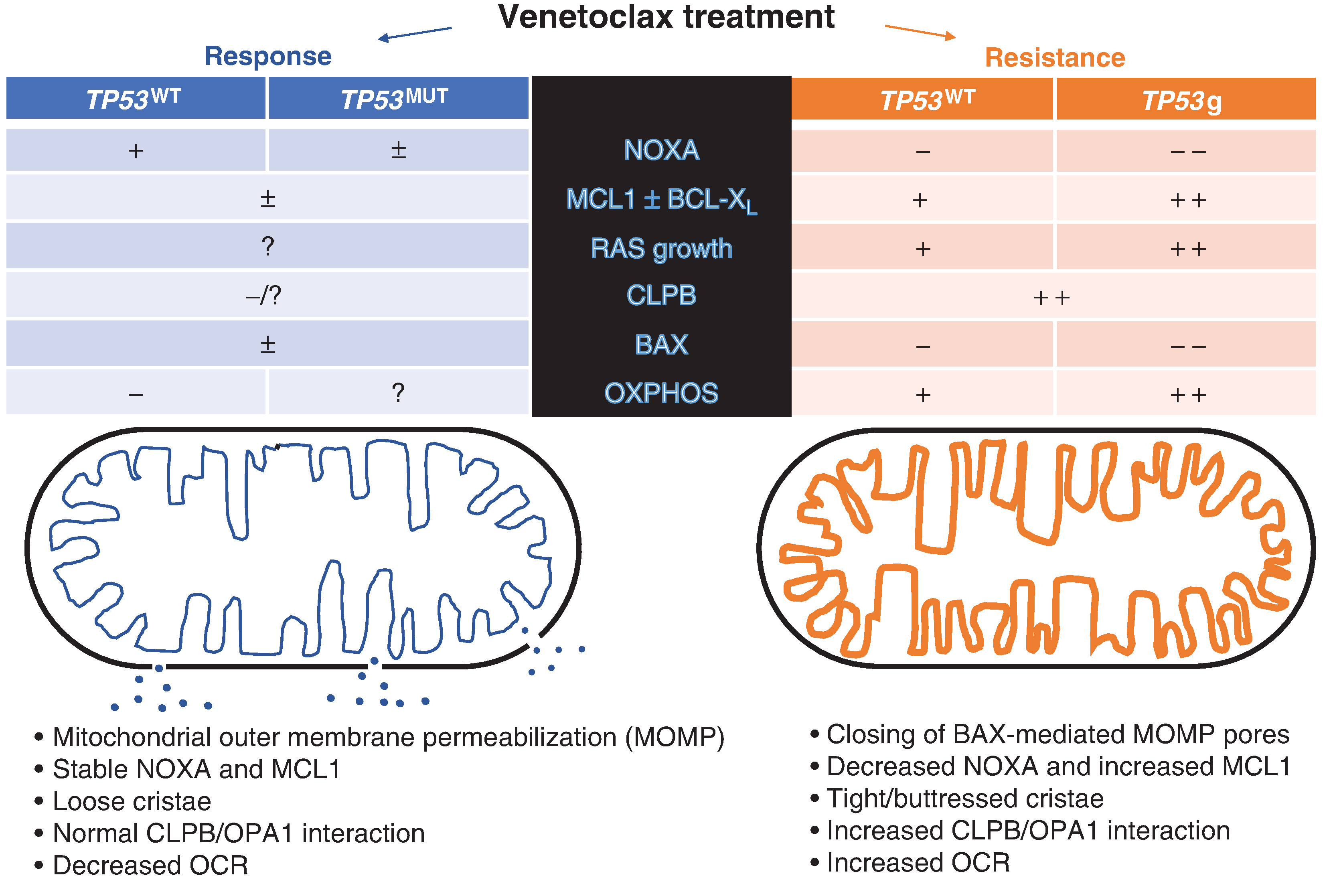
Venetoclax therapy leading to response in AML is noted in blue on the left side of the figure. Response to therapy leads to biochemical changes in both wild-type (TP53WT) and mutant (TP53MUT) TP53, noted in columns in blue. In AML resistant to venetoclax (orange), there are differential changes in metabolism, signaling, and componentry of apoptosis between AML which is TP53WT and TP53g (single-guide RNA inactivation of TP53) as noted in respective columns in orange. On the left, mitochondrial structure influenced by CLPB–OPA1 interaction is normal, and mitochondrial outer membrane is permeabilized via activation of BAX by proapoptotic proteins and lack of compensatory MCL1, which is sequestered by normal and elevated NOXA. The resultant cytochrome C (blue dots) escapes from the mitochondria to activate extra-mitochondrial apoptotic signaling through the caspases. TP53 mutational status does not seem to influence this in responding cells. On the right side of figure (orange), intra-mitochondrial apoptosis signaling is disrupted with increases in other antiapoptotic signaling (e.g., MCL1 and BCL-XL) in the absence of NOXA regulation—BAX-activated mitochondrial pores are reduced or not present, and cytochrome C does not escape the mitochondria. As TP53 regulates NOXA, TP53g cells have less NOXA expression and greater increases in MCL1. However, regardless of TP53 status, cristae are tighter and reinforced via CLPB–OPA1 interactions coinciding with increased oxygen use, which is magnified in the setting of TP53g resistance. OXPHOS, oxidative phosphorylation; OCR, oxygen consumption rate.
Using a similar screening strategy, Chen and colleagues also found that loss of TP53, BAX, and PMAIP conferred resistance (2). In addition, they identified negatively selected genes involved in mitochondria organization and structure which confer resistance to venetoclax. In a series of experiments based on the CRISPR screen results, Chen and colleagues describe venetoclax resistance in the context of transcriptional and post-translational mitochondrial adaptations. Of novel interest, Chen and colleagues identify a gene encoding for a key mitochondrial protein, CLPB, to be negatively selected in the CRISPR/Cas9 screen, preferentially overexpressed in AML CD34+ cells versus normal donor CD34+ cells, and non– core essential in normal homeostasis. CLPB interacts with OPA1 to buttress mitochondrial cristae under stress and is a lethal target in venetoclax resistance. Deletion of CLPB alone restored biosynthesis of amino acids and venetoclax sensitivity in venetoclax-resistant AML and represents a new liability to address in the development of new therapy for AML.
Whereas only selective BCL2 blockade is available in the clinic, MCL1-selective inhibitors are in clinical trials (NCT02979366, NCT02675452, NCT03218683) and BCL-XL– and BCL-W–specific inhibitors are in development. Neither of these studies specifically addresses the consideration of alternating (or combining) direct antiapoptotic protein–selective inhibitors in the face of resistance, nor the role of MCL1 or BCL-XL in contributing to metabolic alterations. Yet, indirectly addressing the consequences of venetoclax resistance by targeting mitochondrial structure and NTRK signaling is novel. Resistance to venetoclax is dependent on alternating energy use (13–15), and the powerful approaches suggested here further improve the current understanding of how targeting energy use and mitochondrial structure may lead to enhancement of venetoclax response. The challenges with the heterogeneity of AML cell metabolism across variable patient samples, the disruptions in mitochondrial homeostasis relevant in response and resistance, and therapeutic windows of emerging approaches to venetoclax resistance remain. The methods by which we wield venetoclax, a powerful new tool in our arsenal against AML, are currently being refined. It will be important in future studies to establish how relationships between energy use and structure drive resistance to venetoclax, and how these events can be targeted with selective therapies.
Footnotes
Disclosure of Potential Conflicts of Interest
M.R. Savona has received research support from Astex, Boehringer Ingelheim, Celgene, Incyte, Millennium, Sunesis, and TG Therapeutics; has equity in Karyopharm; and has consulted for Astex, Celgene, Karyopharm, Millennium, and TG Therapeutics. No potential conflicts of interest were disclosed by the other author.
REFERENCES
- 1.Sarosiek KA, Chi X, Bachman JA, Sims JJ, Montero J, Patel L, et al. BID preferentially activates BAK while BIM preferentially activates BAX, affecting chemotherapy response. Mol Cell 2013;51:751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen X, Glytsou C, Zhou H, Narang S, Reyna DE, Lopez A, et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov 2019;9:890–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nechiporuk T, Kurtz SE, Nikolova O, Liu T, Jones CL, D’Alessandro A, et al. The TP53 apoptotic network is a primary mediator of resistance to BCL2 inhibition in AML cells. Cancer Discov 2019;9:910–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019;133:7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wei AH, Strickland SA Jr, Hou JZ, Fiedler W, Lin TL, Walter RB, et al. Venetoclax combined with low-dose cytarabine for previously untreated patients with acute myeloid leukemia: results from a phase Ib/II study. J Clin Oncol 2019;37:1277–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and biological correlates of response in a phase II study of venetoclax monotherapy in patients with acute myelogenous leukemia. Cancer Discov 2016;6:1106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blombery P, Anderson MA, Gong JN, Thijssen R, Birkinshaw RW, Thompson ER, et al. Acquisition of the recurrent Gly101Val mutation in BCL2 confers resistance to venetoclax in patients with progressive chronic lymphocytic leukemia. Cancer Discov 2019;9: 342–53. [DOI] [PubMed] [Google Scholar]
- 8.Del Gaizo Moore V, Brown JR, Certo M, Love TM, Novina CD, Letai A, et al. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J Clin Invest 2007;117:112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan R, Hogdal LJ, Benito JM, Bucci D, Han L, Borthakur G, et al. Selective BCL-2 inhibition by ABT-199 causes on-target cell death in acute myeloid leukemia. Cancer Discov 2014;4:362–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramsey HE, Fischer MA, Lee T, Gorska AE, Arrate MP, Fuller L, et al. A novel MCL1 inhibitor combined with venetoclax rescues venetoclax-resistant acute myelogenous leukemia. Cancer Discov 2018;8:1566–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang X, Bathina M, Lynch J, Koss B, Calabrese C, Frase S, et al. Deletion of MCL-1 causes lethal cardiac failure and mitochondrial dysfunction. Genes Dev 2013;27:1351–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discov 2015;5:25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones CL, Stevens BM, D’Alessandro A, Reisz JA, Culp-Hill R, Nemkov T, et al. Inhibition of amino acid metabolism selectively targets human leukemia stem cells. Cancer Cell 2018;34:724–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lagadinou ED, Sach A, Callahan K, Rossi RM, Neering SJ, Minhajuddin M, et al. BCL-2 inhibition targets oxidative phosphorylation and selectively eradicates quiescent human leukemia stem cells. Cell Stem Cell 2013;12:329–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018;24:1859–66. [DOI] [PMC free article] [PubMed] [Google Scholar]