

Abstract
Glioblastoma (GBM) is the most lethal primary adult brain tumor and its pathology is hallmarked by distorted neovascularization, diffuse tumor-associated macrophage infiltration, and potent immunosuppression. Reconstituting organotypic tumor angiogenesis models with biomimetic cell heterogeneity and interactions, pro-/anti-inflammatory milieu and extracellular matrix (ECM) mechanics is critical for preclinical anti-angiogenic therapeutic screening. However, current in vitro systems do not accurately mirror in vivo human brain tumor microenvironment. Here, we engineered a three-dimensional (3D), microfluidic angiogenesis model with controllable and biomimetic immunosuppressive conditions, immune-vascular and cell-matrix interactions. We demonstrate in vitro, GL261 and CT-2A GBM-like tumors steer macrophage polarization towards a M2-like phenotype for fostering an immunosuppressive and proangiogenic niche, which is consistent with human brain tumors. We distinguished that GBM and M2-like immunosuppressive macrophages promote angiogenesis, while M1-like pro-inflammatory macrophages suppress angiogenesis, which we coin “inflammation-driven angiogenesis.” We observed soluble immunosuppressive cytokines, predominantly TGF-β1, and surface integrin (αvβ3) endothelial-macrophage interactions are required in inflammation-driven angiogenesis. We demonstrated tuning cell-adhesion receptors using an integrin (αvβ3)-specific collagen hydrogel regulated inflammation-driven angiogenesis through Src-PI3K-YAP signaling, highlighting the importance of altered cell-ECM interactions in inflammation. To validate the preclinical applications of our 3D organoid model and mechanistic findings of inflammation-driven angiogenesis, we screened a novel dual integrin (αvβ3) and cytokine receptor (TGFβ-R1) blockade that suppresses GBM tumor neovascularization by simultaneously targeting macrophage-associated immunosuppression, endothelial-macrophage interactions, and altered ECM. Hence, we provide an interactive and controllable GBM tumor microenvironment and highlight the importance of macrophage-associated immunosuppression in GBM angiogenesis, paving a new direction of screening novel anti-angiogenic therapies.
Keywords: Glioblastoma, angiogenesis, endothelial-macrophage interaction, ECM
1. Introduction
Malignant neovascularization and immunosuppression are two hallmarks of glioblastoma (GBM) [1], the most prevalent and aggressive primary adult brain tumor. Newly-diagnosed GBM patients survive less than 15 months despite combinatorial surgery, chemotherapy and radiation therapy [2], while recurrent GBM patients survive less than 6 months with conventional salvage therapies [3]. GBM has been traditionally defined by histological and genetic alterations, but its tumor microenvironment—aberrant neovascularization, infiltrating tumor-associated macrophage (TAMs), and dynamic extracellular matrix (ECM) alterations—is being increasingly recognized as a critical factor of relapse and therapeutic resistance. Consequently, considerable interest has been directed to engineering biomimetic preclinical models to validate therapeutic efficacy and dissect how tumor-stroma interactions confer a drug-resistant GBM subtype.
A diagnostic feature that distinguishes GBMs from low-grade gliomas is aberrant microvascular proliferation due to high expression levels of diverse proangiogenic factors, indicating angiogenic endothelial cells (ECs) are promising GBM therapeutic targets [4]. However, the effects of currently available anti-angiogenic therapies like vascular endothelial growth factor (VEGF) inhibition result in transient, meager clinical responses due to acquired resistance [5]. Increasing evidence suggests GBM immunity contributes to tumor angiogenesis and therapeutic resistance [1]. GBM casts a proangiogenic and immunosuppressive microenvironment by secreting VEGF and transforming growth factor beta (TGF-β1) [6], promoting tumor vascularization and suppress cytotoxic T cell proliferation and function [7] to impede anti-angiogenic therapy and immunotherapy [8]. GBM patients exhibit marked immunosuppression [9], where tumor-infiltrating TAMs can comprise up to 30% of bulk GBM tumor mass [10]. Our past studies prove increasing numbers of tumor-infiltrating, perivascular TAMs after antiangiogenic therapy correlate to poor survival among recurrent GBM patients [11, 12]. These findings highlight TAMs are critical drivers of anti-angiogenic resistance, but the molecular and intercellular mechanisms remain poorly defined. Therefore, investigating the pro-tumorigenic roles of TAMs and the synergy between tumor immunosuppression and angiogenesis is highly relevant for identifying potential targets to optimize GBM anti-angiogenic therapy.
However, a physiologically-accurate, integrated analysis of GBM tumor angiogenesis requires incorporating tumor-associated immunity to understand their parallel evolution during tumor progression and therapeutic resistance [6]. The intrinsic limitations of conventional methods for studying tumor angiogenesis pose a preclinical challenge. Primary dissociated cultures permit functional in vitro assays, but studying an isolated, single cell population in a two-dimensional (2D) environment does not reflect the true intertumoral heterogeneity and interactions [13]. Three-dimensional (3D) in vitro angiogenesis assays are more physiologically-relevant to in vivo GBM tumors than conventional 2D in vitro capillary network formation assays. Recent studies have distinguished differences in pro-/anti-inflammatory signaling between 2D and 3D environments [14], suggesting distinct regulatory roles for pro-angiogenic factor secretion during 2D and 3D angiogenesis [15, 16]. Furthermore, anti-angiogenic therapeutics are clinically delivered through 3D vasculature until it reaches and diffuses into the GBM tumor site, which can be only replicated in 3D vascularized tumor microenvironments. In vivo murine models are the current “gold standard” for cancer therapeutic screenings, but they exhibit several intrinsic limitations for longitudinal immune cell monitoring. Patient-derived xenografts involve suppressing host immune rejection for implanting and studying patient-specific tumors in vivo, but they lack physiologically-accurate human immune responses. Furthermore, genetically-engineered mice models lack the genetic heterogeneity of patient-specific GBM tumors [17]. Patient explant cultures are advantageous because they can preserve 3D tissue architecture and GBM tumor microenvironment interactions, but they lack ongoing, dynamic extracellular cues like altered cell-ECM interactions and controllable biochemical gradients [18].
Recent advances in microfluidics-based in vitro models provide promising solutions for assessing cancer therapeutics, but they are sufficient to only model pathophysiological features independently without complete multiparametric integration into a single chip. Our previous study established a 3D hydrogel microfluidics-based model and demonstrated the biophysical roles of flow shear stress in regulating angiogenesis, but it did not consider the proangiogenic role of immunity [19]. Furthermore, perivascular interactions have been recently linked to sprouting angiogenesis [20], but GBM studies have yet dissected the proangiogenic roles of TAMs and cell-ECM interactions for regulating tumor angiogenesis and therapeutic efficacy. However, there are no existing in vitro 3D physiologically-accurate systems that reconstruct the unique and complex GBM tumor microenvironment with immunosuppressive and angiogenic signatures to dissect the synergy between tumor immunity and angiogenesis.
This requires constructing a microenvironment that mirrors GBM tumor-stroma crosstalk, where communication is mostly mediated between membrane-bound receptors and soluble (cytokines) and nonsoluble (cell adhesion receptors) ligands. Previous studies demonstrate increasing numbers of TAMs being proximal or in physical contact with tumor vasculature, implicating perivascular macrophage-EC interactions may regulate tumor angiogenesis [21, 22]. Furthermore, ECM adhesion is essential for regulating tumor angiogenesis and immunity [23]. In GBM, promoted integrin expression correlates with increasing vascular density [24] and cytokine production and suppressed tumor-cytotoxicity [25]. Mounting evidence has demonstrated elevated integrin αvβ3 expression on angiogenic ECs [26] and glioma cells [24], which may be upregulated due to perivascular macrophage-EC or cell-ECM interactions. In addition, biomaterial studies have demonstrated polysaccharide-based [27] and silicified collagen [16] hydrogels can activate macrophage/monocytes to secrete pro-angiogenic factors for promoting in vitro and in vivo 3D angiogenesis. Therefore, 3D cell-suspending hydrogels offer an opportunity to tune integrin activation and coupled cellular behaviors like macrophage-mediated inflammation and angiogenesis [21]. Probing the dynamic interactions among angiogenic ECs, GBM TAMs and GBM tumors in a tunable ECM can lead to the development of a promising GBM anti-angiogenic therapeutic strategy [18].
Here, we engineered a biomimetic, microfluidics-based brain tumor angiogenesis model by implanting GBM-like tumors (GL261 or CT-2A) in a 3D artificial vascularized hydrogel of tunable ECM properties and immunosuppression gradients to mimic the in vivo GBM tumor niche. This organotypic platform reconstitutes notable GBM hallmarks, including GBM tumor angiogenesis and macrophage-associated immunosuppression. We have demonstrated that our in vitro GBM tumor microenvironment more closely replicates in vivo pathology by fostering immunosuppressive conditions that steer macrophage polarization towards an alternatively-activated M2-like phenotype promoting proangiogenic activity. We show GBM-induced M2-like macrophages exhibit elevated secretion of anti-inflammatory cytokines TGF-β1 and IL-10 to promote EC capillary, proliferation, and angiogenic sprouting, while classically-activated M1-like macrophages suppress proangiogenic activity, which we termed “inflammation-driven angiogenesis.” We reveal soluble immunosuppressive cytokines, predominantly TGF-β1, and surface EC-macrophage interactions are involved in regulating GBM tumor angiogenesis. Our studies implicate perivascular macrophage-EC interactions regulate in vitro proangiogenic activity through integrin (αvβ3) receptors and Src-PI3K-YAP signaling, which can be facilitated either by cell-cell or cell-matrix interactions. Our biomimetic GBM model with macrophage-associated immunosuppression and GBM tumor angiogenesis enables a high throughput screening for testing novel therapeutic combinations such as dual integrin (αvβ3) and cytokine receptor (TGFβ-R1) blockade to improve GBM therapeutic efficacy and minimize anti-angiogenic resistance.
2. Materials and methods
2.1. Cell culture and reagents
Mouse glioma cell lines, GL261 (National Cancer Institute, Division of Cancer Treatment and Diagnosis, Frederick, MD) and CT-2A (a gift from Dr. Thomas Seyfried, Boston College, MA), with characteristics of GBM phenotypes were used to study GBM angiogenesis. GBM cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Sigma-Aldrich) supplemented with 10% heat-inactivated fetal bovine serum (FBS, Gibco), 2 mM glutamine (Gibco) and 1% penicillin/streptomycin (Gibco) (CT-2A) or 0.25% gentamicin (Gibco) (GL261). GL261 and CT-2A were authenticated using mouse short tandem repeat DNA profiling and tested negative by PCR for Mycoplasma contamination (DDC Medical, Fairfield, OH). Mouse ECs (C166-GFP, ATCC), derived from mouse yolk sac, were cultured in DMEM (Sigma-Aldrich), and supplemented with 10% Fetal Bovine Serum and 0.2mg/mL Geneticin (Gibco). Mouse macrophage cells (RAW264.7, ATCC) were grown in DMEM, supplemented with 10% heat inactivated FBS, and 1% penicillin/streptomycin. All cell lines were grown in a 37 °C (5% CO2) incubator to reach 80% confluence for any further experimentation. All cells were cultured no more than 20 passages.
The following inhibitors were used: integrin αvβ3 inhibitor cilengitide (SML1594, Sigma-Aldrich); TGF-β receptor I inhibitor LY-364947 (L6293, Sigma-Aldrich), VEGFR inhibitor cediranib (HY-10205, MedChemExpress LLC), Src inhibitor PP2 (ab120308, Abcam), PI3K inhibitor wortmannin (W1628, Sigma-Aldrich) and YAP inhibitor verteporfin (SML0534, Sigma-Aldrich). All inhibitor concentrations were prepared as 10 μM, except for verteporfin (5 μM), in complete cell culture media. Control groups were prepared with complete cell culture media supplemented with DMSO (0.01%). Integrin β3 blocking antibody (ab119992, 10 μg/mL, Abcam) was prepared in complete cell culture media, while IgG1 isotype control from murine myeloma (M5284, 10 μg/mL, Sigma-Aldrich) was prepared in complete cell culture media as a control group. Inhibitor media was replenished every 24 hrs for 3-day sprouting or 1-day capillary and proliferation assays.
2.2. Macrophage polarization
Macrophage polarization was induced in vitro as previously reported [28]. Untreated RAW264.7 cells were labeled as M0 macrophages. RAW264.7 cells (106 cells/mL) were seeded in 12-well plates overnight before polarization. M1-like macrophages were obtained by stimulating RAW264.7 cells with lipopolysaccharide (LPS; L9764, 100 ng/mL, Sigma-Aldrich) for 24 hrs. M2c and M2d macrophages were obtained by treating RAW264.7 cells with IL-10 (575802, 50 ng/mL, BioLegend) or IL-6 (575702, 100 ng/mL, BioLegend), respectively. As GL261 or CT-2A cells approached 80% confluence, complete culture media was replenished and supernatants were collected after 24hrs and centrifuged at 2000×g for 10 min at 4°C to remove cell debris. Biomimetic TAMs were obtained by culturing M0 RAW264.7 cells in complete culture media supplemented with conditioned media of GBM-mimicking cells (GL261 or CT-2A) (1:1) for 24 hrs.
2.3. Immunoassays
After macrophage polarization, cytokine secretions were examined by using a Mouse Cytokine Antibody Array membrane-based ELISA kit (ab211069, Abcam) according to manufacturer’s protocols. Briefly, supernatants were collected after 24 hrs of polarization and centrifuged at 2000 × g for 10 min at 4°C to remove cellular debris. Biotinylated Antibody Cocktail (1:1) was incubated with supernatants at 4°C overnight, followed by washing and incubation with HRP-labeled Streptavidin (1:1000) for 2 hrs at room temperature. Detection Buffer C and D mixture (1:1) was then applied to visualize chemiluminescence for 2 min at room temperature. Imaging was obtained by using a ChemiDoc Imaging System (Bio-rad). Mean intensity of each spot was quantified with ImageJ (NIH).
2.4. Microfluidic Device Fabrication
Poly-dimethylsiloxane (PDMS) was used to frame a two-channel microfluidic device to reconstruct a vascularized GBM tumor microenvironment in vitro (Supplemental Fig. S1). Master molds were first fabricated on silicon substrates by employing photolithography with SU-8 negative photoresist (2025, Microchem) at a thickness of 20 μm. To facilitate PDMS release, molds were silanized by trichloro(1H,1H,2H,2H-perfluorooctyl) silane (448931, Sigma-Aldrich) vapor overnight in vacuum desiccation. Thick (5 mm) and thin PDMS (1 mm) layers were fabricated by applying soft lithography to the master molds. Briefly, a mixture of PDMS base and curing agent (0008952166, Sylgard 184 Silicone Elastomer, Dow Corning) with a volume-to-volume (v/v) ratio of 10:1 was prepared using a clean glass rod in a laminar flow hood to avoid dust contamination. Followed by vacuum desiccation, PDMS solution was poured over separate master molds to produce thick (5 mm) and thin layers (1 mm) by curing PDMS in an 80°C oven for 1 hr (thin layer) and 2 hrs (thick layer). Once PDMS was peeled from master molds, 0.5 mm and 1.0 mm holes were punched for inlets and outlets, respectively, and 4 mm holes were punched to produce a collagen loading window. Thick and thin layers were activated with plasma (350W, PlasmaEtch) for 2 min and subsequently aligned using a stereomicroscope (Zeiss SteREO Discovery.V8). Aligned PDMS layers were incubated at 80°C overnight. Glass coverslips (22 × 22mm; Thermo Fischer-Scientific) and the thin-layer surface of aligned PDMS were treated with plasma (350W, 1 min). To anchor Type-I collagen hydrogels onto the PDMS-glass collagen window, devices were sequentially activated with plasma (350W, 2min), coated with 0.1 mg/mL Poly-L-Lysine (P8920, Sigma-Aldrich) for 1 hr, and 1% glutaraldehyde (340855, Sigma-Aldrich) for 2 hrs. Devices were rinsed completely using distilled water twice, and subsequently treated to UV-sterilization in a Type 2 class laminar flow hood for 15 min.
Two parallel circular channels were molded within collagen gels by inserting 400 μm stainless steel needles (HS.40 ×40, Hwato) into pre-designed parallel channels. The final diameter of the lumen is in the range of 400 ±50 μm after removing the needles from the soft gels. Prior to insertion, all needles were coated in 1% BSA solution (A9418, Sigma-Aldrich) at 4 °C overnight to prevent unspecified binding between needles and collagen hydrogel. Type-I collagen hydrogels were prepared on ice with 10× Phosphate-buffered saline (PBS; AM9624, Thermo Fischer-Scientific) solution, 1N NaOH (72068, Sigma-Aldrich), complete cell culture media or labeled macrophage suspension (107 cells/mL) and 3.47 mg/mL collagen type I, rat-tail (354236, Corning). After mixing, collagen pH was measured and adjusted to be within 7.3–7.4, and a final concentration of collagen was ~1.8 mg/mL. After loading 100 ul collagen solution into loading window, devices were incubated for 30min at 37°C (5% CO2) for gelation. Subsequently, collagen hydrogels were dropped with complete cell culture media to prevent dehydration and needles were removed carefully to form 2 parallel 3D collagen channels (Supplemental Fig. S1c).
2.5. Fabrication of integrin-specific collagen
RGD peptide sequence GRGDSPC (H-7245, Bachem), specified for integrin αvβ3 [29], was conjugated to a Type-I collagen backbone by using a modified protocol (Supplemental Fig. S1b) [30]. To ease conjugation of RGD, a carboxylic group present on RGD peptide reacted with a crosslinker. The RGD peptide mixed with the EDC coupling reagent was then covalently bound to primary amines of the Type-I collagen backbone via nucleophilic attack. Briefly, 1 mL of 1 M crosslinker solution, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC; 22980, Thermo Fischer-Scientific), was mixed with 0.5 M N-hydroxysuccinimide (NHS; 24510, Thermo Fischer-Scientific) in 0.1 M MES buffer (pH 2–4; 76039, Sigma-Aldrich) with 0.5 mg of RGD peptide for 15 min at 37°C. The mixture was added to 5 mL of 3.47 mg/mL collagen type I, rat-tail (354236, Corning) in 0.02 N acetic acid (695092, Sigma-Aldrich) on ice. To prevent premature collagen gelation, mixtures were placed on a shaker at 4°C overnight. Pierce protein concentrators with a 10 kDa molecular weight cutoff (88516, Thermo Fischer-Scientific) were used to remove unconjugated peptides. The final solution volume was adjusted to 6 mL in 0.02 N acetic acid to reconstitute a 3.47 mg/mL collagen solution. Infrared spectroscopy was used to evaluate the RGD peptide binding with amine residues of Type-I collagen backbone at an absorption frequency of 1550 cm−1 (Supplemental Fig. S1d). To evaluate the cell viability in the 3D integrin-specific collagen, we suspended RAW264.7 cells (107 cells/mL) in 100 μL engineered collagen gels for 2 hrs, followed by adding Propidium Iodide (1 μM) to distinguish dead cells and Calcein AM (2 μM) (Sigma-Aldrich) in complete cell culture media for 30 min at 37˚C.
2.6. 3D angiogenic sprouting assay
To reconstitute 3D GBM angiogenesis, C166-GFP ECs (107 cells/mL) were seeded onto luminal walls of a 3D collagen channel. Devices were incubated at 37°C for 15 min to allow for cell attachment, and then inverted for another 15 min to achieve complete attachment around channel circumference. Afterwards, hydrogels were covered with complete C166-GFP cell culture media or mixture of C166-GFP and macrophages media (1:1) or mixture of C166, RAW264.7 and GBM cells (1:1:1) for 24 hrs. Devices were then imaged for sprouting at day 0, and subsequently treated with inhibitor-enriched media. It normally takes 72 hrs for vessel formation. However, after seeding EC cells onto the lumen of collagen channel, angiogenic sprouting can be observed after 24 hrs. Five images (0.8 mm × 1 mm) were taken for each device, and 3 regions in each image were quantified using the measurement tool ImageJ (NIH) by averaging sprouting lengths. Sprouting length characterizes 3D sprouting [31] and was defined as the distance between the tips of sprouting cells and the 3D endothelial monolayer.
For GBM co-culture, GL261 or CT-2A cells (107 cells/mL) were seeded onto luminal walls of adjacent collagen channel. Afterwards, devices were checked for cell attachment and imaged with an inverted microscope (Zeiss Axio Observer.Z1). Specifically, in Fig. 1c, GL261 cells spontaneously aggregated after being co-suspended with macrophages of different phenotypes within hydrogel after 24hrs and grew to a diameter of 50–150 μm in the hydrogel. C166-RAW264.7-GBM cell culture media (1:1:1) was supplemented with or without inhibitors and devices were replenished every 24 hrs for 3 days. To evaluate the angiogenic potential of conditioned media from macrophages or GBM cells, cells (107 cells) were cultured in 100mm petri dishes (Falcon) for 24 hrs. Media was collected using sterile 1.5mL centrifuge tubes and centrifuged at 2000 × g for 10 min at 4°C to remove cellular debris. Conditioned media was either stored at −20°C for future use or supplemented with complete culture media (1:1) and loaded directly into the adjacent channel. Sprouting length was recorded every 24 hrs for 3 days after conditioned media treatment.
Figure 1. Microengineered in vitro GBM angiogenesis model.
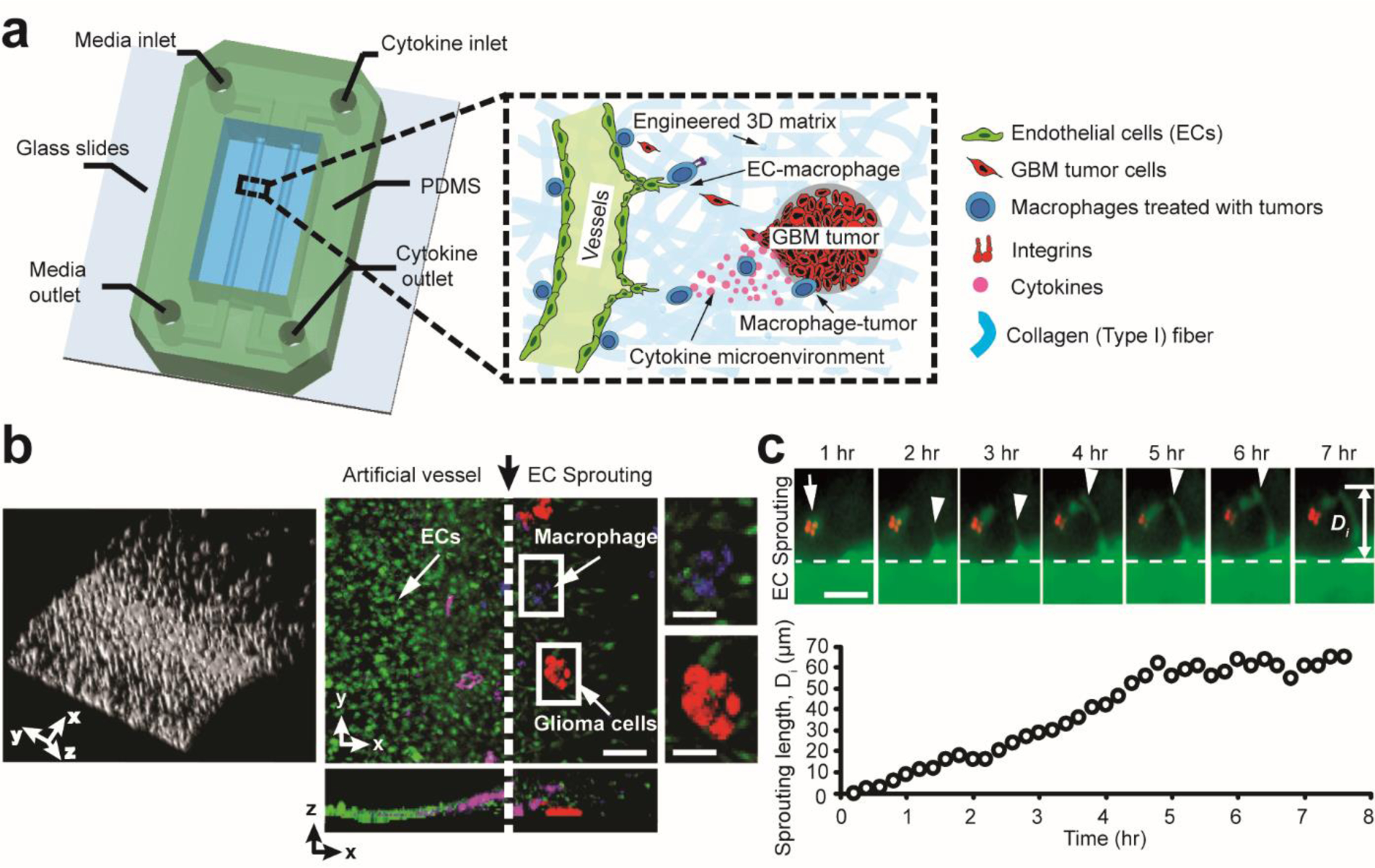
(a) A schematic illustrating the 3D microfluidics-based angiogenesis model for studying 3D inflammation-driven angiogenesis in vitro, engineered with controllable tumor-immune-vascular interactions, pro-/anti-inflammatory macrophage phenotype and cytokine conditions, cell-matrix interactions. (b) 3D reconstruction of 3D luminal EC monolayer invading into hydrogel and confocal image stack projection of directed 3D angiogenic sprouting (green) by GBM tumor cells (red) and GBM-induced TAMs (blue). Note the semi-circular cross-section of artificial vessels in z-x view. Scale bar is 100 μm for left and middle images, and 50 μm for the two zoomed inserts (right). (c) Time-lapse of 3D EC-macrophage dynamics for ~ 8 hrs. Sprouting length () was defined as the distance between the leading protrusions of tip ECs and origin of EC monolayer. Arrow indicates macrophage (M2c) position, and arrow heads represent protruding angiogenic sprouts. Scale bar is 50 μm.
To screen the angiogenic potential of specific cytokines, recombinant mouse pro-inflammatory cytokines, TNF-α (575202, Biolegend) and IL-6 (575702, Biolegend), and anti-inflammatory cytokines, TGF-β1 (763102, Biolegend) and IL-10 (575802, Biolegend), were used and purchased from BioLegend. Different dosages (0–10 ng/mL) of cytokines were reconstituted in complete C166-GFP media and replenished every 24 hrs in devices for a 3-day observation. 3D sprouting was recorded every day and then quantified accordingly.
2.7. 2D and 2.5D capillary assays
Capillary assays were conducted in 2D and 2.5D microenvironments to further confirm the proangiogenic capacity of macrophages or inflammatory cytokines. In 2D assays, a 48-multiwell plate (Falcon) was coated with 50 μl Growth Factor Reduced Matrigel (356231, Corning) per well. For examining capillary network formation with enhanced integrin αvβ3 expression, Matrigel was mixed with αvβ3-specified collagen (v/v ratio of 5:1). Control groups involved mixing Matrigel and unmodified Type-I collagen (5:1). Matrigel-coated wells were incubated at 37°C (5% CO2) for 30 min, and subsequently immersed in 300 μl C166 media, C166-RAW264.7 media (1:1) or C166-RAW264.7-GBM media (1:1:1), supplemented with or without inhibitors. C166-GFP cells, RAW 264.7, GL261 or CT-2A were seeded at 50,000 cells per well. To better mimic in vivo 3D microenvironmental conditions, we adopted a 2.5D assay as previously described [32]. Experimental procedures were consistent with 2D assays, except 4% (v/v) Type-I collagen was supplemented in different cell culture media for experiments.
After 24 hrs of incubation, each well was imaged using an inverted phase contrast microscope (Zeiss Axio Observer.Z1) with a 10× objective. Five representative images per well were recorded and capillary network formation was quantified. Capillary area was designated as the indicator for capillary network formation [33], which was calculated by averaging capillary (connected ECs >3 cells) length multiplied by mean capillary width using ImageJ (NIH).
2.8. 2D and 2.5D proliferation assays
The proliferative capacity of ECs was measured by quantifying the change of cell numbers after 24 hrs. All experimental parameters for 2D and 2.5D proliferation assays are consistent with 2D and 2.5D capillary assays. Briefly, 48-multiwell plates (Falcon) were mostly untreated. To investigate proliferative effects of enhanced integrin αvβ3 expression, 50 μl integrin αvβ3-specified collagen was coated per well prior to experiments.
Before experiments, cells were live-stained with Hoechst 33342 (5 μg/mL; H3570, Thermo Fisher-Scientific) for counterstaining cell nuclei. In 300 μl of different media, 10,000 C166-GFP cells were seeded and allowed to grow for 24 hrs. Five images were taken for each well with an inverted fluorescent microscope (Zeiss Axio Observer.Z1) at a 10× objective. Total cell number was automatically counted using ImageJ (NIH). For macrophage-EC co-culture, 10,000 C166-GFP cells and 10,000 RAW264.7 cells were seeded in a 48-multiwell plate and treated with same media conditions as C166 monoculture for 24 hrs. The macrophage cell number was used as a baseline to calculate the numbers of EC. Finally, the fold-change of ECs was calculated and normalized to that of untreated ECs in normal cell media.
2.9. Confocal microscopy
To monitor 3D angiogenic sprouting, Z-stack images were acquired with a Zeiss LSM 710 or 880 Laser Scanning Confocal Microscopes. All stacked images were reconstructed by using ZEN lite software (Zeiss) and ImageJ (NIH) for 3D visualization. Prior to imaging EC-macrophage-tumor cell interactions, RAW264.7 cells were labeled with Hoechst 33342 (5 μg/mL) and GBM cells were labeled with Cell Tracker Red (5 μM; C34552, Thermo Fisher-Scientific). For other experiments, RAW264.7 cells were labeled with Cell Tracker Red (5 μM; C34552, Thermo Fisher-Scientific).
2.10. Immunofluorescence staining and analysis
To investigate integrin β3 and YAP expressions of C166-GFP and RAW 264.7 macrophages in different conditions, ECs were seeded onto cover glass in 24 well-plates (Falcon) and co-cultured with different macrophage cells (M0, M1, M2c, biomimetic TAMs) for 24 hrs. Cells were fixed with 4% paraformaldehyde (15711, Electron Microscopy Sciences) for 30 min, permeabilized with 0.3% Triton X-100 (11332481001, Sigma-Aldrich) for 10 min, and then blocked with 3% bovine serum for 1 hr on ice to eliminate unspecified binding. Cells were incubated with YAP primary antibodies (sc-101199, Santa-Cruz Biotechnology, 5 μg/mL) for 1 hr, and then visualized with Alexa Fluor 555 conjugated goat anti-mouse IgG secondary antibodies (Invitrogen, 5 μg/mL). Integrin β3 was visualized by anti-integrin beta 3 antibody (ab119992, Abcam, 5 μg/mL) and Alexa Fluor 555 conjugated goat anti-rabbit IgG secondary antibodies (A-21428, Thermo Fisher-Scientific, 5 μg/mL) or directly using PE anti-mouse/rat CD61 antibody (104307, Biolegend, 5 μg/mL). Cells were incubated for 30 min with Hoechst 33342 to counterstain nuclei.
Fluorescent images were obtained using an inverted microscope (Zeiss Axio Observer.Z1) equipped with a digital CMOS camera (ORCA-Flash4.0 LT Digital CMOS camera, Hamamatsu Photonics) and a 40× objective. Integrin β3 expression was quantified by the mean intensity of integrin regions in each cell. Similarly, subcellular localization of YAP in ECs were quantified by the intensity ratios of nuclear and cytoplasmic YAP in grayscale images using ImageJ (NIH) [34].
2.11. Western blot analysis
Cells were lysed in Halt Protease and Phosphatase Inhibitor Cocktail (1:100; 78440, Thermo Fisher-Scientific) diluted in RIPA cell lysis buffer (89900, Thermo Fisher-Scientific). Lysates were centrifuged at 12,000 rpm for 20 min at 4 °C, and the supernatant was collected. Proteins in lysates were separated via polyacrylamide gel (4561021, Mini-PROTEAN TGX Precast Gels, Bio-rad) before transferring to PVDF membranes. PVDF membranes were incubated with 5% (W/V) blocking buffer (1g fat-free milk in 20 mL TBST) for 2 hrs on a shaker, and then primary antibodies (Supplemental Table S3) overnight at 4 °C. After washing for 30 min, goat anti-mouse or anti-rabbit or anti-rat-HRP (1:5000, Bio-rad or Thermo scientific) were used as secondary antibodies for 2 hrs before detection with western ECL substrates (1705060S, Clarity, Bio-rad) according to the manufacturer’s instructions. Chemiluminescence images were obtained by using ChemiDoc Imaging Systems (Bio-rad). Images have been cropped for presentation.
2.12. RNA-sequencing and data analysis
ECs (C166-GFP) were co-cultured with different macrophage subtypes on 2D substrates for 24 hrs, then isolated by using flow cytometry (MoFlo™ XDP cell sorter, Beckman Coulter) for RNA-sequencing analysis. RNA samples were prepared with Qiagen RNA easy plus mini kit (74134, Qiagen) according to the manufacturer’s instructions, and then sequenced on an Illumina HiSeq-4000 High-Output system with single read 50 cycle lane. The sequencing reads were demultiplexed, converted to FASTQ format using Illumina bcl2fastq software, and aligned to the mouse genome (build mm10/GRCm38) using the splice-aware STAR aligner [35]. Gene counts were generated by using a feature Counts program [36], and then normalized before values were log2 transformed, and used to test for differential expression using negative binomial generalized linear models implemented by the DESeq2 R package [37]. We investigated expressions of genes associated with angiogenesis and integrin subtypes. Heat maps are plotted by using Matlab software.
2.13. Statistics
All experiments were repeated at least three times independently. All data are presented as means ± s.e.m unless otherwise specified. The means of groups were compared using one-way one-way analysis of variance (ANOVA) following Tukey’s post hoc test in GraphPad Prism or unpaired, two-tailed Student’s t-test in Excel (Microsoft). Student’s t-test was used when comparing two groups, as shown in the figure legends. Differences (P < 0.05) were considered statistically significant.
3. Results
3.1. Micro-engineered in vitro GBM angiogenesis model with controllable tumor-immune-vascular interactions, inflammatory conditions and ECM properties.
To investigate GBM angiogenesis in a versatile microenvironment that reconstitutes in vivo features, we developed a 3D microfluidics-based angiogenesis platform that integrates 3D artificial vascular vessels and controllable biochemical gradients (e.g. pro-/anti-inflammatory cytokines), intercellular communication (e.g. glioma cells, immune cells, ECs) and cell-matrix interactions (e.g. enhanced cell-adhesion) (Fig. 1 and Supplemental Fig. S1). The microfluidics-based in vitro vascularized tumor microenvironment encloses two parallel channels in a Type I-collagen hydrogel. The lumen of one channel is patterned with ECs (C166-GFP, ATCC) to form 3D endothelial monolayers that develop angiogenic invasion and sprouting (Fig. 1a. left channel), while the adjacent channel is seeded with glioma cells of GBM phenotype (GL261) or serves as a reservoir for cytokines to promote or suppress angiogenesis through the hydrogel (Fig. 1a, right channel). To reproduce pro-/anti-inflammatory settings of gliomas, we suspended different phenotypes of macrophages (RAW 264.7, ATCC) or conditioned the hydrogel with cytokines. Furthermore, cell-matrix interactions were controlled by modifying cell-adhesion ligands present in Type-I collagen hydrogel (Supplemental Fig. S1). Specifically, integrin-specific hydrogels were achieved by reacting RGD peptides with primary amine groups of collagen monomers to form stable amide bonds (Supplemental Fig. S1c). Together, our platform enables the real-time observation (Fig. 1c) of tumor-immune-vascular interactions with tunable cell-matrix and cell-cell interactions, as well as inflammatory conditions in an integrated 3D glioma microenvironment.
3.2. GBM cells polarize macrophages towards an immunosuppressive M2-like phenotype.
Macrophages exhibit a remarkable plasticity in response to different microenvironment cues during inflammatory diseases [38]. They can switch between two major phenotypes, classically-activated (M1) and alternatively-activated (M2) macrophages, and exert anti-tumorigenic or pro-tumorigenic functions depending on surrounding signals [39, 40]. Generally, M1 macrophages are characterized as a pro-inflammatory phenotype that secretes cytokines such as, TNF-α and IL-6, and associated with suppressing tumor progression [28, 39, 40]. In contrast, M2 macrophages are characterized as an anti-inflammatory phenotype that secretes cytokines, such as TGF-β and IL-10, and associated with suppressed immune surveillance [28, 39, 40].
We established in vitro M1 and M2 (M2c or M2d) macrophages by stimulating uncommitted macrophages (M0) with LPS or IL-6/IL-10 (Supplemental Table S1; see Materials and methods 2.2 for details) [28]. Polarized macrophages were classified as either M1 or M2, by profiling expression levels of inducible Nitric Oxide Synthase (iNOS) and Arginase-1 (Arg-1) (Supplemental Fig. S2a) and the secretion levels of pro-inflammatory (TNF-α, IL-6) and anti-inflammatory (TGF-β, IL-10) cytokines using a Mouse Cytokine Antibody Array membrane-based ELISA kit (see Materials and methods 2.3 for details). Using our M1/M2 classification, we examined whether and how GBM tumors alter macrophage phenotype (Fig. 2). To this end, uncommitted macrophages (M0) were co-cultured with GBM cells (GL261) or treated with GBM conditioned-media to produce biomimetic tumor-associated macrophages, ‘TAMs’ (Fig. 2a). Immunostaining results showed polarized M1 macrophages displayed increased expression of iNOS, while M2c, M2d macrophages and TAMs showed elevated expression of Arg-1 (Fig. 2b&c and Supplemental Fig. S2b&c). We then profiled the secretion levels of pro-inflammatory (TNF-α, IL-6) and immunosuppressive (TGF-β, IL-10) cytokines from M1 and M2 macrophage subsets. As expected, our results confirm that M1 macrophages secreted elevated levels of cytokines IL-6 and TNF-α, while M2 macrophages and TAMs produced abundant immunosuppressive cytokines TGF-β and IL-10 (Fig. 2d). Indeed, in vivo TAMs are predominantly shifted towards the M2 phenotype by surrounding anti-inflammatory cytokines such as IL-10 [41] and TGF-β [42], contributing to suppressed immune surveillance and enhanced tumor angiogenesis, invasion, and metastasis [43]. Our in vitro results confirm that GBM tumors distinctly manipulate macrophage phenotype to favor an immunosuppressive microenvironment during tumor progression.
Figure 2. GBM cells promote an immunosuppressive M2-like macrophage phenotype.
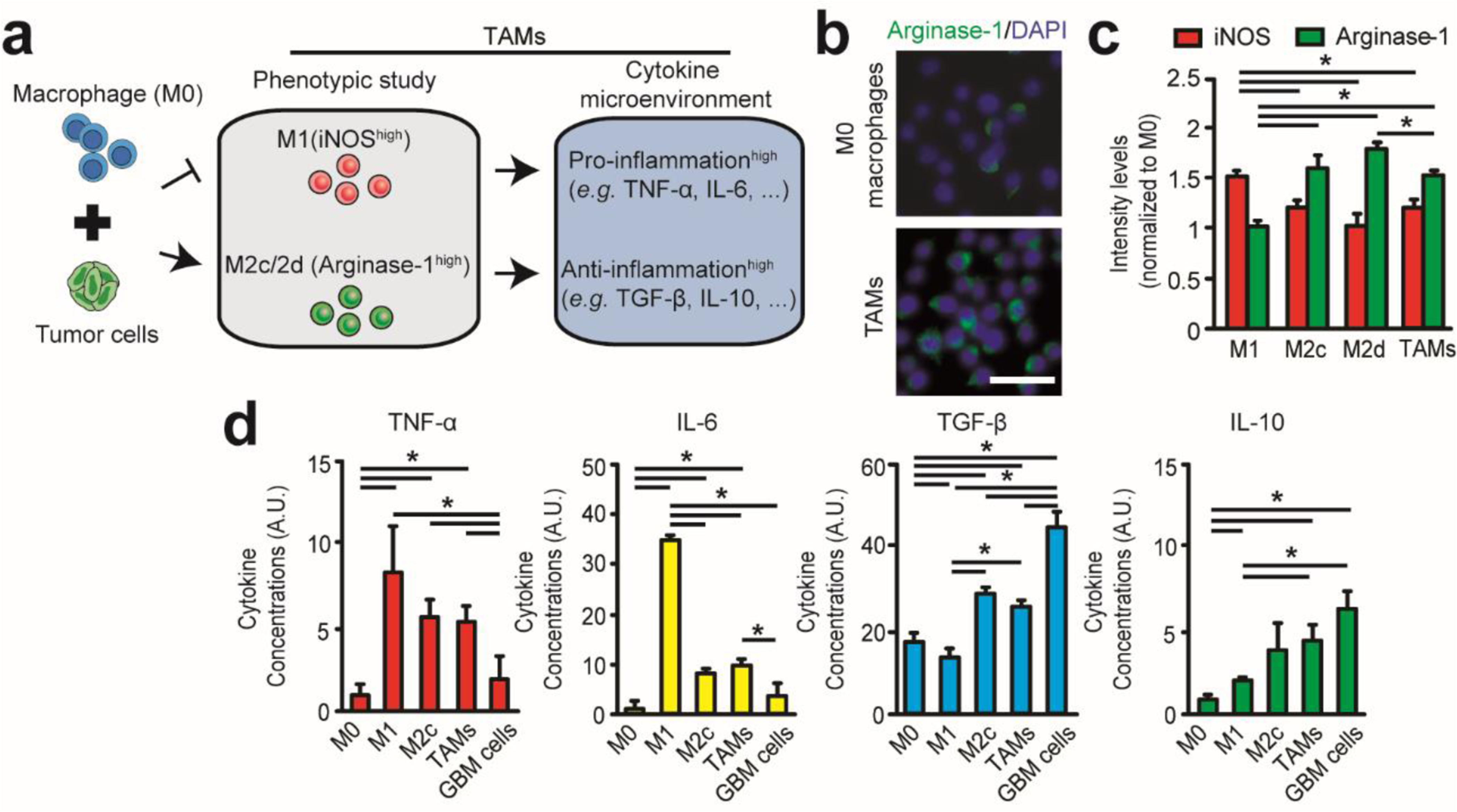
(a) Establishing GBM-induced TAM phenotype after treating M0 macrophages with GBM cells and characterizing polarization markers (iNOS for M1 subsets, Arginase-1 for M2 subsets) and immunoassay profiles of pro-inflammatory (e.g. TNF-α, IL-6) and anti-inflammatory (e.g. TGF-β, IL-10) cytokines. (b) Immunofluorescence images comparing Arginase-1 expressions of uncommitted M0 macrophages and GBM-induced TAMs. Scale bar is 50 μm. (c) Quantified expressions of iNOS (M1 marker) and Arginase-1 (M2 marker) for polarized macrophage subsets. (d) Immunoassay profiles of pro-inflammatory (e.g. TNF-α, IL-6) and anti-inflammatory cytokines (e.g. TGF-β, IL-10) secreted in complete culture media (500 μL per million cells) of M0, M1, M2c, GBM-induced TAMs subsets and GBM cells. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using the one-way ANOVA, followed by the Tukey post hoc test. *, P < 0.05.
3.3. GBM-induced M2-like TAMs promote EC angiogenic activity
To validate a proangiogenic role for TAMs-associated immunosuppression, we conducted multidimensional (2D, 2.5D, and 3D) in vitro angiogenesis assays (Fig. 3). Capillary formation assays were examined in 2D and 2.5D microenvironments, by quantifying area of EC vascular network formation, and measured under the stimuli of different macrophages (M1, M2c, M2d), GBM-induced TAMs, macrophage conditioned media, and pro-/anti-inflammatory cytokines (Fig. 3; see Materials and methods 2.7 for details). In 2D assays, ECs (50,000 cell/well) were seeded with each macrophage subset or their conditioned culture media in a Matrigel-coated (50 μl/well) 48-multiwell plate. In 2.5D assays, experimental procedures were consistent with 2D assays, except 4% (v/v) Type-I collagen was supplemented in different cell culture media for experiments [32]. Culturing ECs in these settings led to the formation of tubular networks after 24 hrs (Fig. 3). Both 2D and 2.5D capillary assays confirmed the co-cultures of GBM-induced M2-like TAMs and M2 macrophage subsets with ECs increased capillary area relative to untreated M0 macrophages, while co-culture with M1 macrophages regressed capillary formation (Fig. 3b).
Figure 3. Direct interactions and immunosuppressive cytokine milieu of GBM-induced M2-like TAMs promote EC angiogenic behaviors.
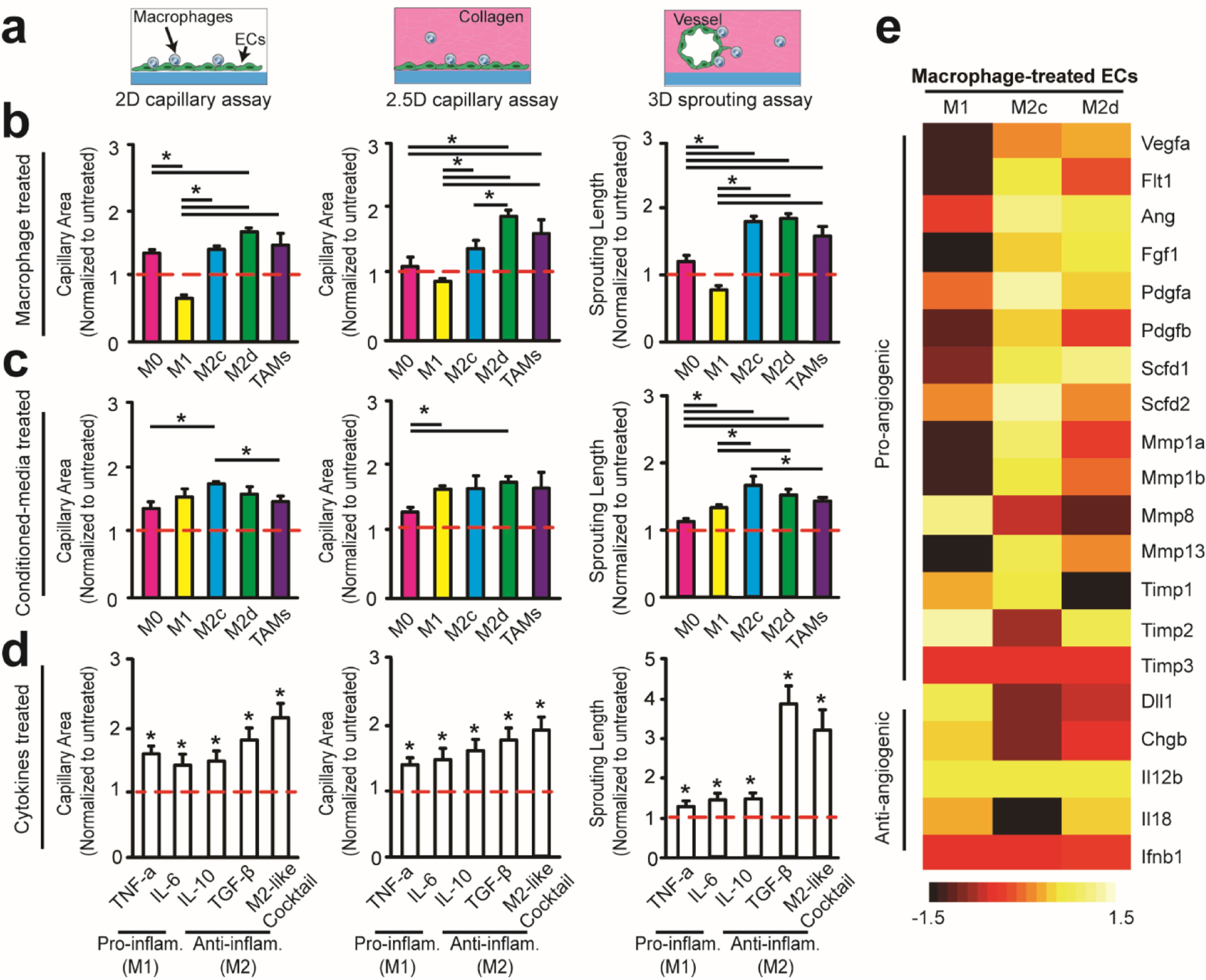
(a) Multidimensional angiogenic behaviors were modeled using 2D and 2.5D capillary assays and a 3D sprouting assay. The proangiogenic capacity of pro-inflammatory and anti-inflammatory macrophage phenotypes and cytokines were assessed with (b) macrophage treated ECs, (c) conditioned media treated ECs and (d) cytokine (5000 pg/mL) treated ECs. Quantified capillary area and sprouting length were normalized to untreated ECs after 24 hrs and 3 days, respectively. (e) Heat map of normalized pro-/anti-angiogenic mRNA expressions in macrophage-treated ECs after 24 hrs of co-culture on cover glass. M0-treated ECs are used as control. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using one-way ANOVA, followed by the Tukey post hoc test. *, P < 0.05.
We investigated the pro-angiogenic capacity of GBM-induced M2-like TAMs in our 3D biomimetic microfluidic device. To achieve this, TAMs were suspended in hydrogel and co-cultured with the sprouting 3D artificial vessel for a 3-day period to examine 3D EC angiogenic sprouting (Fig. 1). Consistent with 2D and 2.5D assays, we determined TAMs promoted 3D EC angiogenic sprouting relative to the effect of uncommitted M0 macrophages, confirming the pro-angiogenic capacity of TAMs (Fig. 3b). From this, our multidimensional in vitro angiogenesis assays confirm that alternatively-activated M2 macrophages (M2c and M2d) promote angiogenesis and classically-activated M1 macrophages suppress angiogenesis (Fig. 3b). It was also determined that GBM cells alone can promote 3D angiogenic sprouting in vitro, but to a lesser extent than GBM-induced TAMs (Supplemental Fig. S4a). Hence, we suspect GBM microenvironments facilitate 3D proangiogenic activity through enhanced pro-angiogenic and immunosuppressive M2-like macrophage polarization. These in vitro results support our previous in vivo observation of increasing numbers of infiltrating M2-like TAMs in recurrent GBM after exposure to antiangiogenic therapy [11, 12].
3.4. TAM-associated inflammatory cytokine milieu regulates GBM proangiogenic activity.
We examined whether promoted angiogenesis was dependent on soluble cytokine secretions of macrophages. To this end, we examined activity of ECs under stimuli of macrophage conditioned media (M1, M2c, M2d, or TAMs) and specific pro-/anti-inflammatory cytokines (Fig. 3c&d). We found that conditioned media of GBM-induced TAMs and M2c/M2d macrophage subsets could promote angiogenesis. However, the pro-angiogenic capacity of conditioned media was less effective compared to the presence of co-cultured TAMs (Fig. 3c). This implies the pro-angiogenic effect of GBM-induced TAMs requires both soluble anti-inflammatory cytokines and direct EC-macrophage interactions.
Isolating specific cytokines found in macrophage secretions, we screened the pro-angiogenic potential of anti-inflammatory cytokines, TGF-β and IL-10, which are predominantly secreted in the GBM microenvironment by M2 macrophages and GBM cells [12]. Our results demonstrated both anti-inflammatory TGF-β and IL-10 can stimulate 2D and 2.5D capillary network formation and 3D sprouting in a dose-dependent manner (Fig. 3d and Supplemental Fig. S3). Remarkably, our cytokine screening (Fig. 2d) and angiogenic assays (Supplemental Fig. S3) indicate that TGF-β is the most potent, in terms of pro-angiogenic capacity, among the anti-inflammatory soluble cytokines due to enhanced sprouting length change (> 4-fold) compared to other tested cytokines. Considering that the GBM microenvironment is enriched with other inflammatory cytokines, we further reconstituted cytokine cocktails to imitate the mixed pro-/anti-inflammatory conditions of the GBM microenvironment in our 3D microfluidic devices. We found that an M2-like cocktail (Supplemental Table S2) can further promote 3D angiogenic activity with (Supplemental Fig. S4b) or without (Fig. 3d) the presence of M2 macrophages.
Interestingly, although co-culture with pro-inflammatory M1 macrophages and ECs significantly suppressed capillary formation and angiogenic sprouting compared to co-culture with TAMs or M2 macrophages (Fig. 3b), M1 macrophage conditioned media and pro-inflammatory (TNF-α, IL-6) cytokines did not show similar anti-angiogenic capacity. Instead, M1 macrophage conditioned media and pro-inflammatory cytokines, in the absence of macrophages, promoted both 2D/2.5D capillary formation and 3D sprouting (Fig. 3c&d, Supplemental Fig. S3). This suggests that the anti-angiogenic effect of M1 macrophages relies on direct EC-macrophage interactions. Our results also indicated that M2 macrophage conditioned media or an M2-like cocktail (Fig. 2d and Supplemental Table S2) is capable of reversing M1-suppressed angiogenesis (Supplemental Fig. S4b&c). Hence, this indicates that TAM anti-inflammatory/immunosuppressive cytokine milieus can indirectly contribute to pro-angiogenic activity by reversing M1-suppressed angiogenesis in GBM.
Furthermore, we performed mRNA sequencing to screen expression levels of genes associated with EC angiogenesis of different macrophage phenotypes (M0, M1, M2c, M2d). To this end, ECs and macrophages were co-cultured on 2D substrates for 24 hrs, then isolated using flow cytometry for EC RNA-sequencing analysis. Overall, there was an evident increase in pro-angiogenic genes Vegfa, Ang, Pdgfa, Mmp13, Flt1, Timp3, compared to anti-angiogenic genes Dll1, Chgb, and Il18 for difference macrophage subsets (Fig. 3e). However, the presence of M1 macrophages limited the expression of pro-angiogenic genes Flt1, Ang, Scfd1, Mmp1a and Mmp13 relative to the presence of M2 macrophages, supporting the anti-angiogenic phenotype of M1 macrophages. Together, these findings strengthen our previous in vivo observation [11, 12] that TAMs act as the main drivers of anti-angiogenic therapeutic resistance in GBM through their immunosuppressive cytokines and EC-macrophage interactions, representing potential therapeutic targets in GBM [12].
3.5. EC-macrophage interaction through αvβ3 integrin regulates inflammation-driven angiogenesis.
Recent studies have confirmed that most TAMs are located near perivascular tumor regions where active angiogenesis occurs, corroborating the significance of EC-macrophage interactions for regulating tumor angiogenesis [21, 22]. Importantly, we observed that macrophages tend to relocate around the 3D artificial vessel and have direct contact with ECs (Fig. 4a), indicating direct EC-macrophage interaction regulates the inflammation-driven angiogenesis. To confirm this notion, different macrophage subsets (M0, M1, M2c, or M2d) were reallocated in the hydrogel platform. Macrophages were suspended in hydrogel to form direct contact from outside vasculature (i), seeded directly onto lumen of the vascularized EC channel to form direct contact from inside vasculature (ii), or cultured in the adjacent channel without direct EC-macrophage contact (iii) for 3 days (Fig. 4a). We found the presence of pro-angiogenic M2 macrophages, from outside (i) and inside (ii) the vessel, significantly promoted 3D angiogenic sprouting, compared to setting without contact (iii) (Fig. 4b). In contrast, the presence of anti-angiogenic M1 macrophages from outside (i) or inside (ii) the vasculature significantly inhibited 3D angiogenic sprouting (Fig. 4b). However, EC-macrophage contact from inside the vessel (ii) further repressed overall sprouting length compared to EC-macrophage contact from outside the vessel (i), implying that secretory gradients may be responsible for promoting angiogenesis (Fig. 4b). In addition, M1 and M2 macrophages seeded in the adjacent channel (iii) generally promoted sprouting, compared to M0 macrophages, confirming the pro-angiogenic effects of soluble cytokines in promoting angiogenesis. Therefore, these findings suggest that inflammation-driven angiogenesis depends on both direct EC-macrophage interactions and soluble inflammatory cytokines. Consequently, we propose these findings may provide novel anti-angiogenic therapeutic targets to suppress inflammation-driven angiogenesis in GBM.
Figure 4. Tuning 3D EC-macrophage interactions induce distinct angiogenic behaviors.
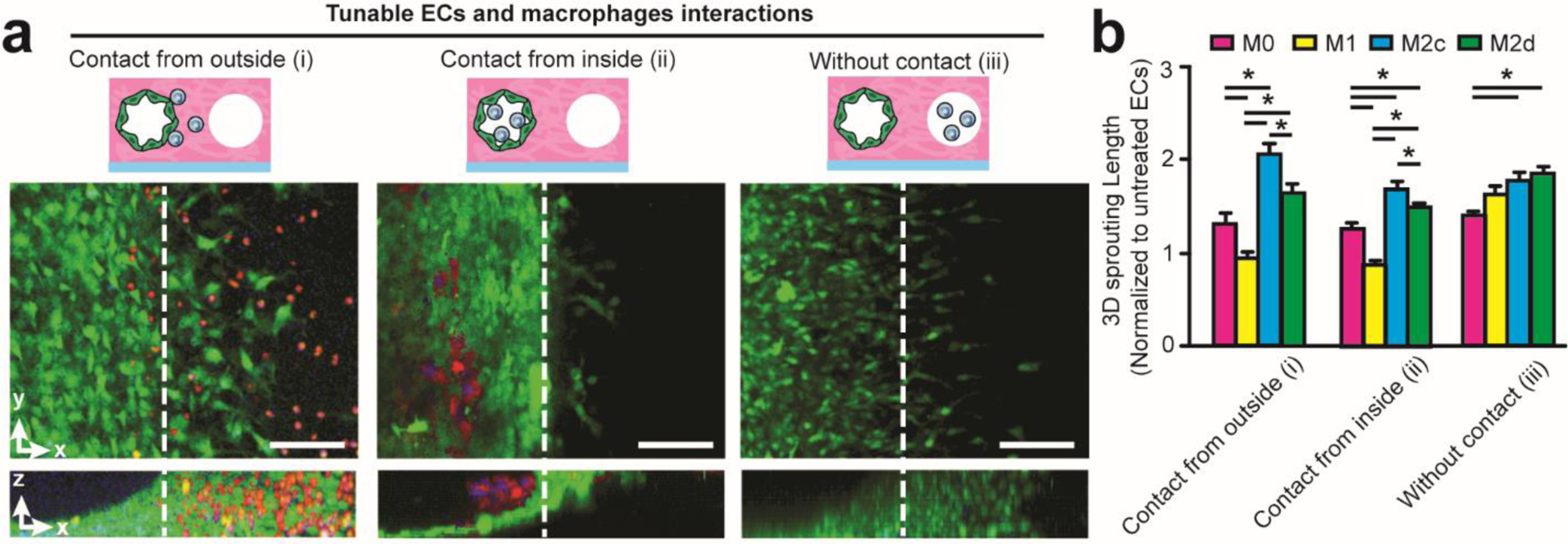
(a) Representative 3D fluorescent projections of confocal image stack showing distinct 3D EC-macrophage interactions with contact from outside (i), contact from inside (ii), and without contact (iii). Noted that TAMs are labelled with Cell Tracker Red. Scale bar is 200 μm. (b) Quantified 3D sprouting length (normalized to untreated ECs) under different EC-macrophage interactions with the presence of different macrophage subsets. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using one-way ANOVA, followed by the Tukey post hoc test. *, P < 0.05.
To further interrogate the proangiogenic mechanism of direct EC-TAM interactions, we demonstrate αvβ3 integrin-mediated signaling regulates inflammation-driven angiogenesis such as EC capillary formation and sprouting (Fig. 5). Emerging evidence suggests that integrin αvβ3 is highly expressed on angiogenic ECs [26] and GBM cells [24]. To determine whether αvβ3 integrin is involved in EC-macrophage interactions, we first investigated β3 integrin expression of different macrophage phenotypes (M0, M1, M2c, M2d). It was found that M2 macrophages exhibit higher β3 integrin expression and M1 macrophages display lower β3 integrin expression, compared to uncommitted M0 macrophages (Fig. 5a&b). Therefore, we hypothesized that αvβ3 integrin expression of ECs may be regulated by EC-macrophage interactions. To confirm this notion, we then conducted mRNA sequencing across a panel of integrin subunits of ECs co-cultured with different macrophage subsets (M0, M1, M2c, M2d) (Fig. 5c). We found that mRNAs of Itga3, Itga5, Itga6, Itgav, Itgb1, Itgb3, Itgb5 were highly expressed in macrophage-treated ECs after 24 hrs. Upon closer examination, we found Itgav (αv integrin) expression was upregulated in ECs co-cultured with M2 macrophages, and Itgb3 (β3 integrin) expression was downregulated in ECs co-cultured with M1 macrophages. Thus, these results confirmed that αvβ3 integrin is a critical participant in EC-macrophage interactions that regulate the inflammation-driven angiogenesis.
Figure 5. Altered β3 integrin expression can regulate EC-macrophage interactions and inflammation-driven angiogenesis.
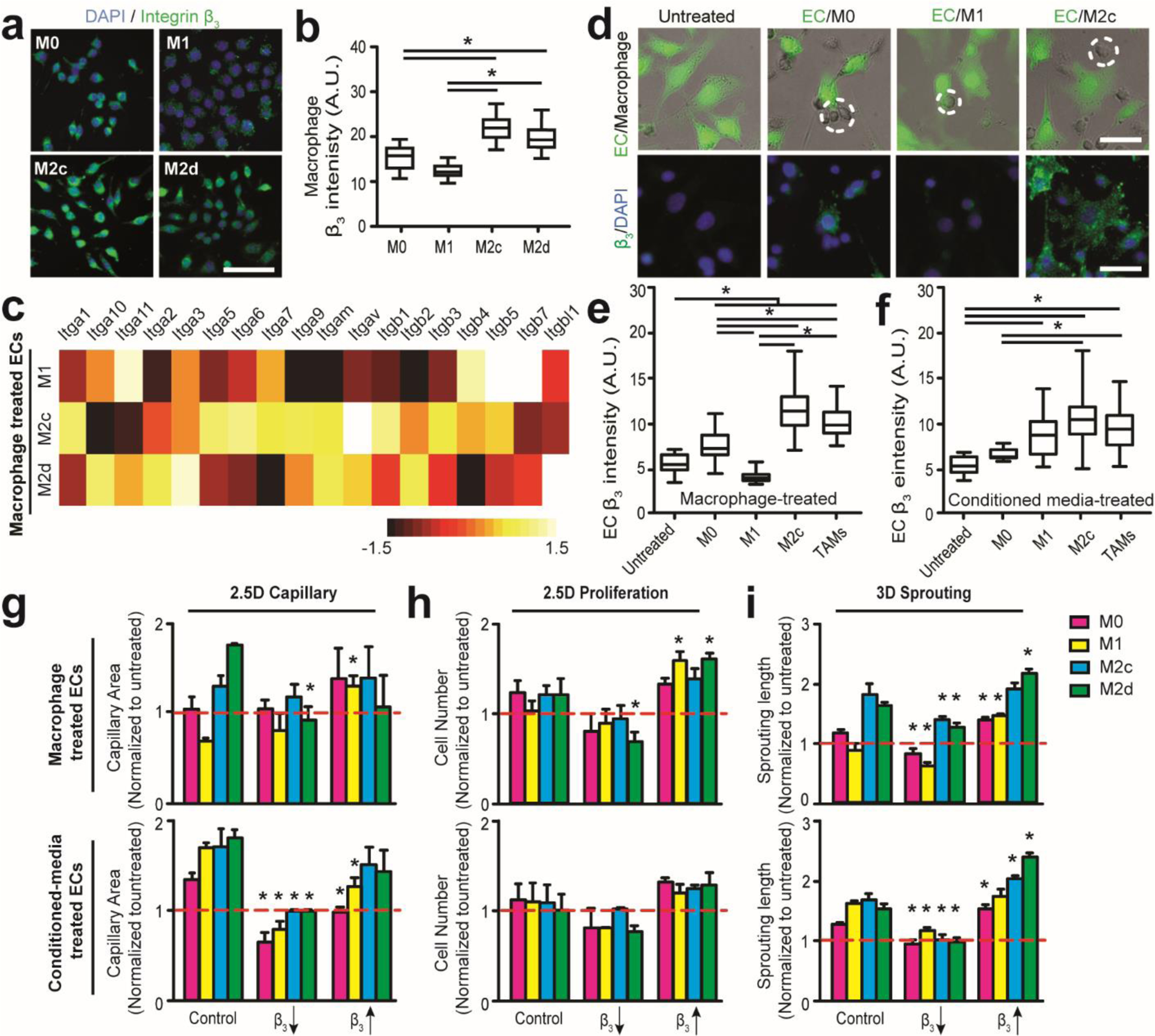
(a) Representative immunofluorescent images comparing β3 integrin subunit expressions in different macrophage subsets on 2D substrates. Scale bar is 100 μm. (b) Quantified β3 integrin subunit expressions of different macrophage subsets on 2D substrates. (c) Heat map of normalized mRNA expressions of different integrin subtypes in macrophage-treated ECs on 2D substrates after 24 hrs. M0-treated ECs are used as control. (d) Representative merged (top) bright field and immunofluorescent images of co-cultured GFP ECs (green) and macrophages (dotted circle), and immunofluorescent staining images (bottom) comparing β3 integrin subunit expressions in EC-macrophage interactions with different macrophage subsets on 2D substrates. Scale bar is 100 μm. (e-f) Quantified expression of integrin subunit β3 in ECs treated with (e) distinct macrophage subsets and (f) conditioned media on 2D substrates. Note the suppressed β3 integrin expression in ECs interacting with M1 macrophages. (g-j) Multidimensional angiogenic behaviors were quantified after suppressing and promoting integrin β3 expression for 24 hrs in (g) 2.5D capillary and 2.5D proliferation (i) assays and (j) 3 days in 3D sprouting assays. Red line represents untreated EC behaviors. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using the one-way ANOVA, followed by the Tukey post hoc test, or Student’s t-test as compared to Control in g-i. *, P < 0.05.
We next evaluated β3 integrin subunit protein levels in ECs after treatment with different macrophage subsets (M0, M1, M2c, TAMs) or their conditioned media for 24 hrs. It was found that M2c macrophages, GBM-induced TAMs, and their conditioned media all enhanced β3 integrin expression levels in ECs, relative to the effects of M0 macrophages (Fig. 5d-f). Interestingly, direct co-culture with M1 macrophages inhibited EC β3 integrin expression, while M1 macrophage conditioned media did not produce the same inhibitory effects. These results indicate that direct EC-macrophage interactions induce distinct β3 expressions in angiogenic ECs, which may distinctly contribute to inflammation-driven angiogenesis. Furthermore, both surface expression of β3 integrin on macrophages and pro-/anti-inflammatory macrophage secretions can regulate EC β3 integrin expression.
3.6. Altered integrin expression can regulate EC-macrophage interactions and angiogenesis through Src-PI3K-YAP signaling.
Not only do ECs interact with macrophages and soluble factors in GBM tumor niche, but ECs also interact with the ECM. Cells can perceive ‘outside’ signals from its microenvironment and bidirectionally transmit information for an adaptive response via integrin receptors that regulate survival, migration, proliferation and differentiation [29]. Integrin αvβ3 is highly expressed on angiogenic ECs [23] but not on quiescent ECs, which we hypothesize is regulated by either EC-macrophage or cell-ECM interactions. In addition, enhanced integrin αvβ3 expressions in ECs has been found within GBM that is characterized with aberrant inflammation-driven angiogenesis, as compared to low-grade of glioma [24]. Hence, we engineered integrin-specified hydrogels and probed the crosstalk among soluble pro-/anti-inflammatory secretions, cell-cell and cell-matrix interactions in the GBM microenvironment that regulate inflammation-driven angiogenesis.
To further dissect the role of integrin in inflammation-driven angiogenesis, we re-engineered a Type-I collagen hydrogel with cell-adhesion sites specific for integrin αvβ3 (Supplemental Fig. S5). Tunable integrin αvβ3 activity was achieved by conjugating RGD peptides onto Type-I collagen monomers for enhanced expression, or incorporating β3-blocking antibodies into hydrogel for suppressed expression. Isotype IgG1 from murine myeloma was used as a control (see Materials and methods 2.1 for details). We first evaluated the cell viability of macrophages in engineered collagen, and found that there were no effects on cell viability (>80%) as compared with that in unmodified collagen (Supplemental Fig. S5d). We then quantified integrin β3 expressions of ECs on different matrix composites, showing improved integrin clustering as an indicator of integrin activation [44], on cover glass coated with modified collagen after 24 hrs. However, applying the β3-blocking antibodies reduced the observed integrin activation (Supplemental Fig. S5). As indicated in Fig. 5g-i, the presence of β3-specific RGD peptides, overall, improved multidimensional angiogenic capillary, proliferation and sprouting for ECs treated with different macrophage phenotypes (M0, M1, M2c, M2d), their conditioned media, or ECs only (Supplemental Fig. S5c). However, the presence of β3-blocking antibodies suppressed multidimensional angiogenesis, which confirms integrin αvβ3 as a prerequisite for inflammation-driven angiogenesis (Fig. 5g-i).
We further explored potential downstream signaling involved in facilitating inflammation-driven angiogenesis. Of interest, it has been well proven that integrin αvβ3 can regulate angiogenesis and glioma progress through Src/phosphoinositide 3-kinase (PI3K) signaling [45, 46]. In addition, PI3K-Akt activation can further regulate pro-angiogenic factor (angiopoietin-2) synthesis via YAP activity [47]. YAP, a transcriptional coactivator in the Hippo pathway, has recently been investigated in endothelial [47] and tumor cells [48], and has been shown to be regulated by nucleocytoplasmic shuttling in a phosphorylation-dependent manner [49]. For instance, YAP phosphorylation at S127 sites can lead to cytoplasmic localization of YAP [49]. Acknowledging these contributions, we proposed and probed Src-PI3K-YAP signaling in angiogenic EC-macrophage interactions (Fig. 6a). YAP immunostaining indicated the presence of GBM-induced TAMs and M2 macrophages with ECs increased the nuclear localization of YAP, one feature of activated ECs [47], compared to the presence of M1 macrophages (Fig. 6b-d). Western blotting confirmed this finding, and phosphorylation of YAP (p-YAP) on its Lats phosphorylation site S127 was lower in ECs after treatment with M2c macrophages relative to M1 macrophages (Fig. 6e), which is consistent with previous studies regarding YAP activity in angiogenic ECs [47, 49]. Inhibiting YAP activity by administering Verteporfin (5 μM) increased cytoplasmic localization of YAP (Supplemental Fig. S6) and inhibited multidimensional capillary, proliferation and sprouting in the presence of macrophages and conditioned media in vitro (Fig. 6f-h, and Supplemental Fig. S7). Furthermore, inhibiting Src and PI3K activity by administering PP2 (10 μM) and Wortmannin (10 μM), respectively, suppressed pro-angiogenic activity of ECs treated with macrophage and conditioned media (Fig. 6f-h). Together, our results imply that GBM-induced M2-like TAMs modulate inflammation-driven pro-angiogenic activity via αvβ3 integrin-Src-PI3K-YAP signaling.
Figure 6. Integrin-mediated Src-PI3K-YAP signaling facilitates inflammation-driven angiogenic behaviors.
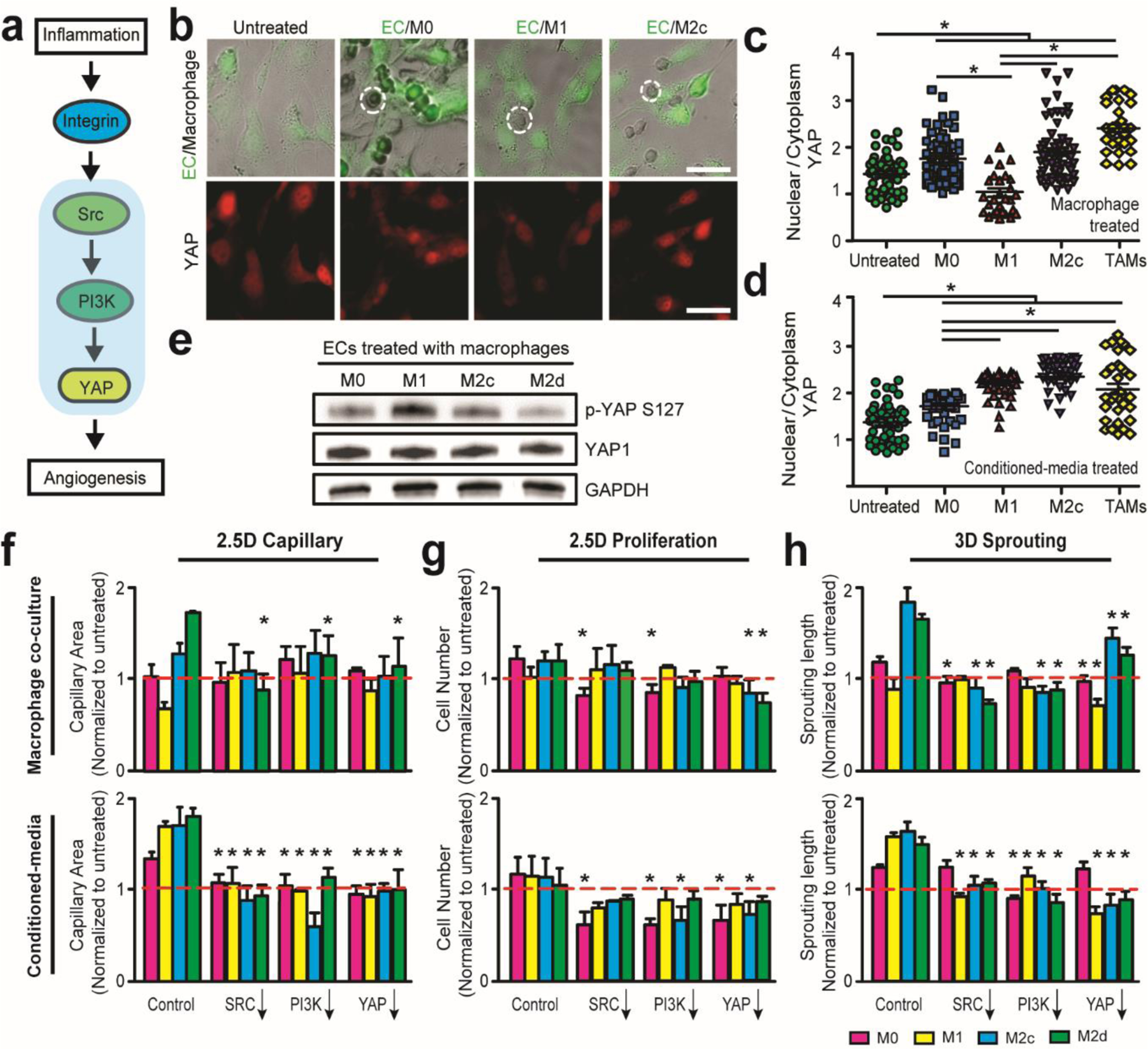
(a) Diagram representing an integrin-mediated Src-PI3K-YAP signaling pathway potentially responsible for distinct inflammation-driven EC behaviors. (b) Representative merged (top) bright field and immunofluorescent images of co-cultured GFP ECs (green) and macrophages (dotted circle), and immunofluorescent images (bottom) of comparing YAP expressions in EC-macrophage interactions with different macrophage subsets on 2D substrates. Scale bar is 100 μm. (c-d) Quantified ratio of nuclear and cytoplasmic YAP expression in ECs treated with (c) distinct macrophage subsets and (d) conditioned media for 24 hrs. (e) Western blot analysis of p-YAP S127 and YAP1 in ECs in the presence of distinct macrophage subsets on 2D substrates after 24 hrs. (f-h) Multidimensional angiogenic behaviors, with or without presence of polarized macrophages, were quantified after administering Src (PP2, 10 μM), PI3K (wortmannin, 10 μM), and YAP (verteporfin, 5 μM) inhibitors for 24 hrs in (f) 2.5D capillary and (g) 2.5D proliferation assays, and 3 days in (h) 3D sprouting assays. Red line represents the untreated EC behaviors. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using the one-way ANOVA, followed by the Tukey post hoc test, or Student’s t-test as compared to Control in f-h. *, P < 0.05.
3.7. Dual integrin αvβ3 and TGFβ-R1 blockade arrest the pro-angiogenic behaviors in GBM and improve anti-angiogenic therapy efficacy.
Anti-angiogenic therapy has become an important target in the treatment of many solid tumors including GBM [50]. Despite promising preclinical assessments, anti-angiogenic agents have largely failed to show clinical benefit in large randomized GBM trials [38]. Our previous study demonstrated a reduction of “rebound” vascularization in anti-VEGF (cediranib) treated GBM patients, but treatment was insufficient to hinder GBM recurrence due to increasing numbers of infiltrating pro-tumorigenic M2 TAMs [11]. Particularly, Cilengitide (EMD121974), an αvβ3 and αvβ5 integrin antagonist, has been assessed in phase III clinical trials for GBM [51]. Unfortunately, current studies display only transient clinical response in GBM patients [52]. Moreover, our data (Fig. 3d) and previous findings have shown that TGF-β is a critical prerequisite in glioma-associated immunosuppression and angiogenesis and facilitates a radiation-resistant GBM phenotype [53]. However, the anti-angiogenic effects of solely TGF-β inhibition are still a matter of debate [54].
Our results suggest that both soluble immunosuppressive cytokine TGF-β (Fig. 3d) and integrin (αvβ3)-dependent EC-macrophage interactions (Fig. 5) contribute to pro-angiogenic activity in GBM. This highlights a potential combinatorial therapeutic blockade of integrin αvβ3 and TGF-β receptor type I (TGFβ-R1) for arresting GBM angiogenesis and improving anti-angiogenic therapeutic efficacy (Fig. 7a). To this end, we assessed the anti-angiogenic efficacy of dual αvβ3 integrin and TGF-β R1 inhibition in EC-TAM interactions. We administered cilengitide (αvβ3 integrin antagonist) and LY-364947 (TGFβ-R1 inhibitor) for 24 hrs in our established in vitro GBM microenvironment model (Fig. 7b-d). We confirmed mono-inhibition suppressed either αvβ3 integrin, YAP or phosphorylated Smad2 (p-Smad2, a downstream TGFβ-R1 signaling marker) in proangiogenic activity [53, 55], while dual-inhibition augmented the suppressive effects on both pathways. Furthermore, cediranib (pan-VEGFR tyrosine kinase inhibitor [11]) was administered as a comparison to confirm dual αvβ3 integrin and TGFβ-R1 inhibitions could optimize anti-VEGFR efficacy in suppressing αvβ3 integrin, YAP, and p-Smad2 (Fig. 7b-d).
Figure 7. Dual αvβ3 integrin and TGFβ-R1 blockade arrests pro-angiogenic EC-TAM interactions in 3D GBM angiogenesis.
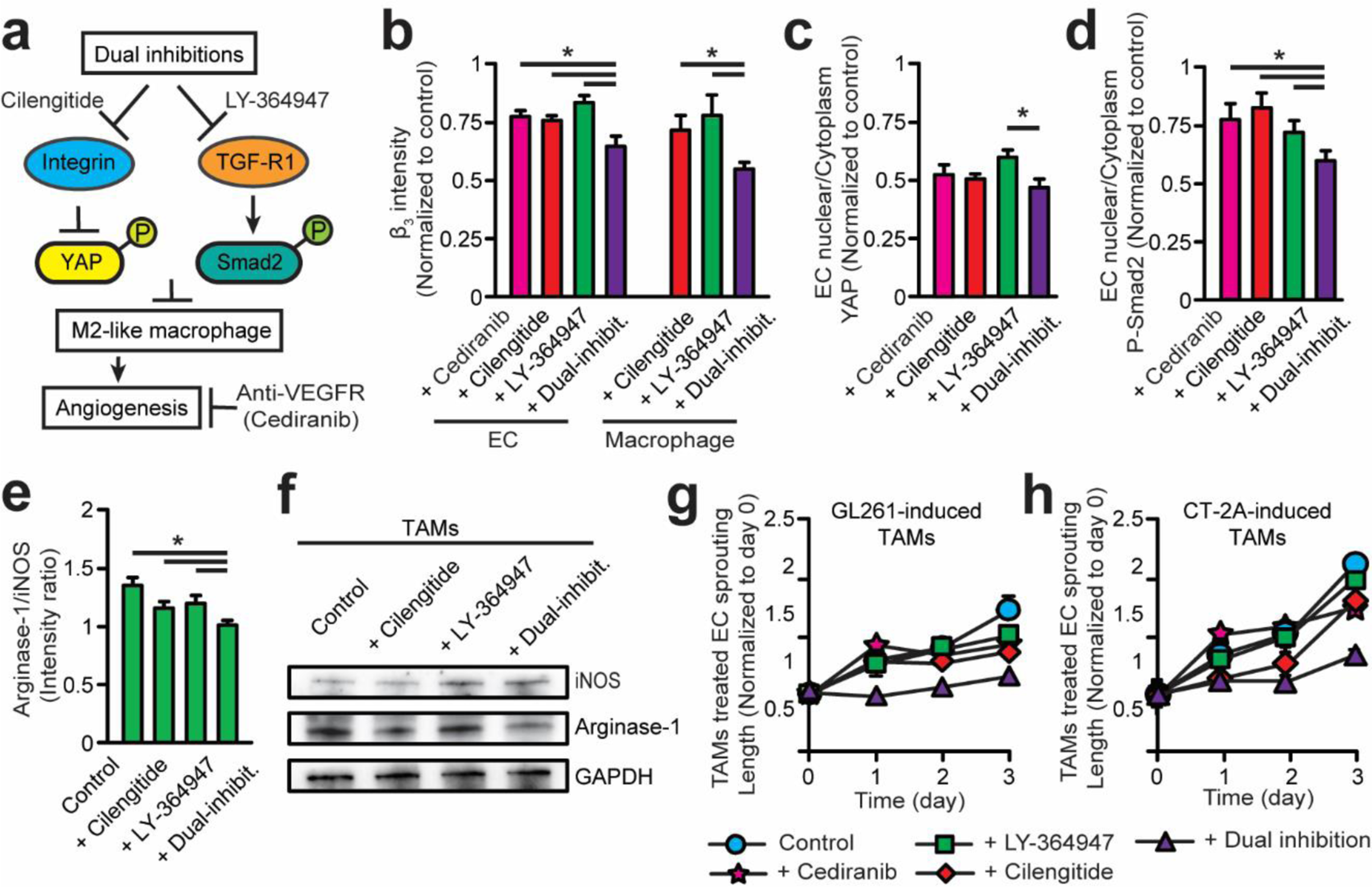
(a) Scheme of administering and comparing combinatorial inhibition of αvβ3 (cilengitide) and TGFβ-R1 (LY-364947) relative to VEGFR (cediranib) inhibition to arrest pro-angiogenic EC-TAM behaviors in GBM angiogenesis. (b) Quantified expression of integrin β3 in TAMs and TAM-treated ECs with mono or dual-inhibitions for 24 hrs on 2D substrates. (c-d) Quantified expression of (c) YAP and (d) p-Smad2, a downstream TGF-β signaling marker, in TAM-treated ECs with mono or dual-inhibitions for 24 hrs on 2D substrates. (e) Quantified expression of stained iNOS and Arginase-1 in GL261-induced TAMs for 24 hrs on 2D substrates. (f) Western blot analysis of iNOS and Arginase-1 levels in GL261-induced TAMs treated with single or dual inhibition after 24 hrs on 2D substrates. (g-h) Quantified 3D sprouting lengths of ECs co-cultured with (g) GL261-induced TAMs and (h) CT-2A-induced TAMs treated with single and dual inhibition for 3 days. Note suppressed 3D sprouting length after dual inhibition. Error bars represent ± s.e.m. from 3 independent experiments. P-values were calculated using the one-way ANOVA, followed by the Tukey post hoc test. *, P < 0.05.
We further explored whether dual-inhibition of integrin αvβ3 and TGFβ-R1 could reverse the proangiogenic nature of GBM-induced M2-like TAMs toward an anti-angiogenic M1 phenotype (Fig. 7e-f), and effectively enhance anti-angiogenic GBM treatment efficacy (Fig. 7g&h). Our results indicate that single blockade of either TGFβ-R1 or αvβ3 integrin with cilengitide or LY-364947, respectively, was insufficient to suppress angiogenesis in GBM. Dual blockade of TGFβ-R1 and αvβ3 integrin (Supplemental Fig. S8a-b) significantly suppressed expression of the M2 marker (Arginase-1) in GBM-induced TAMs (Fig. 7e-f), angiogenic 2D capillary (Supplemental Fig. S8c) and endothelial sprouting in our in vitro 3D GBM microenvironment, compared to the effects of mono-blockade use (Fig. 7g). Particularly, 3D sprouting treated with dual-inhibition at day 3 demonstrated relatively minimal change to sprouting length compared to initial status (day 0). To further assess the functionality of the proposed dual inhibition, we also evaluated the anti-angiogenic efficacy in a more-aggressive GBM tumor cell line (CT-2A) (Fig. 7h) that meet the diagnostic criteria for GBM tumors (pathologic grade IV) and invade the in vivo brain parenchyma more than the GL261 tumors [56]. The anti-angiogenic effects, measured by 2D capillary formation and 3D angiogenic sprouting, of mono- and dual-inhibition therapy was comparable to that of GL261 implantation (Fig. 7h). These results confirm our hypothesis that TAMs may be the main drivers of anti-angiogenic resistance in GBM through their immunosuppressive functions. Given the capability of our 3D GBM angiogenesis model to mimic the clinical hallmarks of in vivo GBM microenvironments including TAM-mediated proangiogenic and immunosuppressive niche, these findings justify a potential combinatorial therapy of co-targeting TGF-β receptors and αvβ3 integrin that may improve the efficacy of current single-target anti-angiogenic GBM therapies by overcoming the immunosuppression and arresting tumor vascularization in the GBM microenvironment.
4. Discussion
Anti-angiogenic therapies represented promising treatment avenues in GBM and their effects were explored in large randomized clinical trials. However, there are significant issues with limited and unpredictable therapeutic efficacy and resistance against anti-angiogenic therapies resulting in GBM tumor progression. Because our previous in vivo study implicated that TAMs may facilitate escape from anti-angiogenic therapies [11, 12], we examined the role of macrophage plasticity in GBM-associated in vitro angiogenesis for improving therapeutic efficacy by using GBM-mimicking glioma cell lines (GL261 or CT-2A). Given the difficulty in examining tumor angiogenesis in vivo, 3D sprouting assays using microfluidic channels within an ECM to mimic blood vessel architecture have been widely used in vitro [31]. Although these studies successfully reconstitute 3D artificial vasculature, most oversimplify in vivo vascularized and immunological microenvironments. Specifically, GBM is characterized by distorted vascularization [57], elevated TAM infiltration [8] and marked immunosuppression [9]. In our 3D angiogenesis assays, EC activity under the stimuli of immunosuppressive factors, TAM phenotype and ECM are investigated altogether. In addition, cell-cell and cell-matrix interactions are controlled independently to reveal their importance in GBM vascularization. Hence, our biomimetic 3D model can emulate accurate features of in vivo GBM microenvironments and local tumor immunity by housing key cellular elements, vascularized ECM, and macrophage-associated immunosuppression in this artificial tumor niche. The clinical applications of such a system can be expanded for high throughput screening and personalizing therapies for GBM.
Our findings suggest that macrophage polarization can be orchestrated by the microenvironment (e.g. GBM tumors) to distinctly regulate EC pro-angiogenic behaviors in vitro [28]. GBM-induced M2-like TAMs and their conditioned media significantly promote angiogenesis. Elevated expressions of placental growth factor (PGF) in M2c macrophages has been suggested to be responsible for promoted angiogenesis [28]. However, our cytokine screening results also show that M2 macrophages enrich the vascularized ECM with anti-inflammatory cytokines such as TGF-β and IL-10 to potentially enhance angiogenesis. Furthermore, macrophages can degrade the ECM by producing proteases, such as metalloproteinase-9 (MMP-9), MMP-2, MMP-3, and excavating routes for EC proliferation and migration, and indirectly promoting angiogenic invasion and sprout formation [58]. Given that TAMs largely have immunosuppressive M2-like phenotype [59], our results further suggest that aberrant vasculature in tumor microenvironments may be induced by the synthesis and secretion of immunosuppressive cytokines, growth factors and proteases together.
Previous studies have demonstrated the importance of EC-macrophage interactions in promoting M2 macrophage polarization, colony formation, and VE-cadherin expressions in endothelial cells [5], as well as the fusion of two sprouts for capillary formation [22]. Indeed, our 3D EC sprouting and 2D capillary assays (Fig. 4a, and Supplemental Fig. S7a) showed that macrophages physically interacted and attached to ECs. Our results further demonstrated the importance of cell-cell contacts in addition to soluble inflammatory cytokine milieus (TGF-β1) by seeding macrophages in different locations relative to ECs (Fig. 4). Further study implied the importance of integrin αvβ3 receptors (Fig. 5) and the downstream YAP signaling (Fig. 6) in the EC-macrophage contacts. Finally, we demonstrated that inhibiting both soluble immunosuppressive cytokine TGF-β receptors and integrin αvβ3 signaling (Fig. 7) can improve the efficiency of single anti-angiogenic drugs.
We also observed that M1 macrophages suppress multidimensional proangiogenic activity when in direct contact with ECs (Fig. 3b). However, conditioned media of M1 macrophages promoted angiogenesis, suggesting that direct interaction between M1 macrophages and ECs impair pro-angiogenic activity [28], but not the M1 macrophage secreted chemokines and anti-inflammatory cytokines. One study suggests that M1-suppressed angiogenesis may depend on elevated thrombospondin-1 (Thbs1) gene expression in M1 macrophages, which can inhibit EC migration, survival and angiogenesis [60]. Our results (Fig. 5) suggest that the blocked αvβ3 integrin expression coupled with the presence of M1-like macrophages may be required for suppressing angiogenesis, because enhanced expression of αvβ3 integrin using RGD-specific collagen significantly abolished the M1-suppressed angiogenesis. Interestingly, soluble anti-inflammatory cytokine cocktails of TGF-β1 and IL-10, not pro-inflammatory cytokines, can also reverse the M1-suppressed angiogenesis in the presence of M1-like macrophages (Supplemental Fig. S4), which implies that anti-inflammatory cytokines in an immunosuppressive GBM microenvironment may override the effects of integrin enhancement and promote angiogenesis in a dose-dependent manner. Hence, normalizing vascularization in GBM anti-angiogenic therapies should redirect TAM polarization towards anti-tumorigenic M1-like phenotypes, and, importantly, relieve immunosuppression simultaneously.
TGF-β1 is a multi-functional cytokine that regulates cell proliferation, differentiation, migration and survival in immunosuppressive tumor microenvironments. Genetic knockdown of TGF-β1 receptor induces severe defects in angiogenesis and embryonic development [61]. Our results (Fig. 3d and Fig. 7) further suggest that TGF-β1 may be responsible for pro-angiogenic M2-like macrophage polarization in GBM microenvironments. It has been reported that inhibiting TGFβ-R1 decreased expression of M2-like polarization genes such as arg1, mcr2, mgl2, and ym1 in macrophages [62]. In addition, our immunoassay results indicate elevated levels of TGF-β present in conditioned media of GBM tumor cells (Fig. 2e). However, solely inhibiting TGFβ-R1 with LY-364947 partially reduced EC activity in our 3D GBM microenvironments (Fig. 7g-h). Given the importance of both soluble immunosuppressive cytokine TGF-β (Fig. 3d) and integrin (αvβ3)-dependent EC-macrophage interaction (Fig. 5) in GBM tumor angiogenesis, a dual inhibition therapy co-targeting TGF-β and αvβ3 integrin shows potential to optimize the efficacy of current mono-target anti-angiogenic GBM therapy. Indeed, dual inhibition of TGFβ-R1 and αvβ3 integrin in GBM microenvironments further suppresses EC proliferation and pro-angiogenic macrophage polarization, emphasizing the significance of integrin expression and immunosuppression in regulating GBM tumor angiogenesis and providing promising adjuvant therapeutic targets in glioblastoma anti-angiogenic therapy.
5. Conclusion
To our knowledge, 3D organotypic GBM angiogenesis models that integrate controlled macrophage-EC interactions, cell-matrix interactions and immunosuppressive conditions have not yet been reported. Using our integrin-specific biomaterials and engineering methods to reproduce a biomimetic microenvironment model, we recapitulated the in vivo GBM proangiogenic and immunosuppressive niche, which are the hallmarks of GBM microenvironments. We further manipulated the EC-macrophage-GBM interaction and immunosuppressive environment in our study to emphasize the roles of macrophage-associated immunosuppression, integrin-mediated EC-macrophage and cell-ECM interactions for regulating malignant 3D angiogenesis in GBM tumors. Hence, our results suggest the development of novel anti-angiogenic therapeutic strategies for GBM should consider macrophage-associated immunosuppression, EC-macrophage interactions and tumor ECM as well as GBM driver mutations. In addition, there are no available biomimetic models currently available to accurately and efficiently predict patient therapeutic efficacy due to the genetic heterogeneity of GBM. Our 3D platform can be easily applied towards phenotyping different GBM tumors by culturing patient-derived GBM cells, ECs, and TAMs in a 3D biomimetic and controllable microenvironment and allows high throughput drug screening. We believe this resource may promote the development and screening of novel therapeutic strategies that incorporate chemotherapy with biologic therapy such as immunotherapy and anti-angiogenic therapy, thereby combining the most promising trends together for anti-GBM therapy. Populating the chip with patient derived tumor cells and immune cells will also enable a truly personalized drug screening, bringing precision medicine another step forward.
Supplementary Material
Funding
This work was supported by the Department of Mechanical and Aerospace Engineering at New York University, the New York University Whitehead Fellowship, the American Heart Association Scientist Development Grant [16SDG31020038], the National Institutes of Health [NCRR S10 RR023704-01A1 for confocal imaging, and P30CA016087 for cell sorting/flow cytometry].
Footnotes
Competing financial interests
The authors declare no competing financial interests.
REFERENCE
- [1].Motz GT, Coukos G, The parallel lives of angiogenesis and immunosuppression: cancer and other tales, Nature reviews. Immunology 11(10) (2011) 702. [DOI] [PubMed] [Google Scholar]
- [2].Swartz AM, Li Q-J, Sampson JH, Rindopepimut: a promising immunotherapeutic for the treatment of glioblastoma multiforme, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Weller M, Cloughesy T, Perry JR, Wick W, Standards of care for treatment of recurrent glioblastoma—are we there yet?, Neuro-oncology 15(1) (2012) 4–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Miletic H, Niclou SP, Johansson M, Bjerkvig R, Anti-VEGF therapies for malignant glioma: treatment effects and escape mechanisms, Expert opinion on therapeutic targets 13(4) (2009) 455–468. [DOI] [PubMed] [Google Scholar]
- [5].Gerstner ER, Batchelor TT, Antiangiogenic therapy for glioblastoma, Cancer journal (Sudbury, Mass.) 18(1) (2012) 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jackson C, Ruzevick J, Phallen J, Belcaid Z, Lim M, Challenges in immunotherapy presented by the glioblastoma multiforme microenvironment, Clinical and Developmental Immunology 2011 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Thomas AA, Ernstoff MS, Fadul CE, Immunotherapy for the treatment of glioblastoma, Cancer journal (Sudbury, Mass.) 18(1) (2012) 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hambardzumyan D, Bergers G, Glioblastoma: defining tumor niches, Trends in cancer 1(4) (2015) 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Agrawal NS, Miller R Jr, Lal R, Mahanti H, Dixon-Mah YN, DeCandio ML, Vandergrift III WA, Varma AK, Patel SJ, Banik NL, Current studies of immunotherapy on glioblastoma, Journal of neurology and neurosurgery 1(1) (2014). [PMC free article] [PubMed] [Google Scholar]
- [10].Vega EA, Graner MW, Sampson JH, Combating immunosuppression in glioma, (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, Andronesi OC, Frosch MP, Wen PY, Plotkin SR, Hedley-Whyte ET, Sorensen AG, Batchelor TT, Jain RK, Glioblastoma Recurrence after Cediranib Therapy in Patients: Lack of “Rebound” Revascularization as Mode of Escape, Cancer Res 71(1) (2011) 19–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lu-Emerson C, Snuderl M, Kirkpatrick ND, Goveia J, Davidson C, Huang Y, Riedemann L, Taylor J, Ivy P, Duda DG, Increase in tumor-associated macrophages after antiangiogenic therapy is associated with poor survival among patients with recurrent glioblastoma, Neuro-oncology 15(8) (2013) 1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Humpel C, Organotypic brain slice cultures: A review, Neuroscience 305 (2015) 86–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].DelNero P, Lane M, Verbridge SS, Kwee B, Kermani P, Hempstead B, Stroock A, Fischbach C, 3D culture broadly regulates tumor cell hypoxia response and angiogenesis via pro-inflammatory pathways, Biomaterials 55 (2015) 110–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ribatti D, Crivellato E, Immune cells and angiogenesis, Journal of cellular and molecular medicine 13(9a) (2009) 2822–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Sun J.-l., Jiao K, Niu L.-n., Jiao Y, Song Q, Shen L.-j., Tay FR, Chen J.-h., Intrafibrillar silicified collagen scaffold modulates monocyte to promote cell homing, angiogenesis and bone regeneration, Biomaterials 113 (2017) 203–216. [DOI] [PubMed] [Google Scholar]
- [17].Daphu I, Sundstrøm T, Horn S, Huszthy PC, Niclou SP, Sakariassen PØ, Immervoll H, Miletic H, Bjerkvig R, Thorsen F, In vivo animal models for studying brain metastasis: value and limitations, Clinical & experimental metastasis 30(5) (2013) 695–710. [DOI] [PubMed] [Google Scholar]
- [18].Shimizu F, Hovinga KE, Metzner M, Soulet D, Tabar V, Organotypic explant culture of glioblastoma multiforme and subsequent single-cell suspension, Curr Protoc Stem Cell Biol Chapter 3 (2011) Unit3 5. [DOI] [PubMed] [Google Scholar]
- [19].Liu C, Cui X, Ackermann TM, Flamini V, Chen W, Castillo AB, Osteoblast-derived paracrine factors regulate angiogenesis in response to mechanical stimulation, Integrative Biology 8(7) (2016) 785–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zheng Y, Chen J, Craven M, Choi NW, Totorica S, Diaz-Santana A, Kermani P, Hempstead B, Fischbach-Teschl C, López JA, In vitro microvessels for the study of angiogenesis and thrombosis, Proceedings of the National Academy of Sciences 109(24) (2012) 9342–9347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Ungefroren H, Sebens S, Seidl D, Lehnert H, Hass R, Interaction of tumor cells with the microenvironment, Cell Communication and Signaling 9(1) (2011) 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Fantin A, Vieira JM, Gestri G, Denti L, Schwarz Q, Prykhozhij S, Peri F, Wilson SW, Ruhrberg C, Tissue macrophages act as cellular chaperones for vascular anastomosis downstream of VEGF-mediated endothelial tip cell induction, Blood 116(5) (2010) 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rape A, Ananthanarayanan B, Kumar S, Engineering strategies to mimic the glioblastoma microenvironment, Advanced drug delivery reviews 79 (2014) 172–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Schnell O, Krebs B, Wagner E, Romagna A, Beer AJ, Grau SJ, Thon N, Goetz C, Kretzschmar HA, Tonn JC, Expression of integrin αvβ3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature, Brain pathology 18(3) (2008) 378–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Overstreet MG, Gaylo A, Angermann BR, Hughson A, Hyun Y-M, Lambert K, Acharya M, Billroth-MacLurg AC, Rosenberg AF, Topham DJ, Inflammation-induced interstitial migration of effector CD4+ T cells is dependent on integrin [alpha] V, Nature immunology 14(9) (2013) 949–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Weis SM, Cheresh DA, αV integrins in angiogenesis and cancer, Cold Spring Harbor perspectives in medicine 1(1) (2011) a006478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feng Y, Li Q, Wu D, Niu Y, Yang C, Dong L, Wang C, A macrophage-activating, injectable hydrogel to sequester endogenous growth factors for in situ angiogenesis, Biomaterials 134 (2017) 128–142. [DOI] [PubMed] [Google Scholar]
- [28].Jetten N, Verbruggen S, Gijbels MJ, Post MJ, De Winther MP, Donners MM, Anti-inflammatory M2, but not pro-inflammatory M1 macrophages promote angiogenesis in vivo, Angiogenesis 17(1) (2014) 109–118. [DOI] [PubMed] [Google Scholar]
- [29].Tian YF, Ahn H, Schneider RS, Yang SN, Roman-Gonzalez L, Melnick AM, Cerchietti L, Singh A, Integrin-specific hydrogels as adaptable tumor organoids for malignant B and T cells, Biomaterials 73 (2015) 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Monteiro GA, Fernandes AV, Sundararaghavan HG, Shreiber DI, Positively and negatively modulating cell adhesion to type I collagen via peptide grafting, Tissue Engineering Part A 17(13–14) (2009) 1663–1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nguyen D-HT, Stapleton SC, Yang MT, Cha SS, Choi CK, Galie PA, Chen CS, Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro, Proceedings of the National Academy of Sciences 110(17) (2013) 6712–6717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Shao Y, Taniguchi K, Gurdziel K, Townshend RF, Xue X, Yong KMA, Sang J, Spence JR, Gumucio DL, Fu J, Self-organized amniogenesis by human pluripotent stem cells in a biomimetic implantation-like niche, Nature materials 16(4) (2017) 419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Khoo CP, Micklem K, Watt SM, A comparison of methods for quantifying angiogenesis in the Matrigel assay in vitro, Tissue Engineering Part C: Methods 17(9) (2011) 895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Sun Y, Yong KMA, Villa-Diaz LG, Zhang X, Chen W, Philson R, Weng S, Xu H, Krebsbach PH, Fu J, Hippo/YAP-mediated rigidity-dependent motor neuron differentiation of human pluripotent stem cells, Nature materials 13(6) (2014) 599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR, STAR: ultrafast universal RNA-seq aligner, Bioinformatics 29(1) (2013) 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features, Bioinformatics 30(7) (2013) 923–930. [DOI] [PubMed] [Google Scholar]
- [37].Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2, Genome biology 15(12) (2014) 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Wang N, Jain RK, Batchelor TT, New Directions in Anti-Angiogenic Therapy for Glioblastoma, Neurotherapeutics (2017) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hambardzumyan D, Gutmann DH, Kettenmann H, The role of microglia and macrophages in glioma maintenance and progression, Nature neuroscience 19(1) (2016) 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sica A, Mantovani A, Macrophage plasticity and polarization: in vivo veritas, The Journal of clinical investigation 122(3) (2012) 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Yang L, Zhang Y, Tumor-associated macrophages: from basic research to clinical application, Journal of hematology & oncology 10(1) (2017) 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Krstic J, Santibanez JF, Transforming growth factor-beta and matrix metalloproteinases: functional interactions in tumor stroma-infiltrating myeloid cells, The Scientific World Journal 2014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A, Macrophage polarization in tumour progression, Seminars in cancer biology, Elsevier, 2008, pp. 349–355. [DOI] [PubMed] [Google Scholar]
- [44].Cluzel C, Saltel F, Lussi J, Paulhe F, Imhof BA, Wehrle-Haller B, The mechanisms and dynamics of αvβ3 integrin clustering in living cells, J Cell Biol 171(2) (2005) 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Somanath PR, Malinin NL, Byzova TV, Cooperation between integrin ανβ3 and VEGFR2 in angiogenesis, Angiogenesis 12(2) (2009) 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Ahluwalia MS, de Groot J, Liu WM, Gladson CL, Targeting SRC in glioblastoma tumors and brain metastases: rationale and preclinical studies, Cancer letters 298(2) (2010) 139–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Choi H-J, Zhang H, Park H, Choi K-S, Lee H-W, Agrawal V, Kim Y-M, Kwon Y-G, Yes-associated protein regulates endothelial cell contact-mediated expression of angiopoietin-2, Nature communications 6 (2015). [DOI] [PubMed] [Google Scholar]
- [48].Mo JS, Park HW, Guan KL, The Hippo signaling pathway in stem cell biology and cancer, EMBO reports 15(6) (2014) 642–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control, Genes & development 21(21) (2007) 2747–2761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Vasudev NS, Reynolds AR, Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions, Angiogenesis 17(3) (2014) 471–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reardon DA, Cheresh D, Cilengitide: a prototypic integrin inhibitor for the treatment of glioblastoma and other malignancies, Genes & cancer 2(12) (2011) 1159–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Ho IA, Shim WS, Contribution of the Microenvironmental Niche to Glioblastoma Heterogeneity, BioMed Research International 2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Han J, Alvarez-Breckenridge CA, Wang Q-E, Yu J, TGF-β signaling and its targeting for glioma treatment, American journal of cancer research 5(3) (2015) 945. [PMC free article] [PubMed] [Google Scholar]
- [54].Neuzillet C, Tijeras-Raballand A, Cohen R, Cros J, Faivre S, Raymond E, de Gramont A, Targeting the TGFβ pathway for cancer therapy, Pharmacology & therapeutics 147 (2015) 22–31. [DOI] [PubMed] [Google Scholar]
- [55].Bayin NS, Ma L, Thomas C, Baitalmal R, Sure A, Fansiwala K, Bustoros M, Golfinos JG, Pacione D, Snuderl M, Patient-specific screening using high-grade glioma explants to determine potential radiosensitization by a TGF-β small molecule inhibitor, Neoplasia 18(12) (2016) 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Binello E, Qadeer ZA, Kothari HP, Emdad L, Germano IM, Stemness of the CT-2A immunocompetent mouse brain tumor model: Characterization in vitro, Journal of Cancer 3 (2012) 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Gilbertson RJ, Rich JN, Making a tumour’s bed: glioblastoma stem cells and the vascular niche, Nature reviews. Cancer 7(10) (2007) 733. [DOI] [PubMed] [Google Scholar]
- [58].Anghelina M, Krishnan P, Moldovan L, Moldovan NI, Monocytes and macrophages form branched cell columns in matrigel: implications for a role in neovascularization, Stem cells and development 13(6) (2004) 665–676. [DOI] [PubMed] [Google Scholar]
- [59].Zhou W, Ke SQ, Huang Z, Flavahan W, Fang X, Paul J, Wu L, Sloan AE, McLendon RE, Li X, Periostin secreted by glioblastoma stem cells recruits M2 tumor-associated macrophages and promotes malignant growth, Nature cell biology 17(2) (2015) 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lawler J, Thrombospondin‐1 as an endogenous inhibitor of angiogenesis and tumor growth, Journal of cellular and molecular medicine 6(1) (2002) 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ferrari G, Cook BD, Terushkin V, Pintucci G, Mignatti P, Transforming growth factor‐beta 1 (TGF‐β1) induces angiogenesis through vascular endothelial growth factor (VEGF)‐mediated apoptosis, Journal of cellular physiology 219(2) (2009) 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Gong D, Shi W, Yi S.-j., Chen H, Groffen J, Heisterkamp N, TGFβ signaling plays a critical role in promoting alternative macrophage activation, BMC immunology 13(1) (2012) 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.