

Background: Immunocompromised persons may be at higher risk for recrudescent infections and reinfections with SARS-CoV-2. However, the 2 entities can be difficult to distinguish, and cases have been infrequently documented.
Objective: To distinguish SARS-CoV-2 reinfection from recrudescence in a liver transplant recipient with 2 distinct episodes of COVID-19 using clinical history and viral genomic sequencing.
Case Report: The patient is a 61-year-old man who has a history of liver transplant due to chronic hepatitis B and C virus infections and is receiving maintenance immunosuppression with tacrolimus and mycophenolate mofetil. He presented to the emergency department (ED) with fever, nausea, vomiting, and cough. He was stable in the ED and was discharged home, and symptoms resolved within 12 days. We detected SARS-CoV-2 by reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) on a nasopharyngeal swab collected during his ED visit. Results were negative on repeated testing for SARS-CoV-2 by RT-qPCR done 48 and 53 days after the initial positive result.
On day 111 after the initial SARS-CoV-2 diagnosis, the patient presented to the ED with several days of increasing confusion, hallucinations, unstable gait, and frequent falls. There was no fever, dyspnea, nausea, abdominal pain, or diarrhea. Magnetic resonance imaging of the patient's brain showed an acute punctate infarction of the right upper pons, and SARS-CoV-2 was detected by RT-qPCR on a nasopharyngeal swab on admission and again 2 days later. A serum IgG assay detecting antibodies to the SARS-CoV-2 receptor-binding domain was negative. The patient's hospital course was complicated by worsening lethargy and hypoxia. He received convalescent plasma, remdesivir, and dexamethasone and was discharged on hospital day 23 (134 days since first infection). One month later, results of repeated testing for SARS-CoV-2 by RT-qPCR were negative (Figure 1)<(1–4)>. Results of an anti–SARS-CoV-2 IgG assay were positive after treatment with convalescent plasma and remained positive 5 months later.
Figure 1. Genetic variation over time observed in SARS-CoV-2 genomes from patient samples.

Five nasopharyngeal specimens were collected spanning 118 d. For viral sequencing, 2 replicate sequencing libraries were prepared from source material for each sample as previously described (1). SARS-CoV-2 genomes were assembled using viral-ngs, v2.1.10.0, assembly pipelines (2). Consensus SARS-CoV-2 genomes were assembled for all positive time points, whereas no genomic data were produced from the negative RT-qPCR test result (T2). The genome from the first time point was 96.6% complete (mean depth: 18 reads), and remaining genomes were 99% complete (mean depth: tens to thousands of reads). Each assembled genome was characterized by comparison to the ancestral reference genome, NC_045512.2 (isolated from one of the first known COVID-19 cases in Wuhan, China). The 3 later time points are nearly identical and share a common set of single-nucleotide variants (SNVs), with T3 having a single additional SNV. Compared with T1, these 3 genomes had more substitutions (11-12 SNVs) than expected from the mean substitution rate of SARS-CoV-2, which is approximately 1 substitution every 2 wk (3). Of note, 5 of the substitutions seen in the first time point were replaced by the ancestral allele in later time points; these apparent reversions strongly suggest that the later genomes reflect an independent infection with a virus from a distinct lineage rather than evolution of the virus of the first time point, especially given the ubiquity of SARS-CoV-2 in the surrounding community. Three amino acid changes present at the first time point were absent from the later time points, and the later time points all bear 3 new amino acid substitutions not seen in the first time point, as well as a deletion. Time points T3–T5 had a notable amino acid substitution in the receptor-binding domain of the spike glycoprotein at position 501 (S:N501T), an amino acid substitution believed to increase affinity for the angiotensin-converting enzyme 2 receptor (4). In the most deeply sequenced later time point, T4, none of the distinguishing variants of the first time point were present in high abundance, and nearly half were absent entirely. For none of the apparent reversions to the ancestral allele did a minor population exist in the most densely sequenced later time point, T4. RT-qPCR = reverse transcriptase quantitative polymerase chain reaction.
Excess material from clinical diagnostic specimens was subjected to whole-genome sequencing to determine if the 2 symptomatic periods represented distinct infections. Genomes from the later time points differed from those at the first time point by 11 to 12 single base substitutions; this exceeds the rate established by the U.S. Centers for Disease Control and Prevention for identifying potential reinfection cases (1). The phylogenetic relationship of the genomes showed that virus from the later infection was more closely related to virus circulating in the community than to that seen in the first infection (Figure 2). Haplotype fingerprinting of residual host genetic material in the samples confirmed that all came from the same patient.
Figure 2. Tree of SARS-CoV-2 genomes generated from this patient and contextual SARS-CoV-2 genomes from surrounding states (MA, CT, RI, VT, NH, ME, and NY) as of 5 February 2021 from the National Center for Biotechnology Information GenBank.
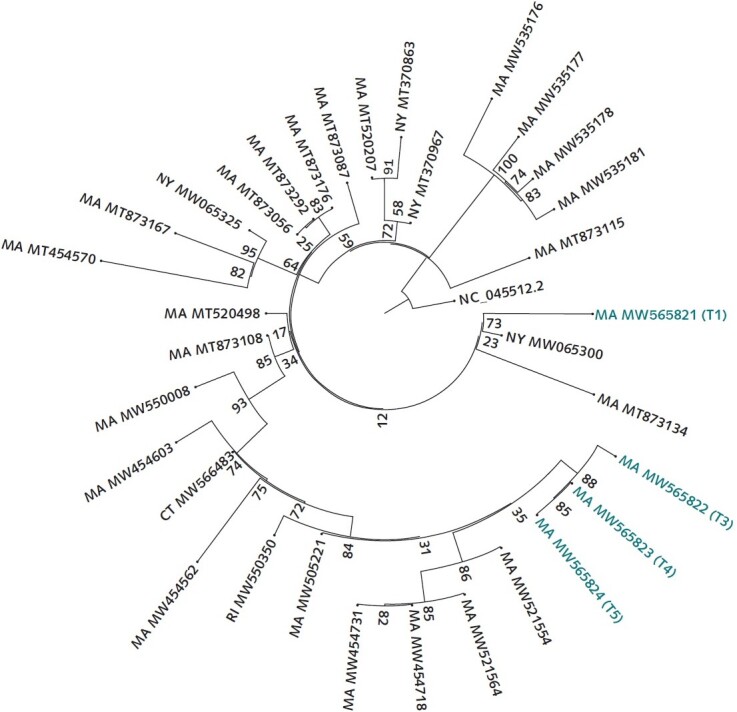
Similarity by genetic distance and placement on the phylogenetic tree show that the second infection is more closely related to and descended from infections circulating in the community than from the viral genome sequenced from the first infection. The sequences were aligned to the reference NC_045512.2 using MAFFT. A maximum likelihood tree was created via IQ-Tree with the general time-reversible model with empirical base frequencies and 3 FreeRate categories, as selected by minimum Akaike information criterion. CT = Connecticut; MA = Massachusetts; ME = Maine; NH = New Hampshire; NY = New York; RI = Rhode Island; VT = Vermont.
Discussion: We present a case of a liver transplant recipient with 2 distinct SARS-CoV-2 infections, separated by 111 days without symptoms and 2 negative test results for SARS-CoV-2 infection. The clinical course suggested reinfection, and viral genomic sequencing was used to distinguish whether the later positive samples were due to SARS-CoV-2 relapse or reinfection. Three lines of genomic evidence support reinfection rather than recrudescence: placement of the 2 infections on distant parts of the phylogenetic tree such that the second infection does not seem to be descended from the first, a preponderance of substitutions in the SARS-CoV-2 genomes relative to the number expected given the published substitution rate of the virus (1, 3), and apparent reversions to the ancestral allele among several of these substitutions. Of note, the substitutions seen in the later time points were present before the selective pressure imposed by administration of remdesivir and convalescent plasma. Taken together with the clinical history, the genomic data are consistent with the second symptomatic period being the result of reinfection. An antibody response after the first infection was not detected, which we speculate contributed to the patient's susceptibility to repeated infection. As shown here, genomic sequencing offers the ability to discern whether subsequent infections may be reinfection. The paucity of reinfection cases in the literature may be due in part to underdetection, highlighting the need for more comprehensive sequencing of possible cases of SARS-CoV-2 reinfection. In addition to helping distinguish between reinfection and recrudescence, genomic sequence data can identify variants circulating in the population that are of high risk because of infectivity, disease severity, or resistance to treatment.
Footnotes
This article was published at Annals.org on 20 April 2021.
References
- 1.Centers for Disease Control and Prevention. Common investigation protocol for investigating suspected SARS-CoV-2 reinfection. 27 October 2020. Accessed at www.cdc.gov/coronavirus/2019-ncov/php/reinfection.html on 23 November 2020.
- 2.viral-pipelines. GitHub. Accessed at https://github.com/broadinstitute/viral-pipelines on 22 March 2021.
- 3. Duchene S , Featherstone L , Haritopoulou-Sinanidou M , et al. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020;6:veaa061. [PMID: ] doi: 10.1093/ve/veaa061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Starr TN , Greaney AJ , Hilton SK , et al. Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell. 2020;182:1295-1310.e20. [PMID: ] doi: 10.1016/j.cell.2020.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]