

Abstract
Purpose of Review
Genome-wide association studies have delineated the genetic architecture of type 2 diabetes. While functional studies to identify target transcripts are ongoing, new genetic knowledge can be translated directly to health applications. The review covers several translation directions but focuses on genomic polygenic scores for screening and prevention.
Recent Findings
Over 400 genomic variants associated with T2D and its related quantitative traits are now known. Genetic scores comprising dozens to millions of associated variants can predict incident T2D. However, measurement of body mass index is more efficient than genetic scores to detect T2D risk groups, and knowledge of T2D genetic risk alone seems insufficient to improve health. Genetically determined metabolic sub-phenotypes can be identified by clustering variants associated with physiological axes like insulin resistance. Genetic sub-phenotyping may be a way forward to identify specific individual phenotypes for prevention and treatment.
Summary
Genomic polygenic scores for T2D can predict incident diabetes but may not be useful to improve health overall. Genetic detection of T2D sub-phenotypes could be useful to personalize screening and care.
Keywords: Genetics, Genomics, Epidemiology, Risk score, Health outcomes, Type 2 diabetes
Introduction
The genome-wide association study (GWAS), a method to reveal genomic variation associated with human traits and diseases, has illuminated much of the genetic architecture of type 2 diabetes (T2D). T2D is a growing scourge, where new prevention approaches are needed, as current efforts seem ineffective at stopping T2D. Deeper insight into T2D pathobiology can come from genetic discovery. In the past 15 years, scientists have discovered hundreds of genomic loci and variants associated with T2D and the underlying biomarker traits, fasting glucose, insulin, and glycated hemoglobin (HbA1c) that define the disease, its antecedent pathophysiology, and its mechanisms of complications [1]. The discoveries define genetic architectural outlines, but much detail remains to be filled in. In this article, we review the most recent T2D GWAS discoveries and consider how this new genetic epidemiological information might translate to better health.
Large-scale GWAS Illuminate T2D Genetic Architecture
The recently published, largest T2D GWAS to date provides a major advance in our understanding of T2D genetic architecture. In “Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps,” the DIAGRAM-DIAMANTE consortium combined data from 74,124 T2D cases and 824,006 controls of European ancestry and used a GWAS array with high-density imputation to test association of ~ 27 million common variants (minor allele frequency, MAF ≥ 1%) to test association with T2D case status [2••]. The sample size represented a > 3-fold increase over any prior T2D GWAS. Results increased the number of loci associated with T2D to 243, 135 of which were new and comprised of 403 distinct variant signals. Because of improved imputation, the study dramatically increased the number of lower-frequency (MAF 1–5%), higher-risk alleles: of 80 lower-frequency variants, 14 had perallele odds ratios > 2. Interestingly, although imputation quality was improved, the much larger sample size contributed most to substantially improved fine-mapping of probable causal variants at loci. For instance, at 51 signals, just one variant accounted for > 80% of the posterior probability of association, substantially narrowing the potential focus of follow-up studies for functional validation. Another innovation was to further improve fine-mapping through integration of tissue-specific epigenomic information that is now becoming available. Considering islet regulatory annotations extended the number of variants with posterior probability of association > 80% from 51 to 73. High posterior probability coding variants were surprisingly uncommon: of 243 loci, just 18 had associations in genes and attributable to coding variants.
Mahajan et al. show that, at the allelic resolution MAF > 1%, the majority of T2D-associated variation is noncoding regulatory, with coding variation making up just 7% of genetic targets. The findings confirmed a concurrent exome-wide study by the same group, where examination of coding variants in 81,412 T2D cases and 370,832 controls of diverse ancestry identified 37 distinct signals, only 16 of which were likely (> 80% of the posterior probability of association) to account for the signal [3]. Interestingly, at 13 signals, the apparently associated coding variant clearly was not the causal variant. The inclination to assume that if a GWAS variant is a coding variant, then it is the causal variant is as likely to be wrong as not. Untangling the mechanisms underlying most T2D GWAS variants will have to consider regulatory function even if there is a protein coding hypothesis also raised by the locus association. Genetic architecture can be thought of in several dimensions, including the allele frequency spectrum distribution and mode of inheritance, as well as the functional elements underlying the locus. We now have firm evidence that T2D is a common polygenic disorder, comprised of hundreds of mostly common variants of modest effect size inherited a complex fashion with underlying variation dominated by non-coding, regulatory functional elements and pathways [4].
Biological Evaluation of GWAS Loci Is Underway
Effective sample sizes of current T2D and quantitative traits GWAS will soon exceed millions of individuals. This promises to reveal fainter, distant outlines of the blueprint of the genetic basis of T2D, but now we have “inked in” its basic forms. Where do we go from here? Laboratory functional validation is required to identify causal transcripts and understand molecular mechanism at GWAS loci. Already, causal transcripts and mechanisms have been illuminated by integration of genomic association data with in vitro genomic functional data from islet, liver, muscle, and fat tissue chromatin regulatory mapping [5–9]. Genomic regulatory features linked to GWAS signals include, for instance, enhancer site disruption [10], stretch enhancer potentiation [11], and lncRNA action in human [12] and mouse beta cell lines [13,14]. Increasingly, genomic data have been further assembled into “knowledge portals” facilitating rapid target evaluation ( [15]; http://www.type2diabetesgenetics.org). Already, new discoveries seem to be pointing to novel mechanisms and potentially tractable T2D therapeutic targets [16, 17].
Genomic Application to Health: Mendelian Randomization and Biomarker Genetics
While in vitro target validation is underway, immediate attempts to translate genetic discoveries to public health and clinical care application are warranted. Translational steps even before molecular identity is known are essential if we are to capitalize on our investment in genomic science, because we will have used new knowledge to improve health even as we await molecular target development. Three general areas have emerged where genetic epidemiology may be employed to improve health and prevent T2D and its complications. In one, genetic variation acquired at meiosis is used as an instrumental variable in “Mendelian randomization” analyses that test casual associations between exposure and disease [18]. Such tests have been used, for instance, to disentangle the causal roles of T2D versus hyperglycemia in development of cardiovascular disease [19–21], to disentangle the role of fat distribution on T2D risk [22], or to evaluate cardiovascular safety of T2D therapeutics [23]. Another area is biomarker genetics, where consideration of common variation may be key to defining correct reference ranges for individuals of varying ancestry. For instance, hemoglobin A1c is a key blood biomarker used to diagnose and monitor T2D. In African Americans, an X-linked variant in the gene encoding glucose-6-phospatase (G6PD G202A) that occurs in about 11% of individuals was associated with an absolute decrease in HbA1c of up to 0.8%-units comparing homozygotes [24•]. This large effect, combined with the commonness of the variant, could cause about 650,000 African American adults with T2D to remain undiagnosed if they were screened once with HbA1c alone and diagnosed at the uniform threshold of 7%-units. While such screening is often accompanied by confirmatory fasting glucose testing, the data show that direct application of genetic knowledge could be used in a precision medicine approach to reduce health disparities in T2D [25].
Genomic Risk Scores for Prediction and Personalization
Another direction for translation of genomics to improve health is the concept of “genomic scores.” These assess aggregate genetic risk burden by identifying groups of risk- or trait-raising variants, then weighting and summing them into a continuously distributed genetic score. No knowledge of causal transcripts or specific molecular function is needed. Information from such scores may be used to screen populations to identify high-risk groups, or to predict future disease in individuals. GWAS is predicated on considering each individual variant across the human genome as the single unit of exposure. Each of these variants has a small effect, and while a common variant may be present in up to half of a population, higher impact variants tend to be less common. The idea of aggregating multiple variants into genetic scores offers a convenient way to scale total genetic risk burden as a disease exposure, and one that is present from conception.
Increasing enthusiasm for genetic scores has kept pace with that of variant discovery [26, 27]. However, although million-variant genetic scores have intuitive appeal over simpler scores made of fewer variants, it is not apparent that more sophisticated scores are better for T2D prediction. Data from Vassy et al. supports this contention, where discriminatory capacity for T2D genetic scores was modest and clinical factors were far better than genetics [28]. Here, receiver operating characteristic (ROC) curves for models predicting incident T2D with and without a 62-variant genetic risk score among the Framingham Heart Study white and CARDIA white and black young adults. Full clinical models were adjusted for age, sex, parental diabetes (yes vs. no), BMI, systolic blood pressure, fasting glucose, HDL cholesterol, and triglyceride levels. Genetic information alone had modest predictive value, clinical information had excellent predictive value that was not materially improved when genetic information was added, and models based on European variant discovery performed more poorly in black than in white CARDIA participants. These data underscore the principle that current genetic scores do not perform well in non-European ancestry groups, where poorer discriminatory capacity in blacks than in whites has potential to exacerbate already large health disparities disfavoring blacks [29]. Further, for T2D, adding more variants to a genetic score had diminishing information return [28]. Figure 1 displays genetic scores for risk for coronary artery disease with a 50-variant genetic score and two genomic polygenic scores comprised of ~ 50,000 and ~ 6.6 million variants [26]. Such an exercise has not yet been published for T2D. However, for multifactorial diseases like T2D and coronary artery disease, one can see that as millions of variants enter a genomic polygenic score, the shape of the risk curve does not materially change, although a few individuals at relatively high risk of disease begin to emerge. This indicates that adding variants will not alter the basic predictive capacity of the score, although a few more extremely high-risk individuals will be identified.
Fig. 1.
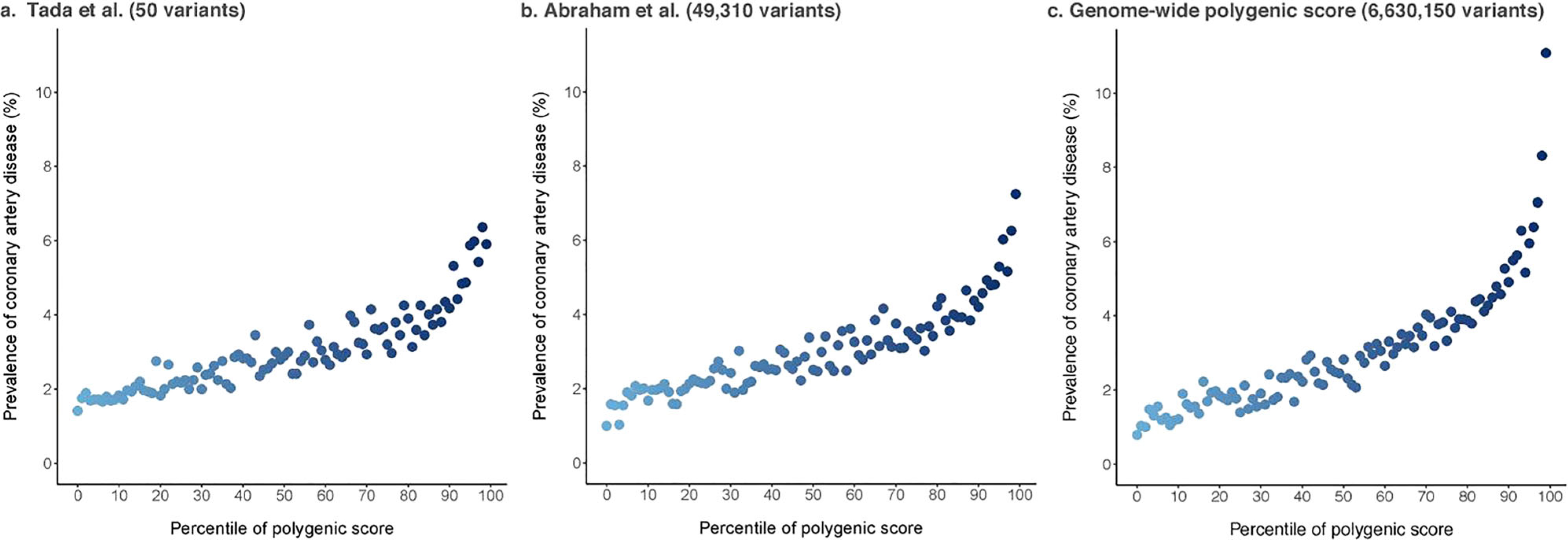
Genomic polygenic risk for coronary artery disease. Three polygenic scores for coronary artery disease were calculated in the UK Biobank (n = 288,978) including 50 (panel a), 49,310 (panel b), and 6,630,150 (panel c) variants. As millions of variants enter a genomic polygenic score, the shape of the risk curve does not change, but a few individuals at relatively high risk of coronary heart disease begin to emerge. While these few additional individuals may be targeted for intervention to reduce risk, sophisticated genomic polygenic scores cannot be expected to improve T2D risk model performance overall (adapted by permission from Springer Nature from Khera et al. Nature Genetics. 2018;50(9):1219–24) [26]
Genomic Polygenic Prediction Is Not as Easily Applied to T2D as It Is to Coronary Artery Disease
Genetic prediction of T2D offers an important contrast to that of coronary artery disease. In coronary artery disease, familial hyperlipidemia (FHL) offers a criterion standard for polygenic prediction, where the 4-fold elevated risk conferred by the monogenic FHL mutation can be used at as cut-point to define “high” polygenic risk. It appears generally agreed that a lipid-based 4-fold elevated risk clearly warrants consideration of statin therapy. T2D has no such monogenic equivalent, either in the form of highly penetrant alleles or in an agreed-upon drug treatment threshold for “high risk”. Despite mutations in many well-known, Mendelian-inherited Maturity Onset Diabetes of the Young (MODY) genes, associated allelic relative risks are variable (but may be high) due to age-related, incomplete penetrance, and that many individuals with MODY mutations do not have a diabetes phenotype [30]. Further, what level of risk constitutes “high” for T2D depends on many factors influencing penetrance, especially age, ancestry, and adiposity. Thus, genomic polygenic scores for T2D, unlike for coronary artery disease, cannot identify a larger proportion of at-risk individuals on the basis of polygenic risk versus monogenic risk. If elevated risk is defined, for instance, as a risk ratio of > 3, current genomic polygenic scores for T2D show that the group of white people with > 3-fold elevated risk of T2D constitutes just 2.5% of a sample. Approximately > 3-fold elevated risk for T2D is also associated with obesity (defined as a BMI > 30 kg/m2) versus not obese in white populations [31]. Figure 2 illustrates that since obesity occurs in about 30% of individuals in the USA, use of an inexpensive stadiometer to assess T2D risk will have an order of magnitude higher efficiency to detect at-risk individuals than will genomic polygenic scores.
Fig. 2.
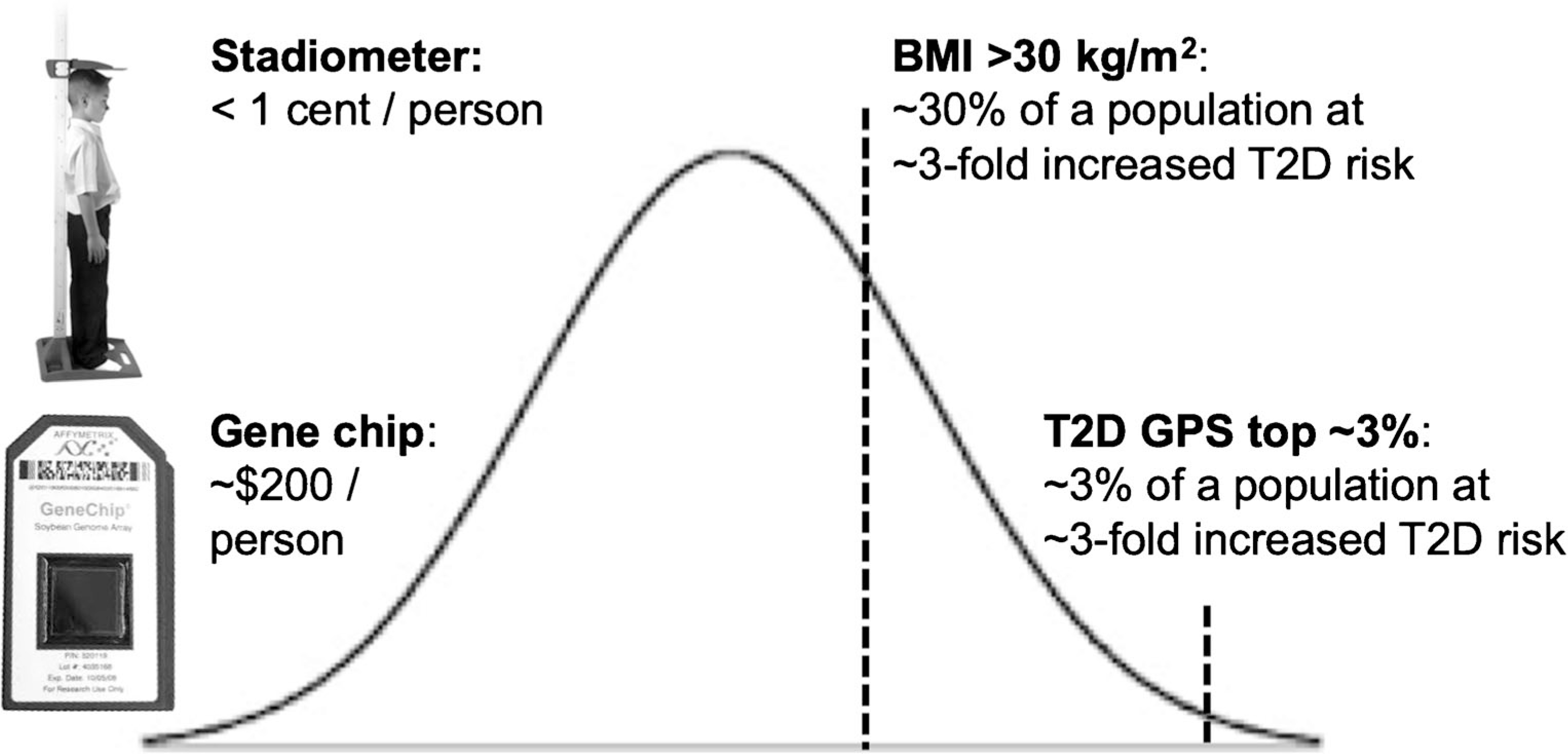
Body mass index outperforms genomic polygenic risk when identifying high—T2D risk individuals. In an example, high genetic risk, defined as the top ~ 3% of a genomic polygenic score, confers 3-fold increased risk versus the rest of the distribution and affects ~ 3% of a screened population, by definition. In 2019, a genomic polygenic score costs about $200/person. BMI exceeding 30 kg/m2, which affects about 30% of the US population, also confers about 3-fold increased risk versus non-obese. While consideration of costs of polygenic scores does not take into account other conditions that may be detected by array genotyping, the cost of a stadiometer is essentially free per patient over time
Knowledge of T2D Genetic Risk Did Not Change Health Behavior in a Randomized Trial
The major problem for clinical application of genomics to T2D screening models is current evidence of the failure of genetic knowledge to change long-term health behavior. For coronary artery disease prevention, people fear a heart attack, and the decision to start or adhere to statin therapy is fairly straightforward. T2D prevention requires people to weigh theoretical future problems while choosing to adhere daily to effective, evidence-based lifestyle and medication interventions. They have to adhere day after day and year after year, following often difficult lifestyle prescriptions and taking medications that may have symptomatic side effects. Although our patient survey indicated that genetic testing would be an incentive to change health behaviors [32], the Genetic Counseling/Lifestyle Change for Diabetes Prevention Study showed in a randomized, controlled trial that T2D genetic screening coupled with medical genetic counselor-provided counseling about the results did not change health behavior or T2D outcomes [33•, 34, 35]. In GC/LC, 108 patients with metabolic syndrome were randomized to T2D genetic testing versus no testing. Participants in the top and bottom quartiles of a 36-variant genetic risk score received individual genetic counseling before being enrolled with untested control participants in a group diabetes prevention program. The primary endpoint was difference between groups in the proportion of participants attending each session of the 12-week program. The study found no significant differences between groups in the primary outcome, nor were there differences in patient self-reported attitudes about health, weight loss, or 6-year incidence of new T2D. Unfortunately, genetic knowledge provided by a genetic risk score alone seemed insufficient as a means to reduce T2D risk, even in very high-risk individuals. Studies using genotype call-back to evaluate specific patients at very high polygenic risk are underway to further elucidate how such information may have clinical utility [35].
Process Polygenic Risk Scores to Define T2D Sub-phenotypes Have Promise for Personalized Medicine
Genomic polygenic scores can also be partitioned into underlying physiological domains, so-called partitioned polygenic scores. While global T2D genetic risk assessment continues to seek a role in health improvement, the idea to use genomic information definition to define metabolic phenotypes and to conduct T2D sub-phenotyping has very attractive promise. Partitioned polygenic scores may be generated by identifying sets of variants that are associated with physiological quantitative traits like, for instance, elevated fasting insulin, triglycerides, and BMI, thus inferring an association with an insulin resistance phenotype. Such a partitioned polygenic score might be called an “insulin resistance genetic score” correlated with underlying insulin resistance physiology [36]. More sophisticated approaches use the entire genome, considering linkage disequilibrium and level of assortation for all variants, or “soft clustering” approaches that consider the correlation of all variants with all evaluated traits to define physiological clusters. In Fig. 3, different clustering approaches produce similar genetically defined phenotypes with convincing and distinct patterns, offering potential for genetically based sub-phenotyping of T2D that reflects growing appreciation for the phenotypic heterogeneity of the T2D risk phenotype [37]. Trait-based sub-phenotyping of at-risk for and newly diagnosed T2D shows promise to discriminate differential outcomes depending on sub-phenotype [38, 39]. Whether genomic-based sub-phenotyping, or combining of several different partitioned polygenic scores, meaningfully discriminates outcomes is now being tested in follow-up studies of incident T2D and T2D complications.
Fig. 3.
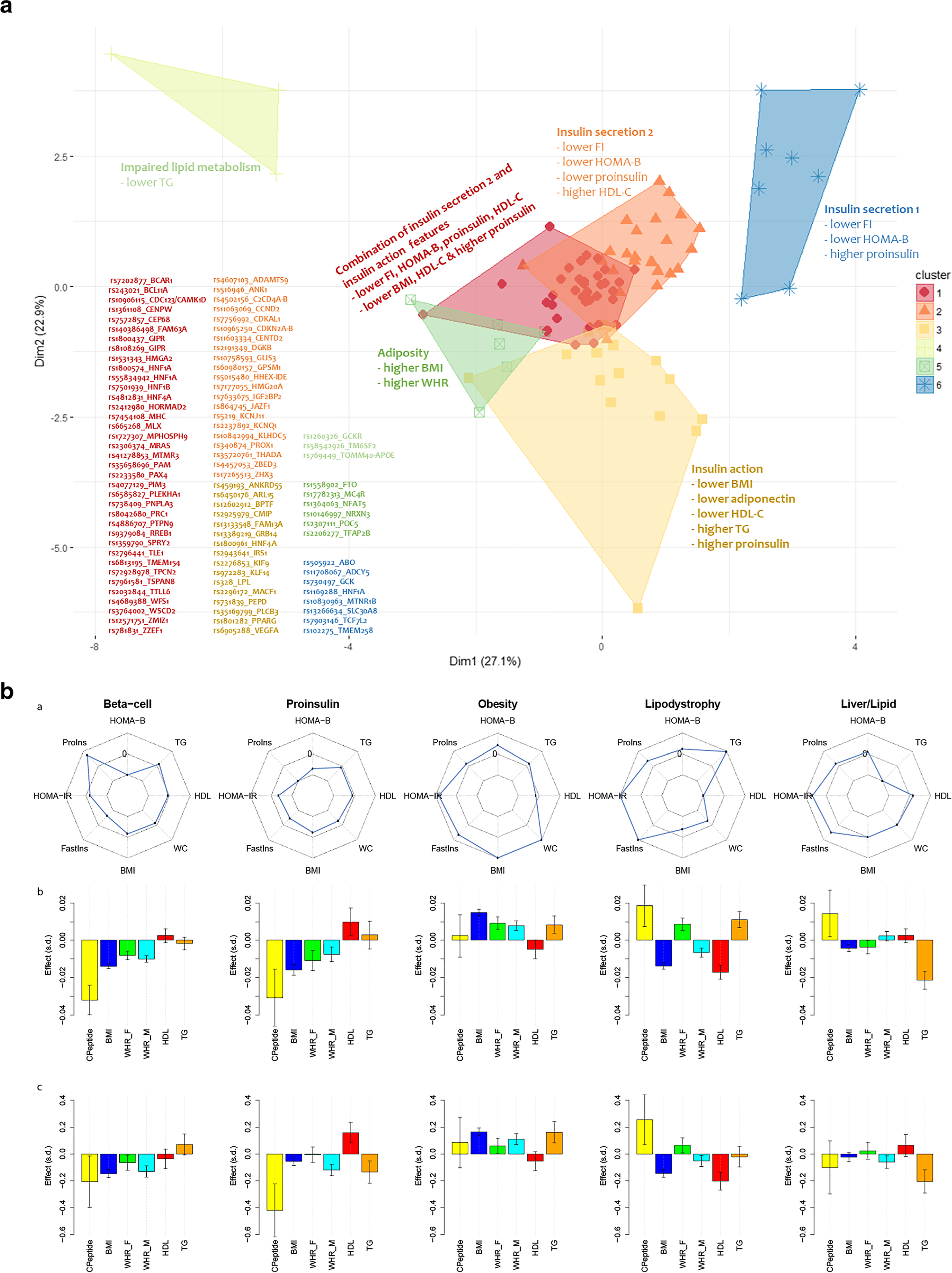
Distinct physiological processes underlying T2D can be defined by genetic variant clusters and “partitioned” polygenic scores. Panel a: clustering variants by direction of effect and magnitude of association with T2D-related metabolic traits defines physiological clusters. Traits like fasting glucose, insulin, lipids or BMI, then further clustering correlated groupings using a fuzzy c-means algorithm that assigns a score to a variant for each cluster (soft clustering, where a variant may be assigned to more than one cluster) produced 5–6 clusters that appeared to represent distinct physiological domains like insulin secretion, insulin action, excess adiposity, or impaired lipid metabolism (panel a is adapted by permission from Springer Nature from Mahajan et al. Nature Genetics. 2018;50(4):559–71) [3]. Panel b: In another study, a similar soft clustering approach defined five distinct physiological domains (columns) with variants combined to generate a “partitioned” polygenic score (PPS). The top row displays spider plots of association of physiological PPSs with metabolic traits (Proins, fasting proinsulin adjusted for fasting insulin; HOMA-B, homeostasis model assessment of beta cell function; TG, serum triglycerides; WC, waist circumference; BMI, body mass index; Fastins, fasting insulin; HOMA-IR, homeostasis model assessment of insulin resistance; WHR-F, waist-hip-ratio in females; WHR-M, waist-hip ratios in males). Points on spider plots in the inner part of the circle show negative association of the PPS and traits and those in the outer circle, positive association. The middle row shows associations of each of five PPSs with metabolic traits in four studies (METSIM, Ashkenazi, Partners Biobank, and UK Biobank, meta-analyzed together), and the bottom row displays the values of individuals with T2D who have each PPS in the highest decile versus all other individuals with T2D. Y-axes are effect size and direction and x-axes are metabolic traits. Soft clustering of genomic data produces genetically defined phenotypes with convincing and distinct patterns, offering potential for genetically based sub-phenotyping of T2D (panel b is adapted from Udler et al. PLoS Medicine. 2018;15(9):e1002654; Creative Commons user license https://creativecommons.org/licenses/by/4.0/)[37]
Conclusions
Outlines of the blueprint of T2D genetic architecture have become clearer over the past 15 years, thanks to society’s investment in large-scale human genomics combined with large-scale international collaborative research teamwork. Now, granular details are being sketched in, and various translational approaches to improve human health are under evaluation. Already, some conclusions can be drawn. First, accurate genetic prediction tools for non-European ancestry individuals have to be developed because current, European-based genomic scores do not perform adequately in minority groups disproportionately affected by T2D. Next, T2D genomic polygenic scores are unlikely, by themselves, to improve individual or societal health, because easily measured and obvious clinical traits like glucose and BMI will always be a more efficient approach to identify T2D risk. Nonetheless, genomic information is here now, and patients are presenting to their physicians with commercially obtained genomic polygenic scores. While such commercial genomic score results seem to promise little direct health value, their availability and increasing market penetration do afford the opportunity elevate society’s conversations around health awareness and general knowledge of genetics. Increased availability of consumer and clinical genomic information pushes doctors and patients towards increased numeracy and literacy around the proper role of modern human genomics in disease prevention and care. For genetic information to effect metabolic health behavior change, it needs to be set in broader clinical and social contexts that support healthy choices. Meanwhile, the coming years will see more and finer detail appear in the T2D genetic blueprint, and more opportunities to use this knowledge to improve health and better prevent and treat T2D.
Footnotes
Compliance with Ethical Standards
Conflict of Interest James B. Meigs reports that he is an Academic Associate for Quest Diagnostics, Inc.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Florez JC, Udler MS, Hanson RL. Genetics of type 2 diabetes. In: Cowie CCCS, Menke A, Cissell MA, Eberhardt MS, Meigs JB, Gregg EW, Knowler WC, Barrett-Connor E, Becker DJ, Brancati FL, Boyko EJ, Herman WH, Howard BV, Narayan KMV, Rewers M, Fradkin JE, editors. Diabetes in America, 3rd ed NIH Pub No. 17–1468 ed. Bethesda: National Institutes of Health; 2018. p. 14.1–25. [Google Scholar]
- 2.Mahajan A, Taliun D, Thurner M, Robertson NR, Torres JM, Rayner NW, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–13•• This genome-wide association study of more than one million people of European ancestry is the current “definitive” framework for T2D common variant genetic architecture. Supplementary Figure 10 shows the distribution of a genomic polygenic score for T2D in individuals of European ancestry.
- 3.Mahajan A, Wessel J, Willems SM, Zhao W, Robertson NR, Chu AY, et al. Refining the accuracy of validated target identification through coding variant fine-mapping in type 2 diabetes. Nat Genet. 2018;50(4):559–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuchsberger C, Flannick J, Teslovich TM, Mahajan A, Agarwala V, Gaulton KJ, et al. The genetic architecture of type 2 diabetes. Nature. 2016;536(7614):41–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pasquali L, Gaulton KJ, Rodriguez-Segui SA, Mularoni L, Miguel-Escalada I, Akerman I, et al. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet. 2014;46(2):136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.van de Bunt M, Manning Fox JE, Dai X, Barrett A, Grey C, Li L, et al. Transcript expression data from human islets links regulatory signals from genome-wide association studies for type 2 diabetes and glycemic traits to their downstream effectors. PLoS Genet. 2015;11(12):e1005694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Varshney A, Scott LJ, Welch RP, Erdos MR, Chines PS, Narisu N, et al. Genetic regulatory signatures underlying islet gene expression and type 2 diabetes. Proc Natl Acad Sci U S A. 2017;114(9):2301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan DZ, Garske KM, Alvarez M, Bhagat YV, Boocock J, Nikkola E, et al. Integration of human adipocyte chromosomal interactions with adipose gene expression prioritizes obesity-related genes from GWAS. Nat Commun. 2018;9(1):1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott LJ, Erdos MR, Huyghe JR, Welch RP, Beck AT, Wolford BN, et al. The genetic regulatory signature of type 2 diabetes in human skeletal muscle. Nat Commun. 2016;7: 11764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Roman TS, Cannon ME, Vadlamudi S, Buchkovich ML, Wolford BN, Welch RP, et al. A type 2 diabetes-associated functional regulatory variant in a pancreatic islet enhancer at the ADCY5 locus. Diabetes. 2017;66(9):2521–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kycia I, Wolford BN, Huyghe JR, Fuchsberger C, Vadlamudi S, Kursawe R, et al. A common type 2 diabetes risk variant potentiates activity of an evolutionarily conserved islet stretch enhancer and increases C2CD4A and C2CD4B expression. Am J Hum Genet. 2018;102(4):620–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Akerman I, Tu Z, Beucher A, Rolando DMY, Sauty-Colace C, Benazra M, et al. Human pancreatic beta cell lncRNAs control cell-specific regulatory networks. Cell Metab. 2017;25(2):400–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnes L, Akerman I, Balderes DA, Ferrer J, Sussel L. betalinc1 encodes a long noncoding RNA that regulates islet beta-cell formation and function. Genes Dev. 2016;30(5):502–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moran I, Akerman I, van de Bunt M, Xie R, Benazra M, Nammo T, et al. Human beta cell transcriptome analysis uncovers lncRNAs that are tissue-specific, dynamically regulated, and abnormally expressed in type 2 diabetes. Cell Metab. 2012;16(4):435–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Flannick J, Florez JC. Type 2 diabetes: genetic data sharing to advance complex disease research. Nat Rev Genet. 2016;17(9): 535–49. [DOI] [PubMed] [Google Scholar]
- 16.Sigma_Type_2_Diabetes_Consortium, Williams AL, Jacobs SB, Moreno-Macias H, Huerta-Chagoya A, Churchhouse C, et al. Sequence variants in SLC16A11 are a common risk factor for type 2 diabetes in Mexico. Nature. 2014;506(7486):97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gusarova V, O’Dushlaine C, Teslovich TM, Benotti PN, Mirshahi T, Gottesman O, et al. Genetic inactivation of ANGPTL4 improves glucose homeostasis and is associated with reduced risk of diabetes. Nat Commun. 2018;9(1):2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23(R1):R89–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ahmad OS, Morris JA, Mujammami M, Forgetta V, Leong A, Li R, et al. A Mendelian randomization study of the effect of type-2 diabetes on coronary heart disease. Nat Commun. 2015;6:7060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merino J, Leong A, Posner DC, Porneala B, Masana L, Dupuis J, et al. Genetically driven hyperglycemia increases risk of coronary artery disease separately from type 2 diabetes. Diabetes Care. 2017;40(5):687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leong A, Chen J, Wheeler E, Hivert MF, Liu CT, Merino J, et al. Mendelian randomization analysis of hemoglobin A1c as a risk factor for coronary artery disease. Diabetes Care. 2019:dc181712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emdin CA, Khera AV, Natarajan P, Klarin D, Zekavat SM, Hsiao AJ, et al. Genetic association of waist-to-hip ratio with cardiometabolic traits, type 2 diabetes, and coronary heart disease. JAMA. 2017;317(6):626–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott RA, Freitag DF, Li L, Chu AY, Surendran P, Young R, et al. A genomic approach to therapeutic target validation identifies a glucose-lowering GLP1R variant protective for coronary heart disease. Sci Transl Med. 2016;8(341):341ra76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wheeler E, Leong A, Liu CT, Hivert MF, Strawbridge RJ, Podmore C, et al. Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 2017;14(9):e1002383.• This genome-wide association study of HbA1c shows how biomarker genetics can be used to illuminate health disparities in T2D. Andrew Paterson’s editorial (reference 25) expands on the health translation implications of biomarker genetics in diabetes.
- 25.Paterson AD. HbA1c for type 2 diabetes diagnosis in Africans and African Americans: personalized medicine NOW! PLoS Med. 2017;14(9):e1002384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Meigs JB, Shrader P, Sullivan LM, McAteer JB, Fox CS, Dupuis J, et al. Genotype score in addition to common risk factors for prediction of type 2 diabetes. N Engl J Med. 2008;359(21):2208–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Vassy JL, Hivert MF, Porneala B, Dauriz M, Florez JC, Dupuis J, et al. Polygenic type 2 diabetes prediction at the limit of common variant detection. Diabetes. 2014;63(6):2172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin AR, Kanai M, Kamatani Y, Okada Y, Neale BM, Daly MJ. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet. 2019;51(4):584–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flannick J, Beer NL, Bick AG, Agarwala V, Molnes J, Gupta N, et al. Assessing the phenotypic effects in the general population of rare variants in genes for a dominant Mendelian form of diabetes. Nat Genet. 2013;45(11):1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilson PW, Meigs JB, Sullivan L, Fox CS, Nathan DM, D’Agostino RB Sr. Prediction of incident diabetes mellitus in middle-aged adults: the Framingham offspring study. Arch Intern Med. 2007;167(10):1068–74. [DOI] [PubMed] [Google Scholar]
- 32.Grant RW, Hivert M, Pandiscio JC, Florez JC, Nathan DM, Meigs JB. The clinical application of genetic testing in type 2 diabetes: a patient and physician survey. Diabetologia. 2009;52(11):2299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grant RW, O’Brien KE, Waxler JL, Vassy JL, Delahanty LM, Bissett LG, et al. Personalized genetic risk counseling to motivate diabetes prevention: a randomized trial. Diabetes Care. 2013;36(1): 13–9• This randomized, controlled trial of genetic testing showed that knowledge of genetic risk for T2D did not change health behavior.
- 34.Vassy JL, He W, Florez JC, Meigs JB, Grant RW. Six-year diabetes incidence after genetic risk testing and counseling: a randomized clinical trial. Diabetes Care. 2018;41(3):e25–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Corbin LJ, Tan VY, Hughes DA, Wade KH, Paul DS, Tansey KE, et al. Formalising recall by genotype as an efficient approach to detailed phenotyping and causal inference. Nat Commun. 2018;9(1):711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lotta LA, Gulati P, Day FR, Payne F, Ongen H, van de Bunt M, et al. Integrative genomic analysis implicates limited peripheral adipose storage capacity in the pathogenesis of human insulin resistance. Nat Genet. 2017;49(1):17–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Udler MS, Kim J, von Grotthuss M, Bonas-Guarch S, Cole JB, Chiou J, et al. Type 2 diabetes genetic loci informed by multi-trait associations point to disease mechanisms and subtypes: a soft clustering analysis. PLoS Med. 2018;15(9):e1002654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wilson PWF, D’Agostino RB Sr, Parise H, Sullivan L, Meigs JB. The metabolic syndrome as a precursor of cardiovascular disease and type 2 diabetes mellitus. Circulation. 2005;112:3066–72. [DOI] [PubMed] [Google Scholar]
- 39.Ahlqvist E, Storm P, Karajamaki A, Martinell M, Dorkhan M, Carlsson A, et al. Novel subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–9. [DOI] [PubMed] [Google Scholar]