

Abstract
Type I interferons (IFN-Is) are emerging as key drivers of inflammation and immunosuppression in chronic infection. Control of these infections requires IFN-I signaling; however, prolonged IFN-I signaling can lead to immune dysfunction. IFN-Is are also emerging as double-edged swords in cancer, providing necessary inflammatory signals, while initiating feedback suppression in both immune and cancer cells. Here, we review the proinflammatory and suppressive mechanisms potentiated by IFN-Is during chronic virus infections and discuss the similar, newly emerging dichotomy in cancer. We then discuss how this understanding is leading to new therapeutic concepts and immunotherapy combinations. We propose that, by modulating the immune response at its foundation, it may be possible to widely reshape immunity to control these chronic diseases.
The Complex Relationships between Inflammation and Immunosuppression
Inflammation and immunosuppression induced by chronic virus infection and cancer can induce a dysfunctional immune state unable to eliminate disease. Why this ‘exhausted’ state has evolved to emerge in these chronic diseases is a matter of debate, but its initial invocation is critical to limit excessive immunopathology while maintaining some level of immunological control. Due to their direct lysis of infected and cancer cells, the majority of effort to understand immune exhaustion has focused on CD8 T cells. However, CD8 T cell functionality represents an endpoint of a complex set of molecular and cellular interactions that broadly reprogram all levels of the immune response leading to a collaborative failure to control disease. Yet, the underlying mechanisms that program and maintain the suppressive environment are less clear.
Somewhat paradoxically, chronic virus infections and many cancers are characterized by simultaneous immunosuppression and inflammation. Seemingly mutually exclusive, these immune states coexist and recent evidence indicates that their regulation is integrally linked. This linkage makes immunological sense because the immune response must initiate counter-regulatory measures to avert excessive or ongoing immune mediated disease once a pathogen is controlled. IFN-Is are emerging as central drivers of inflammation in chronic virus infections; however, IFN-Is also induce many of the suppressive factors that limit immunity to promote chronicity. Thus, emerging evidence places IFN-Is as a common nexus in the pathogenesis of multiple chronic diseases. In this review, we explore the emerging role of IFN-I signaling as a central node underlying the inflammation and the immune dysfunctions in chronic virus infection (Figure 1]. We then discuss how many of these same suppressive mechanisms are being identified to limit immune control of cancer and the potential positive and negative role of IFN-Is in the process. Finally, we highlight the complexity of targeting IFN-Is therapeutically and the potential for enhancing and inhibiting IFN-Is to augment immune function to control chronic viruses and cancer.
Figure 1. Type I Interferons (IFN-Is) Promote and Inhibit Multiple Environmental and Cellular Functions to Modulate All Levels of Immunity during Viral Persistence and Cancer.
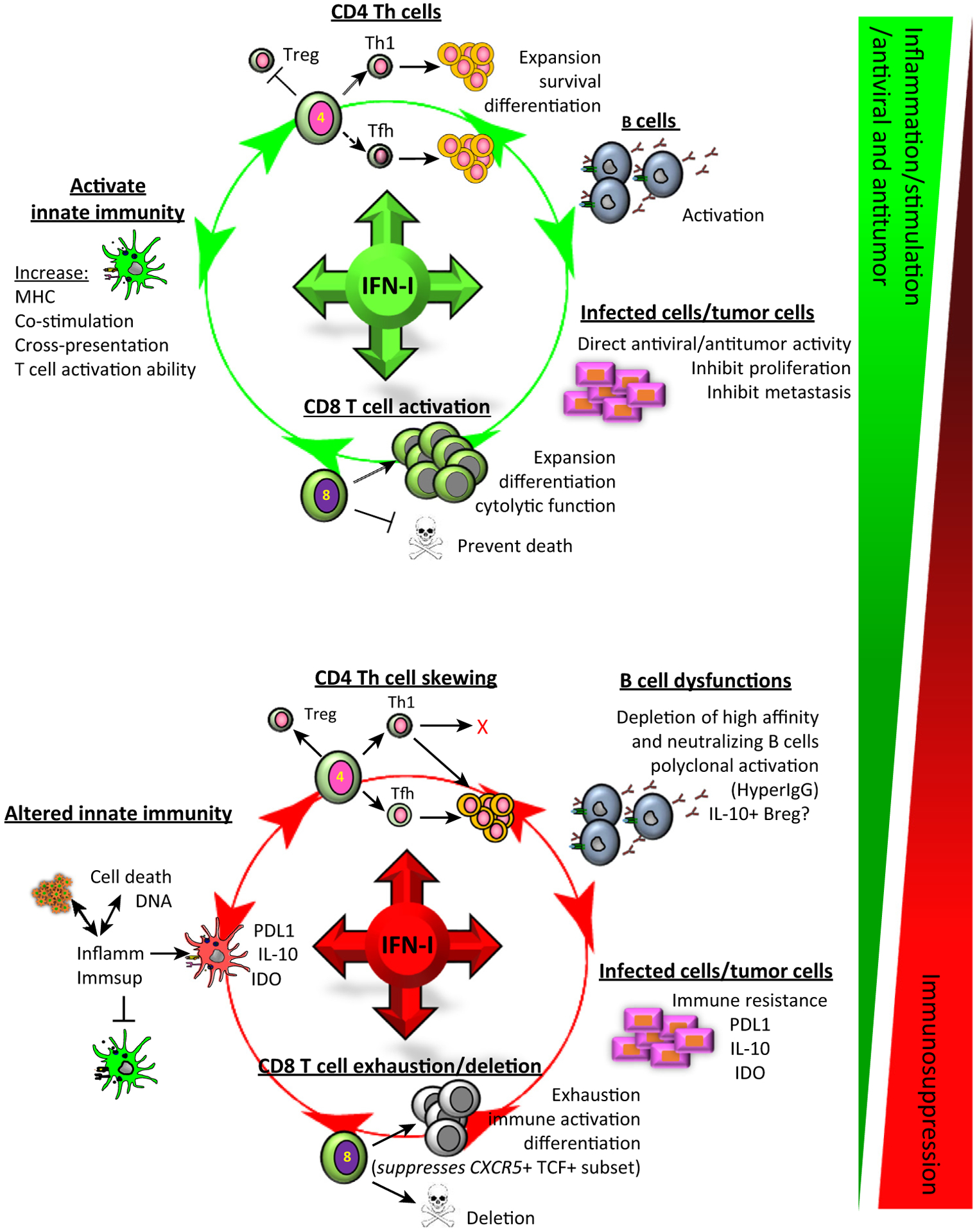
Most studies in chronic virus infections and cancer have focused on CD8 T cells and these cells are undoubtedly important in the inability of the immune system to overcome these diseases. However, CD8 T cells represent an endpoint of a complex set of cellular interactions, alterations in differentiation, and redirection of factors that underlie the global deterioration of multiple components of the immune response and ultimately lead to the attenuation of CD8 T cells and the failure to control these diseases. IFN-Is underlie many of the cellular functions and dysfunctions observed in chronic virus infections and this is also now beginning to come to light in multiple cancer types. IFN-Is promote immune maturation and differentiation from the innate to the adaptive immune response and, in times of chronic disease, also induce many of the immune dysfunctions throughout the immune response that impede virus and cancer control. These range of effects occur simultaneously throughout chronic viral infection and likely cancer, and ultimately represent a sliding scale dependent on many things, including the levels of IFN-Is, type of IFN-Is, duration of signaling, intracellular transcriptional programs, and other signals that cells are receiving. Abbreviations: MHC, major histocompatibility complex; Tfh, follicular helper T cell; Th, T helper cells; Treg, regulatory T cells.
IFN-Is: A Nexus Balancing Inflammation and Suppression
The IFN-I family comprises a single IFNβ gene and 13 or 14 IFNα genes (in human and mouse, respectively). IFN-Is signal through a dimeric receptor comprising IFNαR1 and IFNαR2 (termed here ‘IFNR’) that activates the kinases Janus kinase 1 (Jak1] and tyrosine kinase 2 (Tyk2] to initiate transduction of IFN-I signaling through signal transducer and activator of transcription (Stat)1 and Stat2 phosphorylation. In addition to canonical Stat1/Stat2 signaling, IFN-Is also activate a variety of other Stat proteins (e.g., Stat 3, 5, and 6) as well as phosphatidylinositide 3-kinase (PI3K) and mitogen-activated protein kinases (MAPK) (in particular ERK1/2 and P38] [1]. Furthermore, adding to the complexity of IFN-I signaling, different IFN-I proteins bind the receptor complex with different affinities and IFNβ can bind IFNαR1, propagating diverse signals and transcriptional programs [2]. Together, these signaling networks lead to activation of the multitude of IFN regulatory factors (IRFs) and IFN-stimulated genes (ISGs) promoting an inflammatory environment and initiating antiviral mechanisms associated with IFN-Is. Importantly, under biological conditions, IFN-I signaling does not occur in isolation. As a result, the presence of other inflammatory signals also affect the pathways of IFN signaling and modulate their functions adding even further complexity, via poorly understood signal crosstalk.
Sustained inflammation and subsequent immune activation is associated with worsened disease progression in chronic infections and, although many drivers of inflammation exist, ongoing IFN-I signaling is emerging as a primary mechanism [3]. In monkey models of SIV infection, natural hosts that do not progress to AIDS despite ongoing virus replication have lower IFN-I signaling signatures, while AIDS-progressing non-natural host monkey species exhibit high levels of sustained IFN-I signaling, independent of viral loads [4–6]. This suggests that, in addition to promoting antiviral immunity, chronic inflammation mediated by IFN-I leads to progressive immune dysfunction and disease, separate from virus replication. Moreover, IFNs can induce programmed death-ligand 1 (PD-L1], IL-10, and indoleamine 2,3 deoxygenase (IDO) expression by immune cells, nonhematopoietic cells, and, in some cases tumor cells, driving suppressive circuits [7–10]. Although the ability of IFN-Is to induce counter-regulatory mechanisms has long been known, the biological impact of this feedback in chronic inflammation has only recently come to light. Using the chronic lymphocytic choriomeningitis virus (LCMV) model, several research groups have demonstrated that many of the immune dysfunctions and suppressive programs associated with chronic virus infections were abolished when IFN-I signaling was inhibited, including a decrease in IL-10 and PD-L1 expression by dendritic cells (DCs) and macrophages, lowered chronic inflammation, enhanced multiple antiviral cell populations, and restored lymphoid architecture [7,11,12]. Exactly which of these functions contribute to the ultimate viral control is an area of active investigation, but it is likely that these modifications converge to promote enhanced cellular interactions, allow preservation of antibody-producing B cells, and engender a stimulatory instead of suppressive environment (discussed in greater depth below). The effects of IFN-I signaling do not necessarily transform from ‘good’ to ‘bad’ as chronic infection progresses, but rather aspects of each are present throughout the infection. It is how individual target cells and their intracellular signaling networks are temporally altered as infection proceeds that likely determine responsiveness to IFN-I signaling, immune programming, and consequent virus control.
In cancer, the role of IFN-Is has generally been considered beneficial, necessary to both promote T cell responses and to prevent metastases. However, there have been indications that IFN-Is can also have a negative role by promoting negative feedback and immunosuppression. IFN-Is can increase IDO expression by DCs and macrophages and upregulate expression of checkpoint inhibitors that attenuate antitumor T cell responses [13–17]. Thus, similar to chronic virus infections, ongoing IFN-I signaling may be a key driver of immune dysfunction in some cancers. Yet, in response to foreign pathogens, multiple pattern recognition receptors (PRRs), including toll-like receptors (TLRs) and cytosolic receptors RIG-I and MDA5, lead to IFN-I induction. By contrast, in the tumor setting, IFN-Is are often dependent on STING signaling [18], suggesting that cellular dysregulation and potentially immune recognition of DNA from dying cells underlie IFN-I induction (discussed below). Thus, differences in the basic biology of where and how IFN-I-inducing signals are transduced in the tumor compared with infection could affect the composition and impact of IFN-I-induced inflammatory and suppressive programs.
Direct IFN Effects on Virus Infection and on Tumor Cells
IFN-Is were first identified based upon their profound ability to render cells resistant to virus infection. Almost all cells express IFNRs, which, upon signaling, rapidly induce multiple antiviral response genes to inhibit virus replication in infected cells and send an alert to prevent infection of nearby cells. Testament to the fundamental role of IFN-Is in virus control are the observations in mice that, in the absence of IFN-I signaling, viruses that are normally rapidly controlled instead are either lethal or persist. The specific antiviral factors induced by IFN-Is and how they individually restrict viruses have been described in many reviews, including [19]. Recent evidence in humans demonstrated the pressure exerted by IFN-Is to restrict initial HIV infection. Upon HIV infection, the founder viruses that establish infection are relatively resistant to IFN-Is due, at least in part, to escape from the restriction mediated by the IFN-I-induced antiviral IFITM1 protein [20–22]. However, as HIV infection progresses, sensitivity of HIV to IFN-I-mediated restriction factors and mechanisms increases (despite ongoing IFN-I production and the pressure on the virus that would exert), suggesting an initial immune resistance of chronic IFN-I signaling that allows HIV to subvert its antiviral activity [20,23]. The initial resistance to IFN-Is is not absolute and founder viruses (particularly HIV Clade C) that do not exhibit IFN-I resistance can also be identified [24], indicating that overcoming the initial IFN-I antiviral program is one of the overall tactics HIV can use to establish infection. Although IFN-I dependence was not tested, increased expression of PD-L1 is also observed on multiple lineages of LCMV-infected cells, likely serving to inhibit the ability of cytotoxic T lymphocytes (CTLs) to respond to and kill the infected cells [7,8,25]. Thus, while restrictive to their replication, viruses can also take advantage of the counter-regulation induced by IFN-Is to promote immune suppression and allow the persistence of infected cells.
Similar to virally infected cells, IFN-Is have a direct inhibitory effect on tumor cells, limiting their proliferation and driving senescence and death. Despite the fact IFN-driven antagonism of tumor growth has been known for 40 years, there is not a well-defined understanding of mechanisms responsible for the IFN response of cancer cells; however, it is clear IFN-I-dependent inhibition of tumor cell expansion is a combination of cycle arrest and cell death [26,27]. In melanoma and breast cancer cells, IFN-I-driven expression of the TNFα-family member TRAIL was responsible for caspase 8-dependent apoptotic sensitivity to IFN-Is [28,29], while, in cervical carcinoma, IFN-Is caused apoptosis-independent proliferative arrest and early cytoplasmic accumulation of the antiapoptotic protein cFLIP and caspase 8 [30]. The net effect of the cellular response was initial proliferative arrest and senescence. However, over time, the composition of the death-inducing signaling complex favored caspase 8 activation, resulting in apoptosis, suggesting that initial IFN-I signals were cytostatic, but that prolonged stimulation is required for cell death. Interestingly, in vivo studies in breast cancer models found that metastasis required a loss of IRF7-driven gene signatures [31], while, in patients with breast cancer, reduced STAT1 activation was associated with worse overall outcomes [32]. In addition, acquired resistance to radiation therapy with immune checkpoint inhibition is driven through IFN-dependent STAT activation, which increases the tumor cell-intrinsic expression of immune suppressive receptors, such as PD-L1 [13]. Thus, breaking free from IFN-I-mediated regulation can be critical for cancer progression and, even while still being regulated by IFN-Is, cancer cells can co-opt the normal counter-regulatory mechanisms induced by IFN-Is to prevent immune cell killing.
Altered Innate Immunity
High levels of IFNα and β are rapidly produced in response to virus infections, but expression is curtailed within a few days or so regardless of viral clearance. Multiple studies have noted that, around the same time that IFN-I production is reduced, the immune response and antigen-presenting cells (APCs), in particular, become refractory to subsequent TLR stimulation and IFN production [33–35]. Although inherent culling of IFN-I signaling may serve to prevent excessive immunopathology, it can also promote infection, since sustaining IFN-I signaling through the administration or deletion of the IFN-I inhibitor OASL1 led to clearance of otherwise chronic LCMV infection [36,37]. After the initial robust IFN-I production subsides, individual IFNα and β subtypes are decreased to levels observed in uninfected conditions, although the IFN-I-dependent ISG signature is sustained [12], indicating that smoldering IFN-I production continues. How an almost unmeasurable level of IFN-I production continues to have such a dramatic impact on the antiviral immune response is an important question that may include IFN-I production at points of cell–cell interaction and/or transcriptional and epigenetic reprogramming to enable increased sensitivity to small amounts of IFNR activation.
Initial and chronic IFN-I signaling functions at all levels of the immune response and impacts immunity from the ‘ground up’, from innate APCs to T cell responses and on each individual cell type in between. Inflammatory IFN-I signals promote and modulate macrophage and DC development, maturation, and stimulatory capacity. Not only will these effects have obvious positive impacts on priming and generating the adaptive immune response, but the IFN-I mediated restriction of infection in certain APC subtypes can also be equally important by limiting antigen presentation, such as by CD169+ macrophages, to promote antiviral antibody production [38], or to constrain virus replication to limit systemic viral persistence [39]. How IFN-I negatively impacts the innate response in chronic virus infection is less well understood. In response to chronic LCMV infection, expression of many inhibitory factors, including IL-10, PD-L1, and IDO, is specifically induced by IFN-I on specific populations of CD39+ CD95+ immunoregulatory DCs and macrophages that suppress antiviral T cells [7,40]. Interestingly, conventional DCs (cDCs) did not adopt the IFN-I-mediated suppressive program, but rather IFN-I directly induced the expression of IL-10 and PD-L1 and suppressive activity on monocytederived (mo)DCs [7]. In the absence of IFNR expression on moDCs, these cells remained highly T cell stimulatory in the otherwise suppressive chronic infection. How exactly IFN-Is are induced to specifically target these cells is unclear, but a potential interaction with apoptotic red blood cells has been implicated [41]. In addition to generating immunoregulatory CD39+ DCs, IFN-Is also suppressed cDC numbers [7], effectively shifting the balance toward the suppressive innate immune environment associated with chronic virus infection. A similar CD39+ immunoregulatory DC phenotype was also evident in a mouse model of Mycobacterium tuberculosis, in human cells from HIV-infected humanized mice, and in B16 tumors [7], suggesting that emergence of these same DC populations is a conserved mechanism in chronic disease characterized by chronic inflammation and IFN-I signaling.
IFN-Is are critical during the initial stages of cancer development for the activation of DCs to cross-prime tumor-specific CD8 T cells [42,43]. However, until recently, the role of IFN-Is in innate immune regulation and tumor pathogenesis was thought to end there. Yet, recent data have begun to indicate that IFN-Is continue to modulate the innate immune response both in the tumor and systemically. In cancer plasmacytoid DCs (pDCs), accumulation at the tumor margin and in sentinel lymph nodes has been observed, most notably in melanoma and breast cancer [44,45] and correlates with a lack of mature cDCs [44], increased CTLA-4hi regulatory T cell populations [46], a decrease in proinflammatory cytokine production [47], and a poorer prognosis overall [44]. A question that arises is why an IFN-I-producing cell type would be negatively associated with cancer outcomes. One explanation has been that tumor-associated pDCs are specifically defective for IFN-I production [46] and responsiveness to IFN-I [45], similar to the refractory state observed by pDCs in chronic virus infections [35]. By contrast, pDCs present in sentinel lymph nodes during breast cancer and melanoma express IDO, suppress T cell responses, and promote regulatory T cell (Treg) expansion [48,49]. Although the IDO+ population of pDCs is a minority, they have an outsized impact on immunity, and IDO inhibition dramatically alters antitumor immunity [9]. The bulk of the literature suggests that IFN-I and IDO dynamics in the tumor and lymph nodes drive a balance of pro- and anti-inflammatory effects, providing productive immunity while limiting bystander pathology. Thus, smoldering autocrine/paracrine IFN-I may synergize with other signal pathways in the tumor or draining lymph nodes to drive the IDO+ regulatory pDCs. IDO was recently reported to suppress IFN-I-driven responses to viral infection by an aryl hydrocarbon receptor-dependent mechanism [50]. Thus in an IDOhigh environment driven by sustained IFN-I stimulation, pDCs may be skewed toward a regulatory phenotype with reduced ability to produce IFN-I upon stimulation, promoting immune suppression and tumorigenesis. In this view, tumor-associated PDCs are not ‘dysfunctional’ per se, but rather reflective of a chronically IFN-I+ microenvironment.
Macrophage exposure to IFN-Is primes a proinflammatory state and IFN-I+ macrophages in the tumor or draining tissues drive antitumor effects. This would argue that the primary effect of IFN-Is is to promote a classically inflammatory ‘M1’ phenotype. However, PD-L1 (similar to IDO) is an IFN-I-responsive gene that drives a counter-regulatory response suppressing CTL activity and solidifying a PD-1hi phenotype in FoxP3+ Tregs [51]. Given that PD-L1+ macrophages are found in a range of tumors, the effect of IFN-Is on this group may be an important mechanism of immune suppression. Tissue-resident macrophages exposed to dying cells drive tolerance by mechanisms dependent on PD-L1 [52] and IDO [53], and STING-deficient phagocytes failed to induce IDO, IL-10, PD-L1, or TGF-β after apoptotic cell uptake in vivo [54], an effect that is dependent on autocrine and paracrine STING-dependent IFN-I production (T.L. McGaha, unpublished data, 2017). This is consistent with the ability of DNA from dying tumor cells to drive STING-dependent IFN-I production [55,56]. However, STING-induced IFN-I responses can drive both tolerogenic and inflammatory immunity. How this dichotomy is perpetuated is unclear, but may be due to ‘antigen’ amounts. For example, in the face of large-scale tumor cell death, apoptotic tumor DNA may provide a strong STING agonist, provoking inflammatory immunity that is able to outweigh the counter-regulatory suppressive factors also induced. However, low-level STING activity as a result of tumor cell turnover and phagocytosis by macrophages may drive sustained, comparatively lower IFN-I production, promoting IDO, PD-L1, and IL-10-dependent regulatory mechanisms. This then would suggest that tumors incorporate mechanisms maintaining host equilibrium to persist. It will be interesting to determine whether, similar to chronic virus infections, the high antigen burden that would initially trigger the STING-driven inflammatory response also leads to increasingly potent suppressive signals that attenuate the immune response. Multiple groups are testing targeted STING activation by administration of cyclic di-nucleotides in anticancer therapy [57–59]. While early results are promising, the clear feedback inhibition induced by IFN-Is in general, and STING in particular, must be considered in this methodology.
CD8 T Cells: Activating, Sustaining and Wearing down the Effectors
Although it is well established that the inability to clear virus and cancer leads to the progressive dysfunction of antiviral CD8 T cells, these ‘exhausted’ CD8 T cells maintain some function and are critical to sustain a limited degree of viral or tumor control. Thus, the magnitude and quality of CTL responses are key factors in maintaining control, and are highly regulated by IFN-Is throughout infection. At the onset of viral infection, direct IFN-I signaling on antiviral CD8 and CD4 T cells is required for maximal T cell expansion and protection from natural killer (NK) cell-mediated killing [60–63], although other inflammatory cytokines can compensate during certain infections [64]. In addition, early IFN-I signaling drives the acquisition of CD8 T cell cytolytic function [61,62,65]. In cancer, IFN-Is have also been shown to enhance antitumor CD8 T cell effector function ex vivo by increasing their killing ability, which presumably accounted for the better tumor control upon adoptive transfer [66]. In vivo, IFN-I enhances antitumor CD8 T cell responses indirectly by enhancing cross-presentation by DCs [42,43]. However, direct survival effects of IFN-I have also been reported on intratumoral CD8 T cells [67]. Interestingly, in a colon cancer model, the tumor microenvironment actively downregulated IFNR on CD8 T cells, decreasing their survival and increasing tumorigenesis, while enforced IFNR expression on CTLs alone delayed tumorigenesis [67]. Thus, direct signaling on antiviral and antitumor CD8 T cells is critical for the initial activation and survival of CD8 T cell responses.
Upon systemic blockade of IFN-I signaling in vivo using an anti-IFNR antibody during chronic LCMV infection, antiviral CD8 T cell numbers remain unchanged or were slightly decreased [11,12]. However, IFNR blockade skewed antiviral CD8 T cell subsets, increasing a TCF-1+ CXCR5+ subset [68] that exhibits enhanced proliferative and renewal capacity, and is responsible for sustaining long-term antiviral CD8 T cell activity during chronic virus infections [69–71]. This TCF-1+ CXCR5+ CD8 T cell subset is present in multiple chronic infections, including HIV, HCV, and in cancer [68,69,71], and, importantly, has been reported to be the antiviral CD8 T cell subset that preferentially expands upon PD-L1 blockade and mediates viral control [69–71]. Thus, during chronic infection, IFN-Is suppress the antiviral CD8 T cell subset that sustains long-term viral control, favoring the formation of terminally differentiated antiviral CD8 T cells that do not renew but have enhanced cytotoxic function. This subset skewing likely contributes to the progressive IFN-I-mediated immune dysfunction during chronic infection [11,12] and may also link chronic IFN-I signaling to decreased success of anti-PD-L1 blockade and other checkpoint inhibitor therapies by limiting the progenitor CD8 T cell populations able to respond. Indeed, TCF-1+ CD8 T cells have also been identified in tumor-infiltrating lymphocytes (TIL) [68]; however, the regulation of this population by IFN-I signaling and its capacity to re-expand upon PD-L1 blockade remain to be determined.
CD4 T Cells: The Underappreciated Need for Sustained Help
Robust and sustained CD4 T cell responses are a strong correlate of control of multiple chronic infections. CD4 T cells are critical to maintain CD8 T cell responses when a virus or cancer cannot be controlled acutely and provide help to B cells for antibody responses that also contribute to control. As such, chronic viral infections induce CD4 T helper 1 (Th1] and follicular helper T cell (Tfh) responses, which predominantly help CD8 T cells and B cells, respectively. IFN-I profoundly affects CD4 T cell priming and differentiation dependent on the stage of viral infection [72] and, as a result, the type of help that CD4 T cells are able to provide. At the onset of what will become a chronic virus infection, IFN-I suppresses Tfh formation, but does not alter Th1 differentiation [72,73]. By contrast, virus-specific CD4 T cells primed once the chronic infection has been established yield only CD4 Tfh cells with de novo Th1 priming inhibited by IFN-Is [72,74]. A similar accumulation of CD4 Tfh is observed in multiple chronic virus infections characterized by chronic IFN-I signaling, including HIV, HCV, and SIV [75–77], suggesting a conserved mechanism by which IFN-I limits CD4 Th breadth. The inhibition of new CD4 Th1 cells was not a direct consequence of IFN-I signaling on CD4 Th1 cells, but instead IFN-I-induced IL-10 and PD-L1 expression by CD39+ suppressive DCs prevented Th1 differentiation [74]. Reconstituting the CD4 Th1 cells overcame many aspects of CD8 T cell exhaustion, including their progressive numerical decline, and facilitated enhanced control of the chronic infection [74]. PD-L1 and IL-10 also suppressed CD4 Th1 differentiation at the onset of acute and persistent LCMV infection [74], highlighting a role for IL-10 and PD-L1 as Th1 suppressive factors, and adding another mechanism by which PD-L1 regulates immune responses to virus infection.
In contrast to viral infection, the extent to which IFN-Is regulate or skew CD4 T cell responses in tumors has not been explored in detail. In one study, patients with IFNα-treated, nonprogressing chronic myeloid leukemia (CML) exhibited increased frequency of central and effector memory IFNγ+ and TNFα+ CD4 T cells [78]. Importantly, the patients showed residual leukemia that failed to expand, suggestive of immune surveillance. Other clues come from studies of immunity following chemotherapy. For example, treatment of B cell lymphoma with cyclophosphamide drives a temporary reduction in tumor size associated with infiltration of polyfunctional CD4+ T cells capable of producing IFN-γ and TNF-α [79]. The CD4+ T cells were required for CTL activation and, importantly, diminished in IFNαR−/−mice, suggesting IFN-I-driven differentiation [80]. Interestingly, in this model, the response was not durable and the antitumor CD4 T cells gradually acquired a PD-1hi exhausted phenotype [80]; however, PD-1 inhibition reversed this phenotype and drove long-term remission [81].
Regulatory CD4 T Cells
Although the contribution of Tregs to the suppression of antiviral immune responses and the control of chronic virus infection remains controversial, emerging evidence suggests that IFN-Is are important in determining their impact. The effect of IFN-Is on Tregs in viral infection remains controversial, with one report demonstrating that direct IFN-I signaling on Tregs at the onset of acute LCMV infection suppressed their numbers and activation, leading to increased antiviral CD4 and CD8 T cell responses and a slight lowering of viral titers [82], although another report did not see an effect of IFN-Is on Tregs [83]. Ex vivo depletion of Treg cells or their suppressive factors in peripheral blood mononuclear cells (PBMCs) from patients with HIV and HCV enhanced CD8 T cell activation and function, although how Tregs restrict the antiviral response in vivo (aside from limiting secondary immunopathology or secondary effects due to depletion) remains unclear [84]. In vivo depletion of Tregs in the midst of chronic LCMV infection substantially increased virus-specific CD8 T cell numbers and restored function, but did not change viral titers [85]. Interestingly, the decrease in Tregs in chronic LCMV infection was accompanied by an increase in PD-L1 expression that, when co-blocked with Treg depletion, did enable virus control. Although not analyzed, it will be important to determine whether the increase in PD-L1 following depletion of Tregs is due to increased levels of inflammation and IFN-Is that trigger counter-regulation by PD-L1. This type of counter-regulatory mechanism to therapeutically increased inflammation is beginning to come to light in cancer models wherein IFN-Is induced by radiation therapy increase PD-L1 expression that secondarily inhibits antitumor immunity [13]. Overall, the interplay between Treg-mediated suppression and control of inflammation and the effect that this has on subsequent suppressive factors in chronic virus infections remains to be better defined.
The inherent tumor-promoting role of Tregs in cancer has been extensively examined; however, the effects of acute or chronic IFN-I expression on Treg numbers or function are not well characterized. Similar to the suppressive effects of IFN-Is on antiviral Treg numbers, treatment with IFNα2b in a melanoma model reduced systemic Treg numbers [86], as did the delivery of intratumoral IFNα in a colon cancer model in addition to enhancing functional CD8 T cells and inhibiting tumor growth [87]. Furthermore, exposure to IFNα can functionally paralyze human CD4+ Tregs, inhibiting cyclic AMP (cAMP)-dependent suppression of antitumor responses [88], suggesting that IFN-Is limit both Treg number and function in cancer. By contrast, IFN-Is can also enhance the suppressive effects of Tregs. IL-10 production by tumor-associated, but not systemic, Tregs was reportedly dependent on IFNαR1 signaling in colon cancer, suggesting that local IFN-I production in the tumor drives the suppressive Treg phenotype [89]. IFN-Is likely also regulate Treg function indirectly by induction of downstream regulatory effectors, such as IDO, which, when inhibited, causes a loss of Treg suppression in many tumor types [90]. Recently, it was reported that IDO activation drives a PD-1hi Treg phenotype that is stabilized by interaction with PD-L1 [51], suggesting that multiple targets of IFN-Is work together to drive suppression in the tumor microenvironment (TME) and sentinel lymph nodes. Interestingly, infection of IDO1−/−mice with chronic LCMV did not change the course of infection (D.G. Brooks, work in progress), although suppressive APCs produce IDO during persistent LCMV infection [40] and during HIV infection [91]. Thus, although common programs of suppression are instituted in response to chronic antigen stimulation, their regulatory impact on the immune response and infection and/or tumor control may be specific to the pathogen and/or tumor present and weighted at different levels of importance to suppress immunity.
B Cell Immunity
Although present and necessary, B cell function and dysfunction during chronic infections are less understood than that of T cells. Interestingly, the progressive increase in Tfh and prevention of new Th1 cells represents a clear push by the immune system toward B cell immunity as chronic viral infection progresses [74–77,92], suggesting that, for good or bad, the immune system focuses on this direction in chronic virus infections. Even less well understood than B cell dynamics is the potential role that IFN-Is have toward B cell modulation in chronic infection. In LCMV and HIV infections, high-affinity virus-specific B cells are rapidly deleted from the repertoire and neutralizing antibodies are not generated until late in infection [93]. The B cells that are present often display decreased proliferative capacity, abnormal subpopulations and a terminally differentiated phenotype. IFN-Is are associated with polyclonal B cell activation in response to virus infection [94], and the increased ‘nonspecific’ antibody production, termed ‘hypergammaglobulinemia’, inhibits other antibody effector mechanisms, including antibody-mediated phagocytosis and clearance of infected cells [95,96]. Three papers recently identified a critical role for IFN-Is in the early deletion of high-affinity antibody-producing B cells during viral persistence [65,97,98]. Interestingly, the effect of IFN-Is was not directly on the B cells themselves, but rather IFN-Is directly stimulated CD8 T cells to kill LCMV-specific B cells [65]. Given the role of B cells in the control of chronic infection [99] and that long-term B cell responses were the best correlate with response to anti-PD1 immunotherapy [100], it will be important to determine how IFNR signaling directly affects B cell differentiation and survival, and/or whether it has a role in polyclonal activation in the setting of chronic infection.
In cancer, the role of B cells and antibodies is even less clear. B cells are often observed in conjunction with other TIL populations in multiple tumor types, generating tertiary lymphoid structures that could foster both B cell function and interactions [101,102]. In some cases, the presence of B cells in the tumor correlates with enhanced prognosis; however, how and why is less well understood [103–105]. Neo-antigens might induce antibody-targetable epitopes, but whether this is the case is unclear. B cells could also serve as APCs to modulate T cell responses and/or to generate optimal lymphoid structures and architecture that enable T cell function. T cell-independent stimulation drives the expression of IDO in B cells, limiting survival and functional maturation [106] and, although IDO expression was associated with stimuli that drove significant IFN-I production, it is not clear whether this was the factor driving the IDOhigh phenotype. Likewise, IL-10- and IDO-producing B cells have been identified in some tumors and are associated with Treg development [107]. It will be interesting to determine whether this is driven by IFN-I signaling to balance inflammation, as observed in chronic viral infections.
IFN-I Therapy: Restoring Immunity by Balancing Positive and Negative
IFN-Is as a Therapy
IFN-Is as a monotherapy for chronic viral infection has met with only limited and controversial success (reviewed in-depth in [108]), although, in combination with combination antiretroviral therapy (cART), a decreased latent reservoir and longer time to HIV rebound were observed [109]. A similar variable outcome to IFN-I therapy is also observed in cancer treatment (reviewed in-depth in [110]), although, similar to HIV, the combination of IFN-I with other tumoricidal or immune inducing therapies may prove effective. There are likely many reasons for clinical failure, including inherent biological mechanisms of resistance to further IFN-I signaling, changes in the cell populations that respond to IFN-Is, and institution of counter-regulatory pathways that diminish subsequent responses to IFN-Is. IFN-Is in combination with the antiviral agent ribavirin have long been the main anti-HCV therapy [111]. How IFN-Is in this combination contribute to control of HCV is not entirely clear, but likely includes both direct antiviral mechanisms and immune stimulation. However, in many cases, this therapy is not effective. This lack of effectiveness appears to be associated with a high pre-existing IFN-I signature [112,113] that may be refractory to further IFN-I signaling or may alter how IFN-I signals are interpreted. In the latter case, more IFN-I signaling may further reinforce immune dysfunctions. A question that does arise is whether the natural abrogation of the robust IFN-I production shortly after infection allows for viral persistence and whether initial IFN-I stimulation could be augmented to prevent viral persistence. Although likely difficult to implement in the clinic, when IFN-I signaling was prolonged in the LCMV system through either administration of IFNα and β or knockout of the negative regulator of IFN signaling OASL1, chronic infection was prevented [36,37]. Similarly, administration of IFN-Is at the onset of SIV infection increased resistance to infection [33], although the ability to control virus by supplementing with IFN-Is alone waned within 1 week as the host became refractory to further IFN-I stimulation [33,37].
In cancer, initial excitement about the potential for IFN-I therapy has waned as the realities of the complexities of IFN-I biology and delivery have become apparent. IFN-I administration shows best efficacy in hematological disease, such as in CML, wherein IFNα treatment significantly improved survival when administered as part of a combination therapy [114]. Similarly, data suggest that some patients with myeloma exhibit substantial benefit from IFN-I treatment as a combination or adjuvant therapy [115,116]. However, in solid tumors, the results are more mixed. High-dose IFNα adjuvant therapy was associated with a significant relapse-free response and an overall survival benefit in high-risk patients with melanoma [117]. By contrast, breast and ovarian cancer response rates to IFN-I therapy were low and associated with significant toxicity [118–123]. Overall, the cumulative data suggest that IFN-I therapy is most beneficial against early or disseminated cancer, but much less effective against established or metastatic tumors. Thus, in both virus models and cancer, the efficacy of IFN-Is is highest before the infection and/or cancer has robustly established; however, once established, IFN-I therapy alone is less effective, likely reflective of adaptive resistance and changes in IFN-I signaling outcomes.
Another important approach currently being explored to target cancer cells is the use of oncolytic viruses and induction of a virus infection-like state in cancer cells. Oncolytic viruses target tumor cells due, at least in part, to diminished IFN-I signaling in the cancer cells themselves. Although this is likely to be an escape mechanism on the part of the tumor, it allows the use of oncolytic viruses that are highly susceptible to IFN-I-mediated control and, therefore, preferentially infect cancer cells without affecting nontumor cells [124]. Interestingly, recent data suggest that DNA-demethylating agents are active against colorectal tumors by inducing double-stranded (ds)RNA from retroviral elements and mimicking an IFN-I-induced antiviral state [125]. The activity of 5-AZA-CdR was dependent on an MDA5-MAVS-IRF7 virus-recognition circuit that induced type III interferon and led to a decrease in the self-renewal ability of the cancer-initiating cell population [125]. Thus, therapies can take advantage of the tumor-intrinsic loss of IFN-I sensitivity or induce an antiviral state in tumor cells, mimicking IFN-I signaling, although, as discussed above, this same strategy can also be co-opted by tumor cells to suppress the immune system to improve their survival and tumorigenic potential.
It is important to consider the roles of other pathways that are induced by IFN-Is and whether they could serve as viable targets in conjunction with IFN-I therapy. One obvious target would be IL-10, which has well-documented regulatory functions and is a critical immune-suppressive effector produced in tumors and chronic virus infections [126,127]. However, as is the case with IFN-Is, IL-10 appears to have pleiotropic effects and, in some cases, may impede antitumor responses, while, in others, it may promote tumor clearance by driving CD8 T cell differentiation, IFN-γ production, and APC maturation [128,129]. Surprisingly, PEGylated IL-10 could reduce intratumoral Tregs despite significant IDO expression, suggesting that IL-10 could overcome the IFN-IDO axis of suppression that is problematic in many cancers [130]. Thus, the overall complexity in the IFN-I pathway and its downstream targets suggests that determining how best to modulate IFN-I activity and combinations with other blocking pathways will likely need to be explored based on the cancer type and the composition of cells present to react to IFN-I signaling.
Blocking IFN-Is as a Therapy
The emerging concept that IFN-I-driven chronic inflammation promotes HIV disease progression has spurred the idea that blocking IFN-I signaling could reset the immune response, essentially therapeutically achieving the situation observed in natural SIV hosts that do not progress to AIDS despite ongoing virus replication [4–6]. The concept that blocking IFN-I signaling could be effective was strengthened by the experiments described above in the chronic LCMV system where blocking IFNR decreased levels of immune activation and immunosuppression, allowing immune-mediated control of the chronic infection. To investigate the effect of blocking IFN-I at the onset of SIV infection, Sandler et al. used an engineered high-affinity IFN-α2 mutant (IFN-ant) to diminish IFN-I signaling [33,131]. Interestingly, although IFN-ant decreased levels of global immune activation following infection, the loss of IFN-I signaling increased virus replication (likely due to the loss of IFN-I-induced antiviral activity) and ultimately accelerated AIDS progression. Importantly, this study provided a critical cautionary note, about balancing the temporal regulation of positive and negative aspects of IFN-I networks in virus infections and reminded us of the fundamental antiviral role of IFN-Is in limiting infection.
To explore how ongoing IFN-I signaling during the chronic phase of HIV infection contributed to overall immune activation, T cell dysfunction and HIV replication, researchers administered an anti-IFNR2-blocking antibody to humanized mice 10+ weeks following HIV infection [132]. Strikingly, 1 week of anti-IFNR treatment in these HIV-infected mice reduced the numbers of PD1, Tim3, CD38, and HLA-DR-positive CD4 and CD8 T cells, and decreased the surface level of these markers on the cells that retained expression, indicating that immune activation in HIV infection requires constant IFN-I-dependent stimulation. The decrease in immune activation was accompanied by increased anti-HIV CD8 T cell responses and a reduction in HIV titers and infected cells [132]. In a second set of studies, a similar humanized mouse approach was used to investigate the effect of blocking IFN-I signaling in cART-suppressed HIV infection [133]. As reported in some humans with cART-suppressed HIV replication, IFN-I signaling and immune activation continued at low levels despite undetectable virus replication in the mice. When anti-IFNR1 antibody was given to HIV-infected mice with undetectable virus, the level of immune activation further decreased, the anti-HIV T cell response was enhanced (something that cART alone did not achieve), and the size of the reactivatable latent reservoir was diminished, resulting in a longer time to virus rebound when cART was withdrawn. Interestingly, combination cART plus anti-IFNR1 treatment led to ‘blips’ in virus reactivation that were not observed in cART alone, suggesting that IFN-Is continue to provide some level of antiviral containment to prevent virus reactivation and/or smoldering reservoirs during therapy. The blockade of IFNR and the addition of IFN-Is were both associated with a decrease in the latent reservoir and extension of the time to virus rebound following cART interruption, suggesting that IFN-Is both prevent and stimulate reactivation of latent virus [109,133].
Similar to virus infections, IFN-Is clearly have critical effects required to initiate the antitumor T cell response. However, data are also starting to indicate that these beneficial effects are countered by the induction of suppressive mechanisms in cancer. Administration of an anti-EGFR antibody conjugated to IFNβ using a mouse model in which B16 melanoma was engineered to express EGFR led to rapid activation and cross-priming of CD8 T cells by DCs in a DC-IFNR-dependent mechanism. However, the IFNβ component also increased expression of PD-L1 on the tumor cells and the inclusion of anti-PD-L1 with the antibody-IFNβ conjugate led to tumor clearance [134], indicating that the induced expression of PD-L1 by IFNβ suppressed the antitumor immunity and tumor control. Likewise, recent data demonstrated that IFN-I signals induced as a consequence of combination radiation therapy drive adaptive tumor cell resistance to immunity, at least in part, by increasing PD-L1 expression [13]. Importantly, blocking IFN-I signals in this study (either by genetic ablation of the receptor or administration of JAK inhibitors) was sufficient to prevent this adaptive mechanism and enhanced the response to checkpoint inhibitors. However, inhibiting PD-L1 did not completely account for the suppressive effects of IFN-I following radiation therapy, suggesting that other IFN-I induced pathways, such as IL-10 or IDO, are involved. Together, these observations suggest that sustained IFN-I stimulation could be a key tolerogenic circuit induced by injury that, if its downstream effects can be understood and appropriately targeted, may enable increased responsiveness to immune enhancing therapies.
Overcoming an Initially High IFN Signature
A reason for the failure of IFN-I administration therapy in chronic virus infections and cancer may be what is termed ‘adaptive resistance’, with cells becoming refractory to IFN-I signaling due to chronic exposure. In HCV infection, the failure of IFN-I plus ribavirin therapy is highest in patients with a pre-existing elevated IFN-I signature [113]. A similar refractoriness to additional IFN-I signaling is observed in HIV and LCMV infection [33,37]. Resistance of cancer cells to IFN-I signaling is likely often the result of selective mutational pressure [135]; however, reduced responsiveness will be driven by a variety of additional factors, including: (i) altered/reduced signaling; (ii) epigenetic modification; (iii) regulatory feedback attenuating IFN-I circuits; and (iv) transcription and/or translation responsiveness. Ultimately, regardless of the driving mechanism, once a refractory state is established, additional IFN-Is would not further enhance the antiviral and/or immune stimulatory effects. However, refractory and absent are different states, and, although the antiviral and immune-stimulatory effects of IFN-Is may not be potentiated, IFN-I therapy in the presence of a pre-existing IFN-I signaling signature may further induce the suppressive counter-regulatory signals. Thus, a strategy to measure the interferon signature prior to the initiation of therapy may be beneficial to determine which patients will and will not respond. In situations where the IFN-I signature is high, it might be possible to initially decrease IFN-I signaling for a brief time to allow the immune system to recalibrate itself and once again become responsive to IFN-I in therapy. A total block may not be necessary, but rather it may be sufficient (and beneficial) to partially decrease IFN-I signaling. Following the ‘IFN-I break’, IFN-I therapy may become effective. Understanding how the immune system functions under variable levels of IFN-I signaling and the effect that providing an IFN-I ‘holiday’ has toward resetting IFN-I sensitivity and immunity will be critical to these next therapeutic steps.
Concluding Remarks
The role of IFN-Is in chronic virus infections and cancer are complex, often leading to distinct outcomes depending on the timing, cells present, the cumulative levels of IFN-I signals, and the IFNα/β subtypes mediating the effects. Compared with virus infections, relatively little is known about how IFN-Is modulate the immune environment (and tumor cells themselves) in cancer. However, this is rapidly changing as both the stimulatory and regulatory aspects of IFN-I induction in malignant neoplastic disease are identified, with much of this information being gleaned in the past year. Furthermore, it is becoming clear that the timing of IFN-I administration or blockade can have dramatically different effects, revealing the intricate underlying biology. Thus, the superficially straightforward proinflammatory circuit has given way to an intricate, highly ordered, yet poorly understood, network of feedforward and feedback mechanisms working in a sequential and concurrent fashion impacting immunity at all levels. The enormous complexity of the IFN-I network and its implications in health and disease make it imperative that the full spectrum of regulatory biology be properly explored (see Outstanding Questions). This will reveal key general as well as disease-specific biology promoting more efficient and targeted therapy.
Outstanding Questions.
What are the relevant suppressive mechanisms induced by IFN-Is? While IFN-Is induce a range of regulatory responses, it is likely that some will be more important than others in limiting efficacy. If these pathways (which are potentially different in different contexts) can be identified then the effectiveness of IFN-I therapy is likely to improve tremendously.
What are the signaling and transcriptional networks that differentially induce suppressive versus proinflammatory immune outcomes? Is it possible to functionally separate pro- versus regulatory effects of the IFN-I response? If this could be delineated, it may be possible to harness all aspects of IFN-Is for inflammatory and tolerogenic therapies.
There is a need to understand how cells become resistant or divergently respond to IFN-I therapy and strategies to restore sensitivity.
What about autoimmunity? IFN-I responses are key drivers of autoimmune disease. Since runaway IFN-I activity is a nodal driver of immune dysfunction and pathology, there is a significant risk that adverse autoimmune reactions could be greater than those seen with current checkpoint inhibitors with improved IFN-I responses (i.e., devoid of regulatory feedback). Toxicity is already an issue with IFN-I therapy, thus care must be taken to assess this potential adverse effect.
Trends.
IFN-Is drive multiple feedforward and feedback mechanisms promoting inflammatory immunity in a regulated fashion. However, in response to chronic exposure, these regulatory mechanisms may predominate and suppress immunity, thereby promoting pathogen or tumor persistence.
In viral infection, IFN-Is are induced, often at high levels, by multiple pattern or damage recognition receptors. In cancer, IFN-Is are likely induced by a more restricted set of receptors recognizing tumor cell death. The magnitude and mode of death may ultimately be determinant factors driving the development of functional inflammatory or regulatory immunity.
IFN-I-induced negative regulatory pathways are emerging as key drivers of chronic inflammation in chronic virus infections and barriers to anticancer checkpoint-inhibitor therapy. However, the benefits and risks of therapeutically enhancing or nullifying IFN-Is and their downstream effectors must be carefully weighed, given the role of IFN-Is as both drivers and suppressors of immune responses.
Acknowledgments
We thank the entire Brooks and McGaha laboratories for their help and ideas. The work was supported by Training Grant from the Fonds de la recherche en santé du Québec (to L.M.S.), the National Institutes of Health (AI085043 to D.G.B.; AI105500, AR067763, and CA190449 to T.L.M.), CIHR Foundation Grant FDN148386 (to D.G.B.), and the Medicine by Design Award#C1TPA-2016-20 (to D.G.B. and T.L.M.).
References
- 1.Gonzalez-Navajas JM et al. (2012) Immunomodulatory functions of type I interferons. Nat. Rev. Immunol 12, 125–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Weerd NA et al. (2013) Structural basis of a unique interferon-beta signaling axis mediated via the receptor IFNAR1. Nat. Immunol 14, 901–907 [DOI] [PubMed] [Google Scholar]
- 3.Klatt NR et al. (2013) Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol. Rev 254, 326–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bosinger SE et al. (2009) Global genomic analysis reveals rapid control of a robust innate response in SIV-infected sooty mangabeys. J. Clin. Invest 119, 3556–3572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris LD et al. (2010) Downregulation of robust acute type I interferon responses distinguishes nonpathogenic simian immunodeficiency virus (SIV) infection of natural hosts from pathogenic SIV infection of rhesus macaques. J. Virol 84, 7886–7891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jacquelin B et al. (2009) Nonpathogenic SIV infection of African green monkeys induces a strong but rapidly controlled type I IFN response. J. Clin. Invest 119, 3544–3555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cunningham CR et al. (2016) Type I and Type II interferon coordinately regulate suppressive dendritic cell fate and function during viral persistence. PLoS Pathog. 12, e1005356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mueller SN et al. (2007) Viral targeting of fibroblastic reticular cells contributes to immunosuppression and persistence during chronic infection. Proc. Natl. Acad. Sci. U. S. A 104, 15430–15435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Munn DH and Mellor AL (2016) IDO in the tumor microenvironment: inflammation, counter-regulation, and tolerance. Trends Immunol. 37, 193–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spranger S et al. (2013) Up-regulation of PD–L1, IDO, and T (regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci. Transl. Med 5, 200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teijaro JR et al. (2013) Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 340, 207–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilson EB et al. (2013) Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 340, 202–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benci JL et al. (2016) Tumor interferon signaling regulates a multigenic resistance program to immune checkpoint blockade. Cell 167, 1540–1554 e1512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai W and Gupta SL (1990) Regulation of indoleamine 2, 3-dioxygenase gene expression in human fibroblasts by interferon-gamma. Upstream control region discriminates between interferon-gamma and interferon-alpha. J. Biol. Chem 265, 19871–19877 [PubMed] [Google Scholar]
- 15.Jiang GM et al. (2017) Bortezomib relieves immune tolerance in nasopharyngeal carcinoma via STAT1 suppression and indoleamine 2,3-dioxygenase downregulation. Cancer Immunol. Res 5, 42–51 [DOI] [PubMed] [Google Scholar]
- 16.McGaha TL et al. (2012) Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol. Rev 249, 135–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Terawaki S et al. (2011) IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J. Immunol 186, 2772–2779 [DOI] [PubMed] [Google Scholar]
- 18.Gajewski TF et al. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng CT et al. (2016) Alpha and beta type 1 interferon signaling: passage for diverse biologic outcomes. Cell 164, 349–352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fenton-May AE et al. (2013) Relative resistance of HIV-1 founder viruses to control by interferon-alpha. Retrovirology 10, 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foster TL et al. (2016) Resistance of transmitted founder HIV-1 to IFITM-mediated restriction. Cell Host Microbe 20, 429–442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Parrish NF et al. (2013) Phenotypic properties of transmitted founder HIV-1. Proc. Natl. Acad. Sci. U. S. A 110, 6626–6633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sunshine JE et al. (2015) Fitness-balanced escape determines resolution of dynamic founder virus escape processes in HIV-1 infection. J. Virol 89, 10303–10318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song H et al. (2016) Transmission of multiple HIV-1 subtype C transmitted/founder viruses into the same recipients was not determined by modest phenotypic differences. Sci. Rep 6, 38130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sherwin SA et al. (1982) A multiple-dose phase I trial of recombinant leukocyte A interferon in cancer patients. JAMA 248, 2461–2466 [PubMed] [Google Scholar]
- 26.Hall M et al. (1995) Evidence for different modes of action of cyclin-dependent kinase inhibitors: p15 and p16 bind to kinases, p21 and p27 bind to cyclins. Oncogene 11, 1581–1588 [PubMed] [Google Scholar]
- 27.Sangfelt O et al. (1999) Molecular mechanisms underlying interferon-alpha-induced G0/G1 arrest: CKI-mediated regulation of G1 Cdk-complexes and activation of pocket proteins. Oncogene 18, 2798–2810 [DOI] [PubMed] [Google Scholar]
- 28.Bernardo AR et al. (2013) Synergy between RA and TLR3 promotes type I IFN-dependent apoptosis through upregulation of TRAIL pathway in breast cancer cells. Cell Death Dis. 4, e479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chawla-Sarkar M et al. (2001) Preferential induction of apoptosis by interferon (IFN)-beta compared with IFN-alpha2: correlation with TRAIL/Apo2L induction in melanoma cell lines. Clin. Cancer Res 7, 1821–1831 [PubMed] [Google Scholar]
- 30.Apelbaum A et al. (2013) Type I interferons induce apoptosis by balancing cFLIP and caspase-8 independent of death ligands. Mol. Cell Biol 33, 800–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bidwell BN et al. (2012) Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat. Med 18, 1224–1231 [DOI] [PubMed] [Google Scholar]
- 32.Widschwendter A et al. (2002) Prognostic significance of signal transducer and activator of transcription 1 activation in breast cancer. Clin. Cancer Res 8, 3065–3074 [PubMed] [Google Scholar]
- 33.Sandler NG et al. (2014) Type I interferon responses in rhesus macaques prevent SIV infection and slow disease progression. Nature 511, 601–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wonderlich ER and Barratt-Boyes SM (2013) SIV infection of rhesus macaques differentially impacts mononuclear phagocyte responses to virus-derived TLR agonists. J. Med. Primatol 42, 247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zuniga EI et al. (2008) Persistent virus infection inhibits type I interferon production by plasmacytoid dendritic cells to facilitate opportunistic infections. Cell Host Microbe 4, 374–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee MS et al. (2013) Negative regulation of type I IFN expression by OASL1 permits chronic viral infection and CD8(+) T-cell exhaustion. PLoS Pathog. 9, e1003478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang Y et al. (2012) Timing and magnitude of type I interferon responses by distinct sensors impact CD8 T cell exhaustion and chronic viral infection. Cell Host Microbe 11, 631–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Honke N et al. (2016) Immunoactivation induced by chronic viral infection inhibits viral replication and drives immunosuppression through sustained IFN-I responses. Eur. J. Immunol 46, 372–380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nice TJ et al. (2016) Type I interferon receptor deficiency in dendritic cells facilitates systemic murine norovirus persistence despite enhanced adaptive immunity. PLoS Pathog. 12, e1005684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilson EB et al. (2012) Emergence of distinct multiarmed immunoregulatory antigen-presenting cells during persistent viral infection. Cell Host Microbe 11, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ohyagi H et al. (2013) Monocyte-derived dendritic cells perform hemophagocytosis to fine-tune excessive immune responses. Immunity 39, 584–598 [DOI] [PubMed] [Google Scholar]
- 42.Diamond MS et al. (2011) Type I interferon is selectively required by dendritic cells for immune rejection of tumors. Exp. Med 208, 1989–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fuertes MB et al. (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. J. Exp. Med 208, 2005–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Treilleux I et al. (2004) Dendritic cell infiltration and prognosis of early stage breast cancer. Clin. Cancer Res 10, 7466–7474 [DOI] [PubMed] [Google Scholar]
- 45.Vermi W et al. (2003) Recruitment of immature plasmacytoid dendritic cells (plasmacytoid monocytes) and myeloid dendritic cells in primary cutaneous melanomas. J. Pathol 200, 255–268 [DOI] [PubMed] [Google Scholar]
- 46.Sisirak V et al. (2012) Impaired IFN-alpha production by plasmacytoid dendritic cells favors regulatory T-cell expansion that may contribute to breast cancer progression. Cancer Res. 72, 5188–5197 [DOI] [PubMed] [Google Scholar]
- 47.Munn DH et al. (2004) Expression of indoleamine 2,3-dioxygenase by plasmacytoid dendritic cells in tumor-draining lymph nodes. J. Clin. Invest 114, 280–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mansfield AS et al. (2009) Simultaneous Foxp3 and IDO expression is associated with sentinel lymph node metastases in breast cancer. BMC Cancer 9, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Speeckaert R et al. (2012) Indoleamine 2,3-dioxygenase, a new prognostic marker in sentinel lymph nodes of melanoma patients. Eur. J. Cancer 48, 2004–2011 [DOI] [PubMed] [Google Scholar]
- 50.Yamada T et al. (2016) Constitutive aryl hydrocarbon receptor signaling constrains type I interferon-mediated antiviral innate defense. Nat. Immunol 17, 687–694 [DOI] [PubMed] [Google Scholar]
- 51.Sharma MD et al. (2015) The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci. Adv 1, e1500845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Getts DR et al. (2011) Tolerance induced by apoptotic antigen-coupled leukocytes is induced by PD-L1+ and IL-10-producing splenic macrophages and maintained by T regulatory cells. J. Immunol 187, 2405–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravishankar B et al. (2012) Tolerance to apoptotic cells is regulated by indoleamine 2,3-dioxygenase. Proc. Natl. Acad. Sci. U. S. A 109, 3909–3914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Huang L et al. (2013) Cutting edge: DNA sensing via the STING adaptor in myeloid dendritic cells induces potent tolerogenic responses. J. Immunol 191, 3509–3513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng L et al. (2014) STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity 41, 843–852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Woo SR et al. (2014) STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 41, 830–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Corrales L et al. (2015) Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell Rep. 11, 1018–1030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Luo M et al. (2017) A STING-activating nanovaccine for cancer immunotherapy. Nat. Nanotechnol Published online April 24, 2017. 10.1038/nnano.2017.52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Smith TT et al. (2017) Biopolymers codelivering engineered T cells and STING agonists can eliminate heterogeneous tumors. J. Clin. Invest Published online April 24, 2017. 10.1172/JCI87624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crouse J et al. (2014) Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40, 961–973 [DOI] [PubMed] [Google Scholar]
- 61.Kolumam GA et al. (2005) Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J. Exp. Med 202, 637–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wiesel M et al. (2012) Type-I IFN drives the differentiation of short-lived effector CD8+ T cells in vivo. Eur. J. Immunol 42, 320–329 [DOI] [PubMed] [Google Scholar]
- 63.Xu HC et al. (2014) Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40, 949–960 [DOI] [PubMed] [Google Scholar]
- 64.Thompson LJ et al. (2006) Innate inflammatory signals induced by various pathogens differentially dictate the IFN-I dependence of CD8 T cells for clonal expansion and memory formation. J. Immunol 177, 1746–1754 [DOI] [PubMed] [Google Scholar]
- 65.Moseman EA et al. (2016) Type I interferon suppresses virus-specific B cell responses by modulating CD8+ T cell differentiation. Sci. Immunol 1, eaah3565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hervas-Stubbs S et al. (2010) Effects of IFN-alpha as a signal-3 cytokine on human naive and antigen-experienced CD8(+) T cells. Eur. J. Immunol 40, 3389–3402 [DOI] [PubMed] [Google Scholar]
- 67.Katlinski KV et al. (2017) Inactivation of interferon receptor promotes the establishment of immune privileged tumor microenvironment. Cancer Cell 31, 194–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wu T et al. (2016) The TCF1-Bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci. Immunol 1, eaai8593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.He R et al. (2016) Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature 537, 412–428 [DOI] [PubMed] [Google Scholar]
- 70.Im SJ et al. (2016) Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Utzschneider DT et al. (2016) T cell factor 1-expressing memory-like CD8(+) T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427 [DOI] [PubMed] [Google Scholar]
- 72.Osokine I et al. (2014) Type I interferon suppresses de novo virus-specific CD4 Th1 immunity during an established persistent viral infection. Proc. Natl. Acad. Sci. U. S. A 111, 7409–7414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ray JP et al. (2014) Transcription factor STAT3 and type I interferons are corepressive insulators for differentiation of follicular helper and T helper 1 cells. Immunity 40, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Snell LM et al. (2016) Overcoming CD4 Th1 cell fate restrictions to sustain antiviral CD8 T cells and control persistent virus infection. Cell Rep. 16, 3286–3296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feng J et al. (2012) Patients with chronic hepatitis C express a high percentage of CD4(+)CXCR5(+) T follicular helper cells. Gastroenterol. 47, 1048–1056 [DOI] [PubMed] [Google Scholar]
- 76.Lindqvist M et al. (2012) Expansion of HIV-specific T follicular helper cells in chronic HIV infection. J. Clin. Invest 122, 3271–3280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Petrovas C et al. (2012) CD4 T follicular helper cell dynamics during SIV infection. J. Clin. Invest 122, 3281–3294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ilander M et al. (2014) Enlarged memory T-cell pool and enhanced Th1-type responses in chronic myeloid leukemia patients who have successfully discontinued IFN-alpha monotherapy. PLoS One 9, e87794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ding ZC et al. (2012) Polyfunctional CD4(+) T cells are essential for eradicating advanced B-cell lymphoma after chemotherapy. Blood 120, 2229–2239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ding ZC et al. (2010) Chemotherapy rescues tumor-driven aberrant CD4+ T-cell differentiation and restores an activated polyfunctional helper phenotype. Blood 115, 2397–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ding ZC et al. (2014) Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 74, 3441–3453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Srivastava S et al. (2014) Type I interferons directly inhibit regulatory T cells to allow optimal antiviral T cell responses during acute LCMV infection. J. Exp. Med 211, 961–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Che JW et al. (2015) Regulatory T cells resist virus infection-induced apoptosis. J. Virol 89, 2112–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Veiga-Parga T et al. (2013) Role of regulatory T cells during virus infection. Immunol. Rev 255, 182–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Penaloza-MacMaster P et al. (2014) Interplay between regulatory T cells and PD-1 in modulating T cell exhaustion and viral control during chronic LCMV infection. J. Exp. Med 211, 1905–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yu Y et al. (2016) INFalpha–2b inhibitory effects on CD4(+) CD25(+)FOXP3(+) regulatory T cells in the tumor microenvironment of C57BL/6 J mice with melanoma xenografts. BMC Cancer 16, 397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hashimoto H et al. (2014) Type I IFN gene delivery suppresses regulatory T cells within tumors. Cancer Gene Ther. 21, 532–541 [DOI] [PubMed] [Google Scholar]
- 88.Bacher N et al. (2013) Interferon-alpha suppresses cAMP to disarm human regulatory T cells. Cancer Res. 73, 5647–5656 [DOI] [PubMed] [Google Scholar]
- 89.Stewart CA et al. (2013) Interferon-dependent IL-10 production by Tregs limits tumor Th17 inflammation. J. Clin. Invest 123, 4859–4874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sharma MD et al. (2010) Reprogrammed foxp3(+) regulatory T cells provide essential help to support cross-presentation and CD8(+) T cell priming in naive mice. Immunity 33, 942–954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Boasso A et al. (2007) HIV inhibits CD4+ T-cell proliferation by inducing indoleamine 2,3-dioxygenase in plasmacytoid dendritic cells. Blood 109, 3351–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fahey LM et al. (2011) Viral persistence redirects CD4 T cell differentiation toward T follicular helper cells. J. Exp. Med 208, 987–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Noto A and Pantaleo G (2017) B-cell abnormalities and impact on antibody response in HIV infection. Curr. Opin. HIV AIDS Published online February 15, 2017. 10.1097/COH.0000000000000359 [DOI] [PubMed] [Google Scholar]
- 94.Purtha WE et al. (2008) Early B-cell activation after West Nile virus infection requires alpha/beta interferon but not antigen receptor signaling. J. Virol 82, 10964–10974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wieland A et al. (2015) Antibody effector functions mediated by Fcgamma-receptors are compromised during persistent viral infection. Immunity 42, 367–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yamada DH et al. (2015) Suppression of Fcgamma-receptor-mediated antibody effector function during persistent viral infection. Immunity 42, 379–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fallet B et al. (2016) Interferon-driven deletion of antiviral B cells at the onset of chronic infection. Sci. Immunol 1, eaah6817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sammicheli S et al. (2016) Inflammatory monocytes hinder antiviral B cell responses. Sci. Immunol 1, eaah6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bergthaler A et al. (2009) Impaired antibody response causes persistence of prototypic T cell-contained virus. PLoS Biol. 7, e1000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Velu V et al. (2009) Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 458, 206–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bindea G et al. (2013) Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795 [DOI] [PubMed] [Google Scholar]
- 102.Nielsen JS and Nelson BH (2012) Tumor-infiltrating B cells and T cells: Working together to promote patient survival. Oncoimmunology 1, 1623–1625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Buisseret L et al. (2017) Tumor-infiltrating lymphocyte composition, organization and PD-1/PD–L1 expression are linked in breast cancer. Oncoimmunology 6, e1257452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Castino GF et al. (2016) Spatial distribution of B cells predicts prognosis in human pancreatic adenocarcinoma. Oncoimmunology 5, e1085147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kroeger DR et al. (2016) Tumor-infiltrating plasma cells are associated with tertiary lymphoid structures, cytolytic T-cell responses, and superior prognosis in ovarian cancer. Clin. Cancer Res 22, 3005–3015 [DOI] [PubMed] [Google Scholar]
- 106.Shinde R et al. (2015) B cell-intrinsic IDO1 regulates humoral immunity to T cell-independent antigens. J. Immunol 195, 2374–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rosser EC and Mauri C (2015) Regulatory B cells: origin, phenotype, and function. Immunity 42, 607–612 [DOI] [PubMed] [Google Scholar]
- 108.Bosinger SE and Utay NS (2015) Type I interferon: understanding its role in HIV pathogenesis and therapy. Curr. HIV/AIDS Rep 12, 41–53 [DOI] [PubMed] [Google Scholar]
- 109.Azzoni L et al. (2013) Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J. Infect. Dis 207, 213–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zitvogel L et al. (2015) Type I interferons in anticancer immunity. Nat. Rev. Immunol 15, 405–414 [DOI] [PubMed] [Google Scholar]
- 111.Heim MH (2013) 25 years of interferon-based treatment of chronic hepatitis C: an epoch coming to an end. Nat. Rev. Immunol 13, 535–542 [DOI] [PubMed] [Google Scholar]
- 112.Chen L et al. (2005) Hepatic gene expression discriminates responders and nonresponders in treatment of chronic hepatitis C viral infection. Gastroenterology 128, 1437–1444 [DOI] [PubMed] [Google Scholar]
- 113.Sarasin-Filipowicz M et al. (2008) Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. U. S. A 105, 7034–7039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Guilhot F et al. (1997) Interferon alfa-2b combined with cytarabine versus interferon alone in chronic myelogenous leukemia. French Chronic Myeloid Leukemia Study Group. N. Engl. J. Med 337, 223–229 [DOI] [PubMed] [Google Scholar]
- 115.Mandelli F et al. (1990) Maintenance treatment with recombinant interferon alfa-2b in patients with multiple myeloma responding to conventional induction chemotherapy. N. Engl. J. Med 322, 1430–1434 [DOI] [PubMed] [Google Scholar]
- 116.Osterborg A et al. (1993) Natural interferon-alpha in combination with melphalan/prednisone versus melphalan/prednisone in the treatment of multiple myeloma stages II and III: a randomized study from the Myeloma Group of Central Sweden. Blood 81, 1428–1434 [PubMed] [Google Scholar]
- 117.Gogas H et al. (2015) Who benefits most from adjuvant interferon treatment for melanoma? Am. J. Ther 22, 54–60 [DOI] [PubMed] [Google Scholar]
- 118.Alberts DS et al. (2006) Randomized trial of adjuvant intraperitoneal alpha-interferon in stage III ovarian cancer patients who have no evidence of disease after primary surgery and chemotherapy: an intergroup study. Gynecol. Oncol 100, 133–138 [DOI] [PubMed] [Google Scholar]
- 119.Alberts DS et al. (2008) Randomized phase 3 trial of interferon gamma-1b plus standard carboplatin/paclitaxel versus carboplatin/paclitaxel alone for first-line treatment of advanced ovarian and primary peritoneal carcinomas: results from a prospectively designed analysis of progression-free survival. Gynecol. Oncol 109, 174–181 [DOI] [PubMed] [Google Scholar]
- 120.Hall GD et al. (2004) Maintenance treatment with interferon for advanced ovarian cancer: results of the Northern and Yorkshire gynaecology group randomised phase III study. Br. J. Cancer 91, 621–626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Muss HB et al. (1984) A phase II study of recombinant alpha interferon in patients with recurrent or metastatic breast cancer. J. Clin. Oncol 2, 1012–1016 [DOI] [PubMed] [Google Scholar]
- 122.Nethersell A et al. (1984) Recombinant interferon in advanced breast cancer. Br. J. Cancer 49, 615–620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Sherwin SA et al. (1982) A multiple-dose phase I trial of recombinant leukocyte A interferon in cancer patients. J. Am. Med. Assoc 248, 2461–2466 [PubMed] [Google Scholar]
- 124.Bell J and McFadden G (2014) Viruses for tumor therapy. Cell Host Microbe 15, 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Roulois D et al. (2015) DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ruffell B et al. (2014) Macrophage IL-10 blocks CD8+ T celldependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wilson EB and Brooks DG (2011) The role of IL-10 in regulating immunity to persistent viral infections. Curr. Top. Microbiol. Immunol 350, 39–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Mumm JB et al. (2011) IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 20, 781–796 [DOI] [PubMed] [Google Scholar]
- 129.Vicari AP et al. (2002) Reversal of tumor-induced dendritic cell paralysis by CpG immunostimulatory oligonucleotide and anti-interleukin 10 receptor antibody. J. Exp. Med 196, 541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chan IH et al. (2016) PEG-rIL-10 treatment decreases FoxP3 (+) Tregs despite upregulation of intratumoral IDO. Oncoimmunology 5, e1197458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Levin D et al. (2014) Multifaceted activities of type I interferon are revealed by a receptor antagonist. Sci. Signal 7, ra50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Zhen A et al. (2017) Targeting type I interferon-mediated activation restores immune function in chronic HIV infection. J. Clin. Invest 127, 260–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cheng L et al. (2017) Blocking type I interferon signaling enhances T cell recovery and reduces HIV-1 reservoirs. J. Clin. Invest 127, 269–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Yang X et al. (2014) Targeting the tumor microenvironment with interferon-beta bridges innate and adaptive immune responses. Cancer Cell 25, 37–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Zaretsky JM et al. (2016) Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med 375, 819–829 [DOI] [PMC free article] [PubMed] [Google Scholar]