

Abstract
Phosphoinositide (PPI) lipids are a crucial class of low abundance signaling molecules that regulate many processes within cells. Methods that enable simultaneous detection of all PPI lipid species provide a wholistic snapshot of the PPI profile of cells, which is critical for probing PPI biology. Here we describe a method for the simultaneous measurement of cellular PPI levels by metabolically labeling yeast or mammalian cells with myo-3H-inositol, extracting radiolabeled glycerophosphoinositides, and separating lipid species on an anion exchange column via HPLC.
Keywords: phosphatidylinositol; phosphoinositide lipids; PtdIns; PI3P; PI4P; PI5P; PI3,5P2; PI4,5P2; PI3,4P2; PI3,4,5P3; radioactive metabolic labeling
1. Introduction
Phosphorylated phosphatidylinositol signaling lipids (PPI, also known as phosphoinositides) are a low abundance class of molecules that control a variety of signal transduction pathways and play crucial roles in cellular homeostasis. These phosphoinositides are generated by phosphorylation of phosphatidylinositol (PI) on the 3, 4, or 5 position of its inositol ring in every combination to yield a total of seven PPI species. All seven species can be detected in mammalian cell types, whereas only four are readily detectable in Saccharomyces cerevisiae. The levels of these signaling lipids are dynamically regulated in cells. For example, a dynamic change is observed during hyperosmotic shock in S. cerevisiae, which leads to a transient 20-fold elevation in phosphatidylinositol 3,5 bisphosphate (PI3,5P2) [1]. The changes in PPIs are controlled spatially by the specific localization of lipid kinases and phosphatases, and temporally via specific activation of these enzymes (Figure 1) [2–6].
Figure 1: Interconversion of PPIs by PPI kinases and phosphatases.
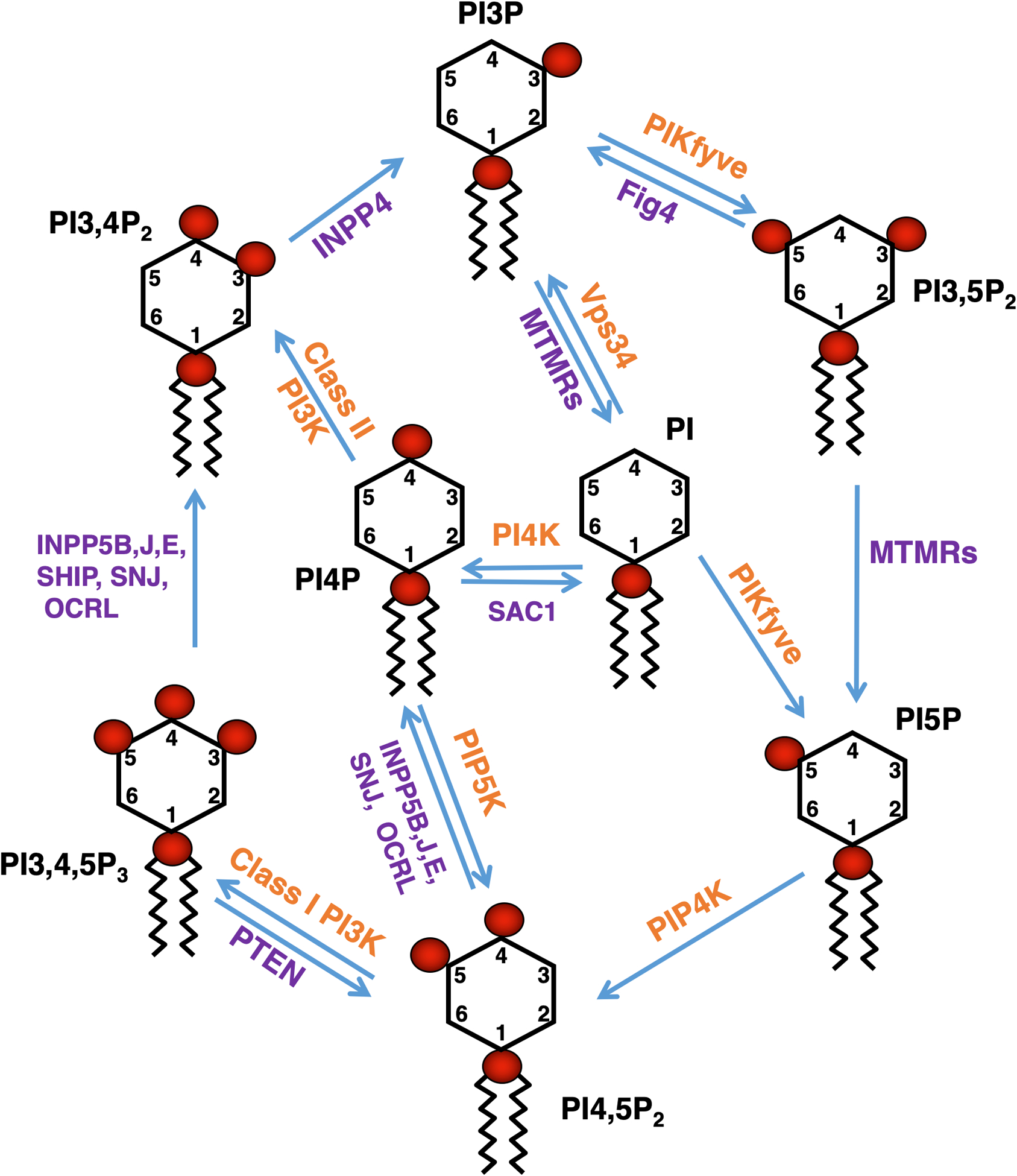
PPIs are generated by the action of phosphoinositide kinases and phosphatases, and these reactions are confined to membrane subdomains where these phosphoinositide modifying enzymes reside (reviewed in [4,2,6,3,5]). These reactions yield mono-phosphates (phosphatidylinositol-3-phosphate (PI3P), phosphatidylinositol-4-phosphate (PI4P), phosphatidylinositol-5-phosphate (PI5P)), bisphosphates (phosphatidylinositol-3,4-bisphosphate (PI3,4P2), phosphatidylinositol-3,5-bisphosphate (PI3,5P2), phosphatidylinositol-4,5-bisphosphate (PI4,5P2)) and tri-phosphate (phosphatidylinositol-3,4,5-triphosphate (PI3,4,5P3)). PI can be phosphorylated by the class III PI3-kinase Vps34 to PI3P, by type-II and type-III PI4-kinases to PI4P, or by PIKfyve to PI5P. PI3P can be phosphorylated by PIKfyve to PI3,5P2 or dephosphorylated by myotubularin-family proteins (MTMRs) to PI. PI3,5P2 can be dephosphorylated to PI5P by MTMRs. PI3,5P2 may also be dephosphorylated to PI3P by Fig4. PI4P can be phosphorylated to PI4,5P2 by PIP5-kinases. Furthermore, PI4,5P2 can be phosphorylated by Class I PI3-kinases to PI3,4,5P3. PI3,4,5P3 can be dephosphorylated to PI3,4P2 by phosphoinositide-5-phosphatases or dephosphorylated to PI4,5P2 by PTEN. PI4,5P2 can be dephosphorylated to PI4P by phosphoinositide-5-phosphatases (INPP5B, INPP5J, INPP5E, Synaptojanin, OCRL). PI4P can be phosphorylated to PI3,4P2 by Class-II PI3-kinases or dephosphorylated to PI by SAC1. PI3,4P2 can be dephosphorylated to PI3P by INPP4 proteins. PI5P can be phosphorylated to PI4,5P2 by PIP4-kinases. Red circles represent phosphate groups. Hydroxyl groups on the inositol ring as well as the ester linkages for the fatty acyl chains are not indicated.
PPI lipids regulate various signaling pathways and various signaling pathways in turn regulate these lipids. Thus, it is important to measure the levels of these lipids. One established method is to use protein-based probes that bind to a specific PPI species. A major advantage of these PPI bioprobes is that they reveal the subcellular localization of the PPI lipid, unlike the metabolic labeling approach described here. However, there are not highly specific bioprobes for every PPI. In addition, there is a concern that an efficient bioprobe would mask the phosphoinositide head group and potentially compete with endogenous PPI effectors. Another concern is that some bioprobes may require both a PPI lipid and specific protein or cellular context. For example, FYVE domains, protein domains that specifically bind PI3P, from Fab1 and EEA1 exhibit localizations dissimilar from each other [7].
PPIs exist in a substrate/product relationship with other PPIs and thus the levels of selected PPI species are linked. For example, during hyperosmotic shock, PI3,5P2 sharply increases by 20-fold [1]. This change in PI3,5P2 also results in a concomitant decrease in the levels of its precursor PI3P. For this reason, changes in a cell’s behavior are likely due to changes in more than one PPI. Moreover, some PPI kinases and phosphatases act on multiple PPI substrates [8]. Thus, drug treatments or knockouts that affect one PPI kinase or phosphatase have the potential to affect the levels of multiple PPIs. Therefore, the ability to simultaneously measure all PPI species within cells provides a critical snapshot of how specific stimuli or treatments impact each PPI lipid. Yet, accurately measuring the levels of these PPIs simultaneously has proven difficult.
PPI lipids were first identified from a phosphatide fraction known as cephalin, defined by their solubility in ether and insolubility in ethyl alcohol [9,10]. These were first separated from phosphatidylethanolamine and phosphatidylserine using a method that took advantage of the lower solubility of PPIs in methanol. Since then, optimization of protocols to extract and detect PPI species has culminated in the current method of metabolic labeling and simultaneous detection of all PPIs by separation on an anion exchange column.
The method described in this chapter starts with metabolic labeling with myo-3H-inositol. Ortho(32P)-phosphate can be used instead [11]. While use of ortho(32P)-phosphate greatly increases sensitivity to specific molecules, in comparison with myo-3H-inositol, there is a much larger cohort of phosphate containing species that co-purify with PPI. Following metabolic labeling, cells are lysed by precipitating all cellular macromolecules using perchloric acid [12,13]. The precipitated material, including phospholipids, is deacylated by mild base hydrolysis using mono-methylamine (Figure 2) [14]. The deacylation reaction generates the corresponding phosphorylated glycero-3-phospho-1-inositol and fatty acid amides. Following deacylation, methylamine is evaporated under vacuum. The phosphorylated glycero-inositol head groups are solubilized in water. Phosphorylated glycero-inositols are further isolated from fatty acid amides and other contaminants by phase separation with butanol/ethyl ether/formic acid. The phosphorylated glycero-inositol fraction is present in the aqueous phase, while fatty acid amides are in the organic phase [14]. This extraction method is distinct from extraction from membranes using acidified chloroform/methanol/HCl [15,16]. In our hands, using the perchloric acid precipitation protocol described here results in detection of higher quantities of PPIs [13]. The resultant phosphorylated glycero-inositol fraction is then separated on an anion exchange column and detected with an in-line flow-through scintillation counter. Anion exchange on an HPLC using ammonium phosphate dibasic was first used to identify PI3P in fibroblasts [17]. The elution gradient described here for yeast was developed to separate the four PPI species in S. cerevisiae [18,19]. A few different gradients are used to separate PPIs in mammalian cells [16]. In addition, either one or two anion exchange columns are used to separate phosphorylated glycero-inositol species [20,21]. Better resolution between PI4P and PI5P peaks is achieved when two anion exchange columns are used in tandem, although utilizing two-columns is more expensive and requires more sample.
Figure 2: The deacylation reaction of PPIs to glycerophosphoinositides.
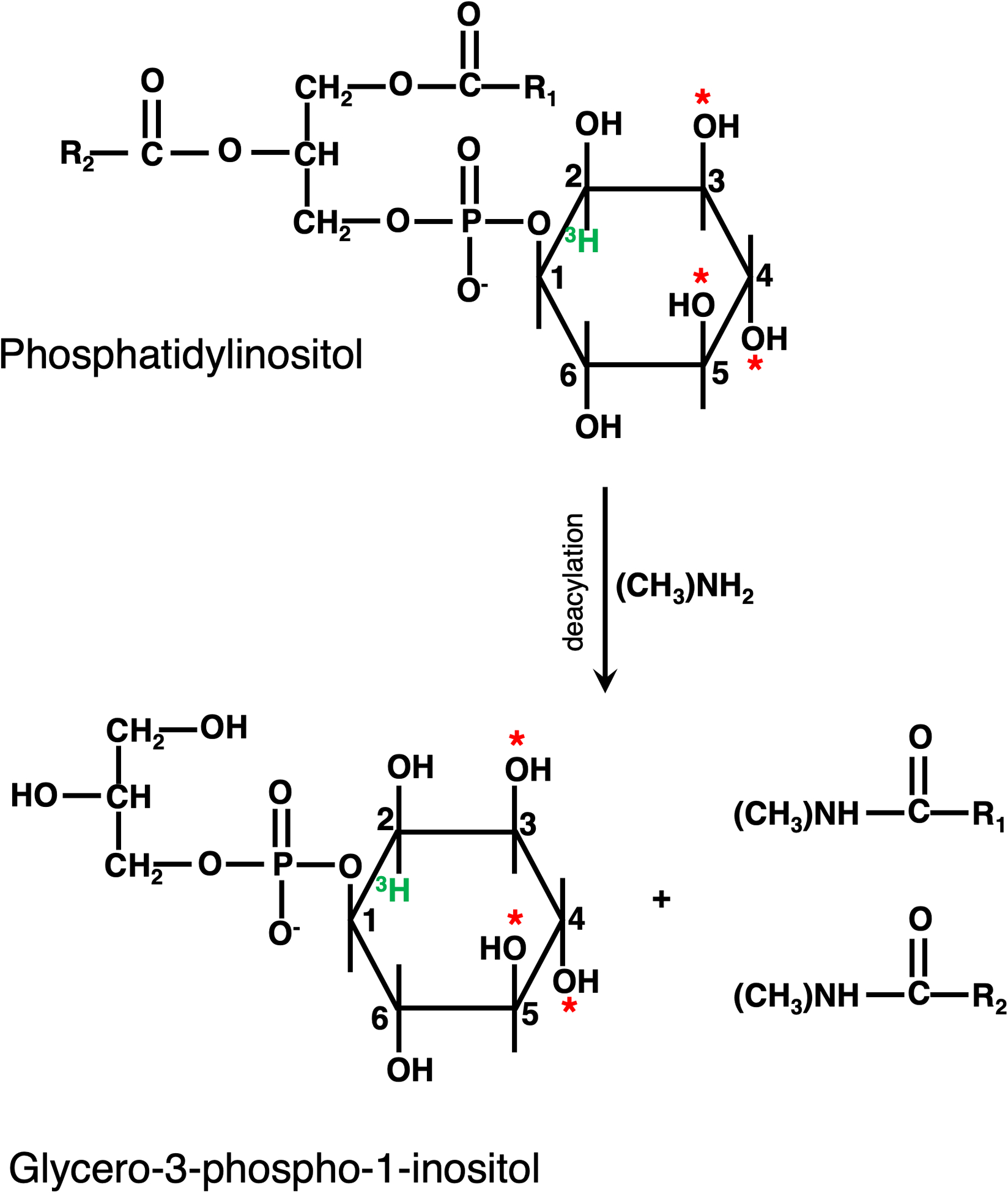
Mild base hydrolysis via methylamine results in the cleavage of the fatty acid chains from the glycero-3-phospho-1-inositol backbone. The resultant phosphorylated glycerol phosphoinositides, can be separated on an anion exchange column. * denotes phosphorylation sites.
New protocols are being developed to measure phospholipids using high-performance liquid chromatography-mass spectrometry [22]. While this new approach can determine the acyl chain structure of PPIs, this method fails to distinguish among monophosphorylated PPIs (PI3P vs PI4P vs PI5P) and among bis-phosphorylated PPIs (PI3,5P2 vs PI4,5P2 vs PI3,4P2).
The method below describes the metabolic labeling and measurement of PPI lipids in S. cerevisiae and some types of mammalian cells. This protocol has been modified for use in other organisms [23,24]. This protocol is a culmination of over 60 years of chemistry and biology research in multiple laboratories and provides a comprehensive snapshot of the PPI lipids in cells.
2. Materials
2.1. General system requirements
Tabletop microcentrifuge
Benchtop centrifuge
Mini-beadbeater (BioSpec Products) (yeast protocol only)
Samco Scientific 233 disposable plastic transfer pipettes with a fine tip (yeast protocol only)
Heating block
Speedvac concentrator
HPLC and flow scintillation system (see Note 1)
4.6 × 250 mm SAX cartridge anion exchange column and anion guard cartridge (Macmod Analytical) (see Notes 2–4). Other anion exchange columns may be appropriate.
Institutional safety approval (see Note 5)
2.2. Materials common to all protocols
myo-3H-inositol (PerkinElmer) dissolved in ddH2O with proprietary stabilizer PT6–271.
4.5% perchloric acid diluted from a 70% stock in ddH2O (see Note 6). Made fresh for each experiment.
100 mM EDTA pH 8.0
Methylamine reagent: Mix methylamine (Alfa Aesar (see Note 7)), methanol, n-butanol and ddH2O to a final concentration of 10.7% methylamine, 45.7% methanol, 11.4% n-butanol. Made fresh for each experiment.
Butanol/ethyl ether/formic acid: Mix n-butanol, ethyl ether, and formic acid in a 20:4:1 ratio. Made fresh for each experiment.
1 M (NH4)2HPO4, pH 3.8 (adjust the pH with o-phosphoric acid)
Liquid scintillation fluid FlowLogic HS (Lab Logic) for high salt samples is used in our lab. Other scintillation liquids may be appropriate.
2.3. Materials specific for Saccharomyces cerevisiae protocol
10x Mg2+, Ca2+, NaCl salts: Prepare 41.5 mM magnesium sulfate, 9.0 mM calcium chloride and 17.1 mM sodium chloride in 500 mL ddH2O and autoclave.
10x potassium salts: Prepare 66.1 mM potassium phosphate monobasic and 8.6 mM potassium phosphate dibasic in ddH2O and autoclave.
10x amino acid stock solution: For 500 mL, use the following mixture of amino acids: 0.1 g L-adenine, 0.1 g L-uracil*, 0.1 g L-histidine-HCL*, 0.1g L-arginine-HCL, 0.1 g L-methionine, 0.15 g L-tyrosine, 0.15 g L-leucine*, 0.15 g L-isoleucine, 0.15 g L-lysine-HCL, 0.25 g L-phenylalanine, 0.5 g L-glutamic acid, 0.5 g L-aspartic acid, 0.750 g L-valine, 2.0 g L-serine, 0.15 g L-cysteine, 0.15 g L-proline. Amino acids are dissolved in 500 mL ddH2O and filter sterilized. To create selective media, the amino acids that are indicated in the recipe with * can be omitted.
100x trace elements: Mix 25 mg boric acid, 2 mg cupric sulfate, 5 mg potassium iodide, 10 mg ferrous sulfate, 20 mg manganese sulfate, 10 mg sodium molybdate, and 20 mg zinc sulfate in 500 mL ddH2O and autoclave.
10x ammonium sulfate: Prepare 378 mM ammonium sulfate in ddH2O and filter sterilize.
40% glucose: Prepare 40% glucose in ddH2O and autoclave.
50x Trp/Thr solution: Mix 10 g threonine and 1 g tryptophan in 500 mL ddH2O and filter sterilize. Store at 4°C.
100x vitamins: Mix 1 mg D-biotin, 100 mg D-pantothenic acid-calcium salt, 0.1 mg folic acid, 20 mg niacinamide, 10 mg p-aminobenzoic acid, 20 mg pyridoxine hydrochloride, 10 mg riboflavin, and 20 mg thiamine hydrochloride in 500 mL ddH2O. Filter sterilize and store at 4°C.
Inositol-free growth media: For 500 mL, mix stock solutions: 50 mL 10x Mg2+, Ca2+, NaCl salts solution, 50 mL 10x potassium salts, 50 mL 10x amino acid stock solution, 5 mL 100x trace elements solution, 50 mL 10x ammonium sulfate solution, 25 mL 40% glucose solution, 10 mL 50x Trp/Thr Solution, 5 mL 100x vitamins solution, and 255 mL ddH2O. Filter sterilize and store at 4°C.
1.8 M NaCl solution (use to induce hyperosmotic shock)
2.4. Materials specific for mammalian cells
Custom Dulbecco’s Modified Eagle Medium (DMEM): Custom made DMEM as the base medium and modified to omit myo-inositol and sodium pyruvate.
Custom Neurobasal-A medium: Custom made Neurobasal-A as the base and modified to omit L-aspartic acid, L-glutamic acid, myo-inositol and L-glutamine.
Phosphate buffered saline (PBS) pH 7.4
Dialyzed fetal bovine serum
1 M HEPES pH: 7.2 to 7.5
Apo-transferrin: Freshly prepare 4 mg/ml of apo-transferrin and filter sterilize before adding to the media.
Insulin
Penicillin-Streptomycin-Glutamine
B27
L-Glutamine
DMEM -inositol labeling medium: inositol-free DMEM with 10 μCi/mL of myo-3H-inositol, 10% dialyzed fetal bovine serum, 20 mM HEPES, 5 μg/mL transferrin, 5 μg/mL insulin, and 1 x penicillin/streptomycin/glutamine.
Neurobasal-A -inositol labeling medium: custom Neurobasal-A medium with 50 μCi/mL myo-3H-inositol, B27, and L-glutamate.
-
Plate the cells and prepare the inositol medium as indicated:
MEF cells: Grow MEF cells on 10 cm dishes to 50–60% confluency. Use one dish per sample and prepare 6 mL of DMEM -inositol labeling medium per dish.
Macrophage: Grow RAW macrophage cells on 35 mm dishes to 20–30% confluency. Use two dishes per sample and prepare 2 mL of DMEM -inositol labeling medium per dish.
Primary neuron culture: Cultured primary rat hippocampal neurons are grown for 21 days on 35 mm dishes. Plate 600,000 cells per 35 mm dish and use two dishes per sample and prepare 2 mL of Neurobasal-A -inositol labeling medium per dish.
3. Methods
While the details of the yeast and mammalian protocols differ, both protocols follow the same general steps: metabolic labeling cells with myo-3H-inositol, harvesting cells followed by addition of perchloric acid to precipitate all macromolecules, mild-base hydrolysis to release and extract phosphorylated glycero-inositol head groups from the PPI lipids, and separation by anion exchange chromatography via HPLC.
3.1. Yeast Protocol
3.1.1. Preparing cells for myo-3H-inositol labeling
-
1
Grow a 10 mL starter culture in appropriate media in a shaker at 24°C, at 200 rpm. Cultures need to reach mid-log phase (5×106 cells/mL) before they are inoculated into myo-3H-inositol-containing media.
-
2
Pellet cells at room temperature at 1,700 × g for 3 min. Wash the cell pellet in 4 mL inositol-free media and pellet again.
-
3
Inoculate 50 mL disposable centrifuge tubes with an appropriate amount of each yeast strain and grow at 24°C, at 200 rpm. In addition, to track the growth of the 3H-containing culture, grow a parallel culture in another 50 mL disposable centrifuge tube containing 5 mL inositol-free media without myo-3H-inositol. Enough cells of the starter culture should be added so that cultures reach mid-log phase (5 × 106 cells/mL) 16 hours following inoculation (see Note 8).
-
4
Following inoculation, add 50 μL of 1 μCi/μL myo-3H-inositol to the appropriate cultures. Loosely tape the lid of the 50 mL disposable centrifuge tube to allow for airflow. Lids are required to prevent aerosol contamination. Allow cells to grow for 16 hours in myo-3H-inositol at 24°C, at 200 rpm.
3.1.2. Cell harvesting and lysis
-
5
Prepare a 2.0 mL screw cap tube for each sample by filling half of it with 0.5 mm zirconia beads and adding 470 μL of 4.5% perchloric acid. Chill the screw cap tubes and the remaining 4.5% perchloric acid on ice.
-
6
At the 16-hour time point, measure the OD600 of the parallel cultures that were grown in inositol-free media without myo-3H-inositol.
-
7
Pellet the cells incubated with myo-3H-inositol at 1,700 × g for 3 min at room temperature.
-
8
Resuspend the cell pellet in 4 mL inositol-free media and pellet the cells at 1,700 × g for 3 min.
-
9
Resuspend the cell pellet in 100 μL inositol-free media and transfer to a microcentrifuge tube.
-
10
To perform hyperosmotic shock, add 100 μL 1.8 M NaCl solution to the samples. Incubate cells for the appropriate time interval. For control cells not undergoing hyperosmotic shock, add 100 μL inositol-free media.
-
11
Add 800 μL ice cold 4.5% perchloric acid and incubate on ice until all samples are ready.
-
12
Transfer the resultant perchloric acid mixture to the prepared 2 mL screw cap tubes containing zirconia beads.
-
13
Lyse the cells by placing each tube into a mini-beadbeater and shaking for 2 min at high speed. Immediately place the cells on ice for 2 min. Repeat this process two more times for a total lysis time of 6 min.
-
14
Using fine tip disposable plastic transfer pipettes, transfer the lysed cells to new microcentrifuge tubes. Make sure to avoid the beads. Centrifuge the lysate at 16,000 × g for 10 min at room temperature.
3.1.3. Deacylation of PPI lipids
-
15
Resuspend each pellet with 1 mL 100 mM EDTA. Ensure that all the cell precipitate is resuspended, including precipitate on the side of the microcentrifuge tube.
-
16
Pellet the cells at 16,000 × g for 10 min at room temperature.
-
17
Resuspend the pellet in 50 μL ddH2O and transfer to a 5 mL glass vial. It is critical to use glass vials with a rubber-lined cap that can make a tight seal.
-
18
Wash the microcentrifuge tube with 1 mL methylamine reagent and add it to the appropriate 5 mL glass vial. Heat the samples at 55°C for 1 hour.
-
19
Allow the tubes to cool for 5 min and transfer to microcentrifuge tubes. Dry the samples in a speedvac concentrator at 55°C for four hours under vacuum.
-
20
Dried samples can be stored at −20°C until the next day. If samples have been stored in −20°C, thaw the samples on the bench for 10 min before the next step.
3.1.4. Butanol/ethyl ether/formic acid extraction
-
21
Resuspend each sample with 300 μL ddH2O and leave at room temperature for 30 min.
-
22
Mix each sample again and vortex for about 30 seconds. Centrifuge at 16,000 × g for 2 min at room temperature.
-
23
Transfer supernatant to a new microcentrifuge tube and add 300 μL butanol/ethyl ether/formic acid solution. Vortex each tube for 15 seconds and centrifuge the samples at 16,000 × g for 2 min at room temperature.
-
24
Transfer the bottom layer (aqueous phase) to a new tube. Add another 300 μL butanol/ethyl ether/formic acid solution. Repeat vortex and centrifugation steps as above.
-
25
Transfer the bottom layer (aqueous phase) to a new tube.
-
26
Dry the sample in a speedvac concentrator at 55°C for four hours under vacuum.
-
27
Samples can be stored at −20°C until loading on HPLC or the protocol can be continued. Samples should be stable at −20°C for several months.
3.1.5. HPLC sample preparation and anion exchange
-
28
Thaw the samples at room temperature for about 5 min and resuspend the sample in 60 μL ddH2O. Incubate the sample at room temperature for 5 min. Centrifuge the sample at 16,000 × g for 5 min to pellet any particulate matter.
-
29
Load 55 μL into the autosampler vial inserts, avoiding any particulate matter at the bottom of the tube (see Note 9).
-
30
Inject 50 μL of sample into the HPLC and separate on a SAX anion exchange column. Buffer A (ddH2O) and Buffer B (1 M (NH4)2HPO4, pH 3.8) are used to generate the following gradients run at a 1 mL/min flow rate: 1% Buffer B for 5 min, 1–20% Buffer B for 44 min, 20–50% Buffer B for 3.75 min, and 50% Buffer B for 8 min. At the end of the run, re-equilibrate the column with ddH2O at 1mL/min for 15 min. Radiolabeled eluate can be detected by an inline flow scintillation analyzer. A 1:2 ratio of eluate to scintillant is used with a 3 mL/min flow rate. Fractions are analyzed by binning counts every 6 seconds. The data is analyzed by Laura-4 software.
-
31
To quantify scintillation counts from each sample, the raw counts in each peak are expressed as a percentage of total phosphatidylinositol-related species, calculated from summation of the counts of the five glycero-inositol peaks present in yeast (PI, PI3P, PI4P, PI3,5P2, and PI4,5P2). Background scintillation counts are calculated from adjacent regions and subtracted from all peaks. Figure 3 illustrates a typical yeast elution profile. The average PPI levels in the SEY6210 yeast strain background are presented in Table 1. It should be noted that PPI levels may vary between different wild-type yeast strains.
Figure 3: Example of anion exchange chromatography of a yeast sample.
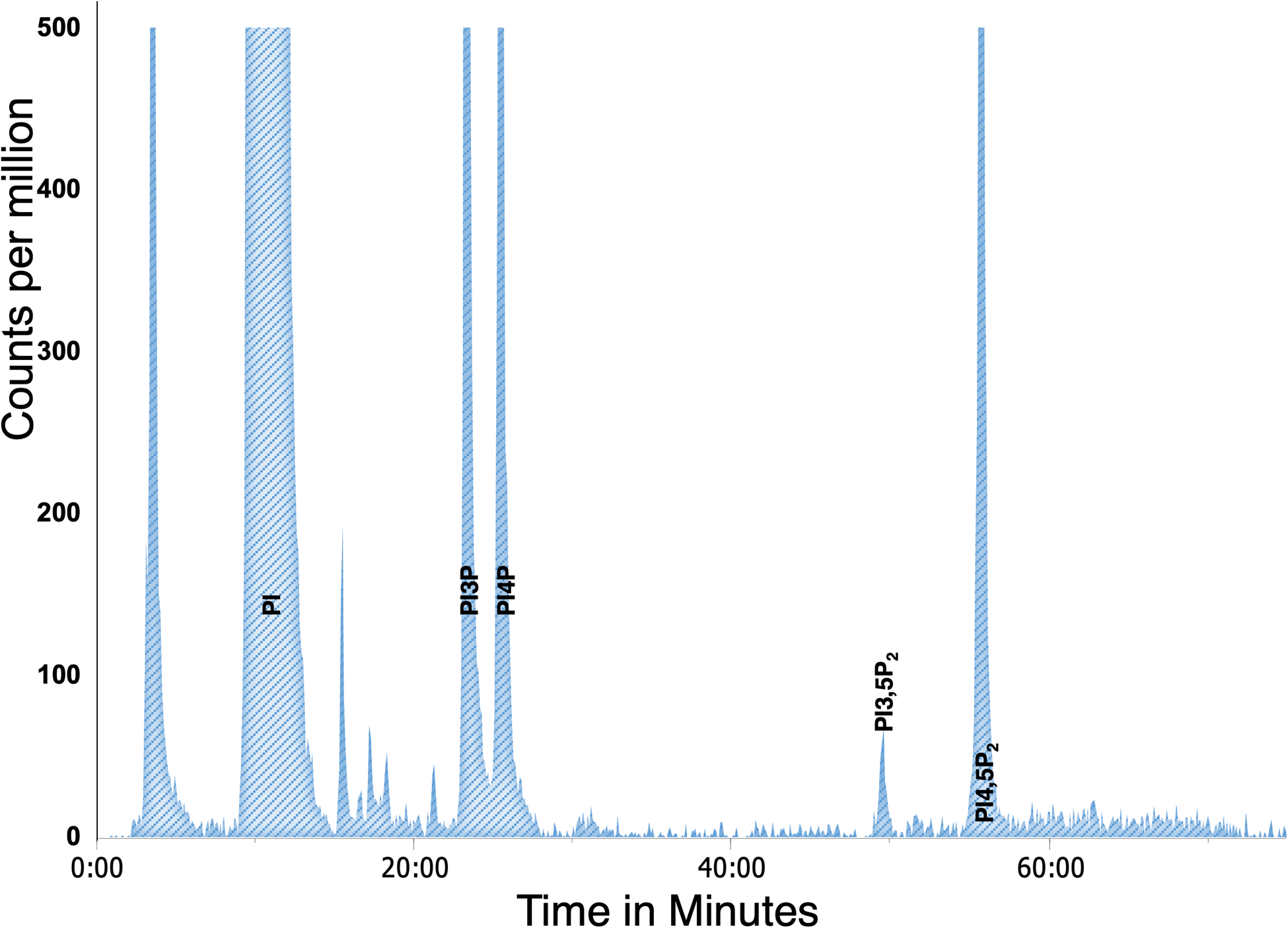
Peaks were truncated at 500 cpm to display all the PPI detected.
Table 1: Levels of PPIs in yeast determined by myo-3H-inositol labelling.
PPI levels were measured in seven wild-type yeast samples (SEY 6210).
| Yeast | |
|---|---|
| PI | 93.45 ± 0.45 |
| PI3P | 2.66 ± 0.17 |
| PI4P | 2.07 ± 0.16 |
| PI5P | Not detectable |
| PI3,5P2 | 0.12 ± 0.01 |
| PI3,4P2 | Not detectable |
| PI4,5P2 | 1.7 ± 0.13 |
| PI3,4,5P3 | Not detectable |
3.2. Mammalian cell protocol
3.2.1. Preparing cells for myo-3H-inositol labeling
-
1
Grow cells to appropriate confluence, which needs to be determined empirically. All cells are grown at 37°C and 5% CO2.
-
2
For all cells described except primary neurons, rinse twice with PBS and incubate for 48 hours with DMEM -inositol labeling medium. For primary neurons, rinse with inositol-free Neurobasal-A medium and incubate with Neurobasal-A -inositol labeling medium for 24 hours.
-
3
To test the effects of small molecule inhibitors or growth factors on PPI levels, cells are treated for an appropriate time during labeling such that the total metabolic labeling time is 48 hours for MEF and macrophages, and 24 hours for primary cortical neurons.
-
4
To test the effect of depleting proteins on PPI levels, cells are infected with lentivirus expressing shRNA to the protein of interest. One day after infection, cells are selected with appropriate antibiotics. After two days of selection, cells are split and plated with an appropriate amount of cells to achieve 50–60% confluency 24 hours later. MEF cells are then washed and incubated with inositol labeling medium for 48 hours. shRNA depletion followed by metabolic labeling has not yet been tested in macrophages or primary neurons.
3.2.2. Cell harvesting and lysis
-
5
Discard growth media and rinse cells two times at room temperature with 5 mL PBS. Neuron cultures are washed with PBS containing 1 mM MgCl2 and 0.1 mM CaCl2.
-
6
Precipitate cells by adding 1 mL of 4.5% perchloric acid. Incubate for 15 min at room temperature, gently rotating plates by hand every 2 min to prevent cells from drying.
-
7
Remove cells from the bottom of the dish using a hard-plastic scraper and transfer to microcentrifuge tubes. Centrifuge at 16,000 × g for 2 min at room temperature.
3.2.3. Deacylation of PPI lipids
-
8
Remove and discard supernatant. Rinse pellet with 1 mL 100 mM EDTA and vortex for 15 seconds. Note that the pellet may not break completely.
-
9
Centrifuge at 16,000 × g for 2 min at room temperature and discard the supernatant.
-
10
Dislodge the pellet in 50 μl of ddH2O. Add 1 mL methylamine reagent to each tube and dissolve the pellet by pipetting. Transfer the contents to a 5 mL glass vial. It is critical to use glass vials with a rubber-lined cap that can make a tight seal.
-
11
Heat the samples at 55°C for 1 hour.
-
12
Allow the glass vials to cool for 5 min and transfer to microcentrifuge tubes. Dry the samples using a speedvac concentrator at 55°C for four hours under vacuum.
-
13
Dried samples can be stored at −20°C until the next day. If samples have been stored at - 20°C, thaw the samples on the bench for 10 min before the next step.
3.2.4. Butanol/ethyl ether/formic acid extraction
-
14
Resuspend each sample in 500 μL ddH2O. Sample will not be fully dissolved. Use a pipette tip to dislodge the pellet and vortex each sample for about 30 seconds. Leave the samples at room temperature for 30 min to let the pellet dissolve.
-
15
Centrifuge at 16,000 × g for 5 min at room temperature.
-
16
Transfer supernatant to a new tube. Add 500 μL butanol/ethyl ether/formic acid solution and vortex each tube for 15 seconds. Centrifuge the samples at 16,000 × g for 5 min.
-
17
Transfer the bottom layer (aqueous phase) to a new microcentrifuge tube.
-
18
Add another aliquot of the 500 μL butanol/ethyl ether/formic acid solution and repeat vortex and centrifugation steps.
-
19
Transfer the bottom layer (aqueous phase) to a new microcentrifuge tube.
-
20
Dry samples using a speedvac concentrator at 55°C for four hours under vacuum.
-
21
Samples can be stored at −20°C prior to anion exchange chromatography by HPLC or the protocol can be continued. Samples should be stable at −20°C for several months.
3.2.5. HPLC sample preparation and anion exchange
-
22
Thaw the samples at room temperature for about 10 min and resuspend each sample in 60–75 μL ddH2O. Vortex and centrifuge the sample at 16,000 × g for 10 min to pellet any particulate matter.
-
23
Load 55 μL into the autosampler vial inserts, avoiding any particulate matter at the bottom of the tube (see Note 9 and 10).
-
24
Inject 50 μL of sample into the HPLC and separate on a SAX anion exchange column. Buffer A (ddH2O) and Buffer B (1 M (NH4)2HPO4, pH 3.8) are used to generate the following gradients run at a 1 mL/min flow rate: 0% Buffer B for 5 min, 0–2% Buffer B for 15 min, 2% Buffer B for 80 min, 2–10% Buffer B for 20 min, 10% Buffer B for 65 min, 10–80% Buffer B for 40 min, 80% Buffer B for 20 min, 80–100% Buffer B for 5 min. At the end of the run, re-equilibrate the column with ddH2O at 1mL/min for 15 min. Radiolabeled eluate can be detected by an inline flow scintillation analyzer. A 1:2 ratio of eluate to scintillant is used with a 3 mL/min flow rate. Fractions are analyzed by binning counts every 6 seconds. The data is analyzed by Laura-4 software.
-
25
To quantify scintillation counts from each sample, the raw counts in each peak are expressed as a percentage of total phosphatidylinositol, calculated from summation of the counts of the eight glycero-inositol peaks (PI, PI3P, PI4P, PI5P, PI3,5P2, PI4,5P2, PI3,4P2, and PI3,4,5P3). Background scintillation counts are calculated from adjacent regions and subtracted from all peaks. Figure 4 illustrates a typical mammalian cell elution profile. The average PPI levels in three mammalian cell types are presented in Table 2.
Figure 4: Example of anion exchange chromatography of a mammalian (MEF) sample.
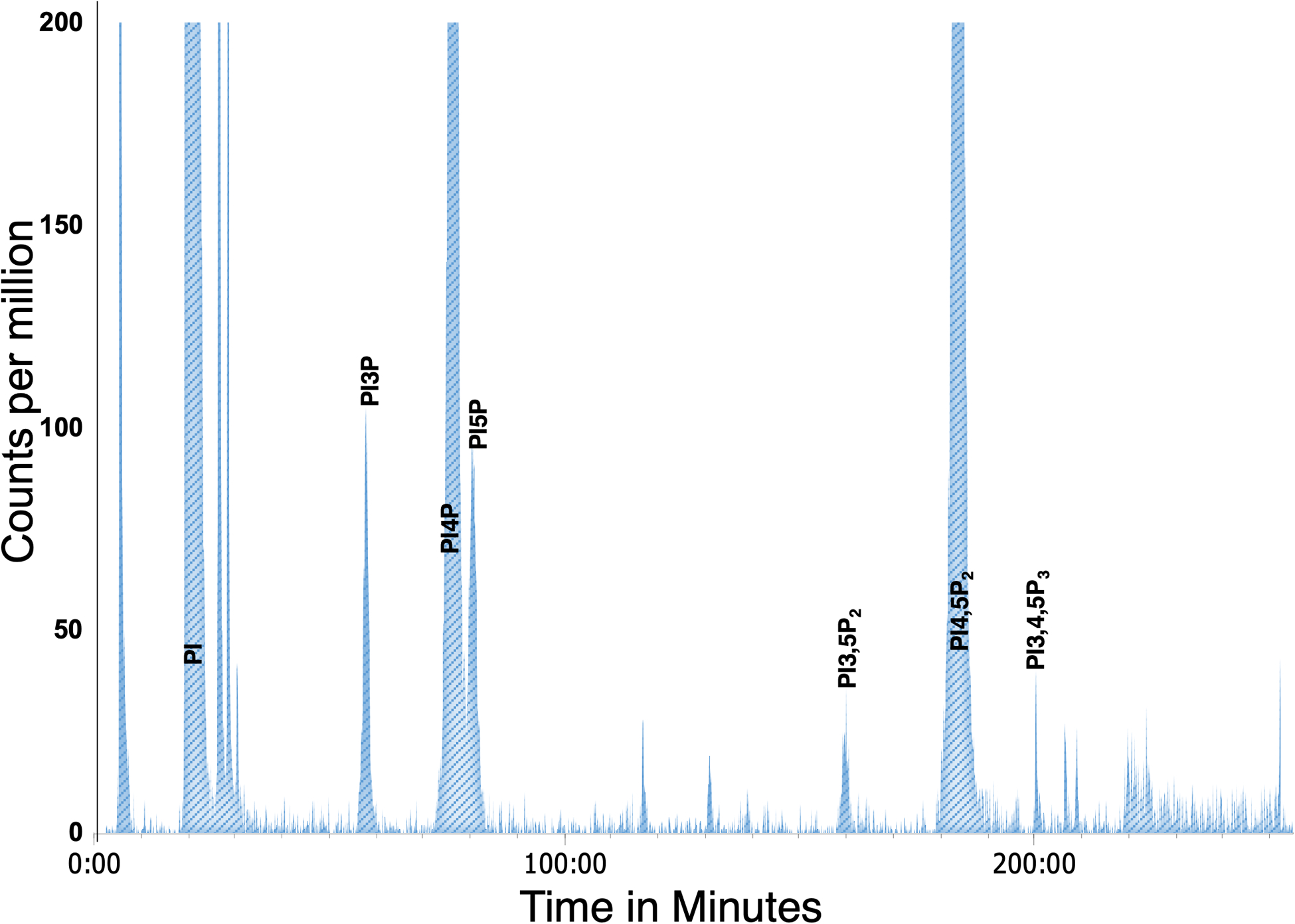
Peaks were truncated at 200 cpm to display all the PPI detected.
Table 2: Levels of PPIs in mammalian cells determined by myo-3H-inositol labelling.
These values were derived from eleven samples from neurons, five samples from mouse embryonic fibroblasts (MEF), and three samples from macrophages [25,26]. Each box is shaded according to the levels of each lipid observed in MEF: less than −50% of MEF - dark blue; 50% to 10% of MEF levels - light blue; within 10% of MEF levels - white; 10% to 50% higher than MEF levels - light red; more than 50% higher than MEF levels - dark red.

|
4. Notes
While other HLPC and flow scintillation systems may be appropriate, our lab uses the Prominence UFLCXR liquid chromatograph (Shimadzu), with SIL-20A auto sampler (Shimadzu) and Beta-RAM model 5 (LabLogic) flow detector. This system requires autosampler vials and vial inserts (Shimadzu). Our lab uses Laura-4 software (LabLogic) for controlling the radiochromatography system.
To protect the anion exchange column, change the guard column and wash the column after running 10 yeast samples or 4 mammalian samples. Each time the guard column is changed, the column is washed at a rate of 1 ml/min for 2 hours with ddH2O warmed to 40°C through all inlet lines. After the wash is completed run a mock sample (water), bypassing the inline flow scintillation analyzer, and using the gradient described for yeast samples, followed by re-equilibration of the column with ddH2O at 1mL/min for 15 min.
Pressure builds up over repeated usage of the column. Replacement of the column is recommended when the pressure of the column is no longer reduced after washing or when the peak separation between PI4P and PI5P is poor. Reversing the orientation of an older column will also help to relieve the column pressure and improve peak separation.
When the HPLC is not in use, the guard column is changed, washed as described above and left in ddH2O.
Radiation safety approval from the institution and proper facilities to monitor radioactive contamination and safely dispose of radioactive waste are necessary before planning this experiment.
Perchloric acid must be stored in a designated acid cabinet because of its strong oxidizing properties. Radioactive samples containing perchloric acid must be disposed separately from the rest of the radioactive waste and neutralized before disposal using sodium carbonate.
Note that our lab has been less successful in deacylation of phosphoinositides with methylamine from other companies.
Yeast tend to grow more slowly in inositol-free media compared to synthetic dropout media so doubling times in inositol-free media should be determined. In a wild-type SEY6210 background, inoculation of 1 × 106 cells will yield a culture with a density of approximately 5 × 106 cells/mL 16 hours after inoculation.
While this protocol should yield an appropriate amount of radioactive glycerol phosphoinositides, if there is concern that a sample does not contain sufficient radioactive material, prior to analyzing the sample on the HPLC, take 2 μL of sample resuspended in ddH2O and perform a scintillation count on the sample using FlowLogic HS scintillant. To obtain good accuracy of each PPI peak, the final sum of the peaks should total 1 to 2 million cpm with not more than 3 million cpm.
In mammalian cells, if PI5P and PI4P are not separated into discrete peaks, loading less sample may help to achieve the separation. On the other hand, if the PI3,5P2 peak is not visible above background signal, loading more sample may help detection of PI3,5P2. Peak separation varies between different batches of the anion exchange column. Individual columns should be tested empirically.
Acknowledgements:
This work was supported by NIH grants R01-NS099340-03 and R01-NS064015-09 to LSW, and LSI Cubed to NS and SSPG. NS was supported in part by NIH T-32-GM007315.
References:
- 1.Duex JE, Nau JJ, Kauffman EJ, Weisman LS (2006) Phosphoinositide 5-phosphatase Fig 4p is required for both acute rise and subsequent fall in stress-induced phosphatidylinositol 3,5-bisphosphate levels. Eukaryot Cell 5 (4):723–731. doi: 10.1128/EC.5.4.723-731.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Balla T (2013) Phosphoinositides: tiny lipids with giant impact on cell regulation. Physiol Rev 93 (3):1019–1137. doi: 10.1152/physrev.00028.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dickson EJ, Hille B (2019) Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem J 476 (1):1–23. doi: 10.1042/BCJ20180022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sasaki T, Takasuga S, Sasaki J, Kofuji S, Eguchi S, Yamazaki M, Suzuki A (2009) Mammalian phosphoinositide kinases and phosphatases. Prog Lipid Res 48 (6):307–343. doi: 10.1016/j.plipres.2009.06.001 [DOI] [PubMed] [Google Scholar]
- 5.Shisheva A, Sbrissa D, Ikonomov O (2015) Plentiful PtdIns5P from scanty PtdIns(3,5)P2 or from ample PtdIns? PIKfyve-dependent models: Evidence and speculation (response to: DOI 10.1002/bies.201300012). Bioessays 37 (3):267–277. doi: 10.1002/bies.201400129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wallroth A, Haucke V (2018) Phosphoinositide conversion in endocytosis and the endolysosomal system. J Biol Chem 293 (5):1526–1535. doi: 10.1074/jbc.R117.000629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Botelho RJ, Efe JA, Teis D, Emr SD (2008) Assembly of a Fab1 phosphoinositide kinase signaling complex requires the Fig4 phosphoinositide phosphatase. Mol Biol Cell 19 (10):4273–4286. doi: 10.1091/mbc.E08-04-0405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malek M, Kielkowska A, Chessa T, Anderson KE, Barneda D, Pir P, Nakanishi H, Eguchi S, Koizumi A, Sasaki J, Juvin V, Kiselev VY, Niewczas I, Gray A, Valayer A, Spensberger D, Imbert M, Felisbino S, Habuchi T, Beinke S, Cosulich S, Le Novere N, Sasaki T, Clark J, Hawkins PT, Stephens LR (2017) PTEN Regulates PI(3,4)P2 Signaling Downstream of Class I PI3K. Mol Cell 68 (3):566–580 e510. doi: 10.1016/j.molcel.2017.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Folch J (1949) Brain diphosphoninositide, a new phosphatide having inositol metadiphosphate as a constituent. J Biol Chem 177 (2):505–519 [PubMed] [Google Scholar]
- 10.Folch J (1949) Complete fractionation of brain cephalin; isolation from it of phosphatidyl serine, phosphatidyl ethanolamine, and diphosphoinositide. J Biol Chem 177 (2):497–504 [PubMed] [Google Scholar]
- 11.Agranoff BW, Murthy P, Seguin EB (1983) Thrombin-induced phosphodiesteratic cleavage of phosphatidylinositol bisphosphate in human platelets. J Biol Chem 258 (4):2076–2078 [PubMed] [Google Scholar]
- 12.Whiteford CC, Best C, Kazlauskas A, Ulug ET (1996) D-3 phosphoinositide metabolism in cells treated with platelet-derived growth factor. Biochem J 319 (Pt 3):851–860. doi: 10.1042/bj3190851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS (2002) Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14p, an activator of the lipid kinase Fab1p. J Cell Biol 156 (6):1015–1028. doi: 10.1083/jcb.200201002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clarke NG, Dawson RM (1981) Alkaline O leads to N-transacylation. A new method for the quantitative deacylation of phospholipids. Biochem J 195 (1):301–306. doi: 10.1042/bj1950301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bligh EG, Dyer WJ (1959) A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37 (8):911–917. doi: 10.1139/o59-099 [DOI] [PubMed] [Google Scholar]
- 16.Hale AT, Clarke BP, York JD (2020) Metabolic Labeling of Inositol Phosphates and Phosphatidylinositols in Yeast and Mammalian Cells. Methods Mol Biol 2091:83–92. doi: 10.1007/978-1-0716-0167-9_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Whitman M, Downes CP, Keeler M, Keller T, Cantley L (1988) Type I phosphatidylinositol kinase makes a novel inositol phospholipid, phosphatidylinositol-3-phosphate. Nature 332 (6165):644–646. doi: 10.1038/332644a0 [DOI] [PubMed] [Google Scholar]
- 18.Stack JH, DeWald DB, Takegawa K, Emr SD (1995) Vesicle-mediated protein transport: regulatory interactions between the Vps15 protein kinase and the Vps34 PtdIns 3-kinase essential for protein sorting to the vacuole in yeast. J Cell Biol 129 (2):321–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schu PV, Takegawa K, Fry MJ, Stack JH, Waterfield MD, Emr SD (1993) Phosphatidylinositol 3-kinase encoded by yeast VPS34 gene essential for protein sorting. Science 260 (5104):88–91 [DOI] [PubMed] [Google Scholar]
- 20.Zhang Y, Zolov SN, Chow CY, Slutsky SG, Richardson SC, Piper RC, Yang B, Nau JJ, Westrick RJ, Morrison SJ, Meisler MH, Weisman LS (2007) Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5-bisphosphate, results in neurodegeneration in mice. Proc Natl Acad Sci U S A 104 (44):17518–17523. doi: 10.1073/pnas.0702275104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarkes D, Rameh LE (2010) A novel HPLC-based approach makes possible the spatial characterization of cellular PtdIns5P and other phosphoinositides. Biochem J 428 (3):375–384. doi: 10.1042/BJ20100129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wakelam MJ (2014) The uses and limitations of the analysis of cellular phosphoinositides by lipidomic and imaging methodologies. Biochim Biophys Acta 1841 (8):1102–1107. doi: 10.1016/j.bbalip.2014.04.005 [DOI] [PubMed] [Google Scholar]
- 23.Jones DR, Ramirez IB, Lowe M, Divecha N (2013) Measurement of phosphoinositides in the zebrafish Danio rerio. Nat Protoc 8 (6):1058–1072. doi: 10.1038/nprot.2013.040 [DOI] [PubMed] [Google Scholar]
- 24.Kanehara K, Yu CY, Cho Y, Cheong WF, Torta F, Shui G, Wenk MR, Nakamura Y (2015) Arabidopsis AtPLC2 Is a Primary Phosphoinositide-Specific Phospholipase C in Phosphoinositide Metabolism and the Endoplasmic Reticulum Stress Response. PLoS Genet 11 (9):e1005511. doi: 10.1371/journal.pgen.1005511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McCartney AJ, Zolov SN, Kauffman EJ, Zhang Y, Strunk BS, Weisman LS, Sutton MA (2014) Activity-dependent PI(3,5)P2 synthesis controls AMPA receptor trafficking during synaptic depression. Proc Natl Acad Sci U S A 111 (45):E4896–4905. doi: 10.1073/pnas.1411117111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Samie M, Wang X, Zhang X, Goschka A, Li X, Cheng X, Gregg E, Azar M, Zhuo Y, Garrity AG, Gao Q, Slaugenhaupt S, Pickel J, Zolov SN, Weisman LS, Lenk GM, Titus S, Bryant-Genevier M, Southall N, Juan M, Ferrer M, Xu H (2013) A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev Cell 26 (5):511–524. doi: 10.1016/j.devcel.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]