

Introduction
Epstein-Barr Virus (EBV) is a ubiquitous human herpesvirus that contributes to the etiology of diverse human cancers and auto-immune diseases. EBV establishes a relatively benign, long-term latent infection in over 90 percent of the adult population. Yet, it also increases risk for certain cancers and auto-immune disorders depending on complex viral, host, and environmental factors that are only partly understood. EBV latent infection is found predominantly in memory B-cells, but the natural infection cycle and pathological aberrations enable EBV to infect numerous other cell types, including oral, nasopharyngeal, and gastric epithelia, B-, T-, and NK-lymphoid cells, myocytes, adipocytes, astrocytes, and neurons. EBV infected cells, free virus, and gene products can also be found in the CNS. In addition to the direct effects of EBV on infected cells and tissue, the effect of chronic EBV infection on the immune system is also thought to contribute to pathogenesis, especially auto-immune disease. Here, we review properties of EBV infection that may shed light on its potential pathogenic role in neurological disorders.
Keywords: Epstein-barr virus, CNS, encephalitis, PCNLS, multiple sclerosis, lytic infection, latent infection
A Brief History of Discovery
The discovery of Epstein-Barr virus (EBV) as the first virus implicated as the causative agent of human cancer was both paradigm shifting and a result of one of the most intriguing, international medical detective stories of the 20th century. This story begins in Africa with Denis Burkitt, a one-eyed, Protestant Irish surgeon who, after his service with the Royal Army Medical Corps in Kenya and Somaliland during World War II, established a medical practice in Uganda to help the people of Uganda “both medically and spiritually” (1). In 1958, Burkitt described an aggressive tumor of the jaw and face that was common to children living across central Africa (2). Although initially the apparent restriction to the jaw suggested that these tumors were sarcomas, Burkitt realized that the tumors of the jaw were associated with tumors at distant sites including the kidneys, ovaries, testes, liver, and occasionally the spinal cord, resulting in paraplegia (2). These findings led Burkitt to conclude that these disparate tumors were all part of the same disease. Subsequent microscopy studies concluded that the tumor tissue comprised a previously undescribed type of lymphoma with larger, paler staining histiocytes in a “starry sky” pattern and the tumor was renamed “Burkitt’s Lymphoma” (3, 4).
Burkitt found that the incidence of Burkitt’s lymphoma was up to 18 per 100,000 children per year, making it the most common childhood tumor in Uganda. Of interest, there was peak incidence in children from 5–6 years of age with a preponderance in boys (Figure 1). Notably, Burkitt defined a geographic “lymphoma belt”, which revealed that the tumors only occurred in areas where the year-round temperature was above 15°C and the annual rainfall was above 20 inches (0.5 M). The prevalence of Burkitt’s lymphoma in other areas of the world, including coastal regions of Papua New Guinea, with other specific environmental conditions suggested that the disease was vector-borne (5). Indeed, the malarial pathogen Plasmodium falciparum is often an important co-factor in the development of Burkitt’s lymphoma and the peak age incidence for the tumor coincides with the age at which children have the highest levels of malaria parasites in their blood (6). However, it was a personal connection that Burkitt made after giving a lecture while on leave in the United Kingdom that would ultimately lead to the discovery of the virus that causes Burkitt’s lymphoma.
Figure 1.
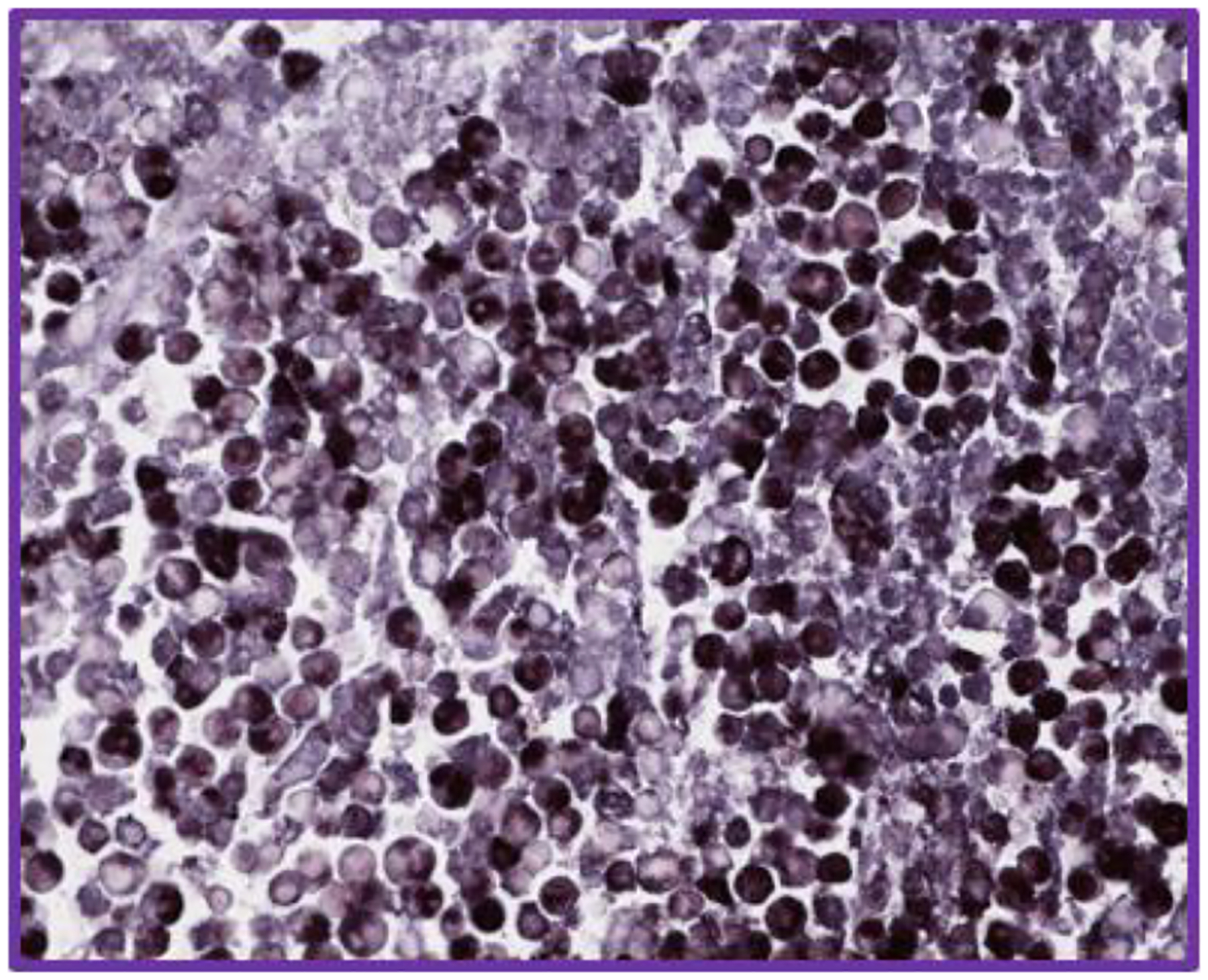
EBV-encoded small RNA in situ hybridization EBER ISH in a murine xenograft model of EBV-associated lymphoma (40X).
When Michael Anthony Epstein attended Burkitt’s lecture at Middlesex Hospital in 1961, his work focused on Rous sarcoma virus—the first oncogenic retrovirus to be described; it causes sarcoma in chickens. After this encounter, Burkitt sent samples of tumor tissue to Epstein who, with Yvonne Barr, was able to generate cell lines from tumor material (7), leading to the identification of herpes-like viral particles in tumor cells by electron microscopy (8). From the UK, the story of viral discovery moved farther west when Epstein sent samples to the laboratory of Werner and Gertrude Henle at the Children’s Hospital of Philadelphia for further characterization. The Henles demonstrated that antibodies to Epstein-Barr virus (EBV) were found in African patients with Burkitt’s lymphoma and were also widespread in healthy individuals in Philadelphia. Moreover, the development of infectious mononucleosis (IM) in the Henles’ laboratory technician, who fortuitously seroconverted to strongly EBV antibody positive after her serum was previously used as a negative control for serologic assays, led to the discovery that EBV causes IM (9). The Henles were also the first to describe a hallmark characteristic of the virus, the ability of EBV infected B cells to transmit the virus to uninfected B cells, leading to their continuous growth transformation (10).
As work on EBV continued in the 1970s and 80s, the virus was linked to nasopharyngeal carcinoma and lymphoproliferative disorders and the association between immunosuppression and EBV-related lymphomas was established (11) (12) (13) (14). More recently, the story of EBV in human disease has expanded to include a major role for EBV in a subtype of lymphoepithelial gastric cancers—it is currently estimated that 1.5% of all human cancers worldwide are attributable to EBV (15). EBV has also been implicated as a contributing etiological agent in various auto-immune diseases and neurological disorders (16) (17) (18). Our understanding of the pathogenic role of EBV in immune dysregulation, especially in combination with environmental factors and genetic susceptibility, continues to expand. This review focuses on the neuropathogenesis of EBV infection.
Virology and Molecular Biology
The systematic name for EBV is human herpesvirus 4 (HHV-4) and it is one of eight known human herpesviruses. EBV is a member of the gammaherpesvirus subfamily and is the prototype member of this group; Kaposi’s sarcoma virus (KSHV) is the other medically important human gammaherpesvirus that has been identified. EBV strains are classified as type 1 or type 2 (formerly known as types A and B, respectively) based primarily on the sequence of their Epstein-Barr Virus Nuclear Antigen 2 (EBNA2) gene (19). Characteristic of all herpesvirus, EBV has a large (172 kilobase pair), double-stranded DNA genome that is linear in the virus particle and contained within an icosahedral capsid that is surrounded by an amorphous tegument, containing both viral mRNAs and viral tegument proteins, including BNRF1, BPLF1, and BGLF2. These tegument proteins enhance EBV reactivation and infection, promote the release of infectious particles, and help the virus evade the innate immune system (20) (21) (22). The tegument is surrounded by a lipid bilayer viral envelope derived from the host cell membrane and containing the viral attachment and entry proteins. There are 13 glycoproteins encoded by EBV; eleven of them are found in the virion envelope and two are nonstructural proteins. These glycoproteins have one or more functions in either facilitating virus entry and spread, virus assembly, and/or manipulating the host immune response (23).
The EBV genome (prototypical strain B95–8) was the first of the herpesviruses to be completely sequenced and encodes over 80 ORFs, non-coding RNAs (EBER1 and EBER2), and 44 mature miRNA (24, 25). There are several internal repeat sequences dispersed throughout the viral genome that can be used to identify EBV strains and to perform molecular epidemiology (25–27). Terminal repeats present at the ends of the linear form of the EBV genome allow for circularization in infected cells, ensuring that latently infected cells contain the viral genome as non-integrated complex mini chromosomes, called episomes. These episomes are located in the host cell nucleus, with approximately 10 – 50 copies present in each infected cell (28). EBV genes are operationally defined by their location on a BamHI restriction map of the viral genome and functionally defined by whether they are expressed during lytic (productive) or latent (no virions produced) replication. Latently infected B-lymphocytes enter one of four latency programs (Latency III, Latency II, Latency I, or Latency 0) with each latency type exhibiting more restrictive expression of a subset of viral proteins and viral RNAs. While Type III latency is characterized by expression of the majority of viral latency factors (9 viral proteins and multiple noncoding RNAs), fewer viral proteins are expressed during Type II latency, and Type I latency is characterized by expression of a single viral protein (EBNAI) along with viral noncoding RNAs. During Latency 0 only the EBER noncoding RNAs are expressed (Figure 2) (29). Five latent genes (EBNA1, EBNA2, EBNA3A and EBNA3C, and LMP1), are required for B-cell transformation (19). Importantly, all EBV-related cancers are associated with latent infection. Lytic genes encode viral transcription factors (e.g. BZLF1), a viral DNA polymerase (BALF5) and associated factors, and viral glycoproteins and structural proteins (25). However, there are exceptions to this pattern, especially in non-lymphoid tumors. For example, the viral glycoprotein BARF1, which is typically expressed as a lytic gene in B-cells, can be detected in nasopharyngeal carcinoma, where it inhibits apoptosis and may enhance immune evasion (30) (23, 31). Furthermore, new genome-wide RNA-sequencing methods reveal expression of viral genes traditionally associated with the lytic cycle in otherwise latently infected tumor cells(32). This raises new possibilities for pathogenic contributions from the many (>100) viral lytic genes that may be expressed in some aberrant latent infections.
Figure 2.
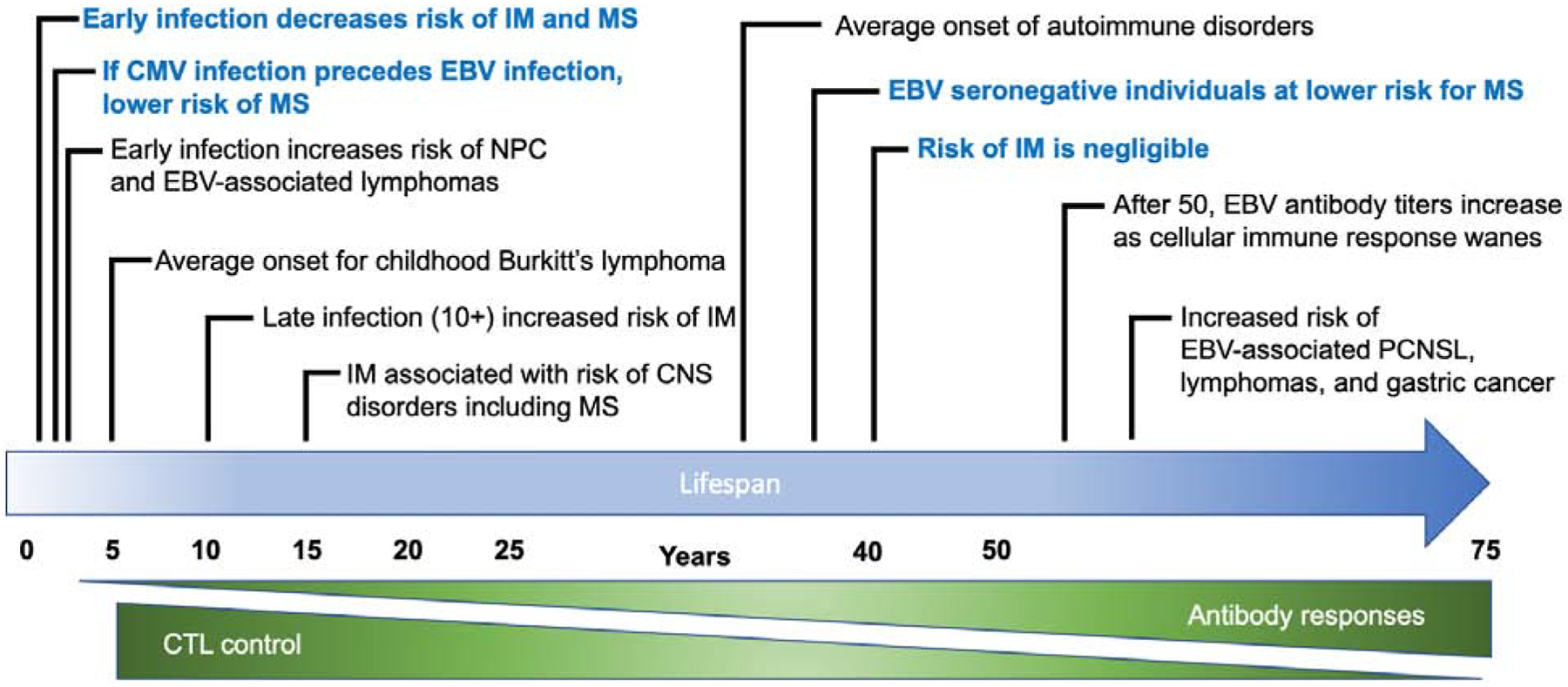
Consequences of EBV Infection over the human lifespan
Natural History of EBV and Infection in the Central Nervous System (CNS)
The natural infection cycle of EBV is important to better understand with respect to the potential role of EBV in CNS disease. However, there are limits to our knowledge of EBV reservoirs in latent and chronically infected individuals. EBV relies primarily on latent replication to proliferate within an individual via clonal replication in dividing B cells. However, abundant sources of viral DNA and virions can be detected in the saliva of chronic shedders(33, 34) (35). Episodic shedding of the virus from the oropharynx occurs without symptoms in many infected individuals(36); in one study, EBV was found in the saliva of 73% of seropositive adults over a period of 14 months (37). Nevertheless, lytically infected cells are rarely detected in organs and tissues that contain latently infected lymphocytes. This tight control of EBV lytic infection is likely due to a robust T-cell immune recognition of EBV lytic proteins, while latently infected cells successfully evade this cellular immune response (38). Cyclical shedding likely reflects a ‘cat and mouse’ game of sporadic viral reactivation with rapid suppression by a healthy immune response.
Viral transmission through salivary contact with permissive epithelial cells in the oral (tonsillar) compartment is thought to be the primary route of primary infection. Although the oral route is the most common route of infection, EBV transmission can occur during organ transplantation and blood transfusion(39–41). Transmission during reactivation occurs through the synaptic interaction of latently infected B-cells circulating with epithelial tissue, typically in the oropharynx(42). EBV uses different attachment protein/host receptor interactions for entry in B cells and epithelial cells. In B cells EBV gp350 binds to CD21 (Complement receptor type 2, also known as complement C3d receptor) (43). Subsequently, EBV gp42 interacts with HLA class II molecules and triggers fusion with the host membrane (43). Alternatively, in epithelial cells, EBV uses its BMRF2 protein to interact initially with b1 integrins; fusion is then triggered by interaction of EBV gH/gL envelope protein with αvβ6/8 integrins (44) (45).
After virus entry and dissolution of the viral nucleocapsid, the EBV genome is transported to the nucleus, initiating virus replication (46). Infection of a naive, resting B-cell results in a complete reprogramming of the B-cell that resembles the germinal center reaction (47). The initial transcription program of EBV Latency III induces naïve B cells to proliferate (48) (49). A subset of these cells enters a germinal center and subsequently switches to the Latency II program where fewer EBV proteins are expressed and the viral latency proteins LMP-1 and LMP2A promote survival and differentiation into memory B cells by mimicking CD40L-mediated signaling and B cell antigen receptor activation, respectively. As these memory B cells exit the germinal center and enter peripheral circulation, EBV gene expression becomes even more restricted (Latency I), allowing the infected cell to escape immune surveillance. Alternatively, direct infection of a resting memory B cells can establish memory phenotype without the requirement of a residency within a germinal center (50) (51).
Infection of cell types other than B cells and endothelial, including T cells, NK cells, and smooth muscle cells can occur (52). However, these cells do not appear to play a major role in the virus lifecycle and the mechanism of entry in these CD21-negative cells is unclear (53). EBV infection of T and NK cells can result in mature NK/T cell lymphomas, which are extremely aggressive and difficult to treat (54) (55). Although EBV is not routinely detected in parenchymal cells of the brain (neurons and glia), EBV infection of primary human fetal neurons and neuroblastoma lines and astrocytes has been demonstrated in vitro(56) (57). Unlike many other herpesviruses, EBV has not been shown to establish latency in neurons.
Most human herpesviruses (HSV-1, HSV-2, CMV, EBV, HHV-6A, HHV-6B, HHV-7) have been detected in the brain and routinely establish lifelong infection of the peripheral (PNS) and/or central nervous system (CNS). Herpesvirus infections of the nervous system can result in a variety of neurological manifestations that range from asymptomatic to fatal, including encephalitis; meningitis; myelitis; vasculopathy; ganglioneuritis; retinal necrosis; cerebellitis; optic neuritis; and congenital CNS disease. Although it is appreciated that herpesviruses neuroinvasion can occur either during primary infection or during subsequent virus reactivation, the molecular mechanisms involved in establishing herpesvirus infection in the CNS are not completely understood—even for the alphaherpesviruses where directional spread from the peripheral nervous system (PNS) to the CNS has been studied extensively (58). In general, the neurotropism of the alphaherpesviruses allows them to remain latent in neural tissue and, upon reactivation, spread to the CNS along axons in a retrograde manner or through endothelial cells of cerebral vessels. Because EBV does not latently infect neurons, CNS infection following EBV reactivation is likely to occur at extraneural sites and enter the CNS via infected lymphocytes (59) (60) (Figure 3). Accordingly, CNS disorders associated with EBV infection can be considered in two groups; those associated with productive (primary or reactivated) infection and those associated with latent infection (61).
Figure 3.
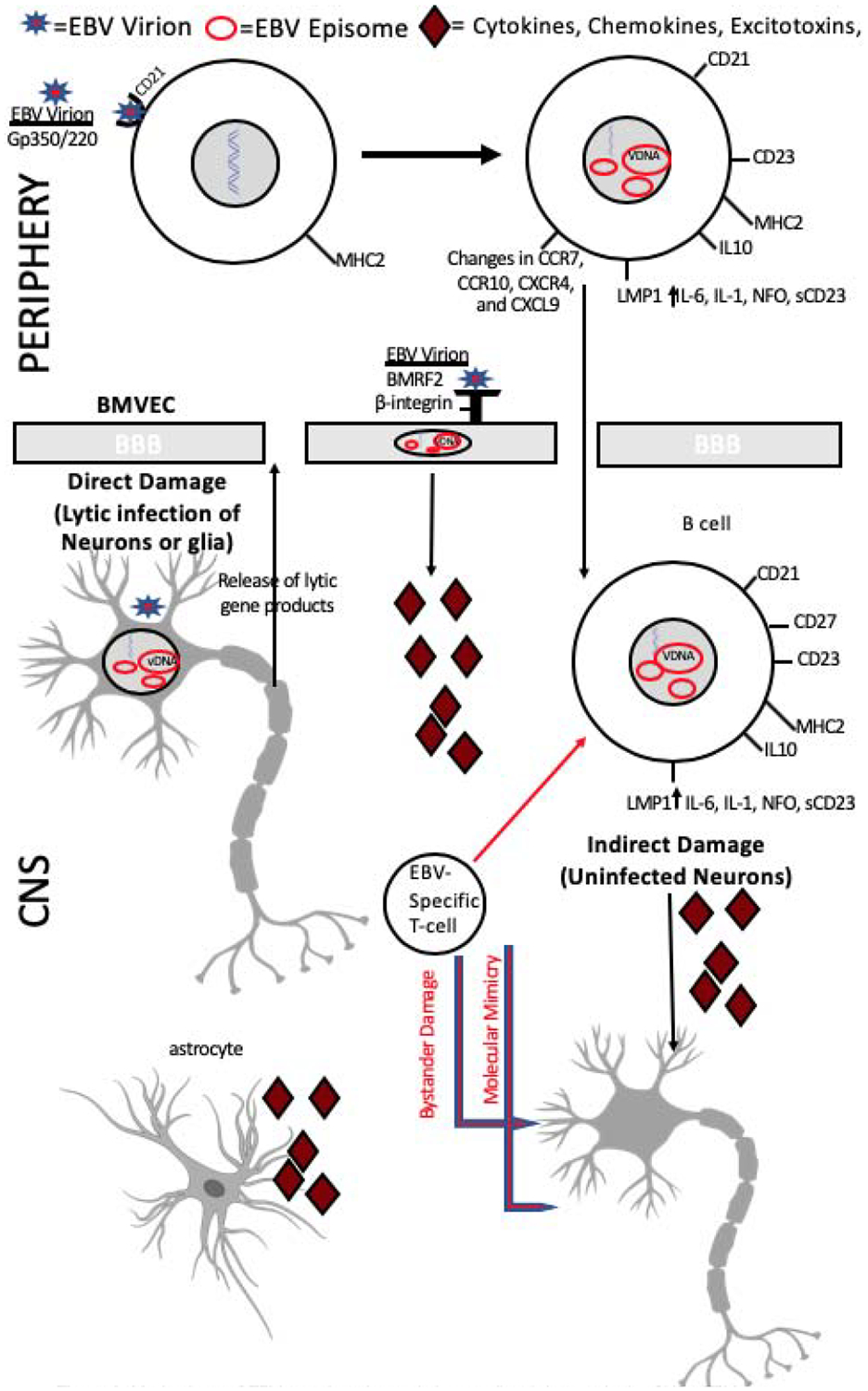
Mechanisms of EBV neuroinvasion and virus mediated damage in the CNS. EBV may enter the brain via normal B cell trafficking or through infection of brain microvasculature endothelial cells (BMVEC). Either endothelial cells of the neurovasculature or infected B cells may be the source of neurotoxicity through the release of inflammatory cytokines and viral proteins. In addition, T-cell mediated responses to infected cells in the brain may lead to bystander damage. Alternatively, molecular mimicry, where similarities between EBV and host-peptides results in the cross-activation of autoreactive T or B cells may be involved in the neuropathogenesis of EBV-associated disorders of the CNS.A less favored hypothesis suggests that EBV infection of neurons and lytic gene expression leads to neuronal damage.
CNS Disorders associated with Productive EBV Infection
Infectious Mononucleosis (IM) and its Reported CNS Complications:
EBV is highly prevalent, infecting more than 90% of the human population worldwide(62). In most developing areas, primary EBV infection usually occurs in early childhood with near universal seroconversion often seen by ages 3–4(63). Low income and crowded family conditions are correlated with the increased likelihood of EBV seropositivity among children (64) (65) (66) (67) (68). In contrast, only 30–50% of children in developed countries are EBV seropositive by age five (Figure 1). In these areas, EBV infection follows a bimodal pattern, with peaks in children below 5 years and again after 10 years of age, frequently delaying exposure until adolescence or early adulthood (69–71) (72, 73). EBV antibody titers in seropositive individuals vary according to age following a U-shaped pattern, with high titers observed among young children during primary infection and in the elderly (above 50 years) as the cellular immune response declines with age (74) (74) (75) (Figure 1).
In infants and small children, primary EBV infection is typically asymptomatic. Primary EBV infection in adolescence and early adulthood, however, causes infectious mononucleosis (IM) in about 50% of individuals who seroconvert at a later age. The incidence of IM peaks around the age of 17 and then begins to decline, becoming extremely rare by age 40 (76, 77). In addition to age of primary infection, host determinants of immune control of EBV infection may determine an individual’s risk of developing IM. For example, an IL-10 promoter polymorphism and an overrepresentation of HLA-B-3501 among IM cases compared to controls have been reported (78). Studies of EBV primary infection have focused on adolescent patients with IM. The incubation of EBV infection before the appearance of symptoms is relatively long (approximately six weeks). EBV DNA can be detected in plasma within two weeks of the onset of symptoms and peak viral loads occur during this time (79). Subsequently, the cellular immune response clears lytically infected cells and viral loads decrease rapidly to low or even undetectable levels in the plasma or serum (80). IM is an acute, though typically self-limiting febrile illness characterized by fatigue, fever, cervical lymphadenopathy, hepatosplenomegaly, hepatitis, and pharyngitis (81). Laboratory findings in IM typically include leukocytosis, hypergammaglobulinemia, increased liver enzyme levels, and occasionally hyperbilirubinemia and hemolytic anemia (82). In most cases, IM lasts for a few weeks and affected individuals make a full recovery soon thereafter. However, sustained levels of EBV DNA are found in saliva for approximately six months after the onset of IM (83). Chronic active EBV infection (CAEBV) is a rare disorder that is characterized by high viral loads, recurrent IM-like symptoms, and an atypical pattern of EBV antibody responses (84); it is more common in Japan and East Asia and should be suspected in IM patients with symptoms that persist for more than three months (82). Fatalities from IM are rare; but when they occur, they are often accompanied by neurological manifestations of EBV infection. The incidence of neurological involvement in IM has been reported to range from 0.37–7.3% (85). In general, neurologic disorders associated with EBV primary infection are diagnosed by the coincidence of EBV seroconversion and the detection of EBV genome in the cerebrospinal fluid (CSF) with the presentation of neurologic symptoms (86).
Mechanisms of EBV-mediated CNS Disease
Among Burkitt’s earliest findings was that most patients with Burkitt’s lymphoma (21/25 in an early autopsy series) had tumors involving the brain or meninges (87). EBV is an important pathogen of the CNS and is associated with diverse diseases of the brain including viral encephalitis and CNS-lymphoma. However, the neuropathogenesis of EBV mediated CNS damage in general is incompletely understood. There are several mechanisms that may lead to damage of brain parenchyma in EBV encephalitis and other CNS disorders resulting from EBV infection and they are not mutually exclusive. First, direct lytic EBV infection of neurons has been suggested to be the source of neuronal damage and lytic gene expression (e.g. BZLF1), in the CNS during EBV encephalitis (59). It has been reported that EBV can be detected in neurons and glia in EBV encephalitis, albeit rarely (88). A murine model for gammaherpesvirus in the brain using murine gammherpesvirus 68 shows widespread infection of glial and neuronal cells and may serve as a model for EBV infection of the brain (89) (90) (91). Second, it is possible that EBV infection of the endothelial cells of the neurovasculature may be the source of neurotoxicity in EBV where the release of viral proteins, inflammatory cytokines, free radicals, and excitatory amino acids from infected endothelial cells lead to neuronal damage (92). Third, lytically infected B cells that enter the brain may also cause inflammation and damage of local neurons, which is likely to be reversable once these cells are eliminated by the immune system. Inflammatory cytokines, including TNF-α, IL-1, and IL-6 are secreted by EBV infected B cells, are associated with many of the symptoms of IM, and are known to have detrimental effects in the CNS (93) (94). Fourth, T-cell responses mounted against CNS infiltrating infected B cells may lead to bystander damage of neuronal cells resulting in encephalitis and other CNS disorders associated with EBV infection. It has been demonstrated that EBV-reactive T-cells in the liver are pathogenic in hepatitis resulting from primary EBV infection and a similarly destructive T-cell response in the brain may contribute to EBV encephalitis (95) (96). Finally, molecular mimicry between EBV and autoantigens of the CNS has been postulated, though not proven, to play a role in autoimmune disorders associated with EBV infection. Molecular mimicry could potentially be involved in disorders associated with either lytic or latent infection (Figures 3 and 4).
Figure 4.
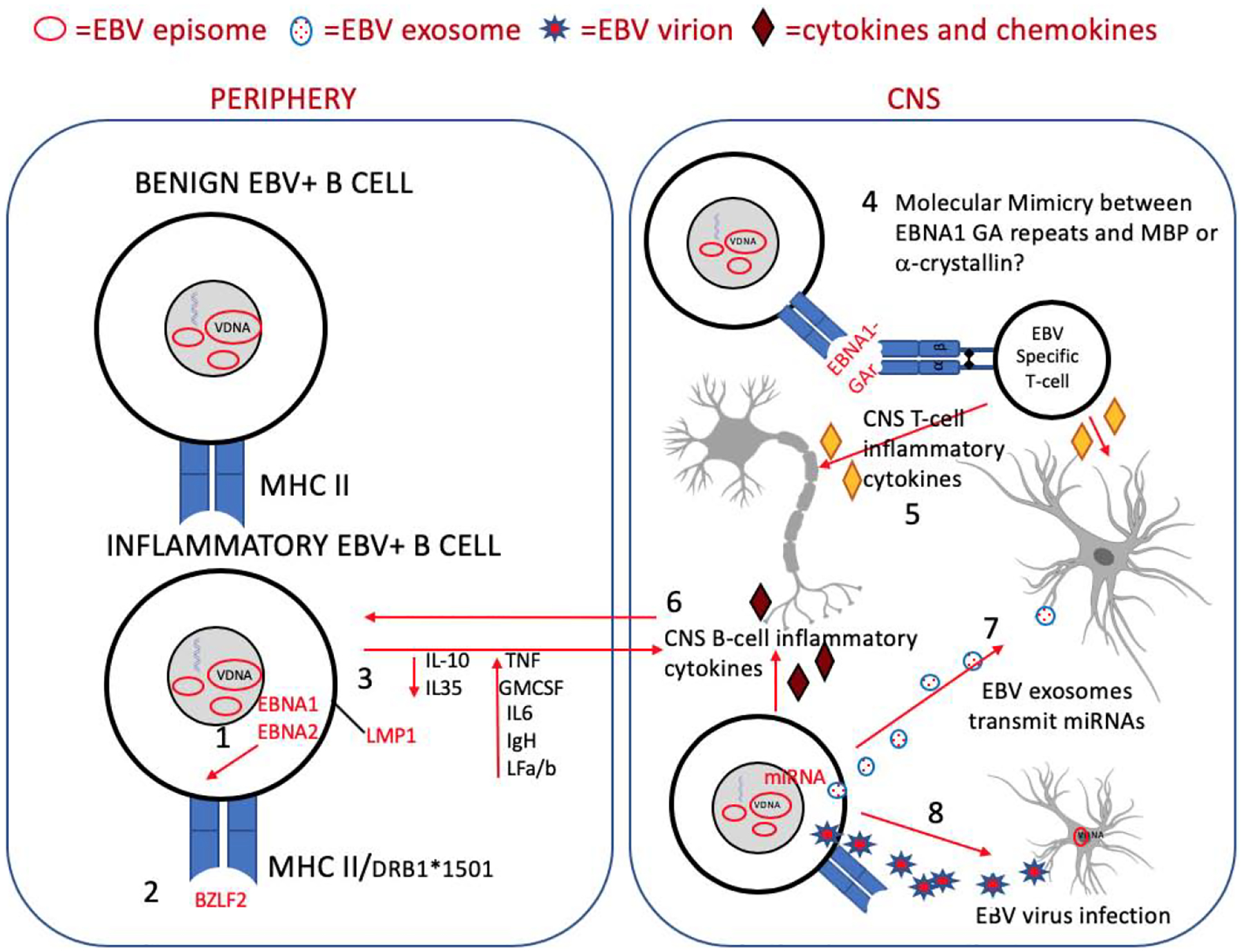
Possible mechanisms for EBV as an etiologic or disease-modifying agent in multiple sclerosis. Several possible hypotheses have been proposed for EBV as a mediator of CNS damage and disease in MS and it is likely that the interplay between host and viral determinants are involved. Viral determinants may include: 1. the EBV latency proteins EBNA1, the master regulator of EBV latency and an important pro-survival factor, and EBNA2, which is involved in B-cell transformation and exerts transcriptional control of HLA disease-risk alleles associated with MS and other autoimmune disorders; 2. the EBV lytic protein, BZLF2, which also interacts with MHC2 and may influence the immunopathogenesis of MS; 3. the latency protein LMP1 that increases expression of proinflammatory cytokines; 7. exosomes released from EBV-infected B cells in the brain containing viral miRNAs, viral transactivators, and inflammatory cytokines that may contribute to virus mediated damage in the CNS; and 8. EBERs that may contribute directly to the inflammatory milieu of the MS lesion by inducing IFN-a and other components of the innate immune system. Host determinants involved in EBV-associated CNS damage may include: 2. Susceptible HLA alleles; 3. Poor CTL control of EBV infection that results in the persistence of inflammatory B cells with increased trafficking to the CNS; 4. molecular mimicry and 5. bystander damage that results from T cell targeting of EBV infection in the CNS and 6. damage from inflammatory B cells present in the brain
EBV Aseptic Meningitis
The association between aseptic meningitis and IM has been long appreciated and probably underreported (86) (97) (98). Headaches are common during acute IM and are likely the result of mild aseptic meningitis. An early review of neurologic complications of IM found that 41% were associated with aseptic meningitis defined by headache, fever, and a stiff neck occurring in the context of the most common manifestations of IM (fever, fatigue, and lymphadenopathy) (98). Moreover, EBV is commonly found by PCR in patients with viral meningitis (99) (100). Fortunately, meningitis associated with IM is almost always self-limiting and disabling sequela are uncommon (101). Because individuals with EBV aseptic meningitis have a generally good prognosis, the histopathology of the disease has not been well described.
EBV Encephalitis
EBV encephalitis is often difficult to diagnose because it shares many signs and symptoms with other forms of viral encephalitis, including headache, fever, confusion, seizures, and paresis. Moreover, non-specific focal features, diffuse slowing, and periodic Electroencephalography (EEG) complexes observed in EBV encephalitis resemble those observed in HSV encephalitis (102) (103). Recognition of EBV encephalitis is further confounded by the observation that while this disorder often occurs in tandem with other manifestations of IM, this is not always the case and EBV encephalitis can occur in the absence of systemic findings (104) (105) (106) (107) (108).
Again, death from EBV encephalitis is rare and, therefore, neuropathological findings are infrequently reported (86). Perivascular infiltrates as well as meningeal and diffuse parenchymal infiltrates consisting of lymphocytes, including EBV-infected B cells, and microglia have been described (86) (109) (110) (111). MRI findings of EBV encephalitis are variable and can include normal parenchyma; edema of gray matter, basal ganglia, cerebellum, limbic cortex and thalamus; white matter involvement correlating with inflammatory findings, particularly in acute disseminated encephalomyelitis (ADEM); glial nodules; neuronophagia; and perivascular infiltrates (86) (105) (109). The CSF of patients with EBV encephalitis is typically remarkable for pleocytosis, mildly increased protein, atypical lymphocytes, and the presence of oligoclonal bands where antibodies to EBV viral capsid antigen (VCA) are frequently detected (105).
EBV has been implicated in Rasmussen’s encephalitis (RE), a rare (incidence of 1.8 per 10 million) pediatric neurologic disorder of unknown etiology distinguished by focal epilepsy and progressive hemisphere atrophy leading to a rapid decline in cognitive function. Surgical intervention (usually a hemispherectomy) is required to prevent intractable epilepsy and severe deterioration of neurological function (112). Pathological characteristics observed in the brains of RE patients mainly include lymphocyte infiltration, neuron loss, vascular cuffing, and microgliosis, which are similar to those observed in viral encephalitis. Recent studies have demonstrated increased detection of EBV latency membrane protein 1 (LMP1) in RE brains compared to controls. Of interest, EBV was localized to RE lesion areas, implicating EBV in the pathogenesis of RE. Further, EBV and HHV-6 antigens were localized in neurons and astrocytes of RE brain and accompanied by a high frequency of CD8+ T-cells, suggesting that a robust CTL response to herpesviruses may be implicated in the pathophysiology of this aggressive form of encephalitis/epilepsy (113) (114).
Acute Disseminated Encephalomyelitis (ADEM)
In addition to meningitis and encephalitis, cranial nerve palsies, transverse myelitis cerebellitis, seizures, acute hemiplegia, and acute demyelinating encephalomyelitis (ADEM) have been associated with either IM or EBV reactivation(86). ADEM is a rare disorder (8 per 1,000,000 per year) that is characterized by an abrupt onset of a demyelinating immune-mediated attack on the brain and spinal cord that is driven by high serum IgG antibodies specific to myelin oligodendrocyte glycoprotein (MOG). The symptoms and pathophysiology of ADEM have similarities with that of multiple sclerosis (MS; discussed later in this chapter), but ADEM is distinguished by its appearance with rapid fever, its preponderance in children, its seasonality (more common in winter and spring), and its specific pattern of MRI abnormalities (115). ADEM is fatal in about 5% of cases, although most children and adolescents will make a full recovery within six months (116) (117) (118). Several viral infectious agents including influenza, dengue, measles, varicella zoster, Borrelia burgdorferi, and EBV have been associated with ADEM (119) (120). ADEM has been reported immediately after IM and after EBV reactivation associated with renal transplantation (121) (122) (123) (124). Cross-reactivity between EBV and myelin proteins, including MOG has been reported (125). Notably, anti-MOG antibodies have been detected in 20% of patients with IM due to primary EBV infection without neurological manifestations, suggesting that the induction of a MOG-specific autoantibody response as a consequence of EBV primary infection is not uncommon (126). While these findings implicate EBV infection as a potential trigger for anti-MOG antibody production, it is unclear if molecular mimicry between antibodies produced in response to EBV antigens and MOG underlies this observation (127). Importantly, children who experience ADEM are more likely to develop MS and the percentage of these children who are ultimately diagnosed with MS ranges from 15–45% (128) (129) (130). A recent study of an Italian cohort of ADEM patients found that children with MRI lesions of the corpus callosum, whose CSF findings were remarkable for the presence of oligoclonal bands, and were EBV positive are more likely to go on to develop MS (127).
Alice in Wonderland Syndrome
An uncommon, though interesting neuropsychiatric syndrome that has been reported to occur in conjunction with IM is Alice in Wonderland syndrome This disorder is named after Lewis Carroll’s (née Charles Dodgson) novel that follows the main character, Alice, into a land of spatial distortions. It has been speculated that Dodgson’s own experiences with migraine headaches and, perhaps, epilepsy inspired some of the novel’s distinctive perspective (131). This syndrome is typically observed in teenagers, though it has been reported in children as young as six (132) (133). Usually coincident with or shortly following acute IM, these patients will have several episodes a day where they experience distortions in the orientation, shape, and size of objects in their environment (dysmetropsia), body image disturbance (aschematia), as well as the tendency for linear objects to appear curvy (metamorphosia) (134). Other neurologic findings are typically normal. Again, the illness is self-limiting and the duration of perception disorders ranges from one week to three months with, ultimately, good neurological outcomes (135) (136). In addition to coinciding with primary EBV infection, Alice in Wonderland syndrome has also been reported to occur in association with influenza H1N1, ADEM, and Lyme neuroborreliosis (137).
EBV and Peripheral Neuropathies
IM has been associated with several forms of peripheral neuropathy including radiculoplexopathy, myeloradiculitis, autonomic neuropathy, and Guillian-Barré syndrome (GBS) (86). In addition, EBV related T-cell lymphoproliferative disorders can affect peripheral nerves (138) (139). GBS is an autoimmune driven peripheral neuropathy that is a rare, but well-documented complication of IM (140) (141) (86). The condition is particularly dangerous when peripheral nerves of the autonomic nervous system are affected and can cause sudden disturbances in blood pressure and heart rate in addition to respiratory failure. The onset of GBS is typically preceded by an infection and several pathogens besides EBV, including Campylbacter jejuni, CMV (HHV-5), Zika virus, Chikungunya virus, Varicella zoster virus (HHV-3), have also been implicated (142) (143) (144, 145) (146).GBS is rare, affecting one per 100,000 per year; of those affected, approximately 7.5% will die (147). The disorder is either marked by demyelination, where damage to myelin sheaths is inflicted by T-cells and macrophages, or antibody-mediated axonal damage. Accordingly, immunotherapy, including plasmapheresis and intravenous immunoglobulins (IVIG) are standard treatment for GBS.
CNS Disorders Associated with Latent EBV Infection
PCNSL
PCNSL is a primary intracranial tumor of B-cell origin that accounts for 1% of all lymphomas and 3–5% of primary brain tumors(148) (149) and is commonly, though not exclusively, observed in patients who are immunocompromised. The incidence of PCNSL has risen steadily over the last 20 years and is approximately 0.51 cases per 100,000 person-years. HIV infection is the highest risk factor for PCNSL, which accounts for 15% of HIV-associated lymphomas(150). PCNSL has a reported incidence of over 1000 times greater in the HIV positive population and the prognosis of PCNSL is better in non-HIV cases than in HIV-related cases(151) (152) (153). Although the advent of cART (combination antiretroviral therapy) has decreased the number of AIDS-related cases of PCNSLs(154), the mortality of CNS complications, including PCNSL, remains high in untreated HIV infected individuals and those unaware of their HIV status(155) (156) (149). In addition, it has been reported that the proportion of minority patients diagnosed with AIDS-related PCNSL has increased compared to the pre-cART era(157). EBV is associated with greater than 90% of PCNSLs in patients who are iatrogenically immunosuppressed and 100% of those who are infected with HIV(158). Moreover, the incidence of PCNSLs has been reported to be on the rise in the non-HIV infected population, and it has been suggested that EBV-associated PCNSLs in the elderly may be underreported(159) (160).
As with all masses in the central nervous system (CNS), the location of PCNSL lesions determines the clinical presentation. Signs and symptoms for PCNSLs include focal neurological deficits (aphasia, hemiparesis, and ataxia); neuropsychiatric symptoms; headache/nausea/vomiting suggestive of increased intracranial pressure; seizures; and ocular symptoms(161). Because of the depth of the tumor, PCNSLs are not typically amenable to surgical resection and the clinical outcomes of PCNSL and other EBV-associated malignancies that have metastasized to the CNS are poor. Most EBVPCNSL are characterized for the expression of EBV non-coding RNA EBERs as the gold standard for EBV positivity. Early reports indicate that EBV expresses a type III latency in PCNSL, similar to diffuse B-cell lymphoma, where most of the latency associated viral gene products are expressed (162) (163). Several inflammatory cytokines, chemoattractants, and adhesion molecules including macrophage inflammatory protein-3alpha (MIP-3alpha, CCL20), stromal cell-derived factor-1 (SDF-1; CXCL12), CXCR4 (SDF-1 receptor), CXCL9, secreted phosphoprotein 1 (SPP1; osteopontin), IL-4, IL4 stimulated genes, and myelin associated glycoprotein (MAG) have been demonstrated to be involved in the CNS tropism, migration, and proliferation of malignant B-cells in PCNSL(164) (165) (166) (167). Collectively, these factors are believed to contribute to perineural CNS invasion by increasing the neuroinvasive potential of malignant B cells and priming them to migrate into the CNS while stimulating the perivascular microenvironment to allow for CNS penetration and angiocentric positioning of these malignant cells(164) (165) (166) (167).
Median survival in AIDS patients with PCNSL is only 4 months with treatment and 2.5 months without(168). Moreover, standard chemotherapeutic regimens for lymphomas, including CHOP (Cyclophosphamide, Hyroxydaunorubicin, Oncovin, and Prednisone) are ineffective in PCNSL, owing in part to poor penetration through the blood brain barrier. In PCNSL, treatment with irradiation and corticosteroids is relatively ineffective with tumor recurrence observed in greater than 90% of patients(168). The inclusion of intravenous rituximab, a monoclonal antibody-based therapy that targets the CD20 phosphoprotein expressed on the surface of B-cells, does not significantly improve treatment outcomes in PCNSL compared to CHOP alone(169). However, less than 1% of systemic rituximab penetrates the leptomeningeal space and a small, recent study suggests that intraventricular delivery of rituximab may overcome the difficulty of penetrating the blood brain barrier, enabling this drug to become a more attractive treatment modality for PCNSLs and underscoring the importance of adequate CNS penetrance in treating this disorder and other disorders of the CNS, like MS, where B-cell depletion has therapeutic benefit (156) (170).
Multiple Sclerosis
MS is the most prevalent demyelinating disease of the central nervous system (CNS) and the most common disabling neurologic disease of young people, affecting an estimated 1,000,000 in the United states and 4–4.5 million people worldwide (171) (172). It is a chronic and incurable disease that significantly detracts from an individual’s quality of life (173) (174). A diverse array of neurological signs and symptoms are associated with MS. These include limb weakness, impaired motor function, sensory symptoms, visual symptoms, eye movement disorders, bladder symptoms, sexual dysfunction, fatigue, ataxia, deafness, spasticity, dementia, and cognitive impairment (175). The life expectancy of a patient with MS is 5–10 years lower than that of the general population (176).
Although the clinical progression of MS is variable and unpredictable, several disease phenotypes have been described; it is critical to distinguish disease phenotypes for both prognosis and decisions regarding treatment strategies. There are four distinct clinical courses in which the majority of MS patients can be classified: relapsing-remitting MS (RRMS); secondary-progressive MS (SPMS); primary-progressive MS (PPMS); and progressive-relapsing MS (PRMS)(177) (178). About 80% of people with MS are initially diagnosed with relapsing-remitting MS (RRMS); this disease phenotype is characterized by disease exacerbations where new symptoms appear or existing symptoms become more severe (171). In RRMS, disease exacerbations last for variable amounts of time and are followed by periods of total or partial recovery. Approximately 65% of individuals who are initially diagnosed with RRMS will develop secondary-progressive MS (SPMS) characterized by the accumulation of progressive disability with or without superimposed relapses. On average, the time between disease onset and conversion from RRMS to SPMS occurs in 19 years (179).
The etiology of MS is unknown. In part, this is owing to the variability of this disease, suggesting that many determinants many be involved in the range of clinical phenotypes that are defined as MS. The prevailing view of the etiology of MS is that it is multifactorial and that genetics and environmental factors contribute collectively to MS susceptibility (180). Over the years, several genes, most of which are associated with immune function, myelin structure, or mitochondria have been tentatively associated with an increased risk of MS (181). (182) (183) (184). Many of these associations have not been demonstrated consistently in different studies. However, a strong association between MS and the major histocompatibility complex (MHC) was first identified in the 1970s (185). Specifically, an increased risk of MS in individuals with MHC class II alleles DR2 and DQw1 has been found consistently in several populations (186); in the preponderance of these studies the primary risk allele for MS is HLA-DRB1*15:01 (187). Carriers of HLA-DRB1*1501 have up to a four-fold increased risk of MS (188). Additional high-risk MHC and non-MHC loci have been identified (189) (190) (191) (192) (193). Interestingly, the majority of the loci associated with MS risk have known immunologic functions and many contribute to the risk of inheriting other autoimmune diseases (194).
Several lines of evidence support a role for viruses as a key environmental component or, perhaps, a trigger in the etiology of MS. (195) Although a number of infectious agents have been associated with MS, none of these viruses have been firmly determined to be the exclusive causative agent. Additionally, mechanism(s) by which virus-host interactions may lead to demyelination are complex, diverse, and incompletely understood (195). Arguably no other candidate infectious agent matches the epidemiology, risk profile, and pathobiology of MS as well as Epstein-Barr virus (EBV) (195). A history of infectious mononucleosis is more common in MS patients and in areas where MS is prevalent and there are high standards of hygiene (196). The overlapping epidemiology of infectious mononucleosis and MS is consistent with the “hygiene hypothesis”, which suggests that decreased exposure to childhood infections predisposes individuals to proinflammatory or autoimmune responses and increases MS risk and may explain the lower incidence of MS in areas where there is an increased prevalence of childhood infections (197) (198). The “hygiene hypothesis” suggests that either 1) MS and infectious mononucleosis arise independently as a consequence of living in conditions of high hygiene in childhood or 2) high hygiene in childhood delays exposure to EBV infection, which subsequently results in an increased risk of MS (199).
An association between EBV and MS is supported by numerous studies demonstrating: MS patients have higher EBV-specific antibody titers against EBNA proteins (EBNA1, EBNA2, EBNA3, EBNA3A, EBNA3B, EBNA3C, EBNA 4, and EBNA6) LMP1, capsid protein VP26, VCA, and the lytic protein BBRF2; increased antibody titers have been observed 15–20 years before onset of neurological symptoms; the presence of EBV infection in brain-infiltrating B cells and plasma cells of 95% of autopsy cases examined; a history of infectious mononucleosis is more common in MS patients and in areas where MS is prevalent; and a reduced risk of MS among individuals who are EBV seronegative, which increases sharply if the same individuals seroconvert (200) (201) (196) (202) (203) (204) (205) (206). In addition, the absence of EBV infection correlates with a much lower risk disease risk in MS (207). EBV-infected B cells and plasma cells have been reported to accumulate in the brains of MS patients, suggesting that neuroinvasion of EBV+ B cells may contribute to the MS pathogenesis (208). Although some studies have not replicated these findings (209, 210). B cells and plasma cells that appear in chronic MS lesions do so in large numbers and are present in areas of demyelination(211). In a detailed study of 17 MS and 9 control brains, EBV LMP1 was found in 93% of MS and 78% of control brains (212). However, EBV lytic gene expression (BZLF1) was only observed in chronic MS lesions (46%). EBER-positive cells were common in MS tissue, but rarely found in non-MS brains (212).
In addition to epidemiologic consistency between the risk of MS and the acquisition of EBV infection, a preponderance of laboratory studies have provided additional support for a role of EBV in both adult and pediatric-onset MS (199). Of interest, it has been demonstrated that there is an increased seroprevalence of EBV for an increased rate of remote EBV infection in patients with pediatric MS compared with controls (213) (214) (215) (216). Moreover, among seropositive children tested for EBV DNA in saliva, increased oral shedding was found for EBV, but not for other herpesviruses, in pediatric MS patients compared with controls. EBV strain variation may also contribute to risk of MS. Of the virus detected in the saliva of these pediatric patients, it was found that type I strain of EBV was detected more frequently than type II, which may reflect the more frequent distribution of type I worldwide, with the exception of Africa and Papua New Guinea where the distribution of the two types appears to be equal (217) (215) (218). It has been suggested that a disruption in the normal sequence of common viral infections, owing to a late primary infection by EBV, may lead to immune dysregulation and ultimately MS in genetically susceptible individuals. Higher frequencies of CD4+ T cells specific for EBNA1 and enhanced IFN-γ production have been demonstrated in MS patients relative to healthy, seropositive individuals (219). In a prospective study following children ADEM, remote EBV infection (defined by the presence of anti-EBNA1 and anti-VCA IgG antibodies) was found to be more common in children with MS than those with monophasic ADEM (220). By contrast, CMV infection was more common in children with monophasic ADEM (220). This study suggests that children with evidence of remote EBV infection without CMV infection were at highest risk of subsequent MS diagnosis and may support previous reports demonstrating a protective role for CMV infection in MS (221) (222).
Because there is limited access to brain tissue, studies on CNS immune responses in MS patients typically focus on CSF samples. The presence of EBV and HHV-6 reactive OCBs in MS CSF has been reported (223) in some studies, but not others (210). In addition, Pfuhl, et al. have correlated the presence of serum antibodies to EBNA1, but not to EBV viral capsid antigen, rubella, or varicella zoster virus, with an elevated intrathecal IgGs in patients with early MS (224), suggesting that EBV infection has a key role in early events in the pathogenesis of MS. In addition to elevated EBV-specific antibodies in the CSF, the presence of CTLs recognizing EBV lytic proteins in the CSF of MS patients was reported (225) complementing earlier studies demonstrating high EBV EBNA1 specific CTLs in MS patients, particularly at the onset of disease (226).
Although EBV is a leading candidate infectious agent for multiple sclerosis (MS) and the increased risk of MS among individuals who have been infected with EBV is increasingly well-accepted, the mechanism by which EBV may either cause or contribute to the pathogenesis of MS has not been established (Figure 4). Several possible hypotheses have been proposed including molecular mimicry, bystander damage, and deficient immune control of EBV infection. The molecular mimicry hypothesis suggests that immune cells specific for EBV antigens, like EBNA1, are cross-reactive for CNS antigens, like myelin basic protein or αB-crystallin, which is induced by B-cells upon EBV infection, leading to a promiscuous TCR that recognizes more than one peptide, ultimately leading to immune cell mediated damage in the CNS(202) (227) (125) (228) (229) (230) (229). Anti-EBNA1 antibodies have been shown to cross-react with epitopes of neuroglial cells as well as transadolase, a protein expressed by oligodendrocytes (231) (232) (233). Bystander damage in the CNS may occur as T-cells target infected B-cells in the brain. In this case, secondary autoimmune responses could manifest as T cells are sensitized to CNS antigens released after bystander damage focused on EBV infected B cells occurs (202). It has also been suggested that deficient CTL control of EBV infected B-cells in MS may be involved in the pathogenesis of MS (234); this theory is supported by increased spontaneous immortalization of EBV infected B-cells of MS patients in culture (235). In humanized mice, it has been shown that adoptive transfer of EBV specific CD8+ T-cell clones are capable of transiently controlling infection (236). Alternatively, proinflammatory B cells may contribute directly to the neuropathogenesis of MS (237). The recent success of B cell depletion therapies (reviewed below) supports an important role for B cells in the pathogenesis of MS. Whether EBNA1 or other EBV genes play direct roles in driving this inflammatory B cell cascade in MS is unknown. Some novel hypotheses for the direct involvement of EBV genes in MS disease pathogenesis have been proposed (238) (239). It has been suggested that exosomes released from EBV-infected B cells contain viral miRNAs, viral transactivators, and inflammatory cytokines that may contribute to virus mediated damage in the CNS (238) (Figure 4). In addition, EBERs may contribute directly to the inflammatory milieu of the MS lesion by inducing IFN-α and other components of the innate immune system (239).
Recent studies may point to a more specific, immunopathogenic role for the interaction between EBV and HLA molecules, both class I and class II, in the pathogenesis of MS (240). EBV viral loads have been reported to be higher in HLAA*02−/B*07+/DRB1*15+ MS patients vs patients HLA-A*A02+/B*07−/DRB1*15, suggesting an important influence of HLA-class I alleles as well (241). Other reports have shown that HLA-B*07 is associated with higher EBV Early Antigen antibodies, higher disability scores, and more active MRI findings in MS patients (242). Individuals with increased EBNA1 IgG levels, presence of DRB1*15 and absence of A*02 have a 16-fold risk increase for MS, indicating that EBV and both MHC class I and class II genotypes collectively influence an individual’s susceptibility to MS (240).
While some studies have demonstrated that the CD8+ T-cell response to EBV infected B cells is deficient in MS and decreases with age (243), others have reported increased EBV-specific CD8+ responses in MS. Studies using HLA class I pentamers have shown that lytic antigen-specific CD8+ T cell responses are detected in fewer inactive MS patients than in active MS patients and controls, while the frequency of CD8+ T cells specific for EBV lytic and latent antigens is higher in both active and inactive MS patients (227). In addition, analysis of post-mortem MS brain samples showed expression of the EBV lytic protein BZLF1 and interactions between cytotoxic CD8+ T cells and EBV lytically infected plasma cells in inflammatory white matter lesions and meninges (227). The authors suggest that that inability to control EBV infection during inactive MS could set the stage for intracerebral viral reactivation and disease relapse (227). A large study comprising 221 MS patients and 218 control subjects from Switzerland used HLA-A2, HLA-B7, and HLA-B8 restricted EBV and CMV specific tetramers to determine the percentage of EBV specific CD8+ T-cells (244). This study determined that there is a higher prevalence of MS patients with HLA HLA-B*0702/EBVRPP-specific CD8+ T cells ex vivo. However, the magnitude of the HLA-B*0702/EBVRPP-specific CD8+ T cell response was lower in MS patients. Upon stimulation with HLA-B7/EBVRPP peptide, MS patients had decreased CD8+ cytotoxic activity compared to controls (244). Pender and colleagues recently described an impaired CD8+ response to EBV, but not CMV lytic antigens in MS at the onset of disease and at all subsequent disease states. Although the frequency of EBV specific CD8+ cells in MS is relatively high, they have reduced cytokine functionality, suggesting that EBV-specific T-cell exhaustion frequently occurs in MS (234). Interestingly, a phase I clinical trial indicated that primary progressive MS patients saw clinical improvement after autologous EBV-specific -T cell therapy targeting EBNA1, LMP1 and LMP2a (245).
It has been suggested, though not conclusively demonstrated, that specific strains of EBV may be associated with MS (246) (247) (248). EBV gene products have the potential play important roles in MS disease pathogenesis. Latency proteins EBNA1, EBNA1, EBNA2, LMP1, and LMP2, in particular, merit additional consideration. As discussed above, EBNA1 aberrant immune responses to EBNA1 are common in MS. The role of EBNA1 in promoting cell survival, driving latency, and maintaining the viral genome suggests the possibility that EBNA1 could affect viral and host gene expression and influence latency programs and the migration of inflammatory B cells in MS (249). Moreover, EBNA1 is required for EBV latency and transcription of viral genes, such as EBNA2 involved in B cell transformation (250). Recent genetic studies have found that disease-risk alleles associated with MS and other autoimmune disorders are enriched for transcription control by EBV latency proteins, especially EBNA2 (251) (252). MS risk genes whose expression is altered by EBV expression have been identified; many of these changes affected the signaling pathway of EBV membrane proteins LMP1 and LMP2 (252). A better understanding of latency phenotypes and the interplay of virus and host genes in the context of CNS inflammation will elucidate the role of EBV in the etiology and neuropathogenesis in MS.
Treatment of EBV in the CNS
Treatment strategies of EBV in the CNS may target either the virus or host cells involved in the pathogenesis of EBV-associated disorders. B-cell depletion strategies, initially developed for lymphomas of the periphery, have shown great promise in the treatment of MS; the success of these therapies coupled with favorable risk-benefit ratios has opened exciting new avenues for treatment modalities in MS and validated the pathogenic role of B-cells, including those latently infected with EBV(253). After the initial success of Rituximab (anti-CD 20), additional anti-CD 20 monoclonals, including ofatumumab and ocrelizumab have been implemented in the treatment of RRMS and PRMS, respectively (254). Additional immune cell targeting therapies, including the purine analog cladribine, which was initially developed for B-cell chronic lymphocytic leukemia and reduces the population of CD19+ B-cells, have been implemented in MS with good clinical outcomes (255). The benefit of these immunomodulatory therapies is derived from the combined effects of the elimination of pathogenic (cancerous or, in the case of MS, immunopathogenic) B cells and the subsequent reconstitution with healthy B-cells. It is unclear if these benefits are also derived from the effect of these drugs on latent EBV within these CD20+ or CD19+ populations. Early studies demonstrating the clinical benefit derived from adaptive transfer of autologous EBV-specific T cells further supports the pathogenic role of EBV+ B-cells in the pathogenesis of MS and suggest that therapies targeting EBV infection in the periphery or CNS may have desirable therapeutic benefit (245).
In addition to targeting pathogenic lymphocytes, therapies that target EBV lytic or latent infection may be beneficial in the protean CNS disorders associated with EBV infection. Valganciclovir, a prodrug for the 2’-deoxy-guanosine analog ganciclovir developed to treat CMV infection, has shown promise in reducing viral loads in IM (256) and in the treatment of EBV encephalitis (101), indicating that antiviral agents targeting EBV lytic infection or reactivation may indeed have therapeutic benefit. However, no selective antiviral approaches to treat PCNSL or other disorders associated with latent EBV infection of the CNS are available. Histone deacetylase inhibitors that induce gene expression in latent EBV infection and sensitize lymphoma cells to antiviral agents are under development for the treatment of EBV associated malignancies and proliferative disorders and may have future applications in EBV-associated CNS and autoimmune disorders (NCT03397706). In addition, small-molecule inhibitors that target EBV latent infection may have potential therapeutic value and several different approaches are under development (257) (258) (259). We and others have developed small molecules that target EBNA1 to selectively inhibit the proliferation of latently infected tumor cells (257) (258, 259). A first-in-human clinical trial using this EBNA1 inhibitor is underway (NCT03682055). Further adaptation of these inhibitors for better CNS penetrance may increase their potential for use in PCNSL, MS, and other EBV-associated disorders of the CNS. As discussed above, EBV-specific adoptive T-cell therapies, modified CTLs, and NK, NKT, and Vγ9Vδ2 T cells in combination are also under investigation for various EBV-associated cancers. Currently, EBV-specific CTL therapies are being adapted and tested in phase I clinical trials for treating RRMS (NCT03283826) (245) (212) (260) (261) (262). These therapies exploit allogenic T-cell libraries comprising EBV-specific T cells characterized across known HLA restrictions. Finally, the development of vaccine strategies to prophylactically prevent primary EBV infection and to prevent reactivation-associated diseases are under development. An EBV vaccine that eliminates or reduces the latent reservoir of EBV would likely reduce the risk of EBV-associated disease (263). Currently, several vaccines are under development and a second-generation candidate targeting the EBV receptor binding site, gp350/220, is entering first-in-human trials (264).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
PML declares financial conflict of interest relationship with Cullinan Apollo, Inc. and ownership interest in Vironika, LLC.
References
- 1.Epstein A, Eastwood MA. 1995. Denis Parsons Burkitt. 28 February 1911–1993., p 90, Biographical memoirs of fellows of the Royal Society, vol 41. [PubMed] [Google Scholar]
- 2.Burkitt D. 1958. A sarcoma involving the jaws in African children. Br J Surg 46:218–23. [DOI] [PubMed] [Google Scholar]
- 3.O’Conor GT, Davies JN. 1960. Malignant tumors in African children. With special reference to malignant lymphoma. J Pediatr 56:526–35. [DOI] [PubMed] [Google Scholar]
- 4.Wright DH. 1963. Cytology and histochemistry of the Burkitt lymphoma. Br J Cancer 17:50–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Booth K, Burkitt DP, Bassett DJ, Cooke RA, Biddulph J. 1967. Burkitt lymphoma in Papua, New Guinea. Br J Cancer 21:657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kafuko GW, Burkitt DP. 1970. Burkitt’s lymphoma and malaria. Int J Cancer 6:1–9. [DOI] [PubMed] [Google Scholar]
- 7.Epstein MA, Barr YM. 1964. Cultivation in Vitro of Human Lymphoblasts from Burkitt’s Malignant Lymphoma. Lancet 1:252–3. [DOI] [PubMed] [Google Scholar]
- 8.Epstein MA, Achong BG, Barr YM. 1964. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1:702–3. [DOI] [PubMed] [Google Scholar]
- 9.Henle W, Henle GE, Horwitz CA. 1974. Epstein-Barr virus specific diagnostic tests in infectious mononucleosis. Hum Pathol 5:551–65. [DOI] [PubMed] [Google Scholar]
- 10.Henle W, Diehl V, Kohn G, Zur Hausen H, Henle G. 1967. Herpes-type virus and chromosome marker in normal leukocytes after growth with irradiated Burkitt cells. Science 157:1064–5. [DOI] [PubMed] [Google Scholar]
- 11.Old LJ, Boyse EA, Geering G, Oettgen HF. 1968. Serologic approaches to the study of cancer in animals and in man. Cancer Res 28:1288–99. [PubMed] [Google Scholar]
- 12.de Schryver A, Friberg S Jr., Klein G, Henle W, Henle G, De-The G, Clifford P, Ho HC. 1969. Epstein-Barr virus-associated antibody patterns in carcinoma of the post-nasal space. Clin Exp Immunol 5:443–59. [PMC free article] [PubMed] [Google Scholar]
- 13.zur Hausen H, Schulte-Holthausen H, Klein G, Henle W, Henle G, Clifford P, Santesson L. 1970. EBV DNA in biopsies of Burkitt tumours and anaplastic carcinomas of the nasopharynx. Nature 228:1056–8. [DOI] [PubMed] [Google Scholar]
- 14.Starzl TE, Nalesnik MA, Porter KA, Ho M, Iwatsuki S, Griffith BP, Rosenthal JT, Hakala TR, Shaw BW Jr., Hardesty RL, et al. 1984. Reversibility of lymphomas and lymphoproliferative lesions developing under cyclosporin-steroid therapy. Lancet 1:583–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Farrell PJ. 2019. Epstein-Barr Virus and Cancer. Annu Rev Pathol 14:29–53. [DOI] [PubMed] [Google Scholar]
- 16.Shibata D, Tokunaga M, Uemura Y, Sato E, Tanaka S, Weiss LM. 1991. Association of Epstein-Barr virus with undifferentiated gastric carcinomas with intense lymphoid infiltration. Lymphoepithelioma-like carcinoma. Am J Pathol 139:469–74. [PMC free article] [PubMed] [Google Scholar]
- 17.Ascherio A, Munger KL. 2015. EBV and Autoimmunity. Curr Top Microbiol Immunol 390:365–85. [DOI] [PubMed] [Google Scholar]
- 18.Bashir R, Luka J, Cheloha K, Chamberlain M, Hochberg F. 1993. Expression of Epstein-Barr virus proteins in primary CNS lymphoma in AIDS patients. Neurology 43:2358–62. [DOI] [PubMed] [Google Scholar]
- 19.Kieff E, Rickinson AB. 2007. Epstein-Barr virus and its replication, p 2603–2654. In Knipe DM, Howley PM (ed), Fields Virology. Lippincott Williams and Wilkins, Philadelphia. [Google Scholar]
- 20.Swaminathan S, Kenney S. 2009. The Epstein-Barr Virus Lytic Life Cycle, p 285–315, DNA Tumor Viruses. [Google Scholar]
- 21.Liu X, Cohen JI. 2016. Epstein-Barr Virus (EBV) Tegument Protein BGLF2 Promotes EBV Reactivation through Activation of the p38 Mitogen-Activated Protein Kinase. J Virol 90:1129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Gent M, Braem SG, de Jong A, Delagic N, Peeters JG, Boer IG, Moynagh PN, Kremmer E, Wiertz EJ, Ovaa H, Griffin BD, Ressing ME. 2014. Epstein-Barr virus large tegument protein BPLF1 contributes to innate immune evasion through interference with toll-like receptor signaling. PLoS Pathog 10:e1003960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hutt-Fletcher LM. 2015. EBV glycoproteins: where are we now? Future Virol 10:1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, et al. 1984. DNA sequence and expression of the B95–8 Epstein-Barr virus genome. Nature 310:207–11. [DOI] [PubMed] [Google Scholar]
- 25.Kanda T, Yajima M, Ikuta K. 2019. Epstein-Barr virus strain variation and cancer. Cancer Sci 110:1132–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gratama JW, Oosterveer MA, Klein G, Ernberg I. 1990. EBNA size polymorphism can be used to trace Epstein-Barr virus spread within families. J Virol 64:4703–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gratama JW, Oosterveer MA, Lepoutre JM, van Rood JJ, Zwaan FE, Vossen JM, Kapsenberg JG, Richel D, Klein G, Ernberg I. 1990. Serological and molecular studies of Epstein-Barr virus infection in allogeneic marrow graft recipients. Transplantation 49:725–30. [DOI] [PubMed] [Google Scholar]
- 28.Shannon-Lowe C, Adland E, Bell AI, Delecluse HJ, Rickinson AB, Rowe M. 2009. Features distinguishing Epstein-Barr virus infections of epithelial cells and B cells: viral genome expression, genome maintenance, and genome amplification. J Virol 83:7749–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanda T. 2018. EBV-Encoded Latent Genes. Adv Exp Med Biol 1045:377–394. [DOI] [PubMed] [Google Scholar]
- 30.Hoebe EK, Le Large TY, Greijer AE, Middeldorp JM. 2013. BamHI-A rightward frame 1, an Epstein-Barr virus-encoded oncogene and immune modulator. Rev Med Virol 23:367–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strockbine LD, Cohen JI, Farrah T, Lyman SD, Wagener F, DuBose RF, Armitage RJ, Spriggs MK. 1998. The Epstein-Barr virus BARF1 gene encodes a novel, soluble colony-stimulating factor-1 receptor. J Virol 72:4015–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakhoul H, Lin Z, Wang X, Roberts C, Dong Y, Flemington E. 2019. High-Throughput Sequence Analysis of Peripheral T-Cell Lymphomas Indicates Subtype-Specific Viral Gene Expression Patterns and Immune Cell Microenvironments. mSphere 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hadinoto V, Shapiro M, Sun CC, Thorley-Lawson DA. 2009. The dynamics of EBV shedding implicate a central role for epithelial cells in amplifying viral output. PLoS Pathog 5:e1000496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thorley-Lawson DA, Hawkins JB, Tracy SI, Shapiro M. 2013. The pathogenesis of Epstein-Barr virus persistent infection. Curr Opin Virol 3:227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thorley-Lawson DA. 2015. EBV Persistence—Introducing the Virus. Curr Top Microbiol Immunol 390:151–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thompson MP, Kurzrock R. 2004. Epstein-Barr virus and cancer. Clin Cancer Res 10:803–21. [DOI] [PubMed] [Google Scholar]
- 37.Ling PD, Lednicky JA, Keitel WA, Poston DG, White ZS, Peng R, Liu Z, Mehta SK, Pierson DL, Rooney CM, Vilchez RA, Smith EO, Butel JS. 2003. The dynamics of herpesvirus and polyomavirus reactivation and shedding in healthy adults: a 14-month longitudinal study. J Infect Dis 187:1571–80. [DOI] [PubMed] [Google Scholar]
- 38.Steven NM, Annels NE, Kumar A, Leese AM, Kurilla MG, Rickinson AB. 1997. Immediate early and early lytic cycle proteins are frequent targets of the Epstein-Barr virus-induced cytotoxic T cell response. J Exp Med 185:1605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gerber P, Walsh JH, Rosenblum EN, Purcell RH. 1969. Association of EB-virus infection with the post-perfusion syndrome. Lancet 1:593–5. [DOI] [PubMed] [Google Scholar]
- 40.Hanto DW, Frizzera G, Purtilo DT, Sakamoto K, Sullivan JL, Saemundsen AK, Klein G, Simmons RL, Najarian JS. 1981. Clinical spectrum of lymphoproliferative disorders in renal transplant recipients and evidence for the role of Epstein-Barr virus. Cancer Res 41:4253–61. [PubMed] [Google Scholar]
- 41.Alfieri C, Tanner J, Carpentier L, Perpete C, Savoie A, Paradis K, Delage G, Joncas J. 1996. Epstein-Barr virus transmission from a blood donor to an organ transplant recipient with recovery of the same virus strain from the recipient’s blood and oropharynx. Blood 87:812–7. [PubMed] [Google Scholar]
- 42.Shannon-Lowe C, Rowe M. 2014. Epstein Barr virus entry; kissing and conjugation. Curr Opin Virol 4:78–84. [DOI] [PubMed] [Google Scholar]
- 43.Speck P, Haan KM, Longnecker R. 2000. Epstein-Barr virus entry into cells. Virology 277:1–5. [DOI] [PubMed] [Google Scholar]
- 44.Xiao J, Palefsky JM, Herrera R, Tugizov SM. 2007. Characterization of the Epstein-Barr virus glycoprotein BMRF-2. Virology 359:382–96. [DOI] [PubMed] [Google Scholar]
- 45.Xiao J, Palefsky JM, Herrera R, Berline J, Tugizov SM. 2009. EBV BMRF-2 facilitates cell-to-cell spread of virus within polarized oral epithelial cells. Virology 388:335–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsurumi T, Fujita M, Kudoh A. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev Med Virol 15:3–15. [DOI] [PubMed] [Google Scholar]
- 47.Thorley-Lawson DA. 2001. Epstein-Barr virus: exploiting the immune system. Nat Rev Immunol 1:75–82. [DOI] [PubMed] [Google Scholar]
- 48.Babcock GJ, Decker LL, Volk M, Thorley-Lawson DA. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395–404. [DOI] [PubMed] [Google Scholar]
- 49.Roughan JE, Thorley-Lawson DA. 2009. The intersection of Epstein-Barr virus with the germinal center. J Virol 83:3968–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kuppers R. 2003. B cells under influence: transformation of B cells by Epstein-Barr virus. Nat Rev Immunol 3:801–12. [DOI] [PubMed] [Google Scholar]
- 51.Kurth J, Spieker T, Wustrow J, Strickler GJ, Hansmann LM, Rajewsky K, Kuppers R. 2000. EBV-infected B cells in infectious mononucleosis: viral strategies for spreading in the B cell compartment and establishing latency. Immunity 13:485–95. [DOI] [PubMed] [Google Scholar]
- 52.Stubbins RJ, Alami Laroussi N, Peters AC, Urschel S, Dicke F, Lai RL, Zhu J, Mabilangan C, Preiksaitis JK. 2019. Epstein-Barr virus associated smooth muscle tumors in solid organ transplant recipients: Incidence over 31 years at a single institution and review of the literature. Transpl Infect Dis 21:e13010. [DOI] [PubMed] [Google Scholar]
- 53.Gru AA, Haverkos BH, Freud AG, Hastings J, Nowacki NB, Barrionuevo C, Vigil CE, Rochford R, Natkunam Y, Baiocchi RA, Porcu P. 2015. The Epstein-Barr Virus (EBV) in T Cell and NK Cell Lymphomas: Time for a Reassessment. Curr Hematol Malig Rep 10:456–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Connor OA, Bhagat G, Ganapathi K, Pedersen MB, D’Amore F, Radeski D, Bates SE. 2014. Changing the paradigms of treatment in peripheral T-cell lymphoma: from biology to clinical practice. Clin Cancer Res 20:5240–54. [DOI] [PubMed] [Google Scholar]
- 55.Somasundaram N, Lim JQ, Ong CK, Lim ST. 2019. Pathogenesis and biomarkers of natural killer T cell lymphoma (NKTL). J Hematol Oncol 12:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jha HC, Mehta D, Lu J, El-Naccache D, Shukla SK, Kovacsics C, Kolson D, Robertson ES. 2015. Gammaherpesvirus Infection of Human Neuronal Cells. MBio 6:e01844–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Menet A, Speth C, Larcher C, Prodinger WM, Schwendinger MG, Chan P, Jager M, Schwarzmann F, Recheis H, Fontaine M, Dierich MP. 1999. Epstein-Barr virus infection of human astrocyte cell lines. J Virol 73:7722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kramer T, Enquist LW. 2013. Directional spread of alphaherpesviruses in the nervous system. Viruses 5:678–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weinberg A, Li S, Palmer M, Tyler KL. 2002. Quantitative CSF PCR in Epstein-Barr virus infections of the central nervous system. Ann Neurol 52:543–8. [DOI] [PubMed] [Google Scholar]
- 60.Meyding-Lamade U, Strank C. 2012. Herpesvirus infections of the central nervous system in immunocompromised patients. Ther Adv Neurol Disord 5:279–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fujimoto H, Asaoka K, Imaizumi T, Ayabe M, Shoji H, Kaji M. 2003. Epstein-Barr virus infections of the central nervous system. Intern Med 42:33–40. [DOI] [PubMed] [Google Scholar]
- 62.Pagano JS, Blaser M, Buendia MA, Damania B, Khalili K, Raab-Traub N, Roizman B. 2004. Infectious agents and cancer: criteria for a causal relation. Semin Cancer Biol 14:453–71. [DOI] [PubMed] [Google Scholar]
- 63.de-The G, Day NE, Geser A, Lavoue MF, Ho JH, Simons MJ, Sohier R, Tukei P, Vonka V, Zavadova H. 1975. Sero-epidemiology of the Epstein-Barr virus: preliminary analysis of an international study - a review. IARC Sci Publ:3–16. [PubMed] [Google Scholar]
- 64.Henle G, Henle W, Clifford P, Diehl V, Kafuko GW, Kirya BG, Klein G, Morrow RH, Munube GM, Pike P, Tukei PM, Ziegler JL. 1969. Antibodies to Epstein-Barr virus in Burkitt’s lymphoma and control groups. J Natl Cancer Inst 43:1147–57. [PubMed] [Google Scholar]
- 65.Mekmullica J, Kritsaneepaiboon S, Pancharoen C. 2003. Risk factors for Epstein-Barr virus infection in Thai infants. Southeast Asian J Trop Med Public Health 34:395–7. [PubMed] [Google Scholar]
- 66.Ozkan A, Kilic SS, Kalkan A, Ozden M, Demirdag K, Ozdarendeli A. 2003. Seropositivity of Epstein-Barr virus in Eastern Anatolian Region of Turkey. Asian Pac J Allergy Immunol 21:49–53. [PubMed] [Google Scholar]
- 67.Biggar RJ, Henle W, Fleisher G, Bocker J, Lennette ET, Henle G. 1978. Primary Epstein-Barr virus infections in African infants. I. Decline of maternal antibodies and time of infection. Int J Cancer 22:239–43. [DOI] [PubMed] [Google Scholar]
- 68.Hesse J, Ibsen KK, Krabbe S, Uldall P. 1983. Prevalence of antibodies to Epstein-Barr virus (EBV) in childhood and adolescence in Denmark. Scand J Infect Dis 15:335–8. [DOI] [PubMed] [Google Scholar]
- 69.Haahr S, Plesner AM, Vestergaard BF, Hollsberg P. 2004. A role of late Epstein-Barr virus infection in multiple sclerosis. Acta Neurol Scand 109:270–5. [DOI] [PubMed] [Google Scholar]
- 70.Henle G, Henle W. 1967. Immunofluorescence, interference, and complement fixation technics in the detection of the herpes-type virus in Burkitt tumor cell lines. Cancer Res 27:2442–6. [PubMed] [Google Scholar]
- 71.Melbye M, Ebbesen P, Bennike T. 1984. Infectious mononucleosis in Greenland: a disease of the non-indigenous population. Scand J Infect Dis 16:9–15. [DOI] [PubMed] [Google Scholar]
- 72.Crawford DH, Swerdlow AJ, Higgins C, McAulay K, Harrison N, Williams H, Britton K, Macsween KF. 2002. Sexual history and Epstein-Barr virus infection. J Infect Dis 186:731–6. [DOI] [PubMed] [Google Scholar]
- 73.Fleisher G, Henle W, Henle G, Lennette ET, Biggar RJ. 1979. Primary infection with Epstein-Barr virus in infants in the United States: clinical and serologic observations. J Infect Dis 139:553–8. [DOI] [PubMed] [Google Scholar]
- 74.Venkitaraman AR, Lenoir GM, John TJ. 1985. The seroepidemiology of infection due to Epstein-Barr virus in southern India. J Med Virol 15:11–6. [DOI] [PubMed] [Google Scholar]
- 75.Wick G, Grubeck-Loebenstein B. 1997. Primary and secondary alterations of immune reactivity in the elderly: impact of dietary factors and disease. Immunol Rev 160:171–84. [DOI] [PubMed] [Google Scholar]
- 76.Rosdahl N, Larsen SO, Thamdrup AB. 1973. Infectious mononucleosis in Denmark. Epidemiological observations based on positive Paul-Bunnell reactions from 1940–1969. Scand J Infect Dis 5:163–70. [DOI] [PubMed] [Google Scholar]
- 77.Auwaerter PG. 1999. Infectious mononucleosis in middle age. JAMA 281:454–9. [DOI] [PubMed] [Google Scholar]
- 78.Helminen ME, Kilpinen S, Virta M, Hurme M. 2001. Susceptibility to primary Epstein-Barr virus infection is associated with interleukin-10 gene promoter polymorphism. J Infect Dis 184:777–80. [DOI] [PubMed] [Google Scholar]
- 79.Mowry SE, Strocker AM, Chan J, Takehana C, Kalantar N, Bhuta S, Shapiro NL. 2008. Immunohistochemical analysis and Epstein-Barr virus in the tonsils of transplant recipients and healthy controls. Arch Otolaryngol Head Neck Surg 134:936–9. [DOI] [PubMed] [Google Scholar]
- 80.Bauer CC, Aberle SW, Popow-Kraupp T, Kapitan M, Hofmann H, Puchhammer-Stockl E. 2005. Serum Epstein-Barr virus DNA load in primary Epstein-Barr virus infection. J Med Virol 75:54–8. [DOI] [PubMed] [Google Scholar]
- 81.Evans AS. 1974. The history of infectious mononucleosis. Am J Med Sci 267:189–95. [DOI] [PubMed] [Google Scholar]
- 82.Fugl A, Andersen CL. 2019. Epstein-Barr virus and its association with disease -a review of relevance to general practice. BMC Fam Pract 20:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fafi-Kremer S, Morand P, Brion JP, Pavese P, Baccard M, Germi R, Genoulaz O, Nicod S, Jolivet M, Ruigrok RW, Stahl JP, Seigneurin JM. 2005. Long-term shedding of infectious epstein-barr virus after infectious mononucleosis. J Infect Dis 191:985–9. [DOI] [PubMed] [Google Scholar]
- 84.Okano M, Kawa K, Kimura H, Yachie A, Wakiguchi H, Maeda A, Imai S, Ohga S, Kanegane H, Tsuchiya S, Morio T, Mori M, Yokota S, Imashuku S. 2005. Proposed guidelines for diagnosing chronic active Epstein-Barr virus infection. Am J Hematol 80:64–9. [DOI] [PubMed] [Google Scholar]
- 85.Silverstein A, Steinberg G, Nathanson M. 1972. Nervous system involvement in infectious mononucleosis. The heralding and-or major manifestation. Arch Neurol 26:353–8. [DOI] [PubMed] [Google Scholar]
- 86.Tselis AC. 2014. Epstein-Barr virus infections of the nervous System, p 286–305. In Tselis AC, Booss J (ed), Handbook of Clinical Neurology, vol 123. [DOI] [PubMed] [Google Scholar]
- 87.Burkitt D. 1970. Lesions outside the jaws. In Burkitt D, Wright DH (ed), Burkitt’s Lymphoma. E. and S. Livingstone, Edinburgh. [Google Scholar]
- 88.Biebl A, Webersinke C, Traxler B, Povysil B, Furthner D, Schmitt K, Weis S. 2009. Fatal Epstein-Barr virus encephalitis in a 12-year-old child: an underappreciated neurological complication? Nat Clin Pract Neurol 5:171–4. [DOI] [PubMed] [Google Scholar]
- 89.Terry LA, Stewart JP, Nash AA, Fazakerley JK. 2000. Murine gammaherpesvirus-68 infection of and persistence in the central nervous system. J Gen Virol 81:2635–43. [DOI] [PubMed] [Google Scholar]
- 90.Rasley A, Bost KL, Marriott I. 2004. Murine gammaherpesvirus-68 elicits robust levels of interleukin-12 p40, but not interleukin-12 p70 production, by murine microglia and astrocytes. J Neurovirol 10:171–80. [DOI] [PubMed] [Google Scholar]
- 91.Hausler M, Sellhaus B, Scheithauer S, Engler M, Alberg E, Teubner A, Ritter K, Kleines M. 2005. Murine gammaherpesvirus-68 infection of mice: A new model for human cerebral Epstein-Barr virus infection. Ann Neurol 57:600–3. [DOI] [PubMed] [Google Scholar]
- 92.Casiraghi C, Dorovini-Zis K, Horwitz MS. 2011. Epstein-Barr virus infection of human brain microvessel endothelial cells: a novel role in multiple sclerosis. J Neuroimmunol 230:173–7. [DOI] [PubMed] [Google Scholar]
- 93.Andersson J. 1996. Clinical and immunological considerations in Epstein-Barr virus-associated diseases. Scand J Infect Dis Suppl 100:72–82. [PubMed] [Google Scholar]
- 94.Foss HD, Herbst H, Hummel M, Araujo I, Latza U, Rancso C, Dallenbach F, Stein H. 1994. Patterns of cytokine gene expression in infectious mononucleosis. Blood 83:707–12. [PubMed] [Google Scholar]
- 95.Petrova M, Muhtarova M, Nikolova M, Magaev S, Taskov H, Nikolovska D, Krastev Z. 2006. Chronic Epstein-Barr virus-related hepatitis in immunocompetent patients. World J Gastroenterol 12:5711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kang MJ, Kim TH, Shim KN, Jung SA, Cho MS, Yoo K, Chung KW. 2009. Infectious mononucleosis hepatitis in young adults: two case reports. Korean J Intern Med 24:381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bernstein TC, Wolff HG. 1950. Involvement of the nervous system in infectious mononucleosis. Ann Intern Med 33:1120–38. [DOI] [PubMed] [Google Scholar]
- 98.Epstein S, Dameshek W. 1931. Involvement of the central nervous system in a case of glandular fever. N Engl J Med:1238–1241. [Google Scholar]
- 99.Kimiya T, Yagihashi T, Shinjoh M, Kai A, Sato Y. 2013. Presence of Epstein-Barr virus in cerebrospinal fluid from patients with aseptic meningitis appears to be common. Infection 41:1045–6. [DOI] [PubMed] [Google Scholar]
- 100.Benjamin LA, Kelly M, Cohen D, Neuhann F, Galbraith S, Mallewa M, Hopkins M, Hart IJ, Guiver M, Lalloo DG, Heyderman RS, Solomon T. 2013. Detection of herpes viruses in the cerebrospinal fluid of adults with suspected viral meningitis in Malawi. Infection 41:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Dyachenko P, Smiianova O, Kurhanskaya V, Oleshko A, Dyachenko A. 2018. Epstein-barr virus-associated encephalitis in a case-series of more than 40 patients. Wiad Lek 71:1224–1230. [PubMed] [Google Scholar]
- 102.Russell J, Fisher M, Zivin JA, Sullivan J, Drachman DA. 1985. Status epilepticus and Epstein-Barr virus encephalopathy. Diagnosis by modern serologic techniques. Arch Neurol 42:789–92. [DOI] [PubMed] [Google Scholar]
- 103.Greenberg DA, Weinkle DJ, Aminoff MJ. 1982. Periodic EEG complexes in infectious mononucleosis encephalitis. J Neurol Neurosurg Psychiatry 45:648–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Walsh FC, Poser CM, Carter S. 1954. Infectious mononucleosis encephalitis. Pediatrics 13:536–43. [PubMed] [Google Scholar]
- 105.Tselis A, Duman R, Storch GA, Lisak RP. 1997. Epstein-Barr virus encephalomyelitis diagnosed by polymerase chain reaction: detection of the genome in the CSF. Neurology 48:1351–5. [DOI] [PubMed] [Google Scholar]
- 106.Schellinger PD, Sommer C, Leithauser F, Schwab S, Storch-Hagenlocher B, Hacke W, Kiessling M. 1999. Epstein-Barr virus meningoencephalitis with a lymphoma-like response in an immunocompetent host. Ann Neurol 45:659–62. [DOI] [PubMed] [Google Scholar]
- 107.Doja A, Bitnun A, Jones EL, Richardson S, Tellier R, Petric M, Heurter H, Macgregor D. 2006. Pediatric Epstein-Barr Virus-Associated Encephalitis: 10-Year Review. J Child Neurol 21:385–391. [DOI] [PubMed] [Google Scholar]
- 108.Thomson DJ. 1975. Focal encephalitis in infectious mononucleosis simulating herpes simplex encephalitis: case report. Mil Med 140:188–9. [PubMed] [Google Scholar]
- 109.Abul-Kasim K, Palm L, Maly P, Sundgren PC. 2009. The neuroanatomic localization of Epstein-Barr virus encephalitis may be a predictive factor for its clinical outcome: a case report and review of 100 cases in 28 reports. J Child Neurol 24:720–6. [DOI] [PubMed] [Google Scholar]
- 110.Caruso JM, Tung GA, Gascon GG, Rogg J, Davis L, Brown WD. 2000. Persistent preceding focal neurologic deficits in children with chronic Epstein-Barr virus encephalitis. J Child Neurol 15:791–6. [DOI] [PubMed] [Google Scholar]
- 111.Sugita Y, Muta H, Ohshima K, Morioka M, Tsukamoto Y, Takahashi H, Kakita A. 2016. Primary central nervous system lymphomas and related diseases: Pathological characteristics and discussion of the differential diagnosis. Neuropathology 36:313–24. [DOI] [PubMed] [Google Scholar]
- 112.Bauer J, Bien CG, Lassmann H. 2002. Rasmussen’s encephalitis: a role for autoimmune cytotoxic T lymphocytes. Curr Opin Neurol 15:197–200. [DOI] [PubMed] [Google Scholar]
- 113.Wang X, Wang Y, Liu D, Wang P, Fan D, Guan Y, Li T, Luan G, An J. 2017. Elevated expression of EBV and TLRs in the brain is associated with Rasmussen’s encephalitis. Virol Sin 32:423–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Liu D, Wang X, Wang Y, Wang P, Fan D, Chen S, Guan Y, Li T, An J, Luan G. 2018. Detection of EBV and HHV6 in the Brain Tissue of Patients with Rasmussen’s Encephalitis. Virol Sin 33:402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang S, Feng J, Shi Y. 2016. Transient widespread cortical and splenial lesions in acute encephalitis/encephalopathy associated with primary Epstein-Barr virus infection. Int J Infect Dis 42:7–10. [DOI] [PubMed] [Google Scholar]
- 116.Menge T, Kieseier BC, Nessler S, Hemmer B, Hartung HP, Stuve O. 2007. Acute disseminated encephalomyelitis: an acute hit against the brain. Curr Opin Neurol 20:247–54. [DOI] [PubMed] [Google Scholar]
- 117.Leake JA, Albani S, Kao AS, Senac MO, Billman GF, Nespeca MP, Paulino AD, Quintela ER, Sawyer MH, Bradley JS. 2004. Acute disseminated encephalomyelitis in childhood: epidemiologic, clinical and laboratory features. Pediatr Infect Dis J 23:756–64. [DOI] [PubMed] [Google Scholar]
- 118.Mohsen H, Abu Zeinah GF, Elsotouhy AH, Mohamed K. 2013. Acute disseminated encephalomyelitis following infectious mononucleosis in a toddler. BMJ Case Rep 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tenembaum S, Chitnis T, Ness J, Hahn JS, International Pediatric MSSG. 2007. Acute disseminated encephalomyelitis. Neurology 68:S23–36. [DOI] [PubMed] [Google Scholar]
- 120.Kamel MG, Nam NT, Han NHB, El-Shabouny AE, Makram AM, Abd-Elhay FA, Dang TN, Hieu NLT, Huong VTQ, Tung TH, Hirayama K, Huy NT. 2017. Post-dengue acute disseminated encephalomyelitis: A case report and meta-analysis. PLoS Negl Trop Dis 11:e0005715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Caucheteux N, Maarouf A, Daelman L, Toupance O, Lavaud S, Tourbah A. 2013. Acute disseminated encephalomyelitis in two renal transplant patients: is there a role for Epstein-Barr virus reactivation? Mult Scler 19:1222–5. [DOI] [PubMed] [Google Scholar]
- 122.Shoji H, Kusuhara T, Honda Y, Hino H, Kojima K, Abe T, Watanabe M. 1992. Relapsing acute disseminated encephalomyelitis associated with chronic Epstein-Barr virus infection: MRI findings. Neuroradiology 34:340–2. [DOI] [PubMed] [Google Scholar]
- 123.Tolly TL, Wells RG, Sty JR. 1989. MR features of fleeting CNS lesions associated with Epstein-Barr virus infection. J Comput Assist Tomogr 13:665–8. [DOI] [PubMed] [Google Scholar]
- 124.Nakamura Y, Nakajima H, Tani H, Hosokawa T, Ishida S, Kimura F, Kaneko K, Takahashi T, Nakashima I. 2017. Anti-MOG antibody-positive ADEM following infectious mononucleosis due to a primary EBV infection: a case report. BMC Neurol 17:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Lang HL, Jacobsen H, Ikemizu S, Andersson C, Harlos K, Madsen L, Hjorth P, Sondergaard L, Svejgaard A, Wucherpfennig K, Stuart DI, Bell JI, Jones EY, Fugger L. 2002. A functional and structural basis for TCR cross-reactivity in multiple sclerosis. Nat Immunol 3:940–3. [DOI] [PubMed] [Google Scholar]
- 126.Kakalacheva K, Regenass S, Wiesmayr S, Azzi T, Berger C, Dale RC, Brilot F, Munz C, Rostasy K, Nadal D, Lunemann JD. 2016. Infectious Mononucleosis Triggers Generation of IgG Auto-Antibodies against Native Myelin Oligodendrocyte Glycoprotein. Viruses 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Papetti L, Figa Talamanca L, Spalice A, Vigevano F, Centonze D, Valeriani M. 2018. Predictors of Evolution Into Multiple Sclerosis After a First Acute Demyelinating Syndrome in Children and Adolescents. Front Neurol 9:1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Banwell B, Bar-Or A, Arnold DL, Sadovnick D, Narayanan S, McGowan M, O’Mahony J, Magalhaes S, Hanwell H, Vieth R, Tellier R, Vincent T, Disanto G, Ebers G, Wambera K, Connolly MB, Yager J, Mah JK, Booth F, Sebire G, Callen D, Meaney B, Dilenge ME, Lortie A, Pohl D, Doja A, Venketaswaran S, Levin S, Macdonald EA, Meek D, Wood E, Lowry N, Buckley D, Yim C, Awuku M, Cooper P, Grand’maison F, Baird JB, Bhan V, Marrie RA. 2011. Clinical, environmental, and genetic determinants of multiple sclerosis in children with acute demyelination: a prospective national cohort study. Lancet Neurol 10:436–45. [DOI] [PubMed] [Google Scholar]
- 129.Verhey LH, Branson HM, Shroff MM, Callen DJ, Sled JG, Narayanan S, Sadovnick AD, Bar-Or A, Arnold DL, Marrie RA, Banwell B, Canadian Pediatric Demyelinating Disease N. 2011. MRI parameters for prediction of multiple sclerosis diagnosis in children with acute CNS demyelination: a prospective national cohort study. Lancet Neurol 10:1065–73. [DOI] [PubMed] [Google Scholar]
- 130.Tantsis EM, Prelog K, Brilot F, Dale RC. 2013. Risk of multiple sclerosis after a first demyelinating syndrome in an Australian Paediatric cohort: clinical, radiological features and application of the McDonald 2010 MRI criteria. Mult Scler 19:1749–59. [DOI] [PubMed] [Google Scholar]
- 131.Woolf J. 2010. The Mystery of Lewis Carroll. St. Martin’s Press. [Google Scholar]
- 132.Copperman SM. 1977. “Alice in Wonderland” syndrome as a presenting symptom of infectious mononucleosis in children: a description of three affected young people. Clin Pediatr (Phila) 16:143–6. [DOI] [PubMed] [Google Scholar]
- 133.Eshel GM, Evov A, Lahat E, Brauman A. 1987. Alice in Wonderland syndrome, a manifestation of acute Epstein-Barr virus infection. Pediatr Infect Dis J 6:68. [DOI] [PubMed] [Google Scholar]
- 134.Weidenfeld A, Borusiak P. 2011. Alice-in-Wonderland syndrome—a case-based update and long-term outcome in nine children. Childs Nerv Syst 27:893–6. [DOI] [PubMed] [Google Scholar]
- 135.Liaw SB, Shen EY. 1991. Alice in Wonderland syndrome as a presenting symptom of EBV infection. Pediatr Neurol 7:464–6. [DOI] [PubMed] [Google Scholar]
- 136.Ho CS, Shen EY, Liaw SB, Huang FY. 1992. Clinical observation and neurological outcomes in “Alice in Wonderland” syndrome. Zhonghua Min Guo Xiao Er Ke Yi Xue Hui Za Zhi 33:89–95. [PubMed] [Google Scholar]
- 137.Mastria G, Mancini V, Vigano A, Di Piero V. 2016. Alice in Wonderland Syndrome: A Clinical and Pathophysiological Review. Biomed Res Int 2016:8243145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Hauptmann S, Meru N, Schewe C, Jung A, Hiepe F, Burmester G, Niedobitek G, Buttgereit F. 2001. Fatal atypical T-cell proliferation associated with Epstein-Barr virus infection. Br J Haematol 112:377–80. [DOI] [PubMed] [Google Scholar]
- 139.Durfey JQ, Allen JE. 1956. The Guillain-Barre syndrome as a manifestation of infectious mononucleosis; report of a case. N Engl J Med 254:279–80. [DOI] [PubMed] [Google Scholar]
- 140.Davie JC, Ceballos R, Little SC. 1963. Infectious Mononucleosis with Fatal Neuronitis. Arch Neurol 9:265–72. [DOI] [PubMed] [Google Scholar]
- 141.Hojberg L, Sondergard E, Pedersen C. 2005. A case of Epstein-Barr virus infection complicated with Guillain-Barre syndrome involving several cranial nerves. Scand J Infect Dis 37:522–4. [DOI] [PubMed] [Google Scholar]
- 142.Parra B, Lizarazo J, Jimenez-Arango JA, Zea-Vera AF, Gonzalez-Manrique G, Vargas J, Angarita JA, Zuniga G, Lopez-Gonzalez R, Beltran CL, Rizcala KH, Morales MT, Pacheco O, Ospina ML, Kumar A, Cornblath DR, Munoz LS, Osorio L, Barreras P, Pardo CA. 2016. Guillain-Barre Syndrome Associated with Zika Virus Infection in Colombia. N Engl J Med 375:1513–1523. [DOI] [PubMed] [Google Scholar]
- 143.Paterson BJ, Durrheim DN. 2012. Guillain-Barre syndrome. N Engl J Med 367:973; author reply 974. [DOI] [PubMed] [Google Scholar]
- 144.Rees JH, Soudain SE, Gregson NA, Hughes RA. 1995. Campylobacter jejuni infection and Guillain-Barre syndrome. N Engl J Med 333:1374–9. [DOI] [PubMed] [Google Scholar]
- 145.Roccatagliata L, Uccelli A, Murialdo A. 2001. Guillain-Barre syndrome after reactivation of varicella-zoster virus. N Engl J Med 344:65–6. [DOI] [PubMed] [Google Scholar]
- 146.Stegmann-Planchard S, Gallian P, Tressieres B, Leparc-Goffart I, Lannuzel A, Signate A, Laouenan C, Cabie A, Hoen B. 2019. Chikungunya, a risk factor for Guillain-Barre syndrome. Clin Infect Dis doi: 10.1093/cid/ciz625. [DOI] [PubMed] [Google Scholar]
- 147.Sejvar JJ, Baughman AL, Wise M, Morgan OW. 2011. Population incidence of Guillain-Barre syndrome: a systematic review and meta-analysis. Neuroepidemiology 36:123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kluin PM, Deckert M, Ferry JA. 2008. Primary diffuse large B-cell lymphoma of the CNS, p 240–1. In Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein, Thiele J, Vardiman JW (ed), WHO classification of tumors of haematopoietic and lymphoid tissues, 4th ed. International Agency for Research on Cancer, Lyon, France. [Google Scholar]
- 149.Paydas S. 2017. Primary central nervous system lymphoma: essential points in diagnosis and management. Med Oncol 34:61. [DOI] [PubMed] [Google Scholar]
- 150.Flinn IW, Ambinder RF. 1996. AIDS primary central nervous system lymphoma. Curr Opin Oncol 8:373–6. [DOI] [PubMed] [Google Scholar]
- 151.Bathla G, Hegde A. 2016. Lymphomatous involvement of the central nervous system. Clin Radiol 71:602–9. [DOI] [PubMed] [Google Scholar]
- 152.Deckert M, Engert A, Bruck W, Ferreri AJ, Finke J, Illerhaus G, Klapper W, Korfel A, Kuppers R, Maarouf M, Montesinos-Rongen M, Paulus W, Schlegel U, Lassmann H, Wiestler OD, Siebert R, DeAngelis LM. 2011. Modern concepts in the biology, diagnosis, differential diagnosis and treatment of primary central nervous system lymphoma. Leukemia 25:1797–807. [DOI] [PubMed] [Google Scholar]
- 153.Villano JL, Koshy M, Shaikh H, Dolecek TA, McCarthy BJ. 2011. Age, gender, and racial differences in incidence and survival in primary CNS lymphoma. Br J Cancer 105:1414–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kadan-Lottick NS, Skluzacek MC, Gurney JG. 2002. Decreasing incidence rates of primary central nervous system lymphoma. Cancer 95:193–202. [DOI] [PubMed] [Google Scholar]
- 155.Matinella A, Lanzafame M, Bonometti MA, Gajofatto A, Concia E, Vento S, Monaco S, Ferrari S. 2015. Neurological complications of HIV infection in pre-HAART and HAART era: a retrospective study. J Neurol 262:1317–27. [DOI] [PubMed] [Google Scholar]
- 156.Fraser E, Gruenberg K, Rubenstein JL. 2015. New approaches in primary central nervous system lymphoma. Chin Clin Oncol 4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gupta NK, Nolan A, Omuro A, Reid EG, Wang CC, Mannis G, Jaglal M, Chavez JC, Rubinstein PG, Griffin A, Abrams DI, Hwang J, Kaplan LD, Luce JA, Volberding P, Treseler PA, Rubenstein JL. 2017. Long-term survival in AIDS-related primary central nervous system lymphoma. Neuro Oncol 19:99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Fine HA, Mayer RJ. 1993. Primary central nervous system lymphoma. Ann Intern Med 119:1093–104. [DOI] [PubMed] [Google Scholar]
- 159.Enblad G, Martinsson G, Baecklund E, Hesselager G, Sundstrom C, Amini RM, Hagberg H. 2017. Population-based experience on primary central nervous system lymphoma 2000–2012: the incidence is increasing. Acta Oncol doi: 10.1080/0284186X.2016.1270465:1–12. [DOI] [PubMed] [Google Scholar]
- 160.Kleinschmidt-DeMasters BK, Damek DM, Lillehei KO, Dogan A, Giannini C. 2008. Epstein Barr virus-associated primary CNS lymphomas in elderly patients on immunosuppressive medications. J Neuropathol Exp Neurol 67:1103–11. [DOI] [PubMed] [Google Scholar]
- 161.Bataille B, Delwail V, Menet E, Vandermarcq P, Ingrand P, Wager M, Guy G, Lapierre F. 2000. Primary intracerebral malignant lymphoma: report of 248 cases. J Neurosurg 92:261–6. [DOI] [PubMed] [Google Scholar]
- 162.Roychowdhury S, Peng R, Baiocchi RA, Bhatt D, Vourganti S, Grecula J, Gupta N, Eisenbeis CF, Nuovo GJ, Yang W, Schmalbrock P, Ferketich A, Moeschberger M, Porcu P, Barth RF, Caligiuri MA. 2003. Experimental treatment of Epstein-Barr virus-associated primary central nervous system lymphoma. Cancer Res 63:965–71. [PubMed] [Google Scholar]
- 163.Amano M, Marutsuka K, Sugimoto T, Todaka T, Setoyama M. 2011. Epstein-Barr virus-associated primary central nervous system lymphoma in a patient with adult T-cell leukemia / lymphoma. J Dermatol 38:575–80. [DOI] [PubMed] [Google Scholar]
- 164.Smith JR, Falkenhagen KM, Coupland SE, Chipps TJ, Rosenbaum JT, Braziel RM. 2007. Malignant B cells from patients with primary central nervous system lymphoma express stromal cell-derived factor-1. Am J Clin Pathol 127:633–41. [DOI] [PubMed] [Google Scholar]
- 165.Rubenstein JL, Fridlyand J, Shen A, Aldape K, Ginzinger D, Batchelor T, Treseler P, Berger M, McDermott M, Prados M, Karch J, Okada C, Hyun W, Parikh S, Haqq C, Shuman M. 2006. Gene expression and angiotropism in primary CNS lymphoma. Blood 107:3716–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Venetz D, Ponzoni M, Schiraldi M, Ferreri AJ, Bertoni F, Doglioni C, Uguccioni M. 2010. Perivascular expression of CXCL9 and CXCL12 in primary central nervous system lymphoma: T-cell infiltration and positioning of malignant B cells. Int J Cancer 127:2300–12. [DOI] [PubMed] [Google Scholar]
- 167.Lim DH, Kim WS, Kim SJ, Yoo HY, Ko YH. 2015. Microarray Gene-expression Profiling Analysis Comparing PCNSL and Non-CNS Diffuse Large B-Cell Lymphoma. Anticancer Res 35:3333–40. [PubMed] [Google Scholar]
- 168.Norden AD, Drappatz J, Wen PY, Claus EB. 2011. Survival among patients with primary central nervous system lymphoma, 1973–2004. J Neurooncol 101:487–93. [DOI] [PubMed] [Google Scholar]
- 169.Rubenstein JL, Combs D, Rosenberg J, Levy A, McDermott M, Damon L, Ignoffo R, Aldape K, Shen A, Lee D, Grillo-Lopez A, Shuman MA. 2003. Rituximab therapy for CNS lymphomas: targeting the leptomeningeal compartment. Blood 101:466–8. [DOI] [PubMed] [Google Scholar]
- 170.Bonnan M, Ferrari S, Bertandeau E, Demasles S, Krim E, Miquel M, Barroso B. 2014. Intrathecal rituximab therapy in multiple sclerosis: review of evidence supporting the need for future trials. Curr Drug Targets 15:1205–14. [DOI] [PubMed] [Google Scholar]
- 171.Markowitz CE. 2013. Multiple sclerosis update. Am J Manag Care 19:s294–300. [PubMed] [Google Scholar]
- 172.Wallin MT, Culpepper WJ, Campbell JD, Nelson LM, Langer-Gould A, Marrie RA, Cutter GR, Kaye WE, Wagner L, Tremlett H, Buka SL, Dilokthornsakul P, Topol B, Chen LH, LaRocca NG, Workgroup USMSP. 2019. The prevalence of MS in the United States: A population-based estimate using health claims data. Neurology 92:e1029–e1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Battaglia MA, Zagami P, Uccelli MM. 2000. A cost evaluation of multiple sclerosis. J Neurovirol 6 Suppl 2:S191–3. [PubMed] [Google Scholar]
- 174.Parise H, Laliberte F, Lefebvre P, Duh MS, Kim E, Agashivala N, Abouzaid S, Weinstock-Guttman B. 2013. Direct and indirect cost burden associated with multiple sclerosis relapses: excess costs of persons with MS and their spouse caregivers. J Neurol Sci 330:71–7. [DOI] [PubMed] [Google Scholar]
- 175.Rodgers MM, Mulcare JA, King DL, Mathews T, Gupta SC, Glaser RM. 1999. Gait characteristics of individuals with multiple sclerosis before and after a 6-month aerobic training program. J Rehabil Res Dev 36:183–8. [PubMed] [Google Scholar]
- 176.Compston A, Coles A. 2008. Multiple sclerosis. Lancet 372:1502–17. [DOI] [PubMed] [Google Scholar]
- 177.Lublin FD, Reingold SC. 1996. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology 46:907–11. [DOI] [PubMed] [Google Scholar]
- 178.Confavreux C, Vukusic S. 2014. The clinical course of multiple sclerosis. Handb Clin Neurol 122:343–69. [DOI] [PubMed] [Google Scholar]
- 179.Rovaris M, Confavreux C, Furlan R, Kappos L, Comi G, Filippi M. 2006. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol 5:343–54. [DOI] [PubMed] [Google Scholar]
- 180.Venkatesan A, Johnson RT. 2014. Infections and multiple sclerosis. Handb Clin Neurol 122:151–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.He B, Yang B, Lundahl J, Fredrikson S, Hillert J. 1998. The myelin basic protein gene in multiple sclerosis: identification of discrete alleles of a 1.3 kb tetranucleotide repeat sequence. Acta Neurol Scand 97:46–51. [DOI] [PubMed] [Google Scholar]
- 182.Kellar-Wood H, Robertson N, Govan GG, Compston DA, Harding AE. 1994. Leber’s hereditary optic neuropathy mitochondrial DNA mutations in multiple sclerosis. Ann Neurol 36:109–12. [DOI] [PubMed] [Google Scholar]
- 183.Reynier P, Penisson-Besnier I, Moreau C, Savagner F, Vielle B, Emile J, Dubas F, Malthiery Y. 1999. mtDNA haplogroup J: a contributing factor of optic neuritis. Eur J Hum Genet 7:404–6. [DOI] [PubMed] [Google Scholar]
- 184.Thompson RJ, Mason CR, Douglas AJ, Hinks LJ, Dwarswaard A, Price SE. 1996. Analysis of polymorphisms of the 2’,3’-cyclic nucleotide-3’-phosphodiesterase gene in patients with multiple sclerosis. Mult Scler 2:215–21. [DOI] [PubMed] [Google Scholar]
- 185.Jersild C, Dupont B, Fog T, Platz PJ, Svejgaard A. 1975. Histocompatibility determinants in multiple sclerosis. Transplant Rev 22:148–63. [DOI] [PubMed] [Google Scholar]
- 186.Cook SD. 1997. Multiple sclerosis and viruses. Mult Scler 3:388–9. [DOI] [PubMed] [Google Scholar]
- 187.Tur C, Ramagopalan S, Altmann DR, Bodini B, Cercignani M, Khaleeli Z, Miller DH, Thompson AJ, Ciccarelli O. 2014. HLA-DRB1*15 influences the development of brain tissue damage in early PPMS. Neurology 83:1712–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sundstrom P, Nystrom L, Jidell E, Hallmans G. 2008. EBNA-1 reactivity and HLA DRB1*1501 as statistically independent risk factors for multiple sclerosis: a case-control study. Mult Scler 14:1120–2. [DOI] [PubMed] [Google Scholar]
- 189.Australia, New Zealand Multiple Sclerosis Genetics C. 2009. Genome-wide association study identifies new multiple sclerosis susceptibility loci on chromosomes 12 and 20. Nat Genet 41:824–8. [DOI] [PubMed] [Google Scholar]
- 190.De Jager PL, Jia X, Wang J, de Bakker PI, Ottoboni L, Aggarwal NT, Piccio L, Raychaudhuri S, Tran D, Aubin C, Briskin R, Romano S, International MSGC, Baranzini SE, McCauley JL, Pericak-Vance MA, Haines JL, Gibson RA, Naeglin Y, Uitdehaag B, Matthews PM, Kappos L, Polman C, McArdle WL, Strachan DP, Evans D, Cross AH, Daly MJ, Compston A, Sawcer SJ, Weiner HL, Hauser SL, Hafler DA, Oksenberg JR. 2009. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet 41:776–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Sawcer S, Hellenthal G. 2011. The major histocompatibility complex and multiple sclerosis: a smoking gun? Brain 134:638–40. [DOI] [PubMed] [Google Scholar]
- 192.International Multiple Sclerosis Genetics C, Hafler DA, Compston A, Sawcer S, Lander ES, Daly MJ, De Jager PL, de Bakker PI, Gabriel SB, Mirel DB, Ivinson AJ, Pericak-Vance MA, Gregory SG, Rioux JD, McCauley JL, Haines JL, Barcellos LF, Cree B, Oksenberg JR, Hauser SL. 2007. Risk alleles for multiple sclerosis identified by a genomewide study. N Engl J Med 357:851–62. [DOI] [PubMed] [Google Scholar]
- 193.Lin X, Deng FY, Lu X, Lei SF. 2015. Susceptibility Genes for Multiple Sclerosis Identified in a Gene-Based Genome-Wide Association Study. J Clin Neurol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Cree BA. 2014. Multiple sclerosis genetics. Handb Clin Neurol 122:193–209. [DOI] [PubMed] [Google Scholar]
- 195.Soldan SS, Jacobson S. Virus Induced Demyelination: The Case for Viruse(s) in Multiple Sclerosis, p 175–220, Neurotropic Viral INfections, vol 2: Neurotropic Retroviruses, DNA Viruses, Immunity and Transmission. Springer. [Google Scholar]
- 196.Warner HB, Carp RI. 1988. Multiple sclerosis etiology—an Epstein-Barr virus hypothesis. Med Hypotheses 25:93–7. [DOI] [PubMed] [Google Scholar]
- 197.Leibowitz U, Antonovsky A, Medalie JM, Smith HA, Halpern L, Alter M. 1966. Epidemiological study of multiple sclerosis in Israel. II. Multiple sclerosis and level of sanitation. J Neurol Neurosurg Psychiatry 29:60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Bach JF. 2002. The effect of infections on susceptibility to autoimmune and allergic diseases. N Engl J Med 347:911–20. [DOI] [PubMed] [Google Scholar]
- 199.Ascherio A, Munger KL. 2010. Epstein-barr virus infection and multiple sclerosis: a review. J Neuroimmune Pharmacol 5:271–7. [DOI] [PubMed] [Google Scholar]
- 200.DeLorenze GN, Munger KL, Lennette ET, Orentreich N, Vogelman JH, Ascherio A. 2006. Epstein-Barr virus and multiple sclerosis: evidence of association from a prospective study with long-term follow-up. Arch Neurol 63:839–44. [DOI] [PubMed] [Google Scholar]
- 201.Ascherio A, Munger KL, Lennette ET, Spiegelman D, Hernan MA, Olek MJ, Hankinson SE, Hunter DJ. 2001. Epstein-Barr virus antibodies and risk of multiple sclerosis: a prospective study. JAMA 286:3083–8. [DOI] [PubMed] [Google Scholar]
- 202.Serafini B, Rosicarelli B, Franciotta D, Magliozzi R, Reynolds R, Cinque P, Andreoni L, Trivedi P, Salvetti M, Faggioni A, Aloisi F. 2007. Dysregulated Epstein-Barr virus infection in the multiple sclerosis brain. J Exp Med 204:2899–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Levin LI, Munger KL, O’Reilly EJ, Falk KI, Ascherio A. 2010. Primary infection with the Epstein-Barr virus and risk of multiple sclerosis. Ann Neurol 67:824–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ruprecht K, Wunderlich B, Giess R, Meyer P, Loebel M, Lenz K, Hofmann J, Rosche B, Wengert O, Paul F, Reimer U, Scheibenbogen C. 2014. Multiple sclerosis: the elevated antibody response to Epstein-Barr virus primarily targets, but is not confined to, the glycine-alanine repeat of Epstein-Barr nuclear antigen-1. J Neuroimmunol 272:56–61. [DOI] [PubMed] [Google Scholar]
- 205.Lindsey JW, Hatfield LM, Vu T. 2010. Epstein-Barr virus neutralizing and early antigen antibodies in multiple sclerosis. Eur J Neurol 17:1263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Cepok S, Zhou D, Srivastava R, Nessler S, Stei S, Bussow K, Sommer N, Hemmer B. 2005. Identification of Epstein-Barr virus proteins as putative targets of the immune response in multiple sclerosis. J Clin Invest 115:1352–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Ascherio A, Munger KL. 2007. Environmental risk factors for multiple sclerosis. Part II: Noninfectious factors. Ann Neurol 61:504–13. [DOI] [PubMed] [Google Scholar]
- 208.Serafini B, Severa M, Columba-Cabezas S, Rosicarelli B, Veroni C, Chiappetta G, Magliozzi R, Reynolds R, Coccia EM, Aloisi F. 2010. Epstein-Barr virus latent infection and BAFF expression in B cells in the multiple sclerosis brain: implications for viral persistence and intrathecal B-cell activation. J Neuropathol Exp Neurol 69:677–93. [DOI] [PubMed] [Google Scholar]
- 209.Willis SN, Stadelmann C, Rodig SJ, Caron T, Gattenloehner S, Mallozzi SS, Roughan JE, Almendinger SE, Blewett MM, Bruck W, Hafler DA, O’Connor KC. 2009. Epstein-Barr virus infection is not a characteristic feature of multiple sclerosis brain. Brain 132:3318–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sargsyan SA, Shearer AJ, Ritchie AM, Burgoon MP, Anderson S, Hemmer B, Stadelmann C, Gattenlohner S, Owens GP, Gilden D, Bennett JL. 2010. Absence of Epstein-Barr virus in the brain and CSF of patients with multiple sclerosis. Neurology 74:1127–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Serafini B, Rosicarelli B, Magliozzi R, Stigliano E, Aloisi F. 2004. Detection of ectopic B-cell follicles with germinal centers in the meninges of patients with secondary progressive multiple sclerosis. Brain Pathol 14:164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Moreno MA, Or-Geva N, Aftab BT, Khanna R, Croze E, Steinman L, Han MH. 2018. Molecular signature of Epstein-Barr virus infection in MS brain lesions. Neurol Neuroimmunol Neuroinflamm 5:e466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Alotaibi S, Kennedy J, Tellier R, Stephens D, Banwell B. 2004. Epstein-Barr virus in pediatric multiple sclerosis. JAMA 291:1875–9. [DOI] [PubMed] [Google Scholar]
- 214.Banwell B, Krupp L, Kennedy J, Tellier R, Tenembaum S, Ness J, Belman A, Boiko A, Bykova O, Waubant E, Mah JK, Stoian C, Kremenchutzky M, Bardini MR, Ruggieri M, Rensel M, Hahn J, Weinstock-Guttman B, Yeh EA, Farrell K, Freedman M, Iivanainen M, Sevon M, Bhan V, Dilenge ME, Stephens D, Bar-Or A. 2007. Clinical features and viral serologies in children with multiple sclerosis: a multinational observational study. Lancet Neurol 6:773–81. [DOI] [PubMed] [Google Scholar]
- 215.Yea C, Tellier R, Chong P, Westmacott G, Marrie RA, Bar-Or A, Banwell B, Canadian Pediatric Demyelinating Disease N. 2013. Epstein-Barr virus in oral shedding of children with multiple sclerosis. Neurology 81:1392–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Pohl D, Krone B, Rostasy K, Kahler E, Brunner E, Lehnert M, Wagner HJ, Gartner J, Hanefeld F. 2006. High seroprevalence of Epstein-Barr virus in children with multiple sclerosis. Neurology 67:2063–5. [DOI] [PubMed] [Google Scholar]
- 217.Kwok H, Chan KW, Chan KH, Chiang AK. 2015. Distribution, persistence and interchange of Epstein-Barr virus strains among PBMC, plasma and saliva of primary infection subjects. PLoS One 10:e0120710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Nasrallah GK, Al Absi ES, Ghandour R, Ali NH, Taleb S, Hedaya L, Ali F, Huwaidy M, Husseini A. 2017. Seroprevalence of hepatitis E virus among blood donors in Qatar (2013–2016). Transfusion 57:1801–1807. [DOI] [PubMed] [Google Scholar]
- 219.Lunemann JD, Edwards N, Muraro PA, Hayashi S, Cohen JI, Munz C, Martin R. 2006. Increased frequency and broadened specificity of latent EBV nuclear antigen-1-specific T cells in multiple sclerosis. Brain 129:1493–506. [DOI] [PubMed] [Google Scholar]
- 220.Makhani N, Banwell B, Tellier R, Yea C, McGovern S, O’Mahony J, Ahorro JM, Arnold D, Sadovnick AD, Marrie RA, Bar-Or A, Canadian Pediatric Demyelinating Disease N. 2015. Viral exposures and MS outcome in a prospective cohort of children with acquired demyelination. Mult Scler doi: 10.1177/1352458515595876. [DOI] [PubMed] [Google Scholar]
- 221.Sundqvist E, Bergstrom T, Daialhosein H, Nystrom M, Sundstrom P, Hillert J, Alfredsson L, Kockum I, Olsson T. 2014. Cytomegalovirus seropositivity is negatively associated with multiple sclerosis. Mult Scler 20:165–73. [DOI] [PubMed] [Google Scholar]
- 222.Waubant E, Mowry EM, Krupp L, Chitnis T, Yeh EA, Kuntz N, Ness J, Chabas D, Strober J, McDonald J, Belman A, Milazzo M, Gorman M, Weinstock-Guttman B, Rodriguez M, Oksenberg JR, James JA, Network USPM. 2011. Common viruses associated with lower pediatric multiple sclerosis risk. Neurology 76:1989–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Virtanen JO, Wohler J, Fenton K, Reich DS, Jacobson S. 2014. Oligoclonal bands in multiple sclerosis reactive against two herpesviruses and association with magnetic resonance imaging findings. Mult Scler 20:27–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pfuhl C, Oechtering J, Rasche L, Giess RM, Behrens JR, Wakonig K, Freitag E, Pache FC, Otto C, Hofmann J, Eberspacher B, Bellmann-Strobl J, Paul F, Ruprecht K. 2015. Association of serum Epstein-Barr nuclear antigen-1 antibodies and intrathecal immunoglobulin synthesis in early multiple sclerosis. J Neuroimmunol 285:156–60. [DOI] [PubMed] [Google Scholar]
- 225.van Nierop GP, Mautner J, Mitterreiter JG, Hintzen RQ, Verjans GM. 2015. Intrathecal CD8 T-cells of multiple sclerosis patients recognize lytic Epstein-Barr virus proteins. Mult Scler doi: 10.1177/1352458515588581. [DOI] [PubMed] [Google Scholar]
- 226.Jaquiery E, Jilek S, Schluep M, Meylan P, Lysandropoulos A, Pantaleo G, Du Pasquier RA. 2010. Intrathecal immune responses to EBV in early MS. Eur J Immunol 40:878–87. [DOI] [PubMed] [Google Scholar]
- 227.Angelini DF, Serafini B, Piras E, Severa M, Coccia EM, Rosicarelli B, Ruggieri S, Gasperini C, Buttari F, Centonze D, Mechelli R, Salvetti M, Borsellino G, Aloisi F, Battistini L. 2013. Increased CD8+ T cell response to Epstein-Barr virus lytic antigens in the active phase of multiple sclerosis. PLoS Pathog 9:e1003220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Holmoy T, Kvale EO, Vartdal F. 2004. Cerebrospinal fluid CD4+ T cells from a multiple sclerosis patient cross-recognize Epstein-Barr virus and myelin basic protein. J Neurovirol 10:278–83. [DOI] [PubMed] [Google Scholar]
- 229.Lunemann JD, Jelcic I, Roberts S, Lutterotti A, Tackenberg B, Martin R, Munz C. 2008. EBNA1-specific T cells from patients with multiple sclerosis cross react with myelin antigens and co-produce IFN-gamma and IL-2. J Exp Med 205:1763–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.van Sechel AC, Bajramovic JJ, van Stipdonk MJ, Persoon-Deen C, Geutskens SB, van Noort JM. 1999. EBV-induced expression and HLA-DR-restricted presentation by human B cells of alpha B-crystallin, a candidate autoantigen in multiple sclerosis. J Immunol 162:129–35. [PubMed] [Google Scholar]
- 231.Vaughan JH, Riise T, Rhodes GH, Nguyen MD, Barrett-Connor E, Nyland H. 1996. An Epstein Barr virus-related cross reactive autoimmune response in multiple sclerosis in Norway. J Neuroimmunol 69:95–102. [DOI] [PubMed] [Google Scholar]
- 232.Esposito M, Venkatesh V, Otvos L, Weng Z, Vajda S, Banki K, Perl A. 1999. Human transaldolase and cross-reactive viral epitopes identified by autoantibodies of multiple sclerosis patients. J Immunol 163:4027–32. [PubMed] [Google Scholar]
- 233.Marquez AC, Horwitz MS. 2015. The Role of Latently Infected B Cells in CNS Autoimmunity. Front Immunol 6:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Pender MP, Csurhes PA, Burrows JM, Burrows SR. 2017. Defective T-cell control of Epstein-Barr virus infection in multiple sclerosis. Clin Transl Immunology 6:e126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Fraser KB, Haire M, Millar JH, McCrea S. 1979. Increased tendency to spontaneous in-vitro lymphocyte transformation in clinically active multiple sclerosis. Lancet 2:175–6. [PubMed] [Google Scholar]
- 236.Antsiferova O, Muller A, Ramer PC, Chijioke O, Chatterjee B, Raykova A, Planas R, Sospedra M, Shumilov A, Tsai MH, Delecluse HJ, Munz C. 2014. Adoptive transfer of EBV specific CD8+ T cell clones can transiently control EBV infection in humanized mice. PLoS Pathog 10:e1004333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, Moore CS, Michel L, Althekair F, Rajasekharan S, Gommerman JL, Prat A, Fillatreau S, Bar-Or A, Canadian BciMST. 2015. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med 7:310ra166. [DOI] [PubMed] [Google Scholar]
- 238.Sampey GC, Meyering SS, Zadeh MA, Saifuddin M, Hakami RM, Kashanchi F. 2014. Exosomes and their role in CNS viral infections. J Neurovirol 20:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Tzartos JS, Khan G, Vossenkamper A, Cruz-Sadaba M, Lonardi S, Sefia E, Meager A, Elia A, Middeldorp JM, Clemens M, Farrell PJ, Giovannoni G, Meier UC. 2012. Association of innate immune activation with latent Epstein-Barr virus in active MS lesions. Neurology 78:15–23. [DOI] [PubMed] [Google Scholar]
- 240.Sundqvist E, Sundstrom P, Linden M, Hedstrom AK, Aloisi F, Hillert J, Kockum I, Alfredsson L, Olsson T. 2012. Epstein-Barr virus and multiple sclerosis: interaction with HLA. Genes Immun 13:14–20. [DOI] [PubMed] [Google Scholar]
- 241.Agostini S, Mancuso R, Guerini FR, D’Alfonso S, Agliardi C, Hernis A, Zanzottera M, Barizzone N, Leone MA, Caputo D, Rovaris M, Clerici M. 2018. HLA alleles modulate EBV viral load in multiple sclerosis. J Transl Med 16:80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Zivadinov R, Weinstock-Guttman B, Zorzon M, Uxa L, Serafin M, Bosco A, Bratina A, Maggiore C, Grop A, Tommasi MA, Srinivasaraghavan B, Ramanathan M. 2009. Gene-environment interactions between HLA B7/A2, EBV antibodies are associated with MRI injury in multiple sclerosis. J Neuroimmunol 209:123–30. [DOI] [PubMed] [Google Scholar]
- 243.Pender MP. 2012. CD8+ T-Cell Deficiency, Epstein-Barr Virus Infection, Vitamin D Deficiency, and Steps to Autoimmunity: A Unifying Hypothesis. Autoimmune Dis 2012:189096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Jilek S, Schluep M, Harari A, Canales M, Lysandropoulos A, Zekeridou A, Pantaleo G, Du Pasquier RA. 2012. HLA-B7-restricted EBV-specific CD8+ T cells are dysregulated in multiple sclerosis. J Immunol 188:4671–80. [DOI] [PubMed] [Google Scholar]
- 245.Pender MP, Csurhes PA, Smith C, Douglas NL, Neller MA, Matthews KK, Beagley L, Rehan S, Crooks P, Hopkins TJ, Blum S, Green KA, Ioannides ZA, Swayne A, Aftab BT, Hooper KD, Burrows SR, Thompson KM, Coulthard A, Khanna R. 2018. Epstein-Barr virus-specific T cell therapy for progressive multiple sclerosis. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Santpere G, Darre F, Blanco S, Alcami A, Villoslada P, Mar Alba M, Navarro A. 2014. Genome-wide analysis of wild-type Epstein-Barr virus genomes derived from healthy individuals of the 1,000 Genomes Project. Genome Biol Evol 6:846–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lay ML, Lucas RM, Toi C, Ratnamohan M, Ponsonby AL, Dwyer DE. 2012. Epstein-Barr virus genotypes and strains in central nervous system demyelinating disease and Epstein-Barr virus-related illnesses in Australia. Intervirology 55:372–9. [DOI] [PubMed] [Google Scholar]
- 248.Brennan RM, Burrows JM, Bell MJ, Bromham L, Csurhes PA, Lenarczyk A, Sverndal J, Klintenstedt J, Pender MP, Burrows SR. 2010. Strains of Epstein-Barr virus infecting multiple sclerosis patients. Mult Scler 16:643–51. [DOI] [PubMed] [Google Scholar]
- 249.Dheekollu J, Malecka K, Wiedmer A, Delecluse HJ, Chiang AK, Altieri DC, Messick TE, Lieberman PM. 2017. Carcinoma-risk variant of EBNA1 deregulates Epstein-Barr Virus episomal latency. Oncotarget 8:7248–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Altmann M, Pich D, Ruiss R, Wang J, Sugden B, Hammerschmidt W. 2006. Transcriptional activation by EBV nuclear antigen 1 is essential for the expression of EBV’s transforming genes. Proc Natl Acad Sci U S A 103:14188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Harley JB, Chen X, Pujato M, Miller D, Maddox A, Forney C, Magnusen AF, Lynch A, Chetal K, Yukawa M, Barski A, Salomonis N, Kaufman KM, Kottyan LC, Weirauch MT. 2018. Transcription factors operate across disease loci, with EBNA2 implicated in autoimmunity. Nat Genet 50:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Afrasiabi A, Parnell GP, Fewings N, Schibeci SD, Basuki MA, Chandramohan R, Zhou Y, Taylor B, Brown DA, Swaminathan S, McKay FC, Stewart GJ, Booth DR. 2019. Evidence from genome wide association studies implicates reduced control of Epstein-Barr virus infection in multiple sclerosis susceptibility. Genome Med 11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Hu Y, Nie H, Yu HH, Qin C, Wu LJ, Tang ZP, Tian DS. 2019. Efficacy and safety of ituximab for relapsing-remitting multiple sclerosis: A systematic review and meta-analysis. Autoimmun Rev 18:542–548. [DOI] [PubMed] [Google Scholar]
- 254.Baker D, Jacobs BM, Gnanapavan S, Schmierer K, Giovannoni G. 2019. Plasma cell and B cell-targeted treatments for use in advanced multiple sclerosis. Mult Scler Relat Disord 35:19–25. [DOI] [PubMed] [Google Scholar]
- 255.Stuve O, Soelberg Soerensen P, Leist T, Giovannoni G, Hyvert Y, Damian D, Dangond F, Boschert U. 2019. Effects of cladribine tablets on lymphocyte subsets in patients with multiple sclerosis: an extended analysis of surface markers. Ther Adv Neurol Disord 12:1756286419854986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Yager JE, Magaret AS, Kuntz SR, Selke S, Huang ML, Corey L, Casper C, Wald A. 2017. Valganciclovir for the Suppression of Epstein-Barr Virus Replication. J Infect Dis 216:198–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Jiang L, Xie C, Lung HL, Lo KW, Law GL, Mak NK, Wong KL. 2018. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics 8:5307–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Hui KF, Yiu SPT, Tam KP, Chiang AKS. 2019. Viral-Targeted Strategies Against EBV-Associated Lymphoproliferative Diseases. Front Oncol 9:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, Tolvinski L, van den Heuvel APJ, Gu BW, Cassel JA, Tran DH, Wassermann BR, Zhang Y, Velvadapu V, Zartler ER, Busson P, Reitz AB, Lieberman PM. 2019. Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth. Sci Transl Med 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Haque T, Wilkie GM, Jones MM, Higgins CD, Urquhart G, Wingate P, Burns D, McAulay K, Turner M, Bellamy C, Amlot PL, Kelly D, MacGilchrist A, Gandhi MK, Swerdlow AJ, Crawford DH. 2007. Allogeneic cytotoxic T-cell therapy for EBV-positive posttransplantation lymphoproliferative disease: results of a phase 2 multicenter clinical trial. Blood 110:1123–31. [DOI] [PubMed] [Google Scholar]
- 261.Munz C. 2017. Epstein-Barr Virus-Specific Immune Control by Innate Lymphocytes. Front Immunol 8:1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Munz C. 2018. Human gamma-Herpesvirus Infection, Tumorigenesis, and Immune Control in Mice with Reconstituted Human Immune System Components. Front Immunol 9:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Cohen JI. 2015. Epstein-barr virus vaccines. Clin Transl Immunology 4:e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kanekiyo M, Bu W, Joyce MG, Meng G, Whittle JR, Baxa U, Yamamoto T, Narpala S, Todd JP, Rao SS, McDermott AB, Koup RA, Rossmann MG, Mascola JR, Graham BS, Cohen JI, Nabel GJ. 2015. Rational Design of an Epstein-Barr Virus Vaccine Targeting the Receptor-Binding Site. Cell 162:1090–100. [DOI] [PMC free article] [PubMed] [Google Scholar]