

Abstract
Introduction:
Toxicity of chemotherapy drugs is the leading cause of poor therapeutic outcome in many cancer patients. Gastrointestinal (GI) toxicity and hepatotoxicity are among the most common side effects of current chemotherapies. Emerging studies indicate that many chemotherapy-induced toxicities are driven by drug metabolism, but very few reviews summarize the role of drug metabolism in chemotherapy-induced GI toxicity and hepatotoxicity. In this review, we highlighted the importance of drug metabolizing enzymes (DMEs) in chemotherapy toxicity.
Areas covered:
Our review demonstrated that altered activity of DMEs play important role in chemotherapy-induced GI toxicity and hepatotoxicity. Besides direct changes in catalytic activities, the transcription of DMEs is also affected by inflammation, cell-signaling pathways and/or by drugs in cancer patients due to the disease etiology.
Expert opinion:
More studies should focus on how DMEs are altered during chemotherapy treatment, and how such changes affect the metabolism of chemotherapy drug itself. This mutual interaction between chemotherapies and DMEs can lead to excessive exposure of parent drug or toxic metabolites which ultimately cause GI adverse effect.
Keywords: Drug metabolizing enzymes, Chemotherapy, Reactive metabolites, Gastrointestinal toxicity, Hepatotoxicity
1. Introduction
Currently, gastrointestinal (GI) toxicity and hepatotoxicity are among the main reasons for the chemotherapeutic failures. Those adverse effects lead to dose reduction and therapy cycle discontinuation, which negatively impact the therapeutic outcomes of chemotherapy (1). Though several management strategies have been developed, the prevalence of these side effects remain high. Serotonin receptor antagonists and peripherally acting μ-opioid receptor agonists (e.g., loperamide) have been primarily used for treating vomiting and diarrhea, respectively. However, for other toxicities such as mucositis, colitis, steatosis, and hepatitis, very few effective prophylaxis treatments are available. Irinotecan (CPT-11), a topoisomerase I inhibitor, not only induces steatohepatitis but also causes grade 3 and 4 diarrhea in 25% of colorectal cancer patients, while the occurrence of 5-fluorouracil-induced (5-FU)-induced diarrhea is around 40% (2,3). HER-2 inhibitors used for breast cancer adjuvant treatment gives concerning safety profile as well: neratinib has extremely high occurrence of diarrhea (~95%) and lapatinib results in severe hepatotoxicity due to reactive metabolism (3). It is acknowledged that deficit of drug metabolizing enzymes (DMEs) contributes to the toxicities of many chemotherapy drugs. For example, cardio- and neurotoxicity of 5-FU, a thymidylate synthase inhibitor, is associated with dihydropyrimidine dehydrogenase (DPD) deficiency (4). Uridine-glucuronosyltransferase (UGT) 2B7 polymorphism has been suggested as a potential predictor of epirubicin-induced cardiotoxicity (5). However, overexpression of DMEs may account for chemotherapy-induced toxicity as well, if reactive metabolites are generated. The role of DMEs in chemotherapy-induced GI toxicity and hepatotoxicity is not well-studied, and it remains unclear how many chemotherapy-induced GI and hepatic toxicities are due to altered DME activity. Tao et al. recently found that irinotecan led to down-regulation of hepatic DME gene expression in mice (6). Understanding the role of DMEs in chemotherapy toxicity may prevent or attenuate the dysregulation of DMEs during chemotherapy treatment and reduce the adverse effects of chemotherapy drugs.
In this review, we describe the role of DMEs in chemotherapy-induced GI and hepatic toxicities, especially focused on reactive metabolism, altered expression of DMEs, polymorphism of DMEs and gut microbiota-mediated drug metabolism (Figure 1). We also discuss the regulation of DMEs by nuclear receptors, inflammatory signaling and Wnt/β-catenin signaling. These regulators and cellular pathways may be affected by chemotherapies. By the end, we provid examples of anti-cancer drugs, of which the GI toxicity and hepatotoxicity are mainly driven by drug metabolism.
Fig 1.
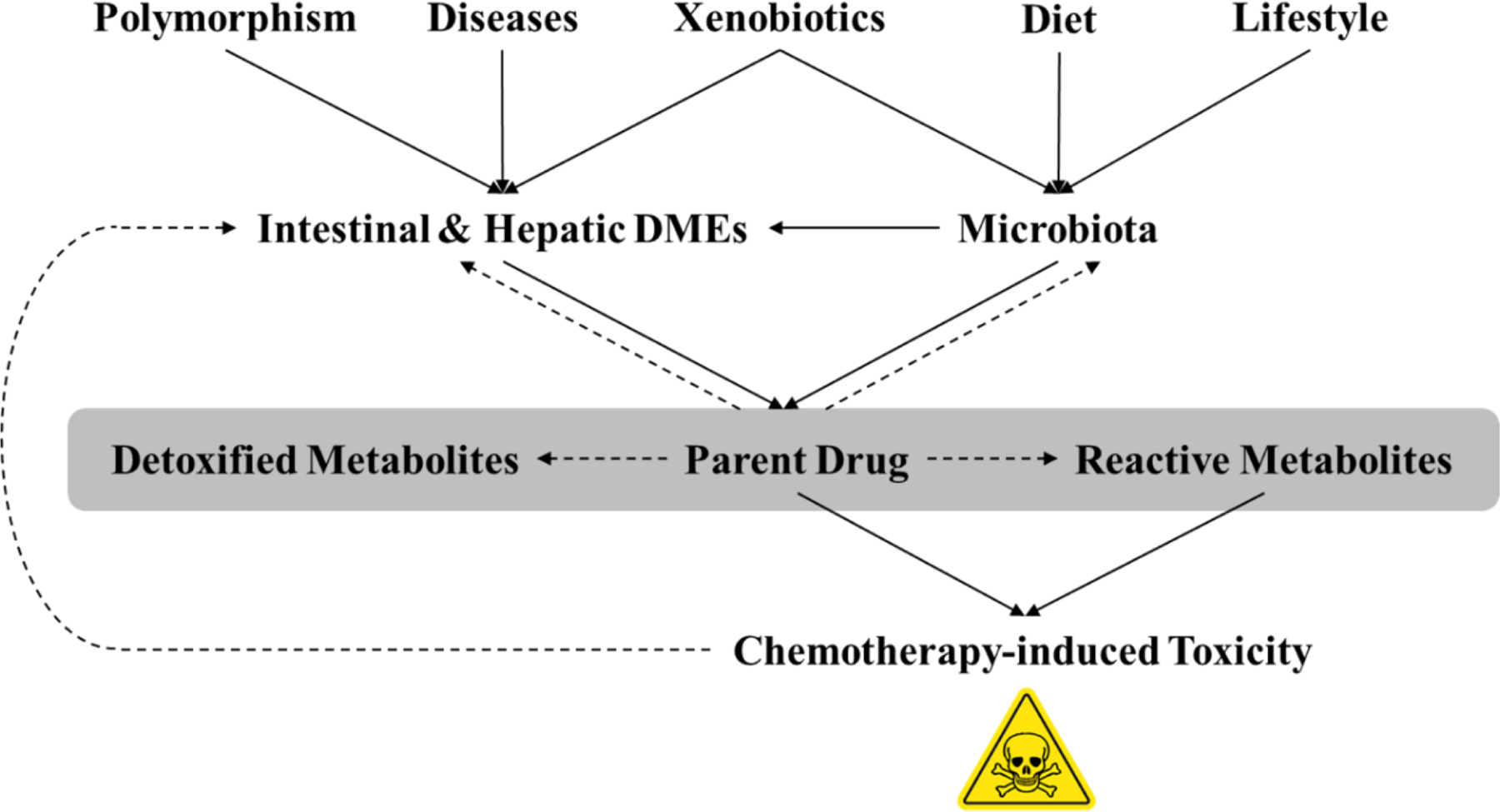
Altered intestinal and/or hepatic drug metabolism mediates chemotherapy-induced toxicity
2. Intestinal DMEs involved in drug metabolism-induced toxicity
Although small intestine in humans has been thought to be the main absorption site for orally administrated drugs, its metabolism function has been neglected. It has been assumed that the majority of first-pass metabolism happens in liver, but the role of intestinal metabolism in drug-drug interaction and GI toxicity is raising more concerns than ever before. It has been recognized that the ATP-binding cassette transporters of enterocytes are involved in the disposition of many chemotherapy drugs (7). However, the role of DMEs in GI tract has been largely overlooked. To the best of our knowledge, cytochromeP450 (CYP) and UDP-glucoronosyltransferase (UGT) families are the predominant DMEs existing in human gut (8).
2.1. Cytochrome P450 (CYP) enzymes in intestine
To date, several studies have been done to quantify the CYP content in human small intestine, which show a mean protein concentration ranging from 20 to 210 pmol/mg. This indicates that the intestinal CYP level is highly variable. The distribution of CYPs along small intestine is not uniform. Total CYP content in humans slightly increases from duodenum to jejunum and then decreases in ileum. Moreover, the expression of CYP enzymes varies within small intestinal villus, where highest concentration is found in differentiated enterocytes and the lower concentration in goblet cells (9). In human enterocytes, CYP1A1, CYP2C, CYP2D6, CYP2E1, CYP3A4, and CYP3A5 mRNAs were detected, whereas other important isoforms including, CYP1A2, CYP2A6, CYP2B6, CYP3A7, and CYP4B1were not detectable.(10) Immunoblotting shows that CYP3A4 and CYP3A5 enzymes are predominantly expressed in human duodenum and jejunum (10). Due to the high expression level, intestinal CYP3A enzyme is important for the metabolism of orally administrated drugs. Generally, the mucosal clearance of midazolam is strongly correlated with CYP3A activity. It is reported that the CYP3A content in duodenum, jejunum and ileum were 44%, 32% and 24% of total hepatic CYP3A content, respectively (11). Likewise, within human small intestine, midazolam clearance was remarkably lower in the distal portion than the proximal part (11). Several studies show that for certain drugs and nutrients, intestinal CYP3A causes more profound impact than transporters in oral bioavailability. It is reported that CYP3A but not P-gp plays a relevant role in the in vivo intestinal clearance of leniolisib, a δ-phosphoinositide-3 kinase inhibitor (12).
2.2. UDP-glucoronosyltransferase (UGT) family in intestine
Human UGT family has generally been classified into four subfamilies, including UGT1, UGT2, UGT3 and UGT8. Although liver is thought to be the main organ harboring most of UGT enzymes, increasing importance has been placed to the role of intestinal UGT in xenobiotic detoxification and first-pass metabolism. Unlike other UGT enzymes predominantly expressed in liver, in human, UGT1A10 and 2B17 were found mainly in GI tract. In human small intestine, UGT1A1, UGT1A8 and UGT1A6 account for 31%, 4% and 5% respectively of total UGT1A mRNA content (13). In colon, the percentage of UGT1A1 and UGT1A8 expression levels are 27% and 9% of total UGT1A level (13). UGT1A7 is only detected in the proximal portion of GI tract such as esophagus and stomach, but barely detectable in distal GI tract like colon.
3. Hepatic DMEs involved in drug metabolism-induced toxicity
It is well-known that hepatic DMEs mainly consists of phase I and phase II enzymes. Phase I enzymes, mainly composed of CYPs, generally catalyze oxidation and reduction, whereas phase II enzymes mediate the conjugation by adding large hydrophilic molecules to facilitate drug elimination. Of the 57 putatively functional human CYPs only the CYP1, 2, and 3 families are involved in the metabolism of xenobiotics, covering 70–80% of all drugs, whereas 2A6, 2D6, 2B6, and 2C19 are less abundant. The UGT enzymes are responsible for the majority of phase II metabolism. UGT enzymes not only catalyze xenobiotics but also endogenous molecules including bilirubin and steroids. This metabolic pathway is particularly variable due to the polymorphisms in the promoter of UGT-encoding genes. It is acknowledged that other than on-target and off-target effects, drug metabolism plays vital role in drug safety issues. Thus, there is a need to consider pharmacogenetics during new drug development. In addition, there are increasing concerns regarding the impact of cancer and associated inflammation on DMEs in patients taking chemotherapy.
3.1. Reactive metabolism and toxicity
Emerging evidence indicates that many of the intermediates generated from phase I metabolism is more active than the parent drug, and such by-products potentially cause undesired consequences.
3.1.1. CYP-mediated reactive metabolism
Acetaminophen (APAP) is one of the most classic examples causing hepatotoxicity due to reactive metabolism. In the cases of overdose, APAP undergoes CYP-mediated oxidation rather than sulfation to generate electrophilic intermediate, N-acetyl-p-benzoquinonimine (NAPQI) (14). Subsequently, NAPQI accumulates and covalently binds to a series of critical proteins leading to mitochondrial dysfunction (14). Nowadays, it is known that reactive quinone imine and quinone are responsible for most of the reactive metabolism-induced toxicity (15). Quinone imines and quinones react as Michael’s acceptors with cell proteins or DNA to promote inflammation and subsequent cell death.(15) Quinone imines and quinones also have high redox potential to form reactive oxygen species (ROS) (15). ROS triggers necrosis which leads to potential gut and liver injury. Mechanism-based inhibition is another CYP-involved reactive metabolism and causes withdrawal of many marketed drugs. Formation of a reactive metabolite in the catalytic pocket of CYP enzyme may lead to the loss of enzyme activity via the modification of CYP apoprotein. The case of tienilic acid and its isomer is an example of such inhibition. Rademacher et al. pointed out that tienilic acid-induced toxicity is initiated with mechanism-based inhibition (16). The 3-thenoyl regioisomer of tienilic acid, 2,3 dichloro-4-(thiophene3-carbonyl)-phenoxy]-acetic acid, is metabolized by CYP2C9 to generate reactive thiophene-S-oxide intermediate (16). Thiophene-S-oxide intermediate forms covalent adduct with CYP2C9 to diminish the enzyme function and initiate immune-mediated liver injury (16).
3.1.2. Flavin containing monooxygenase (FMO)-mediated reactive metabolism
In addition to CYP family, flavin containing monooxygenase (FMO) is also frequently involved in a variety of toxic metabolism. FMOs oxidize drugs containing a soft-nucleophile, usually nitrogen or sulfur. FMO does not require a reductase to transfer electrons from nicotinamide adenine dinucleotide phosphate (NADPH) and the catalytic cycle of FMO is strikingly different from CYPs (17). Recently, Petriello et al. demonstrated a novel diet-toxin interaction that results in high risk of cardiovascular toxicity (18). Exposure to dioxin-like polychlorinated biphenyls (PCBs) is strongly correlated with increased risk of cardiotoxicity, as hepatic FMO3 generates trimethylamine N-oxide (TMAO) poisoning to cardiovascular system. It is reported that PCBs increased FMO3 mRNA level and promoted TMAO plasma concentration in mice. Therefore, high risk of cardiovascular disease associated with the pollution with PCBs is likely due to the induction of FMO3 by PCBs leading to stimulation of TMAO formation.
3.1.3. Sulfotransferases (SULT)-mediated reactive metabolism
Sulfotransferases generate electrophilic metabolites from numerous dietary compounds, environmental pollutants and drugs, often leading to carcinogenicity. Shibutani et al. discovered that α-hydroxytamoxifen was a substrate of hydroxysteroid SULT, resulting in tamoxifen DNA adducts (19). The most well-studied SULT-mediated toxic metabolism is sulfation of furfuryl alcohol. Furfuryl alcohol, existing universally in foods, induces remarkable damage in small intestine. Monien et al. found that furfuryl alcohol was converted by intracellular sulfa-conjugation to 2-sulfooxymethylfuran, an electrophile reacting with DNA (20) The main DNA adducts, N2-(furan-2-yl)-methyl-2’-deoxyguanosine (N2-MFdG) and N6-(furan-2-yl)-methyl-2’-deoxyadenosine (N6-MFdA) were detected in mice dosed with furfuryl alcohol (20). Using transgenic mice, Sachse et al. further reported that the knockout of Sult1a1 led to significant decrease in DNA adducts in all tissues, whereas Sult1a2 depletion slightly attenuated the toxicity of furfuryl alcohol and the effect was limited to kidney and small intestine (21). Besides, SULTs are reported to be involved in 5-hydroxymethylfurfural, methyleugenol and aloe-emodin activation, which causes either hepatocarcinogenesis or in vitro cytotoxicity (22).
3.2. Polymorphism in DMEs and toxicity
It is known that polymorphism in DMEs contributes to interindividual variability in drug metabolism capacity and risk of toxicity (Figure 1). It is reported that genetic polymorphism of inosine triphosphate pyrophosphatase (ITPA) is a determinant factor in mercaptopurine metabolism and related toxicity (23). An et al. reported that patients with N-acetyltransferase 2 (282TT, 590AA and 857GA) alleles had higher susceptibility to anti-tuberculosis drug-induced hepatotoxicity (24).
3.2.1. CYP polymorphism and toxicity
CYP family, especially CYP3A5 CYP2C9, CYP2C19, and CYP2D6 are highly polymorphic genes. Such polymorphisms can cause a higher risk of drug metabolism-associated toxicity. CYP2A6 is a highly polymorphic gene which has currently 38 CYP2A6 alleles and many single nucleotide polymorphisms (SNPs). CYP2A6*2 is a SNP and CYP2A6*4 is a gene deletion variant. Both alleles result in the decreased CYP2A6 catalytic activity. As CYP2A6 is a major catalyst of nicotine metabolism, cigarette smokers of CYP2A6*4 allele are more susceptible to higher nicotine blood concentration. Such correlation was confirmed in a genome-wide association study in which a very significant association of nicotine-induced liver injury and CYP2A6*4 loci was found (25). Likewise, a strong correlation was found between the risk of voriconazole-induced hepatotoxicity and CYP2C19 mutant in Asian population (26).
3.3.2. UGT polymorphism and toxicity
UGT1A1 is the most abundantly expressed UGT isoform in liver. The UGT1A1 promoter contains a functional polymorphic TATA box (A(TA)5/6/7/8TAA) (27). Among these four A(TA)5/6/7/8TAA alleles, A(TA)6TAA and A(TA)7TAA are universal in all populations (28). Phenotype studies showed that the A(TA)7TAA allele is associated with Gilbert’s syndrome, characterized by reduced UGT1A1 activity as well as higher risk of irinotecan-induced toxicity (29). Barbier et al. reported that chenodeoxycholic acid, an endogenous activator of farnesoid X receptor (FXR), elevated UGT2B4 expression level in human primary hepatocytes (30). Promoter deletion studies demonstrated that FXR activators stimulated UGT2B4 transcription via a bile acid response element, termed B4-BARE (30). Agonists of peroxisome proliferator-activated receptor (PPARs) have also been shown to stimulate UGT2B4 expression in HepG2 and Huh7 cells. Further studies showed that PPAR agonists activated UGT2B4 promoter via DR1 PPAR response element (31).
3.3.3. Polymorphism of other DMEs and toxicity
Mammalian FMOs exist as six gene families and the major FMO isoform in human liver, FMO3, is responsible for N-oxygenation of trimethylamine (TMA). Great number of FMO3 mutant alleles have been described and associated with a disease termed trimethylaminuria (TMAU). The TMAU patient excretes large amounts of TMA in urine and sweat (17). The contribution of FMO3-mediated N-oxygenation in drug metabolism may be underestimated, as the N-oxide generated by FMO3 can be reduced back to the parent amine by CYP enzymes or other reductases (17). Tamoxifen, estrogen modulator with tertiary amines is reported to undergo such metabolism. By this mechanism, FMO-mediated N-oxygenation provides a reservoir of parent drug to prolong its action, but for TMAU patients this reservoir effect may be eliminated (17).
Thiopurine methyltransferase (TPMT) metabolizes the thiopurine drug, 6-mercaptopurine (6-MP) that is used as a prodrug to treat leukemia and Crohn’s disease. Aberrantly high level of 6-MP cause myelosuppression and myelotoxicity (32). It is known that TPMT activity can be predicted by genotyping and TPMT polymorphism is correlated to the risk of 6-MP toxicity. Based on several observational studies, heterozygosity or homozygosity for variant TPMT alleles yields odds ratios for myelotoxicity of 4.3 and 20.8 respectively. In fact, TPMT genotyping prior to administration of 6-MP is recommended by the FDA (32).
4. Gut microbiota-mediated chemotherapy toxicity
Several studies have reported that the gut microbiota modulated the host response to anticancer drugs by affecting drug efficacy and/or causing toxicity. Meanwhile, chemotherapies of cancer patients are known to have profound impact on the structure, diversity and abundance of gut microbiome. The complicated network between host immunity, gut microbiome and chemotherapy drugs remains to be fully understood. It is possible that gut microbiome may modulate the drug metabolism of anti-cancer drugs in a mutual manner (Figure 1). Alexander et al. recently summarized five key mechanisms driving such modulation between microbiota and drugs as ‘TIMER’ conceptual framework: Translocation, Immunomodulation, Metabolism, Enzymatic degradation, and Reduced diversity (33). This perspective was supported by Selwyn et al.’s study, which showed that hepatic Cyp3a gene expression was remarkably down-regulated in germ-free mice and treatment with probiotics significantly altered the level of several DMEs in liver (34).
4.1. Gut microbiota-mediated drug metabolism
The gut microbiota has the capability of performing a wide range of metabolic reactions on drugs, including reduction, hydrolysis and acetylation etc. A common example of gut microbiota-mediated drug metabolism is the reduction of antibacterial prodrugs derived from sulfanilamide, like prontosil and neoprontosil that both contain azo-dyes bearing the functional group diazenyl R−N=N−R′(35). Clostridia and Eubacteria were found to perform reduction upon azo-dyes and nitro-polycyclic aromatic hydrocarbons (35). Gut microbiome also shows nitro-reductase activity, which reduces nitro groups to amines. This is important for benzodiazepines derived drugs such as nitrazepam, clonazepam, and bromazepam (35). One of well-studied examples of gut microbiota-mediated nitro reduction is digoxin. It is recently found that the production of the metabolite, dihydrodigoxin, was highly variable inter-individually. The urinary excretion of dihydrodigoxin appeared to be stable over time after either taking single dose or multiple doses. However, when subjects were dosed with erythromycin, a broad-spectrum antibiotic, the renal elimination of dihydrodigoxin was no longer observed. Later, the microbe responsible for the reduction of digoxin was identified as Eubacterium lentum, although it was noted that the correlation between its presence and the production of dihydrodigoxin varies (36). Haiser et al. further revealed the role of a cardiac glycoside reductase (CGR) in such reductive metabolism. This CGR was found in the type strain of Eubacterium lena but not in nonreducing strains and its expression can be inhibited by dietary arginine, which explains the highly variable correlation between bacteria presence and production of metabolites (37).
Both the host and gut microbiota use hydrolytic enzyme to break down large molecules into smaller fragments for further metabolism. Hydrolase catalyzes the addition of a water molecule to the substrate, followed by bond cleavage. The most abundant microbial hydrolases in GI tract are proteases, glycosidases, and sulfatases. Whereas small intestine is dominated by pancreatic serine proteases, the colon mainly contains microbial cysteine- and metalloproteases (38). To improve poor solubility, many drugs are administered as prodrugs containing phosphate or sulfate ester, but the hydrolytic enzymes of gut microbiome readily act on these phosphate or sulfate moieties. In the case of laxative sodium picosulfate, which is administered as a disulfate, its efficacy depends on its conversion to the 4,4’-dihydroxydiphenyl-(2 pyridyl)-methane catalyzed by gut bacteria (39). The de-sulfation appears to be mediated by a novel sulfotransferase, rather than a sulfatase, and required the presence of phenolic compounds like phenol, acetaminophen, tannic acid, or flavonoids (40).
4.2. Chemotherapy toxicity driven by gut microbiota-mediated drug metabolism
It is well recognized that most of chemotherapy toxicity results from the parent drug or hepatic metabolites. However, with increasing evidence revealing the importance of gut microbial metabolism, it is reasonable to evaluate the role of gut microbiome in chemotherapy-induced toxicity. The drug-drug interaction between sorivudine and 5‑FU is a well-studied example. Sorivudine is hydrolyzed to (E)-5-(2‑bromovinyl)-uracil (BVU) through bacterial hydrolysis. Subsequently, BVU inhibits hepatic dihydropyrimidine dehydrogenase (DPD), the main enzyme responsible for detoxifying 5‑FU. The co-administration of sorivudine and 5‑FU leads to robust increase in systemic concentration of 5-FU, which cause fatal toxicity (41). Nakayama et al. demonstrated that the production of BVU is mainly mediated by Bacteroides species and administration of ampicillin, metronidazole, or a cocktail of antibiotics reduced BVU exposure in rats (42). Bacterial mediated de-conjugation is involved in irinotecan-induced GI toxicity like diarrhea. The reactive metabolite of irinotecan, SN-38 is detoxified by hepatic UGT1A1 to generate its glucuronide form SN-38G, and SN-38G is excreted into GI tract via bile duct. In the GI tract, SN-38G can be transformed back to SN-38 via deconjugation by bacterial β-glucuronidases, which results in the enterohepatic circulation of SN-38. It is well-established that the deconjugation of SN-38G in the gut and the enterohepatic circulation of SN-38 is responsible for the GI toxicity of irinotecan (43). It is reported that bacterial β-glucuronidases in colon is produced by specific species of the Enterobacteriaceae family, such as Escherichia coli, Lactobacillus, Streptococcus and Clostridium as well as the Actinobacteria family, like Bifidobacterium dentium (44). Irinotecan-induced dysbiosis is characterized by an increase in the Enterobacteriaceae family including E. coli and Clostridium spp (45). Further studies in rats showed that Clostridium spp. was the main species translocating to mesenteric lymph nodes and spleen accompanied with irinotecan-induced diarrhea (46). Based on these studies, bacterial β-glucuronidases inhibitors are believed to protect patients from irinotecan-induced diarrhea. However, non-specific inhibitors of intestinal bacterial β-glucuronidases cause accumulation of glycosaminoglycan in host tissues which is known as mucopolysaccharidosis type VII (45).
5. Regulation of DMEs
In addition to typical drug-drug interaction, certain pathological condition also leads to altered DMEs activity (47). The most extensively studied mechanisms are inflammatory signaling and Wnt/β-catenin signaling. Inflammatory responses involve toll-like receptors and cytokine receptors, whereas Wnt/β-catenin signaling showcase impact through the crosstalk with transcription factors and nuclear receptors (Figure 2). Such non-canonical regulation potentially leads to undesired effect like reduced drug clearance and higher risk of drug toxicity.
Fig 2. Regulation of DMEs expression by diseases and toxicity.
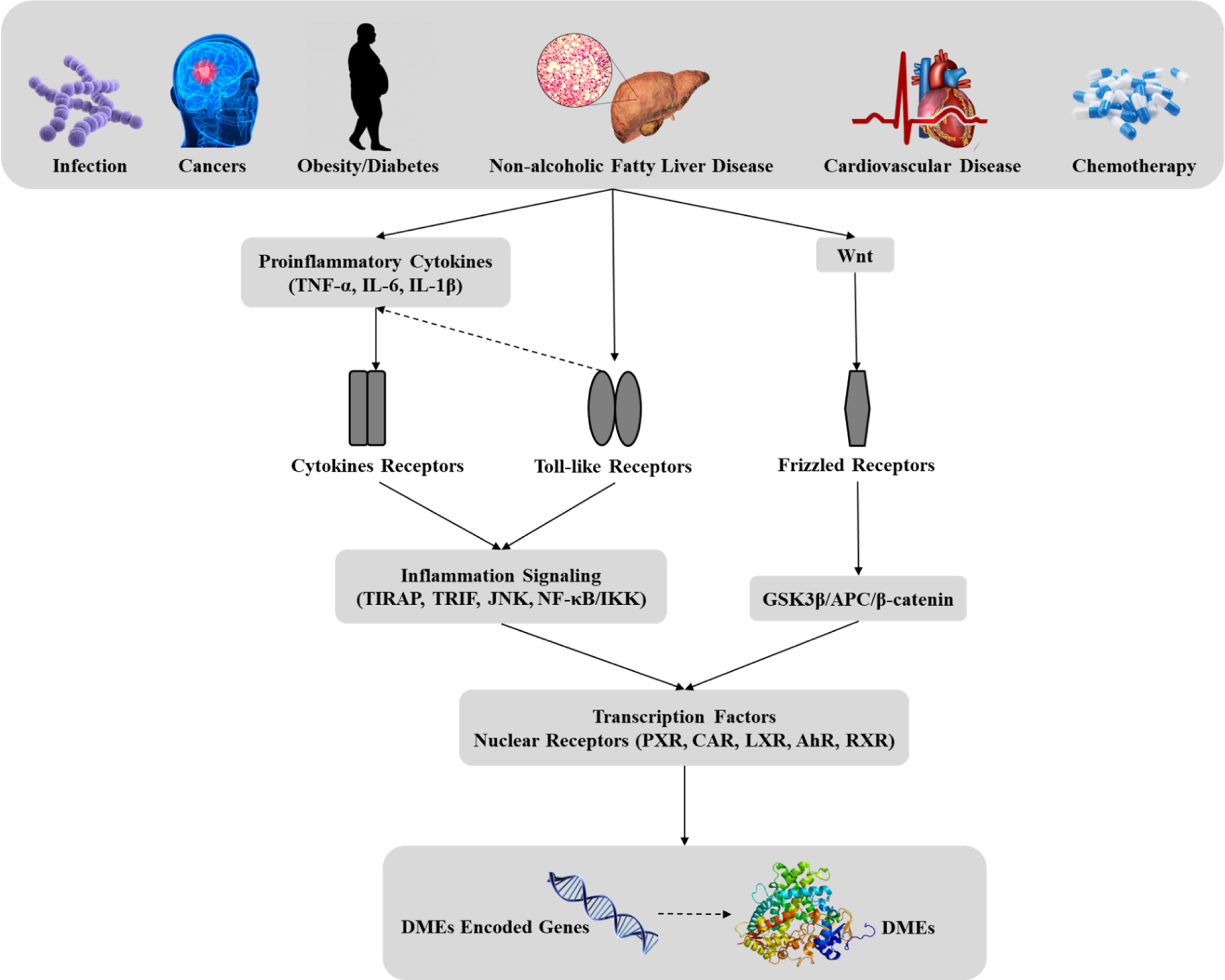
Abbreviations: TNF-α: tumor necrosis factor-α, IL-6: interleukin-6; TIRAP: Toll/interleukin-1-receptor-domain-containing adaptor protein, TRIF: TIR-domain-containing adapter-inducing interferon-β, JNK: c-Jun N-terminal kinases, NF-κB: nuclear factor κ-light-chain-enhancer of activated B cells, IKK: IκB kinase, GSK3β: Glycogen synthase kinase 3β, APC: Adenomatous polyposis coli protein, PXR: Pregnane X receptor, CAR: Constitutive androstane receptor, LXR: Liver X receptor, AhR: Aryl hydrocarbon receptor, RXR: Retinoid X receptor.
5.1. Regulation by nuclear receptors and transcription factors
It is known that DMEs expression is primarily regulated by nuclear receptors and transcription factors. For instance CYP3A family genes is regulated by a large family of nuclear receptors like pregnane X receptor (PXR), constitutive androstane receptor (CAR), retinoic acid receptor (RXR) the glucocorticoid receptor (GR), the vitamin D receptor (VDR), peroxisome proliferator-activated receptors (PPARα and PPARγ), hepatocyte nuclear factor 4α (HNF4α), FXR, and nuclear factor erythroid-related factor 2 (Nrf2) (48). The function of those nuclear receptors not only depends on the binding of endogenous ligands like steroids and bile acids, but also is activated by a wide range of xenobiotics. The classic examples inducing CYP gene expression via nuclear receptors in humans include rifampicin, phenobarbital etc. In liver, the transcription factors regulating phase II enzymes, especially UGT family, are similar to the ones controlling CYP genes. However, recently it was noted that the tissue-specific transcriptional regulation of UGTs involves different mechanism. Gregory et al. reported that caudal-related homeodomain protein 2 (CDX2) controls intestinal expression of UGT1A8, 1A10 and 2B7 (49). The most recent study pointed out that such CDX2-regulating UGT upregulation requires cooperation with HNF4α (50). In addition to regulating CYP families and UGTs, ligand-activated nuclear receptors regulate other DMEs as well including SULT, carboxylesterase (CES) and glutathione S-transferase (GST) (51).
5.2. Regulation by inflammation: Toll-like receptors and proinflammatory cytokines
Increasing studies indicate that most of hepatic DMEs decrease during inflammation, resulting in lower drug clearance. For xenobiotics mainly cleared by hepatic metabolism, this can lead to excessive drug exposure and increased risk of toxicity. Induction of DMEs is much less common during inflammation, but such rare induction will result in drug inefficacy, of which the example is increased CYP2A6 during certain inflammation (52). Emerging evidence shows that toll-like receptors (TLRs) and cytokine receptors play the main role in such regulation. Binding of pathogen-associated molecular patterns (PAMPs) or damage-associated molecular patterns (DAMPs) enable TLRs to activate intracellular kinases that ultimately stimulates the expression of proinflammatory cytokines such as interferons (IFNs), interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF)-α (Figure 2). In the liver, TLRs are widely expressed on monocytes, macrophages like Kupffer cells and hepatocytes (53). TLR4 is the receptor for bacterial lipopolysaccharide (LPS), and acts as the main sensor for gram-negative bacterial infections (54). In mice, LPS administration resulted in the downregulation of several CYP mRNAs as well as decreased Ugt1a1, Sult2a1 and Ces1 and 2 expression (55). Also, retinoid X receptor α (RXRα), one of the most important nuclear receptors in regulating DME expression, was also reduced upon LPS exposure. These effects were diminished in C3H/HeJ mice carrying a naturally null mutation of TLR4 (56). TLR4 is known to activate two distinct downstream pathways, one is through the adaptors, TIRAP (Toll/interleukin-1-receptor-domain-containing adaptor protein) and MyD88 (Myeloid differentiation primary response 88), which leads to the release of pro-inflammatory cytokines, and the second one is mediated by the adaptors, TRIF (TIR-domain containing adaptor protein inducing interferon-β) and TRAM (TRIF related adaptor molecule), which leads to the induction of IFN (57). However, Ghose et al. reported that LPS downregulated Cyp3a11 and Ugt1a1 in TIRAP−/− mice as well as TRIF−/− mice, suggesting that the TIRAP- and TRIF-dependent pathways can function independently in suppressing CYP (56,58). Recently, TLR4 was recently found to play a role in drug-drug interaction, in which paclitaxel upregulates irinotecan-related DMEs in a TLR4-dependent manner (59).
In addition to TLRs, proinflammatory cytokines like IL-6, IL-1 and TNF-α all get involved in the regulation of DMEs. Studies in primary rat hepatocytes demonstrated that IL-1, TNF-α, and IL-6 all downregulated the mRNA of Cyp2c11 (60). In human hepatocytes, CYP1A2, 2C, 2E1, and 3A expression were downregulated by IL-1β, IL-6, or TNF-α respectively (61). Furthermore, Aitken et al. reported that IL-6 downregulated CYP2B6, 2C8, 2C19,and 3A4 with different efficacies in human hepatocytes (62). In vivo evidence showed that in the absence of IL-6, null mice had no decrease in CYP mRNAs upon LPS exposure (61). Injection of polyclonal IL-6 antibodies in arthritic mice reversed the downregulation of hepatic CYP3A enzymes (61). Likewise, Tnfr1 and Tnfr2 double knockout or TNF-α −/− mice exhibited minimal changes in CYP expression with LPS injection (63). Recently, cytokines-mediated CYP inhibition has been observed in chemical-induced colitis. Hu et al. reported that mouse hepatic CYP1A2, 2B1, 2C6, 2C11, 2E1 and 3A1 activities were all reduced in dextran sulfate sodium-induced colitis model (64). Likewise, Erdmann et al. found that in patients with inflammatory bowel diseases, the gene expression of several intestinal DMEs including CYP2C9, UGT1A1 were decreased (65). In addition, it was reported by Mallick et al. that obesity-induced inflammation led to increased reactive metabolites which may cause higher risk of irinotecan toxicity (66). All those studies highlight the role of proinflammatory cytokines in altering drug metabolism and its consequences.
5.3. Regulation by Wnt/β-catenin signaling
The Wnt/β-catenin signaling is an evolutionarily conserved, highly complex pathway and is critical for liver development, differentiation and cellular homeostasis. The protein, β-catenin encoded by gene CTNNB1 is the central player in canonical Wnt pathway. In the absence of Wnt ligands, cytosolic β-catenin is sequestered in the cytoplasm by a protein complex consisting of adenomatous polyposis coli protein (APC) and glycogen synthase kinase 3β (GSK3β) (67). β-catenin is phosphorylated by GSK3β. Once Wnt binds with fizzled receptor on membrane, the activation of fizzled receptor will release β-catenin from the protein complex and facilitate its nuclear translocation (Figure 2) (68). In addition to its role in prenatal liver development, recent work highlighted that Wnt/β-catenin pathway was closely associated with ammonia metabolism, bile acid homeostasis and drug detoxification (69). It is increasingly recognized that Wnt/β-catenin signaling contributes significantly to the regulation of CYP enzymes, independent of inflammatory pathways (70). The initial clue suggesting the role of β-catenin in regulating CYP gene expression came from the studies using a phenobarbital-induced carcinogenesis model. Loeppen et al. first demonstrated that glutamine synthetase was up-regulated in Ctnnb1-mutated mouse liver tumors and served as a carcinogenesis marker. Later, they found that in glutamine synthetase positive tumors, several CYP isoforms increased as well, such as CYP1A, CYP2B, CYP2C and CYP2E1 (71). Recently, Briolotti et al. demonstrated that activation of β-catenin signaling by incubating primary mouse hepatocytes with GSK3β inhibitor led to an increase in Cyp1a1, 2b10, and 2e1 mRNA levels, due to the induction of aryl hydrocarbon receptor (AhR) or PXR (72). In mice with liver-specific knockout of Ctnnb1 gene, Cyp1a2 and Cyp2e1 expression was depleted. Sekine et al. also described reduced level of Cyp2c29 mRNA in Ctnnb1 knockout mice, whereas no changes were seen in the mRNA levels of Cyp1a1 and Cyp3a11 (73).
6. Chemotherapy drugs causing gastrointestinal toxicity and hepatotoxicity
The importance of drug metabolism in chemotherapy-induced GI toxicity and hepatotoxicity has been barely highlighted before. Here we discuss the detailed toxicological mechanism of a series of chemotherapy drugs, of which the GI and hepatic side effect is driven by reactive metabolism (Table 1, Figure 3). Reactive metabolism is found among alkylating agents, antimetabolites, topoisomerase inhibitors and tyrosine kinase inhibitors.
Table 1.
Chemotherapy drugs causing GI toxicity or hepatotoxicity due to reactive metabolisma
| Category | Drug | Involved DMEs | Drug metabolism-driven GI toxicity or hepatotoxicity | Ref. |
|---|---|---|---|---|
| Alkylating agents | Cyclophosphamide | CYP3A5, CYP2B6, CYP2C19 | The identified cytotoxic metabolites derived from cyclophosphamide are chloroacetaldehyde (CAA), acrolein, and phosphoramide mustard (PM), but acrolein and PM are the major toxins contributing to liver toxicity. HCY (4-hydroxy-cyclophosphamide) is formed primarily in the liver and enters cells as its tautomer aldocyclophosphamide (AldoCY). AldoCY is converted to acrolein and PM when decomposing through β-elimination. | (74) |
| Temozolomide | CYP1A1, CYP1A2 | Temozolomide is the prodrug of 3-methyl-(triazen-1-yl)-imidazole-4-carboxamide (MTIC). It degrades to active MTIC after oral administration in the absence of hepatic metabolism, which may contribute to GI toxicity. | (119) | |
| Antimetabolites | 5-Fluorouracil | Dihydropyrimidine dehydrogenase (DPD) | 5-fluorouracil (5-FU) is extensively metabolized in the liver via the microsomal enzyme system, and production of a toxic intermediate may trigger liver injury. While it is widely accepted that the overall toxicity of 5-FU is due to the polymorphism in drug metabolism enzymes, the inhibition of thymidylate synthase also contributes to the associated liver injury. | (78,120) |
| Cytarabine | Deoxycytidine kinase, Pyrimidine kinases | Cytarabine phosphorylation by deoxycytidine kinase is the rate-limiting step in its activation. The resulting cytarabine monophosphate is then further phosphorylated by pyrimidine kinases to the active 5´-triphosphate derivative, cytarabine -cytidine-5´-triphosphate. Such activation is liable for the GI and hepatic toxicity. | (121) | |
| Topoisomerase inhibitors | Etoposide /Teniposide | CYP3A4, CYP3A5 | Etoposide and Teniposide are converted into catechol and o-quinone derivatives by CYP3A enzymes in the liver and in lysosomes respectively. These metabolites are highly oxidative and can directly damage DNA, which may contribute to the adverse effects | (122) |
| Irinotecan | Carboxylesterase (CES), UGT, microbiota | Irinotecan is extensively metabolized in the liver and undergoes modification by CEs, CYP3A and UGT1A1. The UGT1A1 enzyme is responsible for the conjugation of bilirubin to toxic metabolites and is deficit in persons with Gilbert syndrome. As consequence, diarrhea causing by irinotecan is more common in patients with Gilbert syndrome. The cause of steatosis and steatohepatitis from irinotecan therapy is not known. | (33,86) | |
| Mitoxantrone | CYP3A, CYP2E1 | Incubated with liver S9 fraction, mitoxantrone generates five major metabolites, an acetoxy ester derivative, a monocarboxylic acid derivative, glutathione conjugates and the naphtoquinoxaline. Naphtoquinoxaline is responsible for mitoxantrone-induced hepatotoxicity. CYP3A and CYP2E1 inhibitor showed preventive effect against such toxicity. | (123) | |
| Tyrosine kinase inhibitors | Gefitinib | CYP3A, GSH | Gefitinib is designed with a fluorine at the ρ-position of a nitrogen-substituted aromatic ring. Oxidative defluorination, though uncommon in drug metabolism, has been identified as a minor metabolism pathway for gefitinib. GSH adducts were detected in liver microsomal incubations, which accounts for the potential hepatotoxicity. | (93,97) |
| Lapatinib | CYP3A4, CYP3A5 | Lapatinib is metabolized liver largely through the CYP 3A4 and CYP 3A5 pathways to produce protein adducts. Such protein adducts trigger immunogenicity in liver, which is correlated with the HLA alleles DQA1*02:01 and DRB1*07:01. Those mechanisms are responsible for potential diarrhea and hepatotoxicity. | (90,111) | |
| Ponatinib | CYP3A4, CYP3A5, CYP2C8, CYP2D6 | In vitro, ponatinib is mainly metabolized by CYP3A4, followed by CYP2C8, CYP2D6, and CYP3A5. Ponatinib-induced liver injury may be related to production of toxic intermediate. | (93) | |
| Pazopanib | CYP3A, GSH | Trapping studies performed in vitro showing the formation of adducts with methoxylamine and with GSH, suggests a link between reactive metabolites of pazopanib and CYP inhibition. These results concur with the aldehyde intermediates-induced hepatotoxicity in mice. | (93) | |
| Regorafenib | CYP3A, UGT | Arising from oxidative dechlorination and ether hydrolysis, intermediate susceptible for ρ-quinone-imine reactivation is detected in mice plasma. | (93) | |
| Sorafenib | CYP3A, CYP2C8, CYP2C9, CYP2C19 | Sorafenib is primarily metabolized by CYP3A4 and have been reported to cause vomiting, diarrhea, nausea, stomach pain. Recently sorafenib was shown to be a CYP3A, CYP2C8, CYP2C9, and CYP2C19 inactivator. | (93) | |
| Sunitinib | CYP3A4 | Sunitinib causes diarrhea in cancer patients, with an incidence ranging between 29% and 53%. Recently, in vitro studies showed that sunitinib exhibited a strong inhibition of CYP3A4 and a weak inhibition of CYP2C8. | (93,125) |
, Chemotherapy drugs are selected from the database of U.S. FDA, PubChem and DrugBank.ca.
Fig 3.
Chemotherapy drugs causing GI toxicity or hepatotoxicity due to reactive metabolites
6.1. Cyclophosphamide
Cyclophosphamide is an inactive prodrug for leukemia and lymphomas, which causes unpleasant GI symptoms, such as: nausea, vomiting, diarrhea, and mucositis of both intestine and mouth (1). Its hepatic metabolism into active form and other harmful metabolites is directly responsible for both the therapeutic and adverse effects of the drug. The metabolism of cyclophosphamide begins with its hydroxylation to 4-hydroxycyclophosphamide by various hepatic CYP enzymes, mainly CYP2B6, CYP2C9, CYP3A4 and great number of other minor enzymes (74). 4-hydroxycyclophosphamide is in equilibrium with aldophosphamide. Aldophosphamide diffuses out of the liver and enters peripheral tissues, where it spontaneously undergoes β-elimination to form reactive metabolites, phosphoramide mustard and acrolein (75). Phosphoramide mustard is a derivative of nitrogen mustards that are nonspecific DNA alkylating agents. Phosphoramide mustard contains two highly unstable and reactive chloroethyl groups that are readily eliminated to form a cyclic aziridinium ion. This cyclic aziridinium ion is prone to attack nucleophilic DNA molecules such as guanine. Aziridinium can make two linkages to guanine and cross link DNA both within and between strands (76). This results in cell cycle interruption and inhibition of cell growth. Rapidly growing GI mucosal epithelium can become damaged as a result of phosphoramide mustard. The tissue can atrophy and undergo inflammation through production of IL-1 and activation of the NF-κB/IKK pathway (77). The concurrent immunosuppression or destruction of neutrophils and alteration of the gut microbiome also leads to risk for bacterial infection of the GI tract and mouth. Cyclophosphamide-induced diarrhea can be attributed to malabsorption of water and other nutrients due to destruction of the GI barrier. Also, hyperplasia of goblet cells occurs which leads to excessive mucus secretion. The lack of absorption of compounds causes osmotic diuresis, which causes diarrhea in addition to the increased secretions.
6.2. 5‑Fluorouracil
It has been reported that 5‑fluorouracil (5-FU), an antineoplastic agent to treat multiple solid tumors including colorectal, breast and pancreatic cancers, causes severe GI toxicity due to decreased metabolic clearance in some patients, which is primarily caused by dihydropyrimidine dehydrogenase (DPD) deficiency. Wei et al. reported that in lymphocyte DNA, a G to A point mutation at the 5’-splicing site of consensus sequence leads to the skipping of entire exon during RNA transcription and processing (78). Hereby, they suggested that genotyping for the G to A splicing point mutation could be used to predict 5-FU toxicity. Paolo et al. further demonstrated that DPD activity in peripheral blood mononuclear cells (PBMNC) is not necessarily correlated to 5-FU disposition and incidence of 5-FU toxicity, but hepatic DPD activity and pharmacokinetics of 5-FU metabolite, 5-fluoro-5,6-dihydrouracil should be measured to monitor toxicity (4). Schwab et al. reported interesting interaction between gender and the predictability of DPD in 5-FU-induced GI toxicity. A pronounced correlation between DPYD genotype and 5-FU toxicity was found in male patients, but in female population 5-FU toxicity was independent of DPYD genotype (79). In addition to DPD, the same group indicated that methylene tetrahydrofolate reductase (MTHFR) 677C ≥ T was also correlated with better response to 5-FU chemotherapy and higher risk of GI toxicity (79). The mechanism of action of 5-FU is inhibiting thymidylate synthase (TS) which catalyzes the conversion of deoxyuridylate to deoxythymidylate for DNA synthesis (80). Pullarkat et al. found that TS genotype predicts not only TS mRNA level in tumors but also in normal liver tissue. Individuals homozygous for the triple repeat variant (L/L) had higher TS expression than those homozygous for the double repeat variant (S/S). Patients having S/L and S/S genotype are more vulnerable to 5-FU toxicity compared to those with L/L genotype (80). Recently, Alfzal et al. pointed out that combination of polymorphisms in multiple genes were better predictor for 5-FU GI toxicity rather than monitoring polymorphism in single gene (81). Currently, 5-FU dose is determined based on body surface area (BSA). Prado et al. argued that dose adjustment of 5-FU using BSA did not reduce toxicity. They suggested that a cut-off point of 20 mg 5-FU/kg of lean body mass was an effective predictor of 5-FU toxicity (82). Daniele et al. reported that patients orally dosed with glutamine were protected from 5-FU-induced GI toxicity, along with increased intestinal absorption and lower permeability (83,84).
6.3. Irinotecan
Since its first approval as topoisomerase I inhibitor in 1998, irinotecan has been widely used for treating colorectal, pancreatic, and lung cancers (85). Irinotecan is featured by severe GI toxicities and significant interindividual variability in pharmacokinetics. After being administrated via IV infusion, irinotecan generates SN-38 in liver and gut, a much more potent reactive metabolite, to achieve its efficacy. At physiological pH, the lactone-ring of irinotecan and SN-38 is hydrolyzed to carboxylate isoform, but when pH drops below 6 the reaction shifts reversibly (86). As only the lactone form shows anti-cancer activity, minor changes in pH will cause huge difference in the efficacy and pharmacokinetics of irinotecan. Conversion of irinotecan to SN-38 is mainly mediated by hepatic carboxylesterase (CESs1, 2). CES2 is reported to have higher affinity for irinotecan than CES1 and is therefore the major enzyme involved in this metabolism (86). In liver SN-38 is inactivated via glucuronidation to yield SN-38G by UGT1A family enzymes and subsequently excreted into bile (86). UGT1A1, UGT1A7 and UGT1A9 are the major hepatic isoforms detoxifying SN-38 to generate SN-38G. Emerging evidences point out that the polymorphism in UGT family genes and the imbalance of gut microbiota significantly contribute to irinotecan-induced toxicities. UGT1A1, the main enzyme responsible for SN-38 inactivation, is highly variable in different patients. Such unpredictable variability is due to the polymorphism of TATA box in UGT1A1 promoter. The presence of seven TA repeats, which is termed as the A(TA)7TAA allele and known as UGT1A1*28 variant, is associated with lower level of UGT1A1 expression. UGT1A1*28 occurs more frequently in African population and Caucasian population, compared to Asian population (87). Such polymorphism leads to higher risk of irinotecan-induced diarrhea in African-originated population.
Although several attempts have been made to attenuate irinotecan-induced GI toxicity, the clinical benefits of those strategies are not as satisfactory as expected. In a phase I trial conducted by Innocenti et al, a combination of cyclosporine and phenobarbital was tested. Cyclosporine, an ABCC2 (ATP-binding cassette transporter) and ABCB1 inhibitor was used to inhibit SN-38 excretion into bile, and phenobarbital was added to induce UGT1A1 (88). Cyclosporine plus phenobarbital showed 75% reduction of SN-38 AUC but minimal decrease in diarrhea incidence (88). Alternatively, it was reported that activated charcoal and calcium aluminosilicate clay effectively absorbed SN-38 in intestine. The activated charcoal has been found to reduce the incidence of irinotecan-induced diarrhea significantly in a phase II study (89). Since oxidative stress are involved in chemotherapy-induced toxicity, several antioxidant herbal medicines have been evaluated in vivo in attenuating irinotecan-induced mucositis (90,91). Recently, several preclinical studies showed novel drug delivery may diminish irinotecan GI toxicity. Liu et al. reported that in pancreatic cancer mouse model, lipid bilayer-coated mesoporous silica nanoparticle attenuate irinotecan-induced mucositis without compromising efficacy, via decreasing systemic drug leakage and increasing drug concentrations at the tumor sites (92).
6.4. Tyrosine kinase inhibitors
Reactivation and mechanism-based inhibition are the two typical mechanisms involved in drug metabolism-induced toxicity. The most common reactive metabolites derived from tyrosine kinase inhibitors (TKIs) include benzoquinone-imine, nitroso compound and dealkylates. The electrophilic benzoquinone-imine moiety resulting from CYP-mediated oxidation of aniline has been reported in several TKIs metabolism, like dasatinib, erlotinib, gefitinib, and lapatinib (93). Alkylamines are known to undergo CYP-mediated oxidation to generate reactive nitroso compounds that induce toxicity. O- and N-dealkylation are common drug metabolism pathways leading to aldehyde derivatives. Drugs containing primary alcohols and tertiary cyclic amines can also produce iminium and aldehyde metabolites via oxidation (93).
6.4.1. Lapatinib
In 2008, the U.S. FDA issued a black box warning for lapatinib idiosyncratic hepatotoxicity, which was observed during post-market surveillance. Lately, several clinical trials reported that hepatobiliary abnormalities, including grade 3 and 4 ALT/AST elevation (aspartate transaminase and alanine transaminase) were observed in breast cancer patients treated with lapatinib (94). Several research groups reported that inhibition of CYP3A4 by lapatinib was time-, concentration-, and NADPH-dependent. Addition of GSH did not affect the rate of inactivation, which suggests that the reactive metabolite of lapatinib was formed and inactivated by the enzyme prior to its release from the active site (95). Castellino et al. pointed out that incubation of either lapatinib or its dealkylated metabolite with human liver microsomes in the presence of GSH resulted in the formation of a reactive metabolite-GSH adduct (95). Additionally, Hardy et al. showed that a high-activity CYP3A5 genotype resulted in increased formation of the reactive metabolite-GSH conjugate in vitro (96). This suggests that genetic polymorphisms of CYP3A5 could affect the pharmacokinetics of lapatinib and impact the incidence of hepatotoxicity in vivo (96).
6.4.2. Gefitinib
Gefitinib, a first-generation EGFR TKI has been widely used to treat non-small cell lung cancer (NSCLC). However, server GI and pulmonary toxicities limit its therapeutic benefits. Li et al. showed that gefitinib can be bioactivated in hepatic, intestinal, and pulmonary microsomes to form reactive metabolites (97). Two CYP-dependent gefitinib-GSH adducts were detected in vitro, and CYP 1A1 and 3A4 were found to be mainly responsible for formation of adducts. Additionally, when incubating human pulmonary microsomes with gefitinib, a 12-fold increase in gefitinib-GSH adduct was detected in the microsomes from smokers over nonsmokers, which agrees with CYP1A1 being induced by cigarette smoke. Li et al. proposed that the mechanism of reactivation involved oxidative defluorination of gefitinib to form quinone-imine (97). Using recombinant CYP isoforms, CYP3A4 inhibitor, and S9 from Cyp3a-null mice, Liu et al. showed that CYP3A is the major enzyme contributing to the formation of aldehydes, GSH adducts, and primary amines from gefitinib (98). Further studies showed that CYP1A2, CYP2C, CYP2D6 and CYP3A4 all are capable to transform gefitinib into its iminium metabolite (98). Ma et al. reported that in NSCLC patients treated with gefitinib, CYP3A5 (rs776746/CYP3A5*3) was associated with severe diarrhea and hepatotoxicity. As for CYP reductase, the enzyme required for electron transfer to CYP, polymorphism Polymorphisms of cytochrome P450 reductase (the enzyme required for electron transfer to CYP, POR (rs17685/POR*11, rs1057868/POR*37) were associated with severe hepatotoxicity (99).
6.4.3. Other tyrosine kinase inhibitors
Bonvin et al. reported that acute hepatitis induced by dasatinib is due to the mechanism-based inhibition of CYP3A enzymes (100). Another research group showed that dasatinib-GSH adducts were detected when GSH and dasatinib were co-incubated in human liver microsomes (101). Dasatinib is reported to be activated by the formation of quinone-imine and imine-methide intermediates. The quinone-imine is formed via hydroxylation at the para-position of the 2-chloro-6-methylphenyl ring followed by further oxidation (101). More recently, it was reported that reactive intermediates are formed during the metabolism of erlotinib and the intermediates covalently conjugate to the cysteine group of GSH. CYP3A4 was found to be the primary enzyme responsible for the reactivation of erlotinib (102). CYP3A4 and CYP3A5 are irreversibly inactivated by erlotinib in a time- and concentration-dependent manner. The oxidation of erlotinib by CYP generates an epoxide that can react with the sulfhydryl group of cysteine (102). Using a biomimetic catalytic system composed of a metalloporphyrin and a single oxygen atom donor, Paludetto et al. identified several toxic metabolites of sunitinib and pazopanib. The metabolism profile of such metalloporphyrin-based catalyzation is identical with the incubation of human liver microsomes. Among these metabolites, aromatic aldehyde derivatives were unambiguously characterized as the intermediate causing toxicity (103).
7. Chemotherapy drugs altering DMEs
It is increasingly recognized that the chemotherapy induced hepatotoxicity is due to not only DMEs generating reactive metabolites but also owning to inhibition of DMEs by the drug itself. The most well-studied inhibitory mechanism is direct inhibition and mechanism-based inhibition (Table 2). Mechanism-based inhibition is caused by reactive metabolites that have the potential to permanently inactivate DMEs. One of the typical cases is the mechanism-based inhibition of CYP3A4 by lapatinib. Teng et al. suggested that lapatinib is an inactivator of CYP3A4 most likely via the formation and further oxidation of its O-dealkylated metabolite to a quinone imine that covalently modifies the CYP3A4 apoprotein and/or heme moiety (104). Later Barbara et al. showed that mechanism-based inhibition of CYP3A4 by lapatinib involved the formation of quasi-irreversible metabolic intermediate complex (105). Such metabolic intermediate complex formation is mediated via N-hydroxylation of the secondary amine group in lapatinib. Saracatinib is metabolized by CYP3A4 and in vitro experiments showed that saracatinib is a CYP3A time-dependent inhibitor, which may account for saracatinib nonlinear pharmacokinetics via CY3A autoinhibition (106). Recently, Filppula et al. used CYP3A4 and CYP2C8 probe substrate to demonstrate that sunitinib exhibited a strong inhibitory effect on CYP3A4 and a weak inhibition of CYP2C8 (106). In addition to the inhibition of enzymatic catalysis, emerging evidence indicate that chemotherapy may alter DMEs on transcription level via cellular pathways. Harmsen et al. demonstrated that paclitaxel, erlotinib, tamoxifen, ifosfamide, flutamide and docetaxel all activated PXR, while only strong PXR activation leads to increased CYP3A4 expression in human colon cancer cells (107). Mallick et al. reported that dysfunction of TLR4 attenuated paclitaxel-induced Cyp3a11 and Ugt1a1 expression in primary mouse hepatocytes, since paclitaxel is the agonist of both TLR4 and PXR (59). This study reveals the complexity of crosstalk between TLR4 and PXR pathways in regulating DMEs, while TLR4 activation generally reduce the level of DMEs; PXR activation induce the expression levels of DMEs but inhibit TLR4 signaling (108). Most recently, Tao et al. reported that the expression of Ugt1a1 in mouse liver is suppressed during irinotecan-induced steatosis likely in a TLR-dependent manner (6). Although the detailed mechanism underlying this discovery remained to be further explored, it provides a new perspective that DMEs may be influenced by chemotherapy on multiple levels.
Table 2.
Chemotherapy drugs altering DMEs
| Drug | Involved DMEs | Potential mechanism of altering DMEs | Ref. |
|---|---|---|---|
| Lapatinib | CYP3A4/5 | Mechanism-based inhibition arises from the oxidation of lapatinib O-dealkylated metabolite to a quinone imine which forms quasi-irreversible metabolic intermediate complex with CYP3A4. | (105) |
| Crizotinib | CYP3A4/5 | Mechanism-based inhibition. The CYP3A inactivation is likely due to the formation of ο-quinone-imine reactive metabolites. | (93) |
| Erlotinib | CYP3A4/5, CYP2C8 | Time dependent inhibition. CYP3A4/5 inactivation is thought to involved alkyne oxidation (oxirene or ketene reactive metabolites). CYP3A inactivation occur through covalent binding of reactive metabolites to the apoprotein. | (93) |
| Saracatinib | CYP3A4 | Time dependent inhibition. Saracatinib significantly increases (up to eight-fold) the exposure of a CYP3A probe substrate. Inhibition of CYP by saracatinib involves the piperazine ring bioactivation. | (93,126) |
| Sunitinib | CYP3A4, CYP2C8 | Time dependent inhibition. Reactive metabolites formation was reported: a ρ-quinone-imine reactive metabolite arising from oxidative defluorination and an aldehyde intermediate. | (93) |
| Erlotinib | CYP3A4 | Erlotinib induced CYP3A4 gene expression slightly in human colon adenocarcinoma-derived cell line at 10μM but reduced both protein and activity levels of CYP3A4. | (93) |
| Ifosfamide | CYP3A4 | Ifosfamide increased CYP3A4 expression slightly via PXR but did not change CYP3A4 activity. | (107) |
| Flutamide | CYP3A4 | Flutamide increased CYP3A4 at mRNA, protein and activity levels in human colon cancer cell line. | (107) |
| Paclitaxel | CYP3A4, CYP3A11, UGT1A1 | Paclitaxel increased CYP3A4 at both mRNA and protein levels in a PXR-dependent manner in human cancer cells. Deficiency of TLR4 decreased paclitaxel-induced Cyp3a11 and Ugt1a1 expression in mouse hepatocytes. TLR4-PXR crosstalk leads to potential drug-drug interaction. | (59,107) |
| Thiotepa | CYP2B6 | Thiotepa is a potent noncompetitive inhibitor of CYP2B6, with Ki values of 4.8 ± 0.3 and 6.2 ± 0.7μM. | (127) |
| Docetaxel | CYP3A4 | In human liver microsomes, time-dependent inhibition of CYP3A4 activity was reported, as co-administration of docetaxel and midazolam increased the AUC of midazolam by 45.3±21.2%. However, in human cell-based assay docetaxel induced CYP3A4 expression slightly. | (107,128) |
| Sirolimus | CYP3A4 | Time dependent inhibition with IC50= 0.3±0.1μM, it causes 54.8±6.4% shift in AUC of midazolam. | (128) |
| Temsirolimus | CYP3A4 | Time dependent inhibition, it causes 56.7±5.5% shift in AUC of midazolam. | (128) |
8. Conclusion
Drug metabolism-induced GI toxicity and hepatotoxicity have been significant obstacles during the development of chemotherapies for years and remain unresolved yet. Such adverse effects not only hamper therapeutic outcome but also make patients suffer from poor quality of life. Most of drug metabolism-induced toxicity of chemotherapy drugs are attributed to the reactive metabolites. However, increasing studies reveal that intestinal DMEs and gut microbiota also contribute to those toxicities. Intestinal DMEs mainly reduce the bioavailability of oral drugs, whereas microbiota may deactivate or reactivate chemotherapy drugs. Intriguingly, gut microbiota was recently found to affect the gene expression of hepatic DMEs. Those findings indicate that the gut microbiota is involved in chemotherapy drug metabolism and related toxicity. In addition to drug-drug interactions, polymorphisms, diseases states, such as inflammation and tumor burden all alter the expression and activity of DMEs via multiple pathways like TLRs/MyD88 and Wnt/β-catenin signaling. Those changes in DMEs can ultimately lead to chemotherapy GI toxicity and hepatotoxicity. In addition, chemotherapy drug itself affects DMEs profoundly as well, which demonstrates that the interaction between drugs and DMEs is a mutual process. Chemotherapy drugs may inhibit DME activity by adduct formation or mechanism-based inhibition. It may also regulate the gene expression of DMEs via activation of TLRs or nuclear receptors. Among current anticancer drugs, antimicrotubular agents like docetaxel and paclitaxel have the potential to change both expression and activity of DMEs. Tyrosine kinase inhibitors-induced GI and hepatic toxicities are largely owning to benzoquinone-imine moiety mediated DME inhibition. By understanding the role of DMEs in chemotherapy-induced toxicity, we may tailor the dose regimen to avoid excessive drug exposure and develop novel approaches to attenuate the dysregulation of DMEs due to chemotherapies.
9. Expert Opinion
Emerging evidence indicate that DMEs are the central player in many cases of chemotherapy-induced GI toxicity or hepatotoxicity, whereas the impact of altered expression and activity of DMEs on chemotherapy toxicity remains to be fully understood. Such research gap slows the development of new therapeutics and leads to poor therapeutic outcome in cancer patients. Moreover, it is increasingly realized that polymorphism, gut microbiome, diseases states, e.g. inflammation, tumor burden, as well as chemotherapy drug itself all can change the gene expression and activity of DMEs. Given that DMEs is regulated by numerous factors, it is challenging to predict undesired drug metabolism when drug candidates are evaluated in vitro and in laboratory animals that are oversimplified compared to the clinical scenario. Moreover, species differences make it difficult to explore how human DMEs change when cancer patients undergo aggressive chemotherapy. Targeting these changes in DMEs may reduce or prevent undesirable effect of chemotherapy drugs. However, current prophylaxis of chemotherapy-induced GI side effect follows standardized guideline and barely consider the contribution of DME to toxicity response. In general, accurate prediction and effective prevention remain as two main unachieved goals in the area of chemotherapy toxicity. To address those two concerns, the first step is to understand the molecular mechanism driving DME alteration in the cancer patients having chemotherapies.
As to predict toxic drug metabolism, hepatocytes sandwich culture and organ-on-chip are becoming prevalent method nowadays. In recent years, hepatocytes sandwich culture starts being accepted by pharmaceutical industry as supreme tool to detect drug-drug interaction and drug metabolism-related pitfall during drug discovery (109). Kumar et al. recently demonstrated that sandwich cultured hepatocytes showed more similar proteomic profile of transporters with liver tissues than suspended hepatocytes (110). Also, hepatocytes cultured in this way are relatively sensitive to the inducers of CYP and efflux transporters (111). Thanks to the advances in bioengineering, human organ-on-a-chip provides robust platform to simulate drug-DME interaction in vitro. Latest liver-on-a-chip model successfully composed hepatic spheroids, extracellular matrix and biliary system to establish in vitro homeostasis (112). Kim et al. recently fabricated a novel gut-on-a-chip model consisting of intestinal epithelium, peripheral blood mononuclear cells and microbes to mimic intestinal inflammatory bowel disease (113). Such human organ-on-a-chip technology incorporates variable influence of inflammation and microbes on hepatic and intestinal DMEs. It reduces use of animal and provides more confirmatory evidence for clinical study design. Besides in vitro screening platform, recent success in the development of humanized mouse offers unprecedented in vivo model for drug metabolism and toxicity study. Today, humanized CYP3A mice, humanized UGT1A mouse and humanized PXR or PPARγ mice are all commercially available for laboratory research about drug-drug interaction and toxic drug metabolism (114–116). Those humanized mice not only address the species-specific differences between rodents and humans, but also significantly improve the predictability of preclinical results. Such advantages make hepatocyte sandwich culture, organ-on-chip as well as humanized mouse as powerful assets to predict drug metabolism-induced toxicity.
Currently, most anti-diarrhea medicines are empirical therapies and the protective effect in clinical setting is not satisfactory. From our perspective, DMEs can be a promising target for tackling such drug safety issue. Given that intestinal inflammation usually come along with chemotherapy-induced GI toxicity and inflammation itself reduces DME, anti-inflammatory agents are probably a solution to this side effect. As C/EBPα (CCAAT/enhancer-binding protein alpha) is crucial in downregulating DMEs during inflammation, agonist of C/EBPα may attenuate relevant DME deficiency (117). In addition to inflammation, tumor burden, microbiome and drug-drug interaction all have potential to reduce DMEs in cancer patients, so it is difficult to customize prophylactics for each individual case. In recent studies, it was found that modifying diet can increase DME level and showed promising protective effect against irinotecan-induced steatosis (118). Although there are still a lot to be validated, this preliminary study inspires us that dietary approach can be a safe, effective and flexible method to halt chemotherapy toxicity by targeting DMEs.
Here we highlight the importance of DMEs in chemotherapy-induced gastrointestinal toxicity and hepatotoxicity. We demonstrate that the interaction between anticancer drugs and DMEs occur mutually. Thorough understanding of drug-DME interaction enables us to overcome the gap in translating toxicity study from bench to bed. Most importantly deeper insight into the regulation of DMEs in cancer patients help us achieve the goal of personalized medicine and maximize the therapeutic benefit of current chemotherapies.
Article Highlights.
Both intestinal and hepatic drug-metabolizing enzymes (DMEs) contribute to chemotherapy-induced gastrointestinal (GI) toxicity and hepatotoxicity.
Altered activity of DMEs leads to excessive accumulation of parent drug or generation of reactive metabolites and consequently cause GI and hepatic toxicities.
Alterations in DME activities depends on the regulation of DME gene expression and genetic polymorphism of DME.
Gene expression of DMEs is regulated by nuclear receptors, transcription factors that cross talk with inflammatory markers and other intracellular signaling pathways.
Gut microbiota are involved in drug metabolism-induced toxicity by mediating reactive metabolism and by changing the expression of DMEs.
References
Articles of special interest have been highlighted as either of interest (*) or of considerable interest (**) to readers.
- 1.Boussios S, Pentheroudakis G, Katsanos K, et al. Systemic treatment-induced gastrointestinal toxicity: Incidence, clinical presentation and management. Ann Gastroenterol. 2012;25(2):106–118. [PMC free article] [PubMed] [Google Scholar]
- 2.Kehrer DFS, Sparreboom A, Verweij J, et al. Modulation of irinotecan-induced diarrhea by cotreatment with neomycin in cancer patients. Clin Cancer Res. 2001;7(5):1136–41. [PubMed] [Google Scholar]
- 3.Paranjpe R, Basatneh D, Tao G, et al. Neratinib in HER2-Positive Breast Cancer Patients. Ann Pharmacother. 2019;53(6):612–620. [DOI] [PubMed] [Google Scholar]
- 4.Takimoto CH, Lu ZH, Zhang R, et al. Severe neurotoxicity following 5-fluorouracil-based chemotherapy in a patient with dihydropyrimidine dehydrogenase deficiency. Clin Cancer Res. 1996;2(3):477–481. [PubMed] [Google Scholar]
- 5.Innocenti F, Iyer L, Ramírez J, et al. Epirubicin glucuronidation is catalyzed by human UDP-glucuronosyltransferase 2B7. Drug Metab Dispos. 2001;29(5):686–692. [PubMed] [Google Scholar]
- 6.Tao G, Chityala PK, Ghose R. Differential Regulation of Hepatic UDP - glucuronosyltransferase (UGT) 1A1 by Toll - like Receptors during Irinotecan - induced Steatosis. FASEB J. 2020;34(S1):9–11. [Google Scholar]
- 7.Durmus S, Hendrikx JJMA, Schinkel AH. Apical ABC Transporters and Cancer Chemotherapeutic Drug Disposition 1st ed. Advances in Cancer Research: Elsevier Inc.; 2015 [DOI] [PubMed] [Google Scholar]
- 8.Prueksaritanont T, Gorham LM, Hochman JH, et al. Comparative studies of drug-metabolizing enzymes in dog, monkey, and human small intestines, and in Caco-2 cells. Drug Metab Dispos. 1996;24(6):634–642. [PubMed] [Google Scholar]
- 9.Obach RS, Zhang Q, Dunbar D, et al. METABOLIC CHARACTERIZATION OF THE MAJOR HUMAN SMALL INTESTINAL CYTOCHROME P450S. Drug Metab Dispos. 2001;29(3):347–52. [PubMed] [Google Scholar]
- 10.Xie F, Ding X, Zhang QY. An update on the role of intestinal cytochrome P450 enzymes in drug disposition. Acta Pharm Sin B. 2016;6(5):374–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dressman JB, Thelen K. Cytochrome P450-mediated metabolism in the human gut wall. J Pharm Pharmacol. 2009;61(5):541–558. [DOI] [PubMed] [Google Scholar]
- 12.De Buck S, Kucher K, Hara H, et al. CYP3A but not P-gp plays a relevant role in the in vivo intestinal and hepatic clearance of the delta-specific phosphoinositide-3 kinase inhibitor leniolisib. Biopharm Drug Dispos. 2018;39(8):394–402. [DOI] [PubMed] [Google Scholar]
- 13.Wu B, Kulkarni K, Basu S, et al. First-pass metabolism via UDP-glucuronosyltransferase: a barrier to oral bioavailability of phenolics. J Pharm Sci. 2012;101(7):2271–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ramachandran A, Jaeschke H. Acetaminophen toxicity: Novel insights into mechanisms and future perspectives. Gene Expr. 2018;18(1):19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Klopčič I, Dolenc MS. Chemicals and Drugs Forming Reactive Quinone and Quinone Imine Metabolites. Chem Res Toxicol. 2019;32(1):1–34. [DOI] [PubMed] [Google Scholar]
- 16.Rademacher PM, Woods CM, Huang Q, et al. Differential oxidation of two thiophene-containing regioisomers to reactive metabolites by cytochrome P450 2C9. Chem Res Toxicol. 2012;25(4):895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krueger SK, Williams DE. Mammalian flavin-containing monooxygenases: Structure/function, genetic polymorphisms and role in drug metabolism. Pharmacol Ther. 2005;106(3):357–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petriello MC, Hoffman JB, Sunkara M, et al. Dioxin-like pollutants increase hepatic flavin containing monooxygenase (FMO3) expression to promote synthesis of the pro-atherogenic nutrient biomarker trimethylamine N-oxide from dietary precursors. J Nutr Biochem. 2016;33:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shibutani S, Dasaradhi L, Terashima I, et al. α-hydroxytamoxifen is a substrate of hydroxysteroid (alcohol) sulfotransferase, resulting in tamoxifen DNA adducts. Cancer Res. 1998;58(4):647–653. [PubMed] [Google Scholar]
- 20.Monien BH, Herrmann K, Florian S, et al. Metabolic activation of furfuryl alcohol: Formation of 2-methylfuranyl DNA adducts in Salmonella typhimurium strains expressing human sulfotransferase 1A1 and in FVB/N mice. Carcinogenesis. 2011;32(10):1533–1539. [DOI] [PubMed] [Google Scholar]
- 21.Sachse B, Meinl W, Glatt H, et al. The effect of knockout of sulfotransferases 1a1 and 1d1 and of transgenic human sulfotransferases 1A1/1A2 on the formation of DNA adducts from furfuryl alcohol in mouse models. Carcinogenesis. 2014;35(10):2339–2245. [DOI] [PubMed] [Google Scholar]
- 22.Li R, Li W, You Y, et al. Metabolic Activation and Cytotoxicity of Aloe-Emodin Mediated by Sulfotransferases. Chem Res Toxicol. 2019;32(6):1281–1288. [DOI] [PubMed] [Google Scholar]
- 23.Stocco G, Cheok MH, Crews KR, et al. Genetic polymorphism of inosine triphosphate pyrophosphatase is a determinant of mercaptopurine metabolism and toxicity during treatment for acute lymphoblastic leukemia. Clin Pharmacol Ther. 2009;85(2):164–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.An HR, Wu XQ, Wang ZY, et al. NAT2 and CYP2E1 polymorphisms associated with antituberculosis drug-induced hepatotoxicity in Chinese patients. Clin Exp Pharmacol Physiol. 2012;39(6):535–543. [DOI] [PubMed] [Google Scholar]
- 25.Wassenaar CA, Dong Q, Wei Q, et al. Tyndale RF. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J Natl Cancer Inst. 2011;103(17):1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsumoto K, Ikawa K, Abematsu K, et al. Correlation between voriconazole trough plasma concentration and hepatotoxicity in patients with different CYP2C19 genotypes. Int J Antimicrob Agents. 2009;34(1):91–94. [DOI] [PubMed] [Google Scholar]
- 27.Yi J, Zhou HH. Polymorphism of UDP-glucuronosyltransferase 1A gene and drug metabolism. Curr Drug Metab. 2007;38(3):235–238. [PubMed] [Google Scholar]
- 28.Guillemette C, Millikan RC, Newman B, et al. Genetic polymorphisms in uridine diphospho-glucuronosyltransferase 1A1 and association with breast cancer among African Americans. Cancer Res. 2000;60(4):950–956. [PubMed] [Google Scholar]
- 29.Rouits E, Boisdron-Celle M, Dumont A, et al. Relevance of different UGT1A1 polymorphisms in irinotecan-induced toxicity: A molecular and clinical study of 75 patients. Clin Cancer Res. 2004;10(15):5151–5159. [DOI] [PubMed] [Google Scholar]
- 30.Barbier O, Torra IP, Sirvent A, et al. FXR induces the UGT2B4 enzyme in hepatocytes: A potential mechanism of negative feedback control of FXR activity. Gastroenterology. 2003;124(7):1926–1940. [DOI] [PubMed] [Google Scholar]
- 31.Barbier O, Duran-Sandoval D, Pineda-Torra I, et al. Peroxisome proliferator-activated receptor α induces hepatic expression of the human bile acid glucuronidating UDP-glucuronosyltransferase 2B4 enzyme. J Biol Chem. 2003;278(35):3252–3260. [DOI] [PubMed] [Google Scholar]
- 32.Khera S, Trehan A, Bhatia P, et al. Prevalence of TPMT, ITPA and NUDT 15 genetic polymorphisms and their relation to 6MP toxicity in north Indian children with acute lymphoblastic leukemia. Cancer Chemother Pharmacol. 2019;83(2):341–348. [DOI] [PubMed] [Google Scholar]
- 33.Alexander JL, Wilson ID, Teare J, et al. Gut microbiota modulation of chemotherapy efficacy and toxicity. Nat Rev Gastroenterol Hepatol. 2017;14(6):356–365.** The updated and comprehensive review summarizing the role of microbiota in chemotherapy drug metabolim and toxicity.
- 34.Selwyn FP, Cheng SL, Klaassen CD, et al. Regulation of hepatic drug-metabolizing enzymes in germ-free mice by conventionalization and probiotics. Drug Metab Dispos. 2016. February 1;44(2):262–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson ID, Nicholson JK. Gut microbiome interactions with drug metabolism, efficacy, and toxicity. Transl Res. 2017;179:204–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Robertson LW, Chandrasekaran A, Reuning RH, et al. Reduction of digoxin to 20R-dihydrodigoxin by cultures of Eubacterium lentum. Appl Environ Microbiol. 1986;51(6):1300–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Haiser HJ, Seim KL, Balskus EP, et al. Mechanistic insight into digoxin inactivation by Eggerthella lenta augments our understanding of its pharmacokinetics. Gut Microbes. 2014;5(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koppel N, Rekdal VM, Balskus EP. Chemical transformation of xenobiotics by the human gut microbiota. Science (80- ). 2017;356(6344):1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baek MC, Kwon AR, Chung YJ, et al. Distribution of bacteria with the arylsulfate sulfotransferase activity. Arch Pharm Res. 1998;21(4):475–457. [DOI] [PubMed] [Google Scholar]
- 40.Choi MS, Yu JS, Yoo HH, et al. The role of gut microbiota in the pharmacokinetics of antihypertensive drugs. Pharmacol Res. 2018;130:164–71. [DOI] [PubMed] [Google Scholar]
- 41.Ogura K, Nishiyama T, Takubo H, et al. Suicidal inactivation of human dihydropyrimidine dehydrogenase by (E)-5-(2-bromovinyl)uracil derived from the antiviral, sorivudine. Cancer Lett. 1998;122(1–2):107–113. [DOI] [PubMed] [Google Scholar]
- 42.Nakayama H, Kinouchi T, Kataoka K, et al. Intestinal anaerobic bacteria hydrolyse sorivudine, producing the high blood concentration of 5-(E)-(2-bromovinyl)uracil that increases the level and toxicity of 5-fluorouracil. Pharmacogenetics. 1997; 35–43. [DOI] [PubMed]
- 43.Sun R, Zhu L, Li L, et al. Irinotecan-mediated diarrhea is mainly correlated with intestinal exposure to SN-38 : Critical role of gut Ugt. Toxicol Appl Pharmacol. 2020;398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science (80- ). 2010;330(6005):831–835.* First-of-its-kind study to improve the therapeutic outcome of chemotherapy by intervening microbiota.
- 45.Chamseddine AN, Ducreux M, Armand JP, et al. Intestinal bacterial β-glucuronidase as a possible predictive biomarker of irinotecan-induced diarrhea severity. Pharmacol Ther 2019;199:1–15. [DOI] [PubMed] [Google Scholar]
- 46.Nakao T, Kurita N, Komatsu M, Yoshikawa K, et al. Irinotecan injures tight junction and causes bacterial translocation in rat. J Surg Res 2012;173(2):341–347. [DOI] [PubMed] [Google Scholar]
- 47.Gandhi A, Moorthy B, Ghose R. Drug Disposition in Pathophysiological Conditions. Curr Drug Metab. 2012. October 11;13(9):1327–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zollner G, Wagner M, Trauner M. Nuclear receptors as drug targets in cholestasis and drug-induced hepatotoxicity. Pharmacology and Therapeutics. 2010; 228–243. [DOI] [PubMed]
- 49.Gregory PA, Lewinsky RH, Gardner-Stephen DA, Mackenzie PI. Regulation of UDP glucuronosyltransferases in the gastrointestinal tract. Toxicol Appl Pharmacol. 2004;199(3):354–63. [DOI] [PubMed] [Google Scholar]
- 50.Mubarokah N, Hulin JA, Mackenzie PI, et al. Cooperative regulation of intestinal udp-glucuronosyltransferases 1A8, −1A9, and 1A10 by CDX2 and HNF4a is mediated by a novel composite regulatory elements. Mol Pharmacol. 2018;93(5):541–552. [DOI] [PubMed] [Google Scholar]
- 51.Ihunnah CA, Jiang M, Xie W. Nuclear receptor PXR, transcriptional circuits and metabolic relevance. Biochim Biophys Acta - Mol Basis Dis. 2011;1812(8):956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Satarug S, Lang MA, Yongvanit P, et al. Induction of cytochrome P450 2A6 expression in humans by the carcinogenic parasite infection, opisthorchiasis viverrini. Cancer Epidemiol Prev Biomarkers. 1996;5(10): 795–800. [PubMed] [Google Scholar]
- 53.Aitken AE, Richardson TA, Morgan ET. Regulation of Drug-Metabolizing Enzymes and Transporters in Inflammation. Annu Rev Pharmacol Toxicol. 2006;46(1):123–49. [DOI] [PubMed] [Google Scholar]
- 54.Lu YC, Yeh WC, Ohashi PS. LPS/TLR4 signal transduction pathway. Cytokine. 2008;42(2):145–151. [DOI] [PubMed] [Google Scholar]
- 55.Ghose R, Guo T, Vallejo JG, et al. Differential role of toll-interleukin 1 receptor domain-containing adaptor protein in toll-like receptor 2-mediated regulation of gene expression of hepatic cytokines and drug-metabolizing enzymes. Drug Metab Dispos. 2011;39(5):874–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shah P, Omoluabi O, Moorthy B, Ghose R. Role of adaptor protein toll-like interleukin domain containing adaptor inducing interferon b in toll-like receptor 3- and 4-mediated regulation of hepatic drug metabolizing enzyme and transporter genes. Drug Metab Dispos. 2016;44(1):61–67.* Well illustrated study to reveal the detailed mechanism underlying how inflammation decrease DMEs.
- 57.Kagan JC, Su T, Horng T, et al. TRAM couples endocytosis of Toll-like receptor 4 to the induction of interferon-β. Nat Immunol. 2008;9(4):361–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shah P, Guo T, Moore DD, Ghose R. Role of constitutive androstane receptor in toll-like receptor-mediated regulation of gene expression of hepatic drug-metabolizing enzymes and transporters. Drug Metab Dispos. 2014;42(1):172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mallick P, Basu S, Moorthy B, Ghose R. Role of Toll-like receptor 4 in drug-drug interaction between paclitaxel and irinotecan in vitro. Toxicol Vitr. 2017;41:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chen JQ, Strom A, Gustafsson JA, et al. Suppression of the constitutive expression of cytochrome P-450 2C11 by cytokines and interferons in primary cultures of rat hepatocytes: Comparison with induction of acute-phase genes and demonstration that CYP2C11 promoter sequences are involved in the s. Mol Pharmacol. 1995;47(5):940–7. [PubMed] [Google Scholar]
- 61.Siewert E, Bort R, Kluge R, et al. Hepatic cytochrome P450 down-regulation during aseptic inflammation in the mouse is interleukin 6 dependent. Hepatology. 2000;32(1):49–55. [DOI] [PubMed] [Google Scholar]
- 62.Aitken AE, Morgan ET. Gene-specific effects of inflammatory cytokines on cytochrome P450 2C, 2B6 and 3A4 mRNA levels in human hepatocytes. Drug Metab Dispos. 2007;35(9):1687–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ashino T, Oguro T, Shioda S, et al. Involvement of interleukin-6 and tumor necrosis factor α in CYP3A11 and 2C29 down-regulation by bacillus Calmette-Guerin and, lipopolysaccharide in mouse liver. Drug Metab Dispos. 2004;32(7):707–714. [DOI] [PubMed] [Google Scholar]
- 64.Hu N, Huang Y, Gao X, et al. Effects of dextran sulfate sodium induced experimental colitis on cytochrome P450 activities in rat liver, kidney and intestine. Chem Biol Interact [Internet]. 2017;271:48–58. [DOI] [PubMed] [Google Scholar]
- 65.Erdmann P, Bruckmueller H, Martin P, et al. Dysregulation of Mucosal Membrane Transporters and Drug-Metabolizing Enzymes in Ulcerative Colitis. J Pharm Sci. 2019;108(2):1035–1046. [DOI] [PubMed] [Google Scholar]
- 66.Mallick P, Shah P, Gandhi A, Ghose R. Impact of obesity on accumulation of the toxic irinotecan metabolite, SN-38, in mice. Life Sci. 2015;132–138. [DOI] [PubMed]
- 67.Nusse R, Clevers H. Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell. 2017;169(6):985–999. [DOI] [PubMed] [Google Scholar]
- 68.Braeuning A, Köhle C, Buchmann A, et al. Coordinate regulation of cytochrome P450 1a1 expression in mouse liver by the aryl hydrocarbon receptor and the β-catenin pathway. Toxicol Sci. 2011;122(1):16–25. [DOI] [PubMed] [Google Scholar]
- 69.Thompson MD, Monga SPS. WNT/β-catenin signaling in liver health and disease. Hepatology. 2007;45(5):1298–1305. [DOI] [PubMed] [Google Scholar]
- 70.Braeuning A Regulation of Cytochrome P450 Expression by Ras- and β-Catenin-Dependent Signaling. Curr Drug Metab. 2009;10(2):138–158. [DOI] [PubMed] [Google Scholar]
- 71.Loeppen S, Koehle C, Buchmann A, et al. A β-catenin-dependent pathway regulates expression of cytochrome P450 isoforms in mouse liver tumors. Carcinogenesis. 2005;26(1):239–248. [DOI] [PubMed] [Google Scholar]
- 72.Briolotti P, Chaloin L, Balaguer P, et al. Analysis of glycogen synthase kinase inhibitors that regulate cytochrome P450 expression in primary human hepatocytes by activation of β-catenin, aryl hydrocarbon receptor and pregnane X receptor signaling. Toxicol Sci. 2015;148(1):261–275. [DOI] [PubMed] [Google Scholar]
- 73.Sekine S, Lan BYA, Bedolli M, et al. Liver-specific loss of β-catenin blocks glutamine synthesis pathway activity and cytochrome P450 expression in mice. Hepatology. 2006;43(4):817–825. [DOI] [PubMed] [Google Scholar]
- 74.McDonald GB, Slattery JT, Bouvier ME, et al. Cyclophosphamide metabolism, liver toxicity, and mortality following hematopoietic stem cell transplantation. Blood. 2003;101(5):2043–2048. [DOI] [PubMed] [Google Scholar]
- 75.Jain M, Fan J, Baturay NZ, et al. Sulfonyl-containing aldophosphamide analogues as novel anticancer prodrugs targeted against cyclophosphamide-resistant tumor cell lines. J Med Chem. 2004;47(15):3843–3852. [DOI] [PubMed] [Google Scholar]
- 76.Petrillo SK, Desmeules P, Truong TQ, et al. Detection of DNA damage in oocytes of small ovarian follicles following phosphoramide mustard exposures of cultured rodent ovaries in vitro. Toxicol Appl Pharmacol. 2011;253(2):94–102. [DOI] [PubMed] [Google Scholar]
- 77.McQuade RM, Stojanovska V, Abalo R, et al. Chemotherapy-induced constipation and diarrhea: Pathophysiology, current and emerging treatments. Front Pharmacol. 2016;7:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wei X, McLeod HL, McMurrough J, et al. Molecular basis of the human dihydropyrimidine dehydrogenase deficiency and 5-fluorouracil toxicity. J Clin Invest. 1996;98(3):610–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schwab M, Zanger UM, Marx C, et al. Role of genetic and nongenetic factors for fluorouracil treatment-related severe toxicity: A prospective clinical trial by the German 5-FU toxicity study group. J Clin Oncol. 2008;26(13):2131–2138. [DOI] [PubMed] [Google Scholar]
- 80.Pullarkat ST, Lenz HJ. Thymidylate synthase gene polymorphism determines response and toxicity of 5-FU chemotherapy. Pharmacogenomics J. 2001;1(1):65–70. [DOI] [PubMed] [Google Scholar]
- 81.Afzal S, Gusella M, Vainer B, et al. Combinations of polymorphisms in genes involved in the 5-fluorouracil metabolism pathway are associated with gastrointestinal toxicity in chemotherapy-treated colorectal cancer patients. Clin Cancer Res. 2011;17(11):3822–3829. [DOI] [PubMed] [Google Scholar]
- 82.Prado CMM, Baracos VE, McCargar LJ, et al. Body composition as an independent determinant of 5-fluorouracil-based chemotherapy toxicity. Clin Cancer Res. 2007;13(11):3264–3268. [DOI] [PubMed] [Google Scholar]
- 83.Daniele B, Perrone F, Gallo C, et al. Oral glutamine in the prevention of fluorouracil induced intestinal toxicity: A double blind, placebo controlled, randomised trial. Gut. 2001;48(1):28–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Saif MW, Diasio RB. Benefit of uridine triacetate (Vistogard) in rescuing severe 5-fluorouracil toxicity in patients with dihydropyrimidine dehydrogenase (DPYD) deficiency. Cancer Chemother Pharmacol. 2016;78(1):151–156. [DOI] [PubMed] [Google Scholar]
- 85.Xu Y, Villalona-Calero MA. Irinotecan: Mechanisms of tumor resistance and novel strategies for modulating its activity. Ann Oncol. 2002;13(12):1841–1851. [DOI] [PubMed] [Google Scholar]
- 86.de Man FM, Goey AKL, van Schaik RHN, et al. Individualization of Irinotecan Treatment: A Review of Pharmacokinetics, Pharmacodynamics, and Pharmacogenetics. Clin Pharmacokinet 2018;57(10):1229–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Campbell JM, Stephenson MD, Bateman E, Irinotecan-induced toxicity pharmacogenetics: An umbrella review of systematic reviews and meta-analyses. Pharmacogenomics J. 2017;17(1):21–28. [DOI] [PubMed] [Google Scholar]
- 88.Innocenti F, Undevia SD, Ramírez J, et al. A phase I trial of pharmacologic modulation of irinotecan with cyclosporine and phenobarbital. Clin Pharmacol Ther. 2004;76(5):490–502. [DOI] [PubMed] [Google Scholar]
- 89.Michael M, Brittain MA, Nagai J, et al. Phase II study of activated charcoal to prevent irinotecan-induced diarrhea. J Clin Oncol. 2004;22(21):4410–4417. [DOI] [PubMed] [Google Scholar]
- 90.Lam W, Bussom S, Guan F, et al. Chemotherapy: The four-herb Chinese medicine PHY906 reduces chemotherapy-induced gastrointestinal toxicity. Sci Transl Med. 2010;2(45):1–10.* Comprehensive study proved that herbs and diets are effective in attenuating chemotherapy toxicity and regulationg relevant drug metabolism.
- 91.Tao G, Dagher F, Moballegh A, Ghose R. Role of Oxidative Stress in the Efficacy and Toxicity of Herbal Supplements. Curr Opin Toxicol. 2020; 20:36–40 [Google Scholar]
- 92.Liu X, Situ A, Kang Y, et al. Irinotecan Delivery by Lipid-Coated Mesoporous Silica Nanoparticles Shows Improved Efficacy and Safety over Liposomes for Pancreatic Cancer. ACS Nano. 2016;10(2):2702–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paludetto MN, Puisset F, Chatelut E, et al. Identifying the reactive metabolites of tyrosine kinase inhibitors in a comprehensive approach: Implications for drug-drug interactions and hepatotoxicity. Med Res Rev. 2019;39(6):2105–2152. [DOI] [PubMed] [Google Scholar]
- 94.Baselga J, Bradbury I, Eidtmann H, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): A randomised, open-label, multicentre, phase 3 trial. Lancet. 2012;379(9816):633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Castellino S, O’Mara M, Koch K, et al. Human metabolism of lapatinib, a dual kinase inhibitor: Implications for hepatotoxicity. Drug Metab Dispos. 2012;40(1):139–150. [DOI] [PubMed] [Google Scholar]
- 96.Hardy KD, Wahlin MD, Papageorgiou I, et al. Studies on the role of metabolic activation in tyrosine kinase inhibitor-dependent hepatotoxicity: Induction of CYP3A4 enhances the cytotoxicity of lapatinib in HepaRG cells. Drug Metab Dispos. 2014;42(1):162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Li X, Kamenecka TM, Cameron MD. Bioactivation of the epidermal growth factor receptor inhibitor gefitinib: Implications for pulmonary and hepatic toxicities. Chem Res Toxicol. 2009;22(10):1736–1742. [DOI] [PubMed] [Google Scholar]
- 98.Liu X, Lu Y, Guan X, et al. Metabolomics reveals the formation of aldehydes and iminium in gefitinib metabolism. Biochem Pharmacol. 2015;97(1):111–121. [DOI] [PubMed] [Google Scholar]
- 99.Ma Y, Xin S, Huang M, et al. Determinants of Gefitinib toxicity in advanced non-small cell lung cancer (NSCLC): A pharmacogenomic study of metabolic enzymes and transporters. Pharmacogenomics J. 2017;17(4):325–330. [DOI] [PubMed] [Google Scholar]
- 100.Bonvin A, Mesnil A, Nicolini FE, et al. Dasatinib-induced acute hepatitis. Leuk Lymphoma. 2008;49(8):1630–1632. [DOI] [PubMed] [Google Scholar]
- 101.Li X, He Y, Ruiz CH, et al. Correction to “Characterization of dasatinib and its structural analogs as CYP3A4 mechanism-based inactivators and the proposed bioactivation pathways” (Drug Metabolism and Disposition (2009) 37, (1242–1250)). Drug Metab Dispos. 2009;37(10):2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li X, Kamenecka TM, Cameron MD. Cytochrome P450-mediated bioactivation of the epidermal growth factor receptor inhibitor erlotinib to a reactive electrophile. Drug Metab Dispos. 2010;38(7):1238–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Paludetto MN, Bijani C, Puisset F, et al. Metalloporphyrin-Catalyzed Oxidation of Sunitinib and Pazopanib, Two Anticancer Tyrosine Kinase Inhibitors: Evidence for New Potentially Toxic Metabolites. J Med Chem. 2018;61(17):7849–7860. [DOI] [PubMed] [Google Scholar]
- 104.Teng WC, Oh JW, New LS, et al. Mechanism-based inactivation of cytochrome P450 3A4 by lapatinib. Mol Pharmacol. 2010;78(4):693–703. [DOI] [PubMed] [Google Scholar]
- 105.Barbara JE, Kazmi F, Parkinson A, et al. Metabolism-dependent inhibition of CYP3A4 by lapatinib: Evidence for formation of a metabolic intermediate complex with a nitroso/oxime metabolite formed via a nitrone intermediate. Drug Metab Dispos. 2013;41(5):1012–1022. [DOI] [PubMed] [Google Scholar]
- 106.Filppula AM, Neuvonen PJ, Backman JT. In vitro assessment of time-dependent inhibitory effects on CYP2C8 and CYP3A activity by fourteen protein kinase inhibitors. Drug Metab Dispos. 2014;42(7):1202–1209. [DOI] [PubMed] [Google Scholar]
- 107.Harmsen S, Meijerman I, Beijnen JH, et al. Nuclear receptor mediated induction of cytochrome P450 3A4 by anticancer drugs: A key role for the pregnane X receptor. Cancer Chemother Pharmacol. 2009;64(1):35–43. [DOI] [PubMed] [Google Scholar]
- 108.Esposito G, Nobile N, Gigli S, et al. Rifaximin Improves Clostridium difficile Toxin A-Induced Toxicity in Caco-2 Cells by the PXR-Dependent TLR4/MyD88/NF-κB Pathway. Front Pharmacol 2016;7:120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jones HM, Barton HA, Lai Y, et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab Dispos. 2012;40(5):1007–1017. [DOI] [PubMed] [Google Scholar]
- 110.Kumar V, Salphati L, Hop CECA, et al. A comparison of total and plasma membrane abundance of transporters in suspended, plated, sandwich-cultured human hepatocytes versus human liver tissue using quantitative targeted proteomics and cell surface biotinylation S. Drug Metab Dispos. 2019;47(4):350–357. [DOI] [PubMed] [Google Scholar]
- 111.Badolo L, Jensen B, Säll C, Norinder U, et al. Evaluation of 309 molecules as inducers of CYP3A4, CYP2B6, CYP1A2, OATP1B1, OCT1, MDR1, MRP2, MRP3 and BCRP in cryopreserved human hepatocytes in sandwich culture. Xenobiotica. 2015;45(2):177–187. [DOI] [PubMed] [Google Scholar]
- 112.Knowlton S, Tasoglu S. A Bioprinted Liver-on-a-Chip for Drug Screening Applications. Trends in Biotechnology. 2016;34:681–682. [DOI] [PubMed] [Google Scholar]
- 113.Kim HJ, Li H, Collins JJ, et al. Contributions of microbiome and mechanical deformation to intestinal bacterial overgrowth and inflammation in a human gut-on-a-chip. Proc Natl Acad Sci U S A. 2016;113(1):E7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Van Waterschoot RAB, Rooswinkel RW, Sparidans RW, et al. Inhibition and stimulation of intestinal and hepatic CYP3A activity: Studies in humanized CYP3A4 transgenic mice using triazolam. Drug Metab Dispos. 2009;37(12):2305–2313. [DOI] [PubMed] [Google Scholar]
- 115.Cai H, Nguyen N, Peterkin V, et al. A humanized UGT1 mouse model expressing the UGT1A1*28 allele for assessing drug clearance by UGT1A1-dependent glucuronidation. Drug Metab Dispos. 2010;38(5):879–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hasegawa M, Kapelyukh Y, Tahara H, et al. Quantitative prediction of human pregnane X receptor and cytochrome P450 3A4 mediated drug-drug interaction in a novel multiple humanized mouse line. Mol Pharmacol. 2011;80(3):518–528. [DOI] [PubMed] [Google Scholar]
- 117.Abdulla D, Goralski KB, Garcia Del Busto Cano E, et al. The signal transduction pathways involved in hepatic cytochrome p450 regulation in the rat during a lipopolysaccharide-induced model of central nervous system inflammation. Drug Metab Dispos. 2005;33(10):1521–1531. [DOI] [PubMed] [Google Scholar]
- 118.Mallick P, Shah P, Ittmann MM, et al. Impact of diet on irinotecan toxicity in mice. Chem Biol Interact. 2018;291:87–94. [DOI] [PubMed] [Google Scholar]
- 119.Tsang LLH, Quarterman CP, Gescher A, et al. Comparison of the cytotoxicity in vitro of temozolomide and dacarbazine, prodrugs of 3-methyl-(triazen-1-yl)imidazole-4-carboxamide. Cancer Chemother Pharmacol. 1991;27(5):342–6. [DOI] [PubMed] [Google Scholar]
- 120.Petrelli N, Herrera L, Rustum Y, et al. A prospective randomized trial of 5-fluorouracil versus 5-fluorouracil and high-dose leucovorin versus 5-fluorouracil and methotrexate in previously untreated patients with advanced colorectal carcinoma. J Clin Oncol. 1987;5(10):1559–1565. [DOI] [PubMed] [Google Scholar]
- 121.Lamba JK. Genetic factors influencing cytarabine therapy. Pharmacogenomics. 2009;10(10):1657–1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhuo X, Zheng N, Felix CA, et al. Kinetics and regulation of cytochrome P450-mediated etoposide metabolism. Drug Metab Dispos. 2004;32(9):993–1000. [PubMed] [Google Scholar]
- 123.Jacobson P, Green K, Birnbaum A, et al. Cytochrome P450 isozymes 3A4 and 2B6 are involved in the in vitro human metabolism of thiotepa to TEPA. Cancer Chemother Pharmacol. 2002;49(6):461–467. [DOI] [PubMed] [Google Scholar]
- 124.Ho HK, Chan JCY, Hardy KD, et al. Mechanism-based inactivation of CYP450 enzymes: A case study of lapatinib. Drug Metab Rev. 2015;47(1):21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Aparicio-Gallego G, Blanco M, Figueroa A, et al. New insights into molecular mechanisms of sunitinib-associated side effects. Mol Cancer Ther. 2011;10(12):2215–2223. [DOI] [PubMed] [Google Scholar]
- 126.Chen J, Peng Y, Zheng J. Cytochrome P450 Mediated Bioactivation of Saracatinib. Chem Res Toxicol. 2016;29(11):1835–1842. [DOI] [PubMed] [Google Scholar]
- 127.Rae JM, Soukhova NV, Flockhart DA. Triethylenethiophosphoramide is a specific inhibitor of cytochrome P450 2B6: implications for cyclophosphamide metabolism. Drug Metab Dispos. 2002;30(5):525–530. [DOI] [PubMed] [Google Scholar]
- 128.Kenny JR, Mukadam S, Zhang C, et al. Drug-drug interaction potential of marketed oncology drugs: In vitro assessment of time-dependent cytochrome P450 inhibition, reactive metabolite formation and drug-drug interaction prediction. Pharm Res. 2012;29(7):1960–1976. [DOI] [PubMed] [Google Scholar]