

Abstract
Purpose of Review
Low-grade inflammation drives elevations in blood pressure (BP) and consequent target organ damage in diverse experimental models of hypertension. Here, we discuss recent advances elucidating immune-mediated mechanisms of BP elevation and associated target organ damage.
Recent Findings
Inflammatory mediators produced by immune cells or target organs act on the kidney, vasculature, skin, and nervous system to modulate hypertension. For example, cells of the innate immune system, including monocytes, neutrophils, and dendritic cells (DCs), can all promote BP elevation via actions in the vasculature and kidney. Macrophages expressing VEGF-C impact non-osmotic sodium storage in the skin that in turn regulates salt sensitivity. Within the adaptive immune system, activated T cells can secrete tumor necrosis factor-alpha (TNF-α), interleukin-17a (IL-17a), and interferon-gamma (IFN-γ), each of which has augmented BP and renal damage in pre-clinical models. Inversely, deficiency of IL-17a in mice blunts the hypertensive response and attenuates renal sodium retention via a serum- and glucocorticoid-regulated kinase 1 (SGK1)–dependent pathway. Linking innate and adaptive immune responses, dendritic cells activated by augmented extracellular sodium concentrations stimulate T lymphocytes to produce pro-hypertensive cytokines. By contrast, regulatory T cells (Tregs) can protect against hypertension and associated kidney injury.
Summary
Rodent studies reveal diverse mechanisms via which cells of the innate and adaptive immune systems drive blood pressure elevation by altering the inflammatory milieu in the kidney, vasculature, and brain.
Keywords: Inflammation, Immunity, Hypertension, Renal damage, Cytokines
Introduction
High blood pressure (BP) or hypertension is the most prominent risk factor for cardiovascular disease worldwide, with the number of people with hypertension reaching 1.13 billion in 2015 [1]. Current therapies for human hypertension include angiotensin II (Ang II) type 1 receptor blockers (ARBs), angiotensin-converting enzyme inhibitors (ACEIs), calcium channel antagonists, beta-blockers, and diuretics. The effective use of pharmacologic and non-pharmacologic therapies for hypertension has significantly reduced the mortality and morbidity attributable to this disorder. Nevertheless, BP remains poorly controlled in up to half of patients with treated hypertension [2]. While poor access to care or noncompliance accounts for persistent hypertension in many patients, novel therapies are also urgently needed to address biologically resistant hypertension.
Elevations in levels of circulating cytokines and CRP mark hypertension as a low-grade inflammatory condition involving both the innate and adaptive immune systems [3, 4]. Cells of the immune system contribute to the development of hypertension via actions in the kidney, vasculature, skin, and nervous system [5–11]. High-salt intake in susceptible humans and rodents raise blood pressure, reflecting salt-sensitive hypertension [12–15]. While the etiology of salt-sensitive hypertension is complex, experimental models suggest that inappropriate immune activation is a major contributor to salt sensitivity [16–19]. Several laboratories including our own have tried to explore the immune-mediated mechanisms that may underpin salt-sensitive hypertension and related end-organ damage.
Within the innate immune system, myeloid cell populations such as the monocytes, neutrophils, and dendritic cells can all promote BP elevation via actions in the vasculature and kidney [20–22]. Macrophages regulate sodium storage in the skin with consequent downstream effects on salt sensitivity [23, 24]. Within the adaptive immune system, activated B and T lymphocytes have each been shown to increase BP [25, 26] although the effects of B cells on BP appear to require Tcell help. Multiple groups have reported that Tcells augment hypertension by promoting oxidative stress and sodium reabsorption in the kidney [17, 27]. In this review, we will summarize pre-clinical experimental data illustrating how cells of the innate and adaptive immune systems modulate functions of cardiovascular control tissues to drive BP elevation.
Inflammation and Hypertension
Diverse stimuli including infection, Ang II, or a high-salt diet activate innate immune responses leading to the accumulation of inflammatory cells in target organs that regulate BP, including the kidney, vasculature, and brain (Fig. 1). Whereas infection causes vasodilation and hypotension, sterile stimulation of immune responses is associated with BP elevation, possibly reflecting an evolutionary development to prevent circulatory collapse in the face of overwhelming sepsis. More than 50% of elderly populations carry a diagnosis of hypertension [28], associated with elevated markers of inflammation [29]. In general, hypertensive patients have increased circulating levels of inflammatory cytokines, including IL-6, IL-1β, and TNF-α [30–33]. Similarly, in animal studies, feeding a high-salt diet to Dahl salt-sensitive (Dahl-S) rats provokes both hypertension and elevated plasma levels of inflammatory mediators [34]. Inversely, genetic deletion and/or pharmacologic blockade of TNF-α [35], IL-17a [36], IFN-γ [37], and IL-6 [38] in mice attenuates the chronic hypertensive response.
Fig. 1.
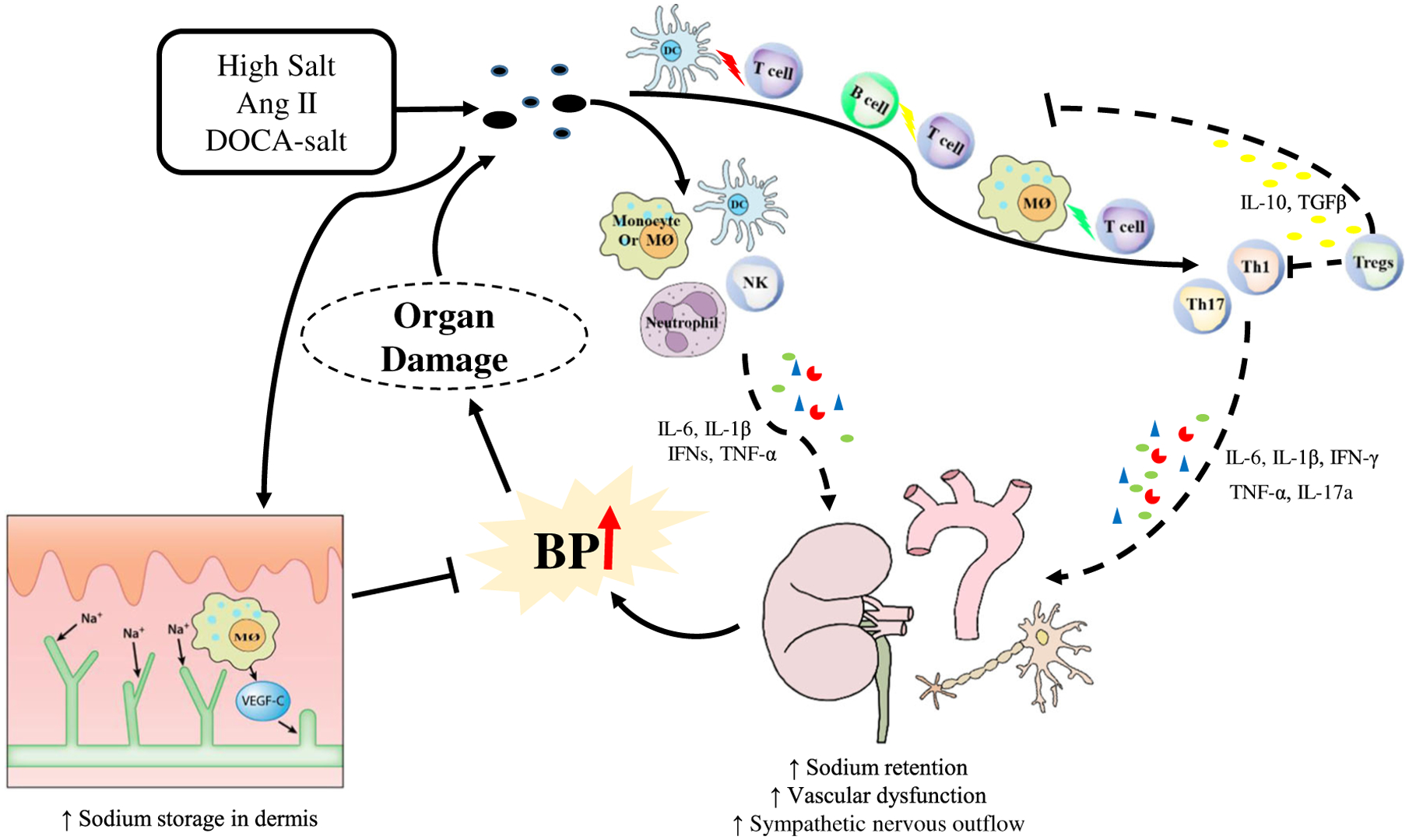
Role of immune cells in inflammation in hypertension. In response to hypertensive stimuli such as high-salt diet, Ang II, and DOCA-salt, neoantigens are generated leading to the activation of the immune system. Innate immune cells such as monocytes, macrophages (MØ), neutrophils, and dendritic cells (DCs) release pro-hypertensive cytokines that promote the BP elevation via actions in the vasculature, kidney, and sympathetic nervous system. However, dermal macrophages can increase expression of vascular endothelial growth factor-C (VEGF-C) during high-salt stress, limiting dermal storage of sodium, and buffering high salt induced BP elevation. Antigen-presenting cells including dendritic cells, macrophages, and B cells can polarize T cells towards pro-inflammatory T helper cells, such as Th1 and Th17, and anti-an inflammatory phenotype known as Tregs. Th1 and Th17 cells produce IL-6, IL-1β, IFN-γ, TNF-α, and IL-17a, increasing sodium retention in the kidney, augmenting vascular dysfunction, and stimulating sympathetic outflow, all of which exacerbate BP elevation and target organ damage. In contrast, Tregs release anti-inflammatory cytokines such as IL-10 and TGFβ to suppress innate and adaptive immune responses
Inflammatory mediators called cytokines are released by immune, endothelial, and epithelial cells following injury or stimulation. Different immune cells serve diverse functions during hypertension or organ damage. At present, more than 120 immune cell subsets have been characterized in humans [39]. Cells of the innate immune system, which include monocytes, macrophage, dendritic cells, and neutrophils, produce several pro-hypertensive cytokines, such as TNF-α, IL-6, and IFNs [40]. Following an injurious stimulus such as Ang II or high-salt diet, the innate immune system engages and activates cells of the adaptive immune system. For example, T lymphocytes undergo activation via antigen presentation on the surface of a macrophage, dendritic cell, and B cell. Among the T cells subsets, CD8+ T cells express IFN-γ, TNF-α, and IL-12 [41]. Among the CD4+ T helper cells, pro-inflammatory Th1 cells produce IFN-γ and TNF-α [42]; Th2 cells release IL-4, IL-5, and IL-13 [43]; Th17 produce pro-hypertensive cytokines IL-17 and IFN-γ [44] whereas regulatory T cells (Tregs) secrete the immunosuppressive cytokines IL-10 and TGF-beta [45]. We will now discuss the role of several immune cell lineages in hypertension, beginning with cells of the innate immune system.
Monocytes
Upon stimulation, circulating monocytes can release pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α [46]. In humans, CD16+ “intermediate” monocytes activated by stretching of the vasculature produce these pro-hypertensive cytokines [47]. On a high-salt diet, Dahl-S rats develop significant hypertension and elevations in total circulating numbers of monocytes compared to Dahl salt-resistant (Dahl-R) rats [48]. Moreover, monocytes contribute to blood pressure elevation and renal damage in several experimental models of hypertension, including high diet, deoxycorticosterone acetate (DOCA)-salt, aldosterone-salt, and Ang II–induced hypertension [49–53]. Regarding underlying mechanisms for these effects, LysM-positive monocytes appear to drive increases in BP during chronic Ang II infusion by instigating oxidative stress in the vasculature [20]. In addition, IL-1 is the prototypical monocyte/macrophage cytokine, and we have found that activation of the receptor for IL-1 potentiates sodium retention and blood pressure elevation by releasing the NKKC2 sodium co-transporter from tonic inhibition by nitric oxide. Thus, monocytes likely act both in the kidney and vasculature to promote hypertension.
Macrophages
As a simplification, macrophages can be dichotomized into pro-inflammatory M1 and anti-inflammatory M2 phenotypes depending on the milieu of activation. Macrophages accumulate in the kidney in various pre-clinical models of hypertension [7, 52–54], and multiple groups have been able to demonstrate a functional role for these macrophages in modulating BP. For example, deleting macrophage colony-stimulating factor (m-CSF) or depleting macrophages blunts the hypertensive response to Ang II or DOCA-salt and attenuates renal scar formation and thereby limits target organ damage [7, 55, 56]. Similarly, depleting macrophages in DOCA-salt-hypertensive rats has analogous favorable effects by restoring appropriate neuronal functions in resistance arteries [57]. Accordingly, just as with their monocyte precursors, tissue macrophages exert effects on blood pressure via actions in the kidney and vasculature. On the other hand, as elucidated in unique studies from Dr. Titze’s group, macrophages in the dermal layer of the skin may mobilize and facilitate clearance of interstitial sodium by stimulating lymphangiogenesis with consequent protection from salt sensitivity [23]. Accordingly depleting these mononuclear phagocytes with clodronate liposomes augments salt-sensitive hypertension and volume retention in the skin [58]. Corroborating these findings, deletion of PGE2 type 4 (EP4) receptor in macrophages increases BP following chronic high-salt exposure both by impairing sodium clearance in the kidney and by permitting increased non-osmotic sodium retention in the skin [24] Thus, macrophages and their precursor monocytes exert complex effects on blood pressure acting both within and outside the kidney.
Neutrophils/NK Cells
The roles of neutrophils and NK cells are less well documented. For example, adoptive transfer of neutrophils cannot restore the hypertensive response in mice depleted of myeloid cells [20]. On the other hand, induction of apoptosis in neutrophils by CD39 on regulatory T cells (Tregs) attenuates hypertension during activation of the renin angiotensin system (RAS), suggesting that neutrophils in this context may foster BP elevation [59]. Moreover, in several human populations, elevated neutrophil levels correlate significantly with the risk of developing hypertension [60, 61]. While NK cells contribute to vascular dysfunction during RAS activation [62], their role in BP regulation requires further study.
Dendritic Cells
As the most potent antigen-presenting cells in the body, dendritic cells (DCs) can invite the participation of the adaptive immune system after injury by stimulating T lymphocytes that carry a receptor specific for the antigen displayed on the DC surface. In 2002, Muller et al. reported that immunosuppression with dexamethasone prevents DC maturation in the injured kidney during Ang II infusion [5]. Interactions between DCs and T cells resulting in the generation of memory effector T cells are required for a full hypertensive response [63, 64], pointing to a vital role for DCs in the pathogenesis of hypertension. In landmark studies, Kirabo and colleagues established that hypertensive stimuli generate oxidant stress within DCs with consequent modification of isolevuglandins that are then presented as pro-hypertensive antigens to T cells [22]. Through this mechanism, DCs can promote vascular dysfunction and BP elevation. Conversely, the sodium retained in the initial stages of hypertension may alter DC function to drive further changes in BP. Excess sodium enters the DC through amiloride-sensitive transporters and sodium hydrogen exchanger 1, activating the DC to produce more IL-1β, ultimately provoking T cell elaboration of the pro-hypertensive cytokines IL-17a and IFN-γ [65]. Accordingly, adoptive transfer of DCs activated by exposure to high levels of extracellular sodium renders the recipient susceptible to hypertension induced by a suppressor dose of Ang II. The importance of DCs to experimental hypertension has been confirmed by a separate group who showed that ablation of myeloid CD11c+ cells prevents the development of hypertension in response to Ang II infusion plus a high-salt diet [66]. These studies indicate that hypertension may be an antigen-specific autoimmune disease which is mediated by DCs. While DCs appear to exert their effects on BP by modulating Tcell function, DCs can also produce cytokines IL-1β and IL-6, which could alter BP independently of T cells. Nevertheless, as detailed below, the adaptive immune system constituted by T and B lymphocytes plays a critical role in the pathogenesis of hypertension.
T Lymphocytes
The specificity of T lymphocytes accrues from their expression of a Tcell receptor (TCR), permitting clonal expansion of precisely targeted T cells following exposure to a putative antigen. Very early studies pointed to a role for lymphocytes in hypertension. For example, in 1976, Svendsen and colleagues showed that nude mice have a blunted elevation of BP with DOCA-salt treatment whereas grafting thymus from wild-type mice into nude mice restores the hypertensive response and intrarenal vascular disease [67]. In experimental hypertension, others and we have shown that T cells infiltrate the kidney, accumulating around the renal vasculature [6, 27, 68]. On susceptible rodent strains, accumulation of T cells in the kidney provokes renal damage as preventing renal T cell infiltration during hypertension ameliorates kidney damage and/or BP elevation [16, 69]. In 2007, conclusive evidence of the role of T cells in the pathogenesis of Ang II–induced hypertension was provided by Guzik et al. In these studies, mice lacking the recombination activating gene-1 (Rag1) and therefore deficient of functional T and B cells do not develop hypertension during Ang II infusion. Adoptive transfer of T but not B cells restores the hypertensive responses [26]. Studies in transgenic rats corroborate this finding and further implicate T cells in engendering salt sensitivity and hypertension-associated renal damage [70]. Similarly, human hypertensive patients feature both renal infiltration of T cells and elevated circulating levels of C-X-C chemokine receptor type 3, suggesting that T cells may be important both in experimental and clinical hypertension [71].
Whereas conventional T cells largely promote hypertension, immunosuppressive regulatory T cells (Tregs) blunt adaptive immunity and have been shown to mitigate hypertension and target organ damage [72–74]. For example, deficiency of Tregs exaggerates RAS-dependent hypertension and organ damage whereas adoptive transfer of Tregs blunts the BP elevation and vascular damage seen in this model [73, 75]. The protective effects of Tregs may accrue from modulation of neutrophil function as discussed above or from alterations in the complement system. In this regard, the attenuated hypertensive response noted in animals lacking the complement receptors C3aR and C5aR is abolished by Treg depletion [76]. Thus, Tregs impart protection from hypertension via unusual actions impacting other arms of the immune system. Nevertheless, as with other cell lineages, retained interstitial sodium may interfere with the protective functions of Tregs and thereby limit their capacity to defend against hypertension [77].
Conventional T cells and also gammadelta T cells [78] can promote hypertension not only by provoking oxidative stress in the brain, kidney, and vasculature but also by elaborating a panel of pro-hypertensive cytokines. The inflammatory mediators propagated by infiltrating T cells can increase sodium reabsorption of the kidney, exacerbate systemic vasoconstriction, and stimulate sympathetic nerve activity. T cells from animals stimulated by Ang II or high salt produce IL-17a, INF-γ, and TNF-α. IL-17a can be produced by several kinds of cells, but more prominently by Th17 cells. Culturing naïve T cells in high-salt media enhances their differentiation into Th17 cells via an SGK1-dependent pathway [79, 80]. Accordingly, mice lacking SGK1 solely on T cells have blunted hypertension and renal injury during chronic Ang II infusion or DOCA-salt treatment [81]. Moreover, genetic deletion or pharmacologic blockade of IL-17a limits the induction of hypertension and consequent renal injury [37, 82]. IFN-γ and TNF-α are produced by CD4+ and CD8+ T cells, but also by macrophages and intrinsic parenchymal cells in target organs. As with IL-17a, IFN-γ and TNF-α deletion or blockade protects against hypertension and/or associated kidney injury [37, 83]. Interestingly, others and we have found that TNF produced in the kidney itself rather than in immune cells may play an important role in renal sodium retention during RAS activation [84, 85], but these effects are highly dependent on local levels of TNF [86]. IL-6 is produced by T cells in the kidney [87] and mediates BP elevation induced by high salt [87] or RAS activation [88], possibly via effects on collecting tubule epithelial function [89]. Finally, immunosuppressive cytokines can have salubrious effects on BP as IL-10 produced by Tregs ameliorates hypertension by mitigating vascular oxidative stress [90]. While all of these studies demonstrate the capacity of T cells to modulate BP via cytokine elaboration, reports of direct interactions between T cells and the kidney epithelium are emerging. For example, CD8+ T cells interact with distal convoluted tubule cells during DOCA-salt treatment, leading to the upregulation and activation of the Na-Cl co-transporter (NCC), with consequent BP elevation [91].
B Lymphocytes
Similar to T cells, B lymphocytes express both a specific B cell receptor (BCR) and a B cell co-receptor. Antigens activate B cells through the BCR triggering their differentiation into plasmablasts, plasma cells, and/or memory B cells. Initiate adoptive transfer of B cells into lymphocyte-deficient recipients suggested that B cells were not critical to the pathogenesis of hypertension [26]. However, in the presence of an other-wise intact immune system, B cells marked by the expression of B cell–activating factor are absolutely required to mount a full hypertensive response and play a vital role in precipitating perivascular fibrosis during RAS activation [25]. Thus, the contributions of B cells to hypertension appear to require help from T cells in the inflammatory milieu.
Conclusions
While the primary instigators of hypertension are inappropriate sodium retention in the kidney and increased vascular resistance driven by intrinsic vascular wall malfunction and/ or increased sympathetic outflow, evidence from pre-clinical and clinical studies indicates that inflammatory responses contribute to the pathogenesis of hypertension by augmenting these defects in the cardiovascular control organs. Mild increases in pro-hypertensive factors like Ang II and high-salt diet activate both the innate and adaptive immune systems. As a result, activated inflammatory monocytes and macrophages promote vasoconstriction and renal sodium retention whereas DCs provoke BP elevation at least in part by engaging T lymphocytes. In turn, T cell subsets can have a variety of effects on BP. For example, Th17 cells stimulate sodium transport in the kidney whereas Tregs blunt vascular reactivity. These effects of inflammatory responses in hypertension are therefore complex as summarized in Fig. 1.
Currently, anti-inflammatory drugs are not approved to treat hypertension, and very selective manipulation of the immune system may be required to protect against organ damage in hypertension without rendering the patient susceptible to dangerous immunosuppression. Nevertheless, MMF, which inhibits the proliferation of T cells and B cells, lowers BP in several experimental hypertension models [6, 16, 54, 92] and even in human hypertensive patients with rheumatic disease [93]. While tacrolimus attenuates hypertension in Dahl-S rats [27], this medication is less attractive as chronic tacrolimus therapy in human transplant patients is associated with renal sodium retention and hypertension [94]. In the aggregate, currently available immunosuppressives appear to raise cardiovascular risk [95]. Thus, further research investigating the anti-hypertensive benefits of targeting specific functions in selected inflammatory cell subpopulations is warranted. Meanwhile, medications developed for other cardiovascular benefits may afford protection in hypertensive patients by diminishing levels of inflammatory cytokines such as TNF-α, IL-1, and IL-6 [96, 97]. Therefore, the use of regular anti-hypertensive medicines in combination with older, adjuvant drugs represents a reasonable therapeutic strategy to control hypertension and mitigate end-organ damage while newer, more incisive immuno-therapies are in development.
Funding Information
This work was supported by NIH grants DK087893, HL128355; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893.
Footnotes
Conflict of Interest The authors declare no conflicts of interest relevant to this manuscript.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Collaboration NCDRF. Worldwide trends in blood pressure from 1975 to 2015: a pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet. 2017;389(10064): 37–55. 10.1016/S0140-6736(16)31919-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Egan BM, Zhao Y, Axon R. US trends in prevalence, awareness, treatment, and control of hypertension, 1988–2008. JAMA. 2010;303(20):2043–50. 10.1001/jama.2010.650. [DOI] [PubMed] [Google Scholar]
- 3.Caillon A, Schiffrin EL. Role of inflammation and immunity in hypertension: recent epidemiological, laboratory, and Clinical Evidence. Curr Hypertens Rep. 2016;18(3):21. 10.1007/s11906-016-0628-7. [DOI] [PubMed] [Google Scholar]
- 4.Norlander AE, Madhur MS, Harrison DG. The immunology of hypertension. J Exp Med. 2018;215(1):21–33. 10.1084/jem.20171773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muller DN, Shagdarsuren E, Park JK, Dechend R, Mervaala E, Hampich F, et al. Immunosuppressive treatment protects against angiotensin II-induced renal damage. Am J Pathol. 2002;161(5): 1679–93. 10.1016/S0002-9440(10)64445-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, et al. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin II exposure. Kidney Int. 2001;59(6):2222–32. [DOI] [PubMed] [Google Scholar]
- 7.Ko EA, Amiri F, Pandey NR, Javeshghani D, Leibovitz E, Touyz RM, et al. Resistance artery remodeling in deoxycorticosterone acetate-salt hypertension is dependent on vascular inflammation: evidence from m-CSF-deficient mice. Am J Physiol Heart Circ Physiol. 2007;292(4):H1789–95. 10.1152/ajpheart.01118.2006. [DOI] [PubMed] [Google Scholar]
- 8.Kossmann S, Hu H, Steven S, Schonfelder T, Fraccarollo D, Mikhed Y, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289(40):27540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiig H, Schroder A, Neuhofer W, Jantsch J, Kopp C, Karlsen TV, et al. Immune cells control skin lymphatic electrolyte homeostasis and blood pressure. J Clin Invest. 2013;123(7):2803–15. 10.1172/JCI60113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, et al. Renal denervation prevents immune cell activation and renal inflammation in angiotensin II-induced hypertension. Circ Res. 2015;117(6): 547–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaldivia MTK, Rivera J, Hering D, Marusic P, Sata Y, Lim B, et al. Renal denervation reduces monocyte activation and monocyte-platelet aggregate formation: an anti-inflammatory effect relevant for cardiovascular risk. Hypertension. 2017;69(2):323–31. [DOI] [PubMed] [Google Scholar]
- 12.O’Donnell M, Mente A, Yusuf S. Sodium intake and cardiovascular health. Circ Res. 2015;116(6):1046–57. [DOI] [PubMed] [Google Scholar]
- 13.Wright JT Jr, Rahman M, Scarpa A, Fatholahi M, Griffin V, Jean-Baptiste R, et al. Determinants of salt sensitivity in black and white normotensive and hypertensive women. Hypertension. 2003;42(6): 1087–92. [DOI] [PubMed] [Google Scholar]
- 14.Chen J, Gu D, Huang J, Rao DC, Jaquish CE, Hixson JE, et al. Metabolic syndrome and salt sensitivity of blood pressure in non-diabetic people in China: a dietary intervention study. Lancet. 2009;373(9666):829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gradin K, Persson B. Chronic salt loading and adrenergic mechanisms in the Sprague-Dawley rat. Pharmacol Toxicol. 1987;60(4): 299–304. [DOI] [PubMed] [Google Scholar]
- 16.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the Dahl salt-sensitive rat. Hypertension. 2006;48(1):149–56. [DOI] [PubMed] [Google Scholar]
- 17.Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P. Lymphocyte responses exacerbate angiotensin II-dependent hypertension. Am J Phys Regul Integr Comp Phys. 2010;298(4):R1089–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, et al. Interleukin-17A regulates renal sodium transporters and renal injury in angiotensin II-induced hypertension. Hypertension. 2016;68(1):167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wade B, Petrova G, Mattson DL. Role of immune factors in angiotensin II-induced hypertension and renal damage in Dahl salt-sensitive rats. Am J Phys Regul Integr Comp Phys. 2018;314(3): R323–R33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wenzel P, Knorr M, Kossmann S, Stratmann J, Hausding M, Schuhmacher S, et al. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124(12):1370–81. [DOI] [PubMed] [Google Scholar]
- 21.Chatterjee M, Saluja R, Tewari S, Barthwal MK, Goel SK, Dikshit M. Augmented nitric oxide generation in neutrophils: oxidative and pro-inflammatory implications in hypertension. Free Radic Res. 2009;43(12):1195–204. [DOI] [PubMed] [Google Scholar]
- 22.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, et al. DC isoketal-modified proteins activate T cells and promote hypertension. J Clin Invest. 2014;124(10):4642–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. 2009;15(5):545–52. 10.1038/nm.1960. [DOI] [PubMed] [Google Scholar]
- 24.Zhang MZ, Yao B, Wang Y, Yang S, Wang S, Fan X, et al. Inhibition of cyclooxygenase-2 in hematopoietic cells results in salt-sensitive hypertension. J Clin Invest. 2015;125(11):4281–94. 10.1172/JCI81550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chan CT, Sobey CG, Lieu M, Ferens D, Kett MM, Diep H, et al. Obligatory role for B cells in the development of angiotensin II-dependent hypertension. Hypertension. 2015;66(5):1023–33. 10.1161/HYPERTENSIONAHA.115.05779. [DOI] [PubMed] [Google Scholar]
- 26.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, et al. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204(10): 2449–60. 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating T lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Ren Physiol. 2011;300(3):F734–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lloyd-Sherlock P, Beard J, Minicuci N, Ebrahim S, Chatterji S. Hypertension among older adults in low- and middle-income countries: prevalence, awareness and control. Int J Epidemiol. 2014;43(1):116–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rafey MA. Resistant hypertension in the elderly. Clin Geriatr Med. 2009;25(2):289–301. [DOI] [PubMed] [Google Scholar]
- 30.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-alpha) and essential hypertension. J Hum Hypertens. 2005;19(2):149–54. [DOI] [PubMed] [Google Scholar]
- 31.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38(3):399–403. [DOI] [PubMed] [Google Scholar]
- 32.Dalekos GN, Elisaf M, Bairaktari E, Tsolas O, Siamopoulos KC. Increased serum levels of interleukin-1beta in the systemic circulation of patients with essential hypertension: additional risk factor for atherogenesis in hypertensive patients? J Lab Clin Med. 1997;129(3):300–8. [DOI] [PubMed] [Google Scholar]
- 33.Yu X, Yang Z, Yu M. Correlation of tumor necrosis factor alpha and interleukin 6 with hypertensive renal damage. Ren Fail. 2010;32(4): 475–9. [DOI] [PubMed] [Google Scholar]
- 34.Hu J, Zhu Q, Xia M, Guo TL, Wang Z, Li P-L, et al. Transplantation of mesenchymal stem cells into the renal medulla attenuated salt-sensitive hypertension in Dahl S rat. J Mol Med. 2014;92(11): 1139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51(5): 1345–51. 10.1161/HYPERTENSIONAHA.107.102152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, et al. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55(2):500–7. 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, et al. Renal transporter activation during angiotensin-II hypertension is blunted in interferon-gamma−/− and interleukin-17A−/− mice. Hypertension. 2015;65(3):569–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang W, Wang W, Yu H, Zhang Y, Dai Y, Ning C, et al. Interleukin 6 underlies angiotensin II-induced hypertension and chronic renal damage. Hypertension. 2012;59(1):136–44. 10.1161/HYPERTENSIONAHA.111.173328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lepone LM, Donahue RN, Grenga I, Metenou S, Richards J, Heery CR, et al. Analyses of 123 peripheral human immune cell subsets: defining differences with age and between healthy donors and cancer patients not detected in analysis of standard immune cell types. J Circ Biomark. 2016;5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Striz I, Brabcova E, Kolesar L, Sekerkova A. Cytokine networking of innate immunity cells: a potential target of therapy. Clin Sci (Lond). 2014;126(9):593–612. [DOI] [PubMed] [Google Scholar]
- 41.Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, et al. CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res. 2015;117(3): 244–53. [DOI] [PubMed] [Google Scholar]
- 42.Gallego A, Vargas JA, Castejon R, Citores MJ, Romero Y, Millan I, et al. Production of intracellular IL-2, TNF-alpha, and IFN-gamma by T cells in B-CLL. Cytometry B Clin Cytom. 2003;56(1):23–9. [DOI] [PubMed] [Google Scholar]
- 43.Kelly BL, Locksley RM. Coordinate regulation of the IL-4, IL-13, and IL-5 cytokine cluster in Th2 clones revealed by allelic expression patterns. J Immunol. 2000;165(6):2982–6. [DOI] [PubMed] [Google Scholar]
- 44.Ma H-L, Napierata L, Stedman N, Benoit S, Collins M, Nickerson-Nutter C, et al. Tumor necrosis factor alpha blockade exacerbates murine psoriasis-like disease by enhancing Th17 function and decreasing expansion of Treg cells. Arthritis Rheum. 2010;62(2):430–40. [DOI] [PubMed] [Google Scholar]
- 45.Haque R, Lei F, Xiong X, Bian Y, Zhao B, Wu Y, et al. Programming of regulatory T cells from pluripotent stem cells and prevention of autoimmunity. J Immunol. 2012;189(3):1228–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ballou SP, Lozanski G. Induction of inflammatory cytokine release from cultured human monocytes by C-reactive protein. Cytokine. 1992;4(5):361–8. [DOI] [PubMed] [Google Scholar]
- 47.Loperena R, Van Beusecum JP, Itani HA, Engel N, Laroumanie F, Xiao L, et al. Hypertension and increased endothelial mechanical stretch promote monocyte differentiation and activation: roles of STAT3, interleukin 6 and hydrogen peroxide. Cardiovasc Res. 2018;114(11):1547–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen K, DeLano FA, Zweifach BW, Schmid-Schonbein GW. Circulating leukocyte counts, activation, and degranulation in Dahl hypertensive rats. Circ Res. 1995;76(2):276–83. [DOI] [PubMed] [Google Scholar]
- 49.Artigues C, Richard V, Roussel C, Lallemand F, Henry JP, Thuillez C. Increased endothelium–monocyte interactions in salt-sensitive hypertension: effect of L-arginine. J Cardiovasc Pharmacol. 2000;35(3):468–73. [DOI] [PubMed] [Google Scholar]
- 50.Beswick RA, Zhang H, Marable D, Catravas JD, Hill WD, Webb RC. Long-term antioxidant administration attenuates mineralocorticoid hypertension and renal inflammatory response. Hypertension. 2001;37(2 Pt 2):781–6. [DOI] [PubMed] [Google Scholar]
- 51.Rocha R, Rudolph AE, Frierdich GE, Nachowiak DA, Kekec BK, Blomme EA, et al. Aldosterone induces a vascular inflammatory phenotype in the rat heart. Am J Physiol Heart Circ Physiol. 2002;283(5):H1802–10. 10.1152/ajpheart.01096.2001. [DOI] [PubMed] [Google Scholar]
- 52.Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats. Kidney Int. 2003;63(5):1791–800. 10.1046/j.1523-1755.2003.00929.x. [DOI] [PubMed] [Google Scholar]
- 53.Vieira JM Jr, Rodrigues LT, Mantovani E, Delle H, Mattar AL, Malheiros DM, et al. Statin monotherapy attenuates renal injury in a salt-sensitive hypertension model of renal disease. Nephron Physiol. 2005;101(4):p82–91. 10.1159/000087576. [DOI] [PubMed] [Google Scholar]
- 54.Tian N, Gu JW, Jordan S, Rose RA, Hughson MD, Manning RD,J. Immune suppression prevents renal damage and dysfunction and reduces arterial pressure in salt-sensitive hypertension. Am J Physiol Heart Circ Physiol. 2007;292(2):H1018–25. 10.1152/ajpheart.00487.2006. [DOI] [PubMed] [Google Scholar]
- 55.De Ciuceis C, Amiri F, Brassard P, Endemann DH, Touyz RM, Schiffrin EL. Reduced vascular remodeling, endothelial dysfunction, and oxidative stress in resistance arteries of angiotensin II-infused macrophage colony-stimulating factor-deficient mice: evidence for a role in inflammation in angiotensin-induced vascular injury. Arterioscler Thromb Vasc Biol. 2005;25(10):2106–13. 10.1161/01.ATV.0000181743.28028.57. [DOI] [PubMed] [Google Scholar]
- 56.Huang L, Wang A, Hao Y, Li W, Liu C, Yang Z, et al. Macrophage depletion lowered blood pressure and attenuated hypertensive renal injury and fibrosis. Front Physiol. 2018;9:473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thang LV, Demel SL, Crawford R, Kaminski NE, Swain GM, Van Rooijen N, et al. Macrophage depletion lowers blood pressure and restores sympathetic nerve alpha2-adrenergic receptor function in mesenteric arteries of DOCA-salt hypertensive rats. Am J Physiol Heart Circ Physiol. 2015;309(7):H1186–97. 10.1152/ajpheart.00283.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Machnik A, Dahlmann A, Kopp C, Goss J, Wagner H, van Rooijen N, et al. Mononuclear phagocyte system depletion blocks interstitial tonicity-responsive enhancer binding protein/vascular endothelial growth factor C expression and induces salt-sensitive hypertension in rats. Hypertension. 2010;55(3):755–61. 10.1161/HYPERTENSIONAHA.109.143339. [DOI] [PubMed] [Google Scholar]
- 59.Fabbiano S, Menacho-Marquez M, Robles-Valero J, Pericacho M, Matesanz-Marin A, Garcia-Macias C, et al. Immunosuppression-independent role of regulatory T cells against hypertension-driven renal dysfunctions. Mol Cell Biol. 2015;35(20):3528–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu X, Zhang Q, Wu H, Du H, Liu L, Shi H, et al. Blood neutrophil to lymphocyte ratio as a predictor of hypertension. Am J Hypertens. 2015;28(11):1339–46. [DOI] [PubMed] [Google Scholar]
- 61.Tatsukawa Y, Hsu W-L, Yamada M, Cologne JB, Suzuki G, Yamamoto H, et al. White blood cell count, especially neutrophil count, as a predictor of hypertension in a Japanese population. Hypertens Res. 2008;31(7):1391–7. [DOI] [PubMed] [Google Scholar]
- 62.Kossmann S, Schwenk M, Hausding M, Karbach SH, Schmidgen MI, Brandt M, et al. Angiotensin II-induced vascular dysfunction depends on interferon-gamma-driven immune cell recruitment and mutual activation of monocytes and NK-cells. Arterioscler Thromb Vasc Biol. 2013;33(6):1313–9. 10.1161/ATVBAHA.113.301437. [DOI] [PubMed] [Google Scholar]
- 63.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, et al. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122(24):2529–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, et al. CD70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118(8): 1233–43. 10.1161/CIRCRESAHA.115.308111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, et al. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21(4):1009–20. 10.1016/j.celrep.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hevia D, Araos P, Prado C, Fuentes Luppichini E, Rojas M, Alzamora R, et al. Myeloid CD11c(+) antigen-presenting cells ablation prevents hypertension in response to angiotensin II plus high-salt diet. Hypertension. 2018;71(4):709–18. 10.1161/HYPERTENSIONAHA.117.10145. [DOI] [PubMed] [Google Scholar]
- 67.Svendsen UG. Evidence for an initial, thymus independent and a chronic, thymus dependent phase of DOCA and salt hypertension in mice. Acta Pathol Microbiol Scand A. 1976;84(6):523–8. [DOI] [PubMed] [Google Scholar]
- 68.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, et al. Stimulation of lymphocyte responses by angiotensin II promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295(2):F515–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.De Miguel C, Das S, Lund H, Mattson DL. T lymphocytes mediate hypertension and kidney damage in Dahl salt-sensitive rats. Am J Physiol Regul Integr Comp Physiol. 2010;298(4):R1136–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abais-Battad JM, Lund H, Fehrenbach DJ, Dasinger JH, Mattson DL. Rag1-null Dahl SS rats reveal that adaptive immune mechanisms exacerbate high protein-induced hypertension and renal injury. Am J Physiol Regul Integr Comp Physiol. 2018;315(1):R28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Youn JC, Yu HT, Lim BJ, Koh MJ, Lee J, Chang DY, et al. Immunosenescent CD8+ T cells and C-X-C chemokine receptor type 3 chemokines are increased in human hypertension. Hypertension. 2013;62(1):126–33. 10.1161/HYPERTENSIONAHA.113.00689. [DOI] [PubMed] [Google Scholar]
- 72.Kvakan H, Kleinewietfeld M, Qadri F, Park JK, Fischer R, Schwarz I, et al. Regulatory Tcells ameliorate angiotensin II-induced cardiac damage. Circulation. 2009;119(22):2904–12. 10.1161/CIRCULATIONAHA.108.832782. [DOI] [PubMed] [Google Scholar]
- 73.Barhoumi T, Kasal DA, Li MW, Shbat L, Laurant P, Neves MF, et al. T regulatory lymphocytes prevent angiotensin II-induced hypertension and vascular injury. Hypertension. 2011;57(3):469–76. 10.1161/HYPERTENSIONAHA.110.162941. [DOI] [PubMed] [Google Scholar]
- 74.Kasal DA, Barhoumi T, Li MW, Yamamoto N, Zdanovich E, Rehman A, et al. T regulatory lymphocytes prevent aldosterone-induced vascular injury. Hypertension. 2012;59(2):324–30. 10.1161/HYPERTENSIONAHA.111.181123. [DOI] [PubMed] [Google Scholar]
- 75.Mian MO, Barhoumi T, Briet M, Paradis P, Schiffrin EL. Deficiency of T-regulatory cells exaggerates angiotensin II-induced microvascular injury by enhancing immune responses. J Hypertens. 2016;34(1):97–108. 10.1097/HJH.0000000000000761. [DOI] [PubMed] [Google Scholar]
- 76.Chen XH, Ruan CC, Ge Q, Ma Y, Xu JZ, Zhang ZB, et al. Deficiency of complement C3a and C5a receptors prevents angiotensin II-induced hypertension via regulatory T cells. Circ Res. 2018;122(7):970–83. 10.1161/CIRCRESAHA.117.312153. [DOI] [PubMed] [Google Scholar]
- 77.Hernandez AL, Kitz A, Wu C, Lowther DE, Rodriguez DM, Vudattu N, et al. Sodium chloride inhibits the suppressive function of FOXP3+ regulatory T cells. J Clin Invest. 2015;125(11):4212–22. 10.1172/JCI81151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caillon A, Mian MOR, Fraulob-Aquino JC, Huo K-G, Barhoumi T, Ouerd S, et al. Gammadelta T cells mediate angiotensin II-induced hypertension and vascular injury. Circulation. 2017;135(22):2155–62. [DOI] [PubMed] [Google Scholar]
- 79.Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, et al. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature. 2013;496(7446):518–22. 10.1038/nature11868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, et al. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature. 2013;496(7446):513–7. 10.1038/nature11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Norlander AE, Saleh MA, Pandey AK, Itani HA, Wu J, Xiao L, et al. A salt-sensing kinase in T lymphocytes, SGK1, drives hypertension and hypertensive end-organ damage. JCI Insight. 2017;2(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Saleh MA, Norlander AE, Madhur MS. Inhibition of interleukin 17-a but not interleukin-17F signaling lowers blood pressure and reduces end-organ inflammation in angiotensin II-induced hypertension. JACC Basic Transl Sci. 2016;1(7):606–16. 10.1016/j.jacbts.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, et al. Lymphocyte adaptor protein LNK deficiency exacerbates hypertension and end-organ inflammation. J Clin Invest. 2015;125(3):1189–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ramseyer VD, Hong NJ, Garvin JL. Tumor necrosis factor alpha decreases nitric oxide synthase type 3 expression primarily via Rho/Rho kinase in the thick ascending limb. Hypertension. 2012;59(6): 1145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zhang J, Patel MB, Griffiths R, Mao A, Song YS, Karlovich NS, et al. Tumor necrosis factor-alpha produced in the kidney contributes to angiotensin II-dependent hypertension. Hypertension. 2014;64(6):1275–81. 10.1161/HYPERTENSIONAHA.114.03863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferreri NR, Escalante BA, Zhao Y, An SJ, McGiff JC. Angiotensin II induces TNF production by the thick ascending limb: functional implications. Am J Phys. 1998;274(1 Pt 2):F148–55. [DOI] [PubMed] [Google Scholar]
- 87.Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in Dahl salt-sensitive rats. Am J Physiol Ren Physiol. 2016;311(3):F555–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Brands MW, Banes-Berceli AKL, Inscho EW, Al-Azawi H, Allen AJ, Labazi H. Interleukin 6 knockout prevents angiotensin II hypertension: role of renal vasoconstriction and janus kinase 2/signal transducer and activator of transcription 3 activation. Hypertension. 2010;56(5):879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, et al. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299(2):R590–5. 10.1152/ajpregu.00207.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kassan M, Galan M, Partyka M, Trebak M, Matrougui K. Interleukin-10 released by CD4(+)CD25(+) natural regulatory T cells improves microvascular endothelial function through inhibition of NADPH oxidase activity in hypertensive mice. Arterioscler Thromb Vasc Biol. 2011;31(11):2534–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, et al. CD8+ T cells stimulate Na-Cl co-transporter NCC in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun. 2017;8:14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Rodriguez-Iturbe B, Quiroz Y, Nava M, Bonet L, Chavez M, Herrera-Acosta J, et al. Reduction of renal immune cell infiltration results in blood pressure control in genetically hypertensive rats. Am J Physiol Ren Physiol. 2002;282(2):F191–201. 10.1152/ajprenal.0197.2001. [DOI] [PubMed] [Google Scholar]
- 93.Herrera J, Ferrebuz A, MacGregor EG, Rodriguez-Iturbe B. Mycophenolate mofetil treatment improves hypertension in patients with psoriasis and rheumatoid arthritis. J Am Soc Nephrol. 2006;17(12 Suppl 3):S218–25. [DOI] [PubMed] [Google Scholar]
- 94.Hoorn EJ, Walsh SB, McCormick JA, Furstenberg A, Yang CL, Roeschel T, et al. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med. 2011;17(10):1304–9. 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bamoulid J, Staeck O, Halleck F, Khadzhynov D, Brakemeier S, Durr M, et al. The need for minimization strategies: current problems of immunosuppression. Transpl Int. 2015;28(8):891–900. [DOI] [PubMed] [Google Scholar]
- 96.Kinlay S, Schwartz GG, Olsson AG, Rifai N, Leslie SJ, Sasiela WJ, et al. High-dose atorvastatin enhances the decline in inflammatory markers in patients with acute coronary syndromes in the MIRACL study. Circulation. 2003;108(13):1560–6. 10.1161/01.CIR.0000091404.09558.AF. [DOI] [PubMed] [Google Scholar]
- 97.Strazzullo P, Kerry SM, Barbato A, Versiero M, D’Elia L, Cappuccio FP. Do statins reduce blood pressure?: a meta-analysis of randomized, controlled trials. Hypertension. 2007;49(4):792–8. [DOI] [PubMed] [Google Scholar]