

Loss of two hepatic nuclear receptors muddles the Golgi and the sugarcoat on proteins.
Abstract
Nuclear receptors farnesoid X receptor (FXR) and small heterodimer partner (SHP) are key regulators of metabolism. Here, we report a previously unknown function for the hepatic FXR-SHP axis in controlling protein N-linked glycosylation. Transcriptome analysis in liver-specific Fxr-Shp double knockout (LDKO) livers revealed induction of genes encoding enzymes in the N-glycosylation pathway, including Mgat5, Fut8, St3gal6, and St6gal1. FXR activation suppressed Mgat5, while Shp deletion induced St3gal6 and St6gal1. Increased percentages of core-fucosylated and triantennary glycan moieties were seen in LDKO livers, and proteins with the “hyperglycoforms” preferentially localized to exosomes and lysosomes. This up-regulation of N-glycosylation machinery was specific to the Golgi apparatus and not the endoplasmic reticulum. The increased glycan complexity in the LDKO correlated well with dilated unstacked Golgi ribbons and alterations in the secretion of albumin, cholesterol, and triglycerides. Our findings demonstrate a role for the FXR-SHP axis in maintaining glycoprotein diversity in the liver.
INTRODUCTION
N-Glycosylation is a key posttranslational modification that decides the fate of several liver proteins, including their targeting to the plasma membrane or for secretion. Briefly, various combinations of sugar molecules (glycan moieties) attach covalently to the nitrogen of asparagine residues of a protein (1, 2), including mannose and glucose in the endoplasmic reticulum (ER) lumen (3–6). Further glycan processing occurs in cis- and medial-Golgi, including the addition of GlcNAc (N-acetylglucosamine) by the MGAT (Mannosyl-glycoprotein N-acetylglucosaminyltransferase) family of proteins resulting in “antennae” (or branch) glycan moieties. These branched glycans are finally modified in trans-Golgi via the addition of fucose by fucosyltransferase (Futs), N-acetylgalactosamine by GalNAc transferases, and sialic acid by sialyltransferases (Stgals and Stsias) (5, 7, 8). These complex glycan structures not only allow for structural and functional heterogeneity in the tissue glycoproteome but also are important for maintaining appropriate liver function (9–11). However, the regulation of this essential protein modification in the liver remains understudied.
Farnesoid X receptor (FXR; NR1H4) and small heterodimer partner (SHP; NR0B2) belong to a superfamily of nuclear receptors that regulate transcription of several genes involved in intermediary metabolism. Global deletion of FXR and SHP induces liver injury (12) and ultimately results in spontaneous hepatic carcinogenesis in mice (13). Here, we set out to identify molecular networks coordinated by hepatic FXR and SHP, in addition to their conventional metabolic targets. Using high-throughput RNA sequencing (RNA-seq), glycomics, glycoproteomics analyses, and electron microscopy, we found a key role for this axis in regulating the liver N-glycome.
RESULTS
Liver-specific Fxr-Shp double-knockout mice exhibit defective bile acid response
We generated liver-specific Fxr-Shp double-knockout (LDKO) and double-floxed homozygous Fxr-Shp (control) mice as shown in fig. S1A. The knockout was validated by genotyping and quantitative reverse transcription polymerase chain reaction (RT-PCR) assays (fig. S1, B and C).
Bile acids (BAs), the cholesterol metabolites synthesized in the liver, are responsible for fat digestion. By activating FXR and SHP in the liver, BAs autoregulate their concentrations (14–17). LDKO mice exhibited poor response to cholic acid (CA; a primary bile acid in humans) supplemented diet. Focal inflammation and ballooned hepatocytes were observed in chow-fed LDKO livers, which were more evident in CA-fed LDKO livers (fig. S2A). Markers of liver injury, alanine aminotransferase (ALT) and aspartate aminotransferase (AST), were significantly elevated in LDKO + CA (Fig. 1A). Compared to controls, LDKO mice accumulated pathological levels of BAs in the plasma, liver (Fig. 1B), and gall bladder (fig. S2B) in response to the CA diet. However, ileal BA levels were unchanged (fig. S2C), indicating that the intact intestinal FXR-SHP axis in LDKO mice was sufficient to maintain the ileal BA concentrations.
Fig. 1. LDKO mice exhibit a unique induction of N-glycosylation gene machinery.
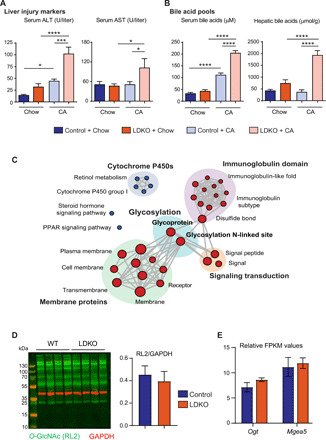
Biochemical characterization revealed (A) severe liver injury (marked by serum ALT and AST levels) and (B) bile acid accumulation in LDKO + CA mice compared to the controls (n = 6 to 11 mice per group, one-way ANOVA, *P < 0.05, ***P < 0.001, and ****P < 0.0001). (C) RNA-seq was performed on the livers of chow-fed control and LDKO mice. Functional clusters display enrichment of glycosylation upon Fxr-Shp deletion. Blue circles: down-regulated; red circles: up-regulated in LDKO mice (n = 3 mice per group). (D) Neither intracellular O-GlcNAcylation levels nor (E) the genes regulated O-GlcNAcylation, Ogt and Mgea5 were enhanced in LDKO mice (n = 3 to 4 mice per group; Student t test, P < 0.05 was considered significant). GAPDH, glyceraldehyde-3-phosphate dehydrogenase. FPKM, fragments per kilobase million; PPAR, peroxisome proliferator–activated receptor.
We next investigated whether the genes involved in maintaining BA homeostasis were altered in LDKO mice (fig. S2D). SHP is known to turn off genes responsible for BA synthesis (14, 18–20), including Cyp7a1 (cholesterol 7 alpha-hydroxylase) and Cyp8b1 (sterol 12-alpha-hydroxylase). Both these genes were poorly repressed in LDKO livers upon CA challenge, unlike the controls. We found that LDKO livers showed reduced basal expression of canalicular transporter Bsep (Abcb11) and lost the CA-mediated up-regulation of Bsep and Mdr1a (Abcb1a) expression compared to control livers. Genes responsible for BA uptake into hepatocytes Ntcp was up-regulated in LDKO + CA but not in control + CA, whereas Oatp1 was suppressed in both groups. Although, CA-fed LDKO livers revealed pronounced induction of BA efflux transporters Mrp3 (Abcc3) and Mrp4 (Abcc4) (fig. S2E), it was insufficient to compensate for the loss in canalicular transport, and the increase in hepatic uptake led to excess hepatic BAs.
LDKO mice exhibit increases in N-glycosylation pathway
To decipher the molecular networks coordinately regulated by hepatic FXR-SHP, we performed RNA-seq analysis and found more than 2600 significantly altered transcripts in LDKO livers compared to the controls. Of these, 1253 were up-regulated, while 865 were down-regulated in LDKO livers (fig. S3, A and C). Gene network analysis pinpointed several known signaling and metabolic pathways including response to nutrients, cell adhesion, and drug clearance. Unexpectedly, we found an enrichment of genes involved in the glycosylation pathway (Fig. 1C and fig. S3B). Upon further analysis, we found that these alterations were unique for N-linked, but not O-linked, glycosylation (fig. S3D). Consistent with this observation, intracellular O-GlcNAcylation (Fig. 1D) and the expression of genes regulating O-GlcNAcylation, Ogt, and Mgea5 (21, 22) were unchanged (Fig. 1E).
Instead, the RNA-seq data showed up-regulation of essential N-linked glycosyltransferases (7, 8, 23) (Fig. 2A), such as Mgat5 (N-acetylglucosaminyl-transferase V) that promotes glycan branching, Fut8 (α-1,6-fucosyltransferase) that adds fucose to the glycan core, and St3gal6 and St6gal1 (sialyltransferases) that add sialyl groups (Fig. 2B). These glycosyltransferases “decorate” proteins associated with the cell-matrix and cell-cell contact and promote tumorigenesis (9, 24–26). Higher expression of Mgat5, St3gal6, and St6gal1 are associated with poor survival of patients with liver cancer (fig. S3E), suggesting that stringent control of their expression is critical to maintain normal liver function.
Fig. 2. Up-regulation of N-glycosylation machinery in LDKO livers is dictated by FXR and SHP and not by BA overload.
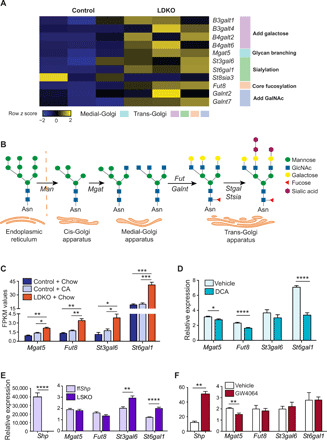
(A) Heatmap indicates up-regulation of key glycosyltransferase genes that are involved in “decorating” glycoproteins in LDKO livers. (n = 3 mice per group, Student t test, fold change > 1.5, P < 0.05). (B) Schematic highlighting the steps of cellular protein processing in ER and Golgi apparatus. (C) Bar graphs comparing the FPKM values indicate up-regulation of glycosylation genes in LDKO livers is not a consequence of BA overload (n = 3 mice per group; one-way ANOVA, *P < 0.05, **P < 0.01, and ***P < 0.001). (D) Incubation of primary hepatocytes with a toxic BA, such as DCA, suppresses the N-glycosylation genes instead (n = hepatocytes cultured in triplicates from two mice per group; Student t test, *P < 0.05 and ****P < 0.0001). Qualitative RT-PCR analysis of (E) liver-specific SHP knockout (LSKO) livers and (F) FXR-activated (treated with GW4064, FXR agonist) livers reveal that FXR and SHP individually regulate some of these glycosylation genes (n = 5 to 8 mice per group; Student t test, **P < 0.01 and ****P < 0.0001).
FXR and SHP, but not BA, overload regulate the induction of N-glycosyltransferases
To test whether BA overload seen in the LDKO livers was sufficient to induce expression of N-linked glycosyltransferases, we analyzed the RNA-seq data of CA-treated control livers. We found no significant differences in transcript levels of glycosyltransferases (Fig. 2C). Then, we tested whether a toxic hydrophobic BA, such as deoxycholic acid (DCA), can alter the expression of glycosyltransferases. When we treated primary hepatocytes with DCA, we found a modest suppression in Mgat5, Fut8, and St6gal1 expression (Fig. 2D) rather than the induction observed in LDKO livers. These findings demonstrate that elevated BAs are not responsible for the induction of glycosyltransferases.
As global Fxr-Shp double-knockout mice show elevated CYP17A1 expression and its enzymatic product 17 hydroxy progesterone (17OHP) (12), we investigated and found similar up-regulation of Cyp17a1 mRNA expression in LDKO livers (fig. S4A). However, serum 17OHP levels in LDKO were below the detectable limit (fig. S4A). Exogenous 17OHP administration to the wild-type (WT) mice (fig. S4, B and C) led to a decrease in mRNA levels of N-glycosylation genes (fig. S4D) with an increase in certain glycan decorations but not fucosylation (fig. S4E). These results indicate that 17OHP is not sufficient to explain the N-glycosylation phenotype of LDKO mice.
Next, we assessed whether Shp alone can regulate expression of these genes and found St3gal6 and St6gal1 up-regulation in the liver-specific Shp knockout mice compared to their respective controls (Fig. 2E). We also tested the role for FXR in controlling the expression of glycosyltransferases by activating it with its agonist GW4064 in WT mice. FXR activation led to a modest but significant down-regulation of Mgat5 but did not alter the expression of Fut8, St3gal6, and St6gal1 levels (Fig. 2F). This result indicates that FXR can suppress Mgat5 expression, whereas SHP can suppress Stgal expression and together coordinate the expression of several glycosyl transferases. We next analyzed the publicly available hepatic chromatin immunoprecipitation sequencing (ChIP-seq) datasets for FXR (27) and SHP (28) to determine whether these nuclear receptors bind to their respective target genes, Mgat5, and St3gal6 and St6gal1. Although no SHP binding was observed in St3gal6 and St6gal1 DNA sequences (fig. S5A), we did find a robust FXR-binding peak along with its core motif (ER2, “TGACCTCAAACTCA”) within the intron 4 of Mgat5 gene (fig. S5B). These data support the hypothesis that FXR may directly regulate Mgat5 transcription, whereas SHP may indirectly regulate transcription of St3gal6 and St6gal1.
Ablation of hepatic Fxr and Shp results in higher hyperbranching and glycan core fucosylation
To assess the functionality of the induction of N-glycosylation genes, we performed glycan profiling on control and LDKO liver lysates (fig. S6A). We found about twofold increase in core fucosylation (Fig. 3A) and hyperbranching (tertiary glycans) in LDKO livers compared to the controls (Fig. 3B), whereas both sialylation and mannosylation were unaltered (fig. S6B). To identify the proteins that were being targeted for altered N-glycan processing, we performed glycoproteomics on control and LDKO liver lysates. Figure S6C illustrates an example of the mass spectrum of a glycoform of a liver protein. Gene Ontology analysis of the transcripts encoding for these altered glycoproteins revealed a metabolic or secretory function and localization to the extracellular exosome, ER, and lysosome (Fig. 3C). These proteins including, but not limited to, cathepsin Z, lysosomal membrane protein, endoplasmin, haptogloblin, and epoxide hydrolase exhibited increases in more complex glycoforms in LDKO livers (Fig. 3D and fig. S6D). For example, we found that HexNAc(5)Hex(5)Fuc(1)NeuAc(1) glycoform of lysosomal membrane protein is completely absent and replaced by a more complex HexNAc(5)Hex(7)Fuc(1)NeuAc(2) glycoform in LDKO livers. These data are in line with gene expression changes, and consistently, we find complex N-glycan decorations in LDKO.
Fig. 3. Fxr-Shp ablation results in hyperbranched glycosylations of liver proteins.
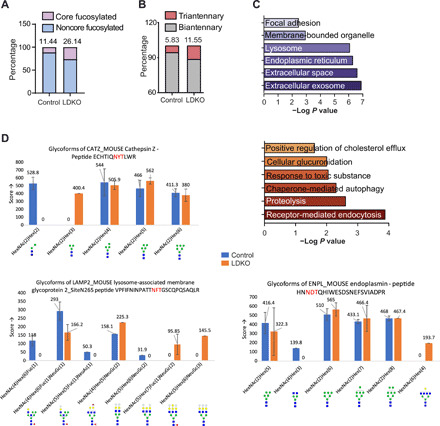
LDKO liver glycoproteins were isolated and analyzed for their glycan profiles. LDKO livers display increased percentage of (A) core fucosylated and (B) triantennary glycans in LDKO liver compared to controls (n = 5 mice averaged per group). (C) Gene Ontology analysis of proteins with increased glycosylation reveals their cellular localization and function (n = 5 livers pooled per group). (D) Relative ratio of complex, sialylated and fucosylated glycoforms for CATZ, EMPL, and lysosomal membrane protein 2 (LAMP2) indicate enrichment of hyperglycoforms in LDKO livers compared to controls (n = 5 mice averaged per group).
Metabolic and functional consequence of increased glycosylation in LDKO mice
To determine the consequence of elevated glycosylation in LDKO livers, we examined the different cellular fates of glucose, such as glycolysis, pentose phosphate pathway, glycosylation, glycogenesis, and gluconeogenesis (Fig. 4A) (29, 30). In agreement with increased glycosylation, LDKO mice exhibited lower circulating glucose levels (Fig. 4B). Furthermore, the genes regulating glycolysis, pentose phosphate pathway, and gluconeogenesis remained unaltered in LDKO livers (Fig. 4, C to E). However, the low glucose correlated with the severe loss of glycogen stores in LDKO livers as evident by periodic acid–Schiff (PAS) staining (Fig. 4F). Next, we analyzed the intermediates of the hexosamine biosynthesis pathway such as uridine 5′-diphosphate (UDP)–GlcNAc and UDP-GalNAc (31–33) in LDKO livers, and their levels were comparable despite the increase in demand for N-glycosylation (Fig. 4G). While the hexose intermediates are not limiting, the liver glycogen levels are diminished, circulating glucose is reduced, and N-glycosylation is increased, when the FXR-SHP axis is ablated in the liver.
Fig. 4. LDKO livers exhibit any depleted glycogen stores.
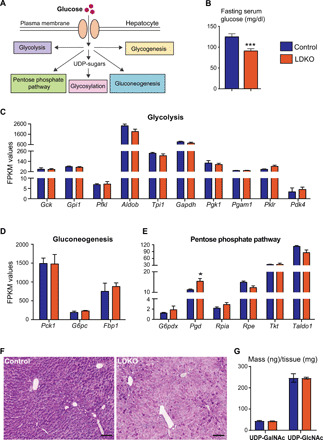
(A) Schematic figure displaying the fate of glucose in a hepatocyte. (B) LDKO mice have reduced fasting blood glucose levels (n = 12 to 15 mice per group; Student t test, ***P < 0.005). Bar graphs comparing FPKM values of genes regulating (C) glycolysis, (D) gluconeogenesis, and (E) pentose phosphate pathway show no significant difference between control and LDKO livers (n = 3 mice per group; Student t test, *P < 0.05). (F) PAS staining indicates lower glycogen content in LDKO livers (n = 5 mice per group). Scale bars, 50 μm). (G) UDP-glycan sugars are not altered in LDKO livers (n = 5 mice per group, Student t test).
LDKO livers exhibit structural and functional defects in the Golgi apparatus
As N-glycan processing occurs in both the ER and the Golgi apparatus (3, 5, 8), we examined and found that the N-linked glucosyl transferases up-regulated in the LDKOs were specifically associated with the Golgi and not the ER (34) except for Alg10b, which catalyzes the final step in the synthesis of N-glycan moiety (fig. S7A). As unfolded protein response can regulate N-glycome (35–38), we quantified ER stress in LDKO livers by XBP-1 (X-box binding protein 1) splicing assays (39–41) and gene expression profiling (42). We found no changes in them compared to control (fig. S7, B and C). These data confirm that the N-glycosylation and the stress pathways in ER are not affected, but several N-glycosylation genes in the Golgi apparatus are induced in LDKO livers (Fig. 2, A and B). So, using electron microscopy, we examined the Golgi structure and observed unstacked and dilated Golgi in LDKO livers, unlike well-stacked Golgi ribbons in controls (Fig. 5A).
Fig. 5. LDKO hepatocytes exhibit Golgi structural defects.
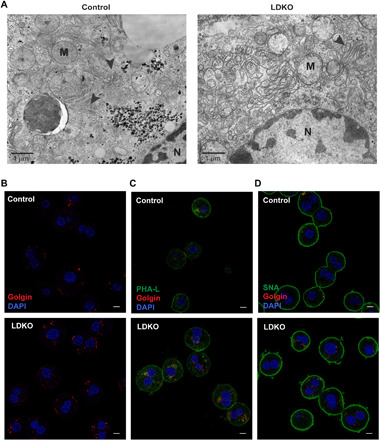
(A) Electron microscopy imaging shows unstacked Golgi apparatus (arrowheads) in LDKO livers compared to controls. Scale bar, 1 μm. (B) Immunofluorescence staining for a Golgi resident protein, Golgin reveals increased expression in LDKO hepatocytes. DAPI, 4′,6-diamidino-2-phenylindole. This Golgin labeling colocalizes with lectins binding to (C) complex and (D) sialylated glycans in LDKO proteome (hepatocytes cultured from n = 3 mice per group). Scale bars, 10 μm.
To validate that the loss of FXR-SHP–induced hepatic Golgi expansion and accumulation of N-glycoproteins, we isolated primary hepatocytes from control and LDKO mice and stained them for the resident Golgi-protein, Golgin. LDKO hepatocytes displayed higher Golgin expression than controls (Fig. 5B). In addition, Phaseolus Vulgaris Leucoagglutinin (PHA-L) and Sambucus nigra agglutinin (SNA) were used to stain-branched (complex) glycans and sialic acid moieties, respectively. Both PHA-L and SNA stained the plasma membrane and some of the Golgi as expected (7). The amount of PHA-L and SNA staining and its colocalization with Golgin were enhanced in LDKO hepatocytes compared to controls (Fig. 5, C and D). Last, we measured plasma profiles of the secreted products trafficked through the Golgi (43, 44) from LDKO livers. We found that, in the LDKO mice, while hepatic albumin levels were unaffected, serum albumin levels were significantly reduced (Fig. 6A). In addition, the circulating levels of cholesterol and triglycerides were significantly altered in LDKO plasma, compared to controls (Fig. 6, B and C). Together, these findings reveal that loss of hepatic FXR-SHP axis results in up-regulation of N-glycosylation transcripts, increases in complex glycoforms, defective Golgi structure, and altered secretion in hepatocytes (Fig. 6D), which likely also affects lipoprotein clearance indirectly.
Fig. 6. Hepatic secretory ability is altered upon the loss of FXR and SHP.
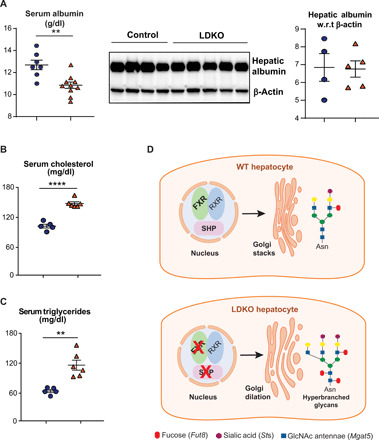
(A) Circulating levels of albumin are reduced in LDKO mice, with no significant change in hepatic albumin levels (n = 4 to 9 mice per group, Student t test, **P < 0.01). Whereas serum levels of (B) cholesterol and (C) triglycerides are higher in LDKO mice (n = 5 to 9 mice per group; Student t test, **P < 0.01 and ****P < 0.0001). (D) Schematic figure displaying unstacked Golgi and hyperbranched N-glycans in an LDKO hepatocyte.
DISCUSSION
Unlike global Fxr-Shp double-knockout mice (12), the LDKOs did not accumulate excessive bile acids under basal conditions. However, they were defective in their response to a bile acid overload (CA-enriched diet), indicating that the hepatic FXR-SHP axis is necessary for this function.
We serendipitously found a role for FXR and SHP in regulating the N-glycosylation status of liver proteins through RNA-seq data of control and LDKO livers. Only the N-glycosylation specific to Golgi was induced, with no difference in the levels of intracellular O-GlcNAcylation or ER N-glycosylation. Particularly, the “branching” glycosyltransferase Mgat5, core fucosyltransferase Fut8, and sialyltransferases St3gal6 and St6gal1 (7, 23, 45) were induced in the absence of hepatic FXR-SHP axis. We validated the gene expression data and found increases in core fucosylation and triantennary complex branching of glycan moieties on LDKO N-glycoproteins by glycomics.
Challenging hepatocytes with high bile acid concentrations in vitro and 17OHP in vivo both resulted in suppression of genes involved in N-glycosylation. This BA-mediated decrease in glcosylation was also observed in previously published studies (46–48). Thus, it is unlikely that the increase in BA or 17OHP directly caused the observed up-regulation of glycosyltransferases in LDKO livers.
Next, we tested whether FXR and SHP can individually alter the expression profiles of these glycosyltransferases. Loss of hepatic SHP induced expression of sialyltransferases, whereas FXR activation suppressed Mgat5 gene, which is responsible for glycan branching, indicating that FXR and SHP coordinate the N-linked glycan landscape at the transcript level in the liver.
To further probe how LDKO mice fuel the increased N-glycosylation in the liver, we examined various glucose metabolic pathways and found no alterations in glycolysis, pentose phosphate pathway, and gluconeogenesis. However, we observed reduced serum glucose levels and hepatic glycogen pools in LDKO mice that could be used through the hexosamine biosynthetic pathways to increase N-glycosylation. Despite increase N-glycans in the LDKO mice, we found no differences in the levels of UDP-GlcNAc and UDP-GalNac in the livers, indicating similar flux through the hexosamine biosynthetic pathway between the LDKO and WT livers and that the hexosamine pathway is not rate limiting in this process.
Since the altered glycosyltransferases in the LDKO mice—Mgat5, Fut8, and St3gals—are localized only in medial- and trans-Golgi apparatus (23, 49), this indicates a spatial specificity of the hepatic FXR-SHP regulation. To this effect, we did not observe any ER stress in the LDKO livers. The structural defect was localized to the Golgi apparatus in LDKO livers. Furthermore, many of the N-linked glycoproteins present in the exosomes, lysosomes, and ER were up-regulated in the LDKO livers. Not only more complex glycoforms are expressed in LDKO livers but also glycoforms that were not detected in controls. These “hyperglycoforms” in LDKO may alter the function of the protein, resulting in defective signaling pathways.
Last, we observed an altered secretome of LDKO livers, which is consistent with a recent study that predicted that FXR may control hepatocyte secretome (50). Our findings provide a mechanism and additional evidence of how the secretory ability of the liver may be altered when the hepatic FXR and SHP axis is deleted. Overall, this study defines a previously unidentified role for FXR and SHP in regulating glycosylation, a less understood posttranslational modification, which is distinct from their conventional role in nutrient homeostasis. Understanding how hepatic FXR-SHP axis specifically regulates Golgi dynamics and N-glycosylation will open new avenues to target various cellular pathways.
MATERIALS AND METHODS
Animals and experimental design
Fxr-floxed and Shp-floxed mice, obtained from K. Schoonjans laboratory, were intercrossed to obtain double-floxed homozygous Fxr-Shp mice (Fxrfl/fl Shpfl/fl). Furthermore, these Fxrfl/fl Shpfl/fl (control) mice were bred with mice expressing the Cre-recombinase under the control of albumin promoter (AlbCre mice, Envigo/Harlan laboratories) to obtain liver-specific double knockouts (Fxrfl/fl Shpfl/fl AlbCre+, LDKO). The mice were maintained on a C57BL/6J background and housed on a standard 12-hour light/12-hour dark cycle. Both the control and LDKO mice were born in regular Mendelian ratios. To study how LDKO mice respond to BA-induced stress, we fed LDKO and control mice with normal chow or 1% CA diet for 5 days. Plasma, liver, gall bladder, and ileum were collected between Zeitgeber times ZT4 to ZT6 (n = 6 to 12 mice per group). Fasted C57BL/6J mice (Envigo/Harlan laboratories) were gavaged with either vehicle [1% methylcellulose and 1% TritonX-100 in phosphate-buffered saline (PBS)] or FXR agonist GW4064 (50 mg/kg). Mice were gavaged twice, first in the evening and second in the morning, and euthanized 3 hours after second gavage (n = 5 mice per group). To examine the role for 17OHP, we injected WT mice with 17OHP (125 mg/kg, i.p) or corn oil twice on the first day, followed by once daily injections for four additional days. All the experiments were carried out on 10- to 12-week-old male mice, fasted for 8 to 10 hours unless specified as outlined in the Guide for the Care and Use of Laboratory Animals, the National Academy of Sciences (NIH publication 86-23, revised 1985), and approved by the Institutional Animal Care and Use Committee at University of Illinois at Urbana-Champaign.
Genotyping
Mouse tails were clipped during weaning to obtain genomic DNA. Tails were lysed using DirectPCR tail lysis buffer (Viagen Biotech) and proteinase K by an overnight incubation at 55°C. Proteinase K was heat-inactivated for 1 to 2 hours, and PCR was performed on the DNA isolated using primers listed in table S1.
Chemicals
Methylcellulose was obtained from Sigma-Aldrich (catalog no. 0262), GW4064 was obtained from Cayman Chemicals and 17OHP (Sigma-Aldrich).
Primary hepatocytes isolated from C57BL/6J mice were incubated with 100 μM DCA (Sigma-Aldrich) for 6 hours (n = 2 mice per group, three technical replicates).
Serum biochemistry
Blood was collected and centrifuged at 7000 to 8000g for 10 min. The plasma samples collected were tested for ALT and AST enzymes using Infinity ALT/GPT (glutamic-pyruvic transaminase) and AST/GOT (glutamic-oxaloacetic transaminase) kits (Thermo Fisher Scientific). Plasma BA levels were measured using a colorimetric BA assay kit (Genway) according to the manufacturer’s protocol. The circulating albumin levels were quantitated using the Albumin [BCG (Bromocresol Green)] Colorimetric Assay Kit (Biovision). Control and LDKO mice (n = 5 to 6 mice per group) were fasted for four hours before collecting blood. Fresh plasma was analyzed for lipid metabolites by fast-protein liquid chromatography at Perelman School of Medicine, University of Pennsylvania. For fasting blood glucose measurements, control and LDKO mice were fasted for 12 hours, and blood glucose was measured by tail bleeds using one-touch glucometer. Serum 17OHP levels were measured by mass spectroscopy at Roy J Carver Biotechnology Center Metabolomics Facility at University of Illinois, Urbana-Champaign.
Tissue BA assay
For hepatic and intestinal BA analysis, about 100 mg of liver tissue or whole ileum was homogenized in 75% ethanol, followed by a 2-hour incubation at 50°C and centrifugation at 15,000g for 10 min. The supernatant was diluted with 1× PBS and used to measure BA content in the liver or intestine, respectively, using the colorimetric BA Assay Kit (GenWay) according to the manufacturer’s protocol. The BA levels were normalized to the weight of the tissue used. For biliary BA measurements, intact gall bladders from mice were punctured to obtain the content, which was further diluted with 1× PBS and analyzed for BA pools.
Histology
Liver tissues from all the mouse cohorts were fixed in 10% neutral-buffered formalin overnight, subsequently dehydrated in increasing concentrations of ethanol (70 to 100%) and xylene, and lastly embedded in paraffin. Liver sections (5 μm thick) were cut, deparaffinized, and stained with hematoxylin and eosin using standard histological protocols. These sections were also stained with periodic acid and Schiff’s reagent for PAS staining. Briefly, the deparaffinized sections were stained with 0.5% periodic acid for 10 min, followed by wash in water and incubation in Schiff’s solution for 30 min. Next, slides are rinsed in 0.55% potassium metabisulfite for 30 s and washed in running warm tap water for 10 min to develop the pink color. Last, the sections were counterstained with hematoxylin, mounted, and imaged.
RNA-seq analysis
RNA was isolated from control and LDKO livers (n = 3 per group) using the RNeasy Tissue Mini Kit (Qiagen). RNA quality was tested using an Agilent bioanalyzer by the Functional Genomics Core at the Roy J. Carver Biotechnology Center, UIUC (University of Illinois, Urbana-Champaign). HiSeq libraries were prepared, and 100–base pair (bp) paired-end Illumina sequencing was performed on a HiSeq 4000 at the high-throughput sequencing and genotyping unit, UIUC. Reads were processed for quality using Trimmomatic (51) and aligned to the mouse genome (mm10) using STAR (52). Gene expression was determined as fragments per kilobase million using count and differential expression values obtained from DESeq2 (53) and HTseq (54). Fold change of >1.5 and <0.67 was considered significant. Gene Ontology pathway and cellular component analysis were performed using DAVID (Database for Annotation, Visualization, and Integrated Discovery) and mapped using Enrichment maps plugin in Cytoscape. Mouse reference gene set served as background. The RNA-seq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus (GEO) (55) and are accessible through GEO Series accession number GSE164447 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164447).
Identifying DNA binding motifs
The gene sequences were retrieved from the Ensembl genome browser, release 102 (56). These sequences were then run using FIMO (Find Individual Motif Occurrences) software (57). FIMO uses a position-dependent scoring matrix that describes the score of each possible nucleotide at each position in the given sequence. Motifs containing or resembling the canonical “IR1 (AGGTCAnTGACCT) and ER2 (TGACCTnnAGGTCA) motifs” were identified and compared with the binding peaks identified using the ChIP-seq dataset (27, 28).
Quantitative real-time PCR and splicing assay analysis
RNA was isolated from snap-frozen livers (n = 4 to 5 per group) using TRIzol (Invitrogen) and reverse-transcribed for gene expression analysis by SYBR Green–based Real-Time Quantitative Reverse Transcription PCR (qRT-PCR) using mouse-specific primer sets listed in table S2. Relative gene expression was calculated by delta-delta Ct method and was normalized to 36B4 as loading control. For Xbp-1 splicing assay, the complementary DNA samples from both control and LDKO livers were run on a 5% nondenaturing polyacrylamide gel electrophoresis (PAGE) gel to examine its spliced isoforms. Percentage (%) exclusion for the variably spliced region was calculated with ImageLab software (Bio-Rad) as [(exon inclusion band intensity)/(exon inclusion band intensity + exon exclusion band intensity) × 100].
Detection of O-GlcNAc by Western blot
Liver tissues were homogenized in Tissue Protein Extraction Reagent (Thermo Fisher Scientific) using the gentleMACS dissociator (Miltenyi Biotec). Protein (30 μm) was run out on a PAGE gel and transferred to a nitrocellulose membrane. Membranes were incubated overnight with antibodies to O-GlcNAc (RL2, Thermo Fisher Scientific) and glyceraldehyde-3-phosphate dehydrogenase (Abcam). Visualization was done using the Li-Cor Odyssey and analysis using the Li-Cor Image Studio Software.
Extraction of proteins from mouse liver for glycomics, glycoproteomics, and proteomics
Control and LDKO liver tissues (n = 5 mice per group) were tested for glycan structural diversity in glycoproteins using matrix-assisted laser desorption/ionization–time-of-flight (45) and mass spectrometry (MALDI-TOF-MS) at Complex Carbohydrate Research Center, University of Georgia. Briefly, about 100 mg of liver tissue was homogenized in a 1:1 ratio of ice-cold methanol/water. Chloroform:methanol:water at the ratio of 4:8:3 was added, mixed vigorously, and left to incubate at room temperature for approximately 2 hours on a bench rocker. The sample was then centrifuged at 4°C, 3000 rpm for 15 min to separate the precipitated tissue proteins from the supernatant glycolipids layer. The chloroform:methanol:water extraction was repeated two more times. The protein pellet was blown for a few minutes under a stream of nitrogen, and 1 ml of ice-cold water, along with 4 ml of ice-cold acetone, was added for acetone precipitation at −20°C. After about 4 hours, the sample was centrifuged at 4°C, 3000 rpm for 15 min, and the supernatant was discarded. Acetone precipitation was repeated one more time, and lastly, the pellets were processed using only 4 ml of ice-cold acetone, following the same procedure. The pellets were then dried under the nitrogen stream and stored at −80°C for further analysis.
Glycomics analysis
About 5 mg of protein pellet was resuspended in 50 mM ammonium bicarbonate buffer and digested with trypsin for 24 hours at 37°C, followed by purification by passing through a C18 solid-phase cartridge after inactivating trypsin by heating at 95°C for 5 min. The eluted peptides were treated twice with peptide N-glycosidase F (PNGaseF) for a total incubation time of 18 hours. The released N-glycans were separated from the peptides by passing through a C18 solid-phase cartridge and were dried by lyophilization. These N-glycans were permethylated with methyl iodide for 10 min at room temperature with shaking. The reaction was quenched by adding water, and the permethylated glycans were extracted in dichloromethane, dried under a stream of nitrogen, and analyzed by MALDI-TOF-MS by using DHB (2,5-dihydroxybenzoic acid) as the matrix (58).
For 17OHP experiments, about 50 mg of liver tissue was washed with PBS two times and homogenized in 50 mM ammonium bicarbonate buffer using a 20-gauge needle and syringe. The sample was probe-sonicated for 2 min, followed by ultrafiltration thrice using 10-kDa centrifuge filters (MilliporeSigma Amicon, UFC501024). The protein solution from the filter was collected in the ammonium bicarbonate buffer with PNGaseF and incubated at 37°C for 48 hours. The N-glycans were then purified, permethylated, and analyzed by MALDI-MS as mentioned above.
Glycoproteomics analysis
About 1 mg of proteins extracted was dissolved in 50 mM ammonium bicarbonate buffer. Dithiothreitol solution (17 mg/ml) was added to the samples and incubated at 37°C for 90 min, followed by incubation with iodoacetamide solution (65 mg/ml) at room temperature for 90 min in the dark for carbamidomethylation. The sample was dialyzed against double-distilled water and digested by trypsin (0.4 μg/μl; Promega) at a trypsin to protein ratio of 1:40 and incubated at 37°C for 12 hours. The digestion was terminated by heating at 95°C for 5 min, desalted by C18 solid-phase extraction cartridges, and then samples were dried under speed vacuum. The peptides and glycopeptides were subsequently redissolved in 0.1% formic acid in water. The digests were enriched for the glycopeptides by using the ProteoExtract Glycopeptide Enrichment Kit (Millipore, MA) and stored at −80°C until analysis by nano–liquid chromatography–tandem MS (LC-MS/MS).
Desalted liver protein digests were analyzed on an Orbitrap Fusion Tribrid mass spectrometer (Thermo Fisher Scientific, MA) equipped with a nanospray ion source and connected to a Dionex binary solvent system. Prepacked nano-LC columns of 15 cm length with 75 μm internal diameter, filled with 3-μm C18 material, were used for chromatographic separation of digests. Precursor ion scan was performed at 120,000 resolution in an Orbitrap analyzer, and precursors at a time frame of 3 s were selected for subsequent fragmentation using higher-energy collisional dissociation (HCD) at a normalized collision energy of 28 and collision-induced dissociation (CID) at a normalized collision energy of 40. The threshold for triggering an MS/MS event was set to 500 counts. The fragment ions were analyzed on Orbitrap after HCD and CID fragmentation at a resolution of 30,000. Charge-state screening was enabled, and precursors with unknown charge state or a charge state of +1 were excluded (positive ion mode). Dynamic exclusion was enabled with an exclusion duration of 30 s.
The LC-MS/MS spectra of the tryptic digest of proteins were searched against mouse protein database in.fasta format (downloaded from uniport) using Byonic 2.3 software with trypsin as the digestion enzyme. Carbamidomethylation of cysteine and oxidation of methionine were selected as variable modifications. Common mammalian N-glycans were used as variable modifications on asparagines. The LC-MS/MS spectra were analyzed manually for the glycopeptide fragmentation on HCD and CID with the support of Xcalibur 3.0.63 software. The spectra of glycopeptides were evaluated for the glycan neutral loss pattern, oxonium ions, and the glycopeptide fragmentations and identified the glycans. For the relative ratio calculation, the peak area of the glycoforms was extracted manually using the Xcalibur 3.0.63 software. Unannotated glycopeptides were identified on the basis of the presence of oxonium ions and neutral losses on the spectra of glycopeptides.
Nucleotide sugar extraction and detection
Sugars from liver tissues were extracted as described previously (59). Approximately 50 mg of mouse liver tissue was homogenized in 1 ml of cold 80% ethanol in a gentleMACS (Miltenyi Biotec) dissociator. Tissue lysates were centrifuged at 8000g for 20 min at 4°C, and the supernatant was evaporated in a vacuum centrifuge overnight. The evaporated samples were dissolved in 9% butanol, and lipids were removed by three extractions with 90% butanol. The lower aqueous fraction was evaporated overnight by vacuum centrifuge. Samples were resuspended in 5 mM ammonium bicarbonate, and sugar nucleotides were extracted using EnviCarb graphitized carbon columns (Supelco). Columns were prepared by washing with 3 ml of 80% acetonitrile and 0.1% trifluoroacetic acid, followed by 2 ml of water. Samples were loaded, and the column was washed with 2 ml of water and 2 ml of 25% acetonitrile, followed by 2 ml of 50 mM TEAA (Triethylammonium acetate) buffer. Samples were eluted with 2 ml of 25% acetonitrile and 50 mM TEAA. Eluate was evaporated overnight in a vacuum centrifuge and dissolved in 240 μl of 40 mM sodium phosphate buffer.
A Dionex (Thermo Fisher Scientific) HPIC (High Pressure Ion Chromatography) system consisting of a Dionex ICS-6000, CarboPac PA1 column 2x250, and PA1 guard column 2x50 under the control of Chromeleon 7 software was used for analysis. The program involved a gradient made of the following solutions: A = dionized water; B = 1 mM NaOH; C = 1 mM NaOH/1 M NaOAC. The gradient was isocratic B followed by a 20-min gradient up to 60% C. A step up to 100 B was used to clean the column before an equilibration back to 100 A. Ultraviolet absorbances were collected at 260 nm. Concentrations of nucleotide sugars detected were normalized to wet weight of tissues.
Lectin staining
Liver sections (5 μm thick) from control and LDKO mice were cut, deparaffinized, and stained for lectins such as UEA (Ulex Europaeus Agglutinin) (binds fucose), PHA-L (binds complex glycans), and SNA (binds sialic acid) from Vector Laboratories. Briefly, sections were blocked with Carbo-free Blocking solution (SP-5040, Vector Laboratories) for 30 min at room temperature, followed by fluorescently labeled lectin in PBS for 30 min at room temperature. The slides were then washed twice in PBST (Phosphate Buffered Saline with Triton X-100) and stained, mounted with Vectashield Hardset mounting media (H-1400, Vector Laboratories), and imaged using confocal microscope LSM 710 at Institute for Genomic Biology Core facility, UIUC using 488-nm laser, and a red psuedocolor was assigned to it.
Immunostaining of primary hepatocytes
Primary hepatocytes from control and LDKO mice were isolated using a two-step collagenase perfusion technique using 3000 U collagenase type I from Worthington. Cells were layered onto a 30% Percoll gradient and centrifuged at 600 rpm for 10 min. The pellet, rich in live hepatocytes, was then washed, cultured in growing medium (Williams E media supplemented with 1× insulin-transferrin-selenium solution from Gibco, 1× penicillin/streptomycin, and 1× l-glutamine), and plated in collagen-coated fluorodishes. Media was changed 2 hours after plating to remove dead, nonadherent cells, and the cells were fixed in 4% paraformaldehyde for 10 to 15 min after two more hours, followed by lectin staining as mentioned above. These cells are further permeabilized with 1× PBS with 0.2% Triton X-100 and 1% normal goat serum for 10 min at room temperature and then incubated in the blocking buffer (1× PBS + 1% normal goat serum) for 30 min. The primary antibody for Golgin 97 (catalog no. 13192, Cell Signaling Technology) was applied to the sections at 1:100 concentration and incubated overnight at 4°C. Next, the sections were washed in the blocking buffer, and secondary fluorescent antibodies (anti-rabbit Alexa Fluor 594) were applied for 1 hour at room temperature. Last, the cells were incubated with NucBlue nuclear staining probes (R37606, Thermo Fisher Scientific) for 15 min at room temperature and then imaged on a Zeiss LSM 710 microscope at Institute for Genomic Biology core facility, UIUC.
Transmission electron microscopy
Liver sections (1 mm thick) were fixed in 2.5% glutaraldehyde (pH 7) for 2 to 3 hours, followed by postfixation with 1% osmium tetroxide (OsO4) in 0.1 M cacodylate buffer for 60 min. These sections are then dehydrated through ethanol series and embedded using epoxy resin. Ultrathin sections are cut and left to dry, followed by imaging using transmission electron microscopy.
Western blot
Total proteins were isolated from ~50 mg of snap-frozen liver tissue by homogenizing in 600 μl of cold radioimmunoprecipitation assay buffer containing 10 mM tris-Cl (pH 8), 1 mM EDTA, 0.5 mM EGTA, 1% Triton X-100, 0.5% sodium deoxycholate, 140 mM NaCl, 0.1% SDS, and phosphatase and protease inhibitors (one tablet per 10 ml of buffer; catalog nos. 4906837001 and 4693159001). Samples were sonicated in a water bath to shear DNA and clarified by centrifugation. Protein concentration was measured using the DC (detergent compatible) Protein Assay Kit (Bio-Rad, 5000112). Proteins (~20 μg) were resolved on a 4 to 20% TGX (Tris-Glycine eXtended) gel (Bio-Rad) and transferred semidry using “Mixed MW” program on the Bio-Rad Trans Blot Turbo System onto a nitrocellulose membrane with 0.45-μm pore size (Bio-Rad). Membranes were blocked for 1 hour at room temperature using tris-buffered saline containing 5% nonfat dry milk and 0.1% Tween 20 (TBST). Membranes were then incubated with primary antibody against albumin (1:1000; Cell Signaling Technology, no. 4929) overnight at 4°C. The membranes were then washed thrice with TBST, followed by incubation with an anti-rabbit horseradish peroxidase–conjugated secondary antibody (1:5000; Bio-Rad, no. 1706515) for 1 hour. Membranes were then visualized on the ChemiDoc MP using the Clarity Western ECL Kit (Bio-Rad).
Statistical analysis
One-way analysis of variance (ANOVA) with post hoc Bonferroni test was used for comparison of multiple groups or unpaired Student’s t test for comparison between two groups. P < 0.05 was considered significant.
Acknowledgments
We thank W. Zhou and J. Liu in Anakk laboratory for the technical help. We are thankful to P. A.B. Orlean for the valuable feedback on glysolyation. We would like to thank the High-Throughput Sequencing Core and the Microscopy Core at UIUC. We would also like to thank the Comparative Biosciences Histology Laboratory for performing PAS staining and L. A. Miller from Material Sciences Laboratory for Electron microscopic analysis. We thank D. S. Rouhani for creating the art for the cover image. We thank D. Cromley and C. Vitali from Radar laboratory for expert technical assistance and helpful discussions. Funding: This research was supported through the Startup funds from University of Illinois at Urbana-Champaign, R01 DK113080 NIDDK; American Cancer Society grant RSG ACS132640 to S.A.; and in part through the NIH, National Glycoscience Resource, CCRC Service and Training (NIH grant no. R24GM137782-01) and the NIH–funded Orbitrap Fusion Tribrid Mass Spectrometer” (NIH grant no. 1S10OD018530) to P.A. Work in the Kalsotra laboratory is supported by the NIH (R01HL126845 and R01AA010154). Work in the Rader laboratory was supported by NIH grants HL134853, HL109489, and DK114291. Author contributions: B.M. and S.A. conceived the project and designed the experiments. B.M., A.S., W.A., Q.C., N.J.H., L.K.A., and S.A. performed experiments and data analysis. K.S. shared the individual Fxrfl/fl and Shpfl/fl mice. B.M. generated the liver-specific Fxr-Shp knockout and control mice and characterized them. A.S., Q.C., and P.A. performed glycomics and glycoproteomics analyses. W.A. performed RNA-seq analysis. N.J.H. and D.J.R. generated and analyzed liver secretome plasma profiles. L.K.A. and J.A.H. quantitated the O-GlcNAc and hexosamine metabolites levels in the livers. B.M. and S.A. interpreted the results and wrote the manuscript. All authors discussed the results and edited the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The RNA-seq data discussed in this publication have been deposited in NCBI’s GEO (55) and are accessible through GEO Series accession number GSE164447 (www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE164447). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabf4865/DC1
REFERENCES AND NOTES
- 1.M. E. Taylor, K. Drickamer, Introduction to Glycobiology (Oxford Univ. Press, ed. 2, 2006), p. 1733. [Google Scholar]
- 2.G. W. Hart, C. M. West, Nucleocytoplasmic Glycosylation, in Essentials of Glycobiology, A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, Eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, ed. 2, 2009), Chapter 17. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1915/?report=classic.
- 3.Schachter H., Complex N-glycans: The story of the yellow brick road. Glycoconj. J. 31, 1–5 (2014). [DOI] [PubMed] [Google Scholar]
- 4.Cherepanova N., Shrimal S., Gilmore R., N-linked glycosylation and homeostasis of the endoplasmic reticulum. Curr. Opin. Cell Biol. 41, 57–65 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz F., Aebi M., Mechanisms and principles of N-linked protein glycosylation. Curr. Opin. Struct. Biol. 21, 576–582 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Aebi M., N-linked protein glycosylation in the ER. Biochim. Biophys. Acta Mol. Cell Res. 1833, 2430–2437 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Stanley P., Golgi glycosylation. Cold Spring Harb. Perspect. Biol. 3, a005199 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.H. Lodish, A. Berk, S. L. Zipursky, P. Matsudaira, D. Baltimore, J. Darnell, Molecular Cell Biology (W. H. Freeman, New York, ed. 4, 2000), Section 17.7, Protein Glycosylation in the ER and Golgi Complex. Available from: https://www.ncbi.nlm.nih.gov/books/NBK21744/.
- 9.Tanichi N., Korekane H., Branched N-glycans and their implications for cell adhesion, signaling and clinical applications for cancer biomarkers and in therapeutics. BMB Rep. 44, 772–781 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Feizi A., Gatto F., Uhlen M., Nielsen J., Human protein secretory pathway genes are expressed in a tissue-specific pattern to match processing demands of the secretome. npj Syst. Biol. Appl. 3, 22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J., Sun Z., Cheng K., Chen R., Ye M., Xu B., Sun D., Wang L., Liu J., Wang F., Zou H., Comprehensive mapping of protein N-glycosylation in human liver by combining hydrophilic interaction chromatography and hydrazide chemistry. J. Proteome Res. 13, 1713–1721 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Anakk S., Watanabe M., Ochsner S. A., McKenna N. J., Finegold M. J., Moore D. D., Combined deletion of Fxr and Shp in mice induces Cyp17a1 and results in juvenile onset cholestasis. J. Clin. Invest. 121, 86–95 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anakk S., Bhosale M., Schmidt V. A., Johnson R. L., Finegold M. J., Moore D. D., Bile acids activate YAP to promote liver carcinogenesis. Cell Rep. 5, 1060–1069 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goodwin B., Jones S. A., Price R. R., Watson M. A., McKee D. D., Moore L. B., Galardi C., Wilson J. G., Lewis M. C., Roth M. E., Maloney P. R., Willson T. M., Kliewer S. A., A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 6, 517–526 (2000). [DOI] [PubMed] [Google Scholar]
- 15.Chiang J. Y. L., Bile acids: Regulation of synthesis. J. Lipid Res. 50, 1955–1966 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.P. A. Dawson, in Physiology of the Gastrointestinal Tract (2012), vol. 2, pp. 1461–1484.
- 17.Russell D. W., Fifty years of advances in bile acid synthesis and metabolism. J. Lipid Res. 50, S120–S125 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perino A., Schoonjans K., Another Shp on the horizon for bile acids. Cell Metab. 20, 203–205 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Makishima M., Okamoto A. Y., Repa J. J., Tu H., Learned R. M., Luk A., Hull M. V., Lustig K. D., Mangelsdorf D. J., Shan B., Identification of a nuclear receptor for bile acids. Science 284, 1362–1365 (1999). [DOI] [PubMed] [Google Scholar]
- 20.Hoeke M. O., Heegsma J., Hoekstra M., Moshage H., Faber K. N., Human FXR regulates SHP expression through direct binding to an LRH-1 binding site, independent of an IR-1 and LRH-1. PLOS ONE 9, e88011 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.G. W. Hart, Y. Akimoto, The O-GlcNAc modification, in Essentials of Glycobiology, A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, Eds. (Cold Spring Harbor Laboratory Press, ed. 2, 1999), pp. 183–193. [Google Scholar]
- 22.I. Brockhausen, H. Schachter, P. Stanley, O-GalNAc Glycans, in Essentials of Glycobiology, A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, Eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, ed 2, 2009). Chapter 9. PMID: 20301232. [PubMed] [Google Scholar]
- 23.Zhang X., Wang Y., Glycosylation quality control by the golgi structure. J. Mol. Biol. 428, 3183–3193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X., Wang X., Tan Z., Chen S., Guan F., Role of glycans in cancer cells undergoing epithelial–mesenchymal transition. Front. Oncol. 6, 33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taniguchi N., Kizuka Y., Glycans and cancer: Role of N-glycans in cancer biomarker, progression and metastasis, and therapeutics. Adv. Cancer Res. 126, 11–51 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Blomme B., Van Steenkiste C., Callewaert N., Van Vlierberghe H., Alteration of protein glycosylation in liver diseases. J. Hepatol. 50, 592–603 (2009). [DOI] [PubMed] [Google Scholar]
- 27.Thomas A. M., Hart S. N., Kong B., Fang J., Zhong X. B., Guo G. L., Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 51, 1410–1419 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim Y. C., Byun S., Zhang Y., Seok S., Kemper B., Ma J., Kemper J. K., Liver ChIP-seq analysis in FGF19-treated mice reveals SHP as a global transcriptional partner of SREBP-2. Genome Biol. 16, 268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Watford M., What is the metabolic fate of dietary glucose? Trends Biochem. Sci. 13, 329–330 (1988). [DOI] [PubMed] [Google Scholar]
- 30.Bouché C., Serdy S., Kahn C. R., Goldfine A. B., The cellular fate of glucose and its relevance in type 2 diabetes. Endocr. Rev. 25, 807–830 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Buse M. G., Hexosamines, insulin resistance, and the complications of diabetes: Current status. Am. J. Physiol. Endocrinol. Metab. 290, E1–E8 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasconcelos-Dos-Santos A., Oliveira I. A., Lucena M. C., Mantuano N. R., Whelan S. A., Dias W. B., Todeschini A. R., Biosynthetic machinery involved in aberrant glycosylation: Promising targets for developing of drugs against cancer. Front. Oncol. 5, 138 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall S., Bacote V., Traxinger R. R., Discovery of a metabolic pathway mediating glucose-induced desensitization of the glucose transport system: Role of hexosamine biosynthesis in the induction of insulin resistance. J. Biol. Chem. 266, 4706–4712 (1991). [PubMed] [Google Scholar]
- 34.Veit C., König J., Altmann F., Strasser R., Processing of the terminal alpha-1,2-linked mannose residues from oligomannosidic n-glycans is critical for proper root growth. Front. Plant Sci. 9, 1807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ng D. T. W., Spear E. D., Walter P., The unfolded protein response regulates multiple aspects of secretory and membrane protein biogenesis and endoplasmic reticulum quality control. J. Cell Biol. 150, 77–88 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wong M. Y., Chen K., Antonopoulos A., Kasper B. T., Dewal M. B., Taylor R. J., Whittaker C. A., Hein P. P., Dell A., Genereux J. C., Haslam S. M., Mahal L. K., Shoulders M. D., XBP1s activation can globally remodel N-glycan structure distribution patterns. Proc. Natl. Acad. Sci. U.S.A. 115, E10089–E10098 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dewal M. B., DiChiara A. S., Antonopoulos A., Taylor R. J., Harmon C. J., Haslam S. M., Dell A., Shoulders M. D., XBP1s links the unfolded protein response to the molecular architecture of mature N-glycans. Chem. Biol. 22, 1301–1312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stevens K. L. P., Black A. L., Wells K. M., Yeo K. Y. B., Steuart R. F. L., Stirling C. J., Schulz B. L., Mousley C. J., Diminished Ost3-dependent N-glycosylation of the BiP nucleotide exchange factor Sil1 is an adaptive response to reductive ER stress. Proc. Natl. Acad. Sci. U.S.A. 114, 12489–12494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshida H., Matsui T., Yamamoto A., Okada T., Mori K., XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell 107, 881–891 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Calfon M., Zeng H., Urano F., Till J. H., Hubbard S. R., Harding H. P., Clark S. G., Ron D., IRE1 couples endoplasmic reticulum load to secretory capacity by processing the XBP-1 mRNA. Nature 415, 92–96 (2002). [DOI] [PubMed] [Google Scholar]
- 41.Walter P., Ron D., The unfolded protein response: From stress pathway to homeostatic regulation. Science 334, 1081–1086 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Bambino K., Zhang C., Austin C., Amarasiriwardena C., Arora M., Chu J., Sadler K. C., Inorganic arsenic causes fatty liver and interacts with ethanol to cause alcoholic liver disease in zebrafish. Dis. Model. Mech. 11, dmm031575 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Schulze R. J., Schottm M. B., Casey C. A., Tuma P. L., McNiven M. A., The cell biology of the hepatocyte: A membrane trafficking machine. J. Cell Biol. 218, 2096–2112 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukherjee S., Maxfield F. R., Cholesterol: Stuck in traffic. Nat. Cell Biol. 1, E37–E38 (1999). [DOI] [PubMed] [Google Scholar]
- 45.Roth Z., Yehezkel G., Khalaila I., Identification and quantification of protein glycosylation. Int. J. Carbohydr. Chem. 2012, 1–10 (2012). [Google Scholar]
- 46.Sharma R., Quilty F., Gilmer J. F., Long A., Byrne A.-M., Unconjugated secondary bile acids activate the unfolded protein response and induce golgi fragmentation via a src-kinase- dependant mechanism. Oncotarget 8, 967–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Byrne A. M., Sharma R., Duggan G., Kelleher D., Long A., Deoxycholic acid impairs glycosylation and fucosylation processes in esophageal epithelial cells. Glycobiology 22, 638–648 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Byrne A.-M., Foran E., Sharma R., Davies A., Mahon C., O’Sullivan J., O’Donoghue D., Kelleher D., Long A., Bile acids modulate the Golgi membrane fission process via a protein kinase Cη and protein kinase D-dependent pathway in colonic epithelial cells. Carcinogenesis 31, 737–744 (2010). [DOI] [PubMed] [Google Scholar]
- 49.P. Stanley, H. Schachter, N. Taniguchi, N-Glycans, in Essentials of Glycobiology, A. Varki, R. D. Cummings, J. D. Esko, H. H. Freeze, P. Stanley, C. R. Bertozzi, G. W. Hart, M. E. Etzler, Eds. (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, ed. 2, 2009), Chapter 8. PMID: 20301244. [PubMed] [Google Scholar]
- 50.Preidis G. A., Kim K. H., Moore D. D., Nutrient-sensing nuclear receptors PPARα and FXR control liver energy balance. J. Clin. Invest. 127, 1193–1201 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Anders S., Pyl P. T., Huber W., HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edgar R., Domrachev M., Lash A. E., Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yates A. D., Achuthan P., Akanni W., Allen J., Allen J., Alvarez-Jarreta J., Amode M. R., Armean I. M., Azov A. G., Bennett R., Bhai J., Billis K., Boddu S., Marugán J. C., Cummins C., Davidson C., Dodiya K., Fatima R., Gall A., Giron C. G., Gil L., Grego T., Haggerty L., Haskell E., Hourlier T., Izuogu O. G., Janacek S. H., Juettemann T., Kay M., Lavidas I., Le T., Lemos D., Martinez J. G., Maurel T., Dowall M. M., Mahon A. M., Mohanan S., Moore B., Nuhn M., Oheh D. N., Parker A., Parton A., Patricio M., Sakthivel M. P., Salam A. I. A., Schmitt B. M., Schuilenburg H., Sheppard D., Sycheva M., Szuba M., Taylor K., Thormann A., Threadgold G., Vullo A., Walts B., Winterbottom A., Zadissa A., Chakiachvili M., Flint B., Frankish A., Hunt S. E., IIsley G., Kostadima M., Langridge N., Loveland J. E., Martin F. J., Morales J., Mudge J. M., Muffato M., Perry E., Ruffier M., Trevanion S. J., Cunningham F., Howe K. L., Zerbino D. R., Flicek P., Ensembl 2020. Nucleic Acids Res. 48, D682–D688 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Grant C. E., Bailey T. L., Noble W. S., FIMO: Scanning for occurrences of a given motif. Bioinformatics 27, 1017–1018 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shajahan A., Heiss C., Ishihara M., Azadi P., Glycomic and glycoproteomic analysis of glycoproteins—A tutorial. Anal. Bioanal. Chem. 409, 4483–4505 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Oikari S., Venäläinen T., Tammi M., Borate-aided anion exchange high-performance liquid chromatography of uridine diphosphate-sugars in brain, heart, adipose and liver tissues. J. Chromatogr. A 1323, 82–86 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/17/eabf4865/DC1