

Abstract
Epigenetic information affects gene function by interacting with chromatin, while not changing the DNA sequence itself. However, it has become apparent that the interactions between epigenetic information and chromatin can, in fact, indirectly lead to DNA mutations and ultimately influence genome evolution. This review evaluates the ways in which epigenetic information affects genome sequence and evolution. We discuss how DNA methylation has strong and pervasive effects on DNA sequence evolution in eukaryotic organisms. We also review how the physical interactions arising from the connections between histone proteins and DNA affect DNA mutation and repair. We then discuss how a variety of epigenetic mechanisms exert substantial effects on genome evolution by suppressing the movement of transposable elements. Finally, we examine how genome expansion through gene duplication is also partially controlled by epigenetic information. Overall, we conclude that epigenetic information has widespread indirect effects on DNA sequences in eukaryotes and represents a potent cause and constraint of genome evolution.
This article is part of the theme issue ‘How does epigenetics influence the course of evolution?’
Keywords: histone, DNA methylation, isochore, mutation, molecular evolution, transposable element
1. Epigenetic information as a mediator of molecular evolution
The success of multicellular organisms stems from the ability of genetically identical cells within individuals to undertake different functions. Epigenetics is the study of how different cellular functions can arise from cells that possess the same genotype [1,2]. The field of epigenetics has evolved over time [3]. Traditionally, epigenetics specifically focuses on heritable information that does not lead to changes in DNA sequence. Indeed, the very term epigenetics means ‘above the genome’. Thus, epigenetic systems represent modes of heritable information that operated in conjunction with, but distinct from, the traditional DNA-based system of inheritance.
Remarkably, however, epigenetic marks can lead to changes in DNA sequences through a variety of mechanisms. Epigenetic information, in the form of DNA methylation, modifications to histone proteins or non-coding RNAs, directly affects chromatin, which is the DNA–protein complex that makes up chromosomes in eukaryotic taxa [4]. The interactions and modifications to chromatin open the door to mutational processes that lead to alterations in DNA sequence and, potentially, evolutionary changes. The effects of epigenetic marks (i.e. changes to the molecular structure of DNA or histone proteins that impart epigenetic information) on genome sequences are underappreciated specifically because epigenetic information is defined as operating without changes to DNA sequences. This review aims to bring together evidence of how epigenetic marks directly and indirectly lead to DNA sequence changes.
2. DNA methylation is a pervasive mutagenic agent in diverse taxa
DNA methylation is a phylogenetically widespread chemical modification of genomic DNA. Two of the four DNA nucleotides, adenine and cytosine, can be modified by the addition of a methyl group. Cytosine methylation is extremely widespread in both eukaryotes and prokaryotes, and has been extensively studied for its roles in gene regulation and genome evolution (e.g. [5–8], figure 1). Adenine methylation is relatively common in prokaryotes [11] and also observed in several eukaryotic species [12]. The function and evolutionary consequences of adenine methylation are currently poorly understood [12], especially in comparison to cytosine methylation. Consequently, in this review, we focus on cytosine methylation, and refer to it simply as ‘DNA methylation’.
Figure 1.

DNA methylation in the DNA double helix. Methylcytosine (yellow) with methyl group circled, alongside unmethylated cytosine (dark blue), guanine (light blue), adenine (red) and thymine (orange) bases. The methylation of DNA is one of the most important factors affecting mutation rate in animals. DNA structure [9] drawn in PyMOL [10].
DNA methylation can expand the information content encoded in genomic sequences and is associated with regulatory functions in diverse taxa [5–7]. Interestingly, DNA methylation is also highly mutagenic [13]. Methylated cytosines undergo spontaneous deamination and can thus transform into thymine. Since thymine is a naturally occurring nucleotide in DNA, these mutations may remain unrepaired, ultimately resulting in a C to T mutation [14]. Therefore, paradoxically, methylation of cytosine can deplete cytosines themselves due to the propensity of methylcytosines to mutate to thymines.
(a). The effects of DNA methylation on CpG content in the genome
In animal genomes, the majority of DNA methylation occurs at cytosines followed by guanines, or ‘CpG’ dinucleotides. Because methylated cytosines in CpG dinucleotides are prone to mutations to TpG dinucleotides, methylated sequences gradually lose CpGs. In the simplest case, the frequency of CpGs in a sequence may be determined solely by the frequencies of C and G nucleotides in the sequence of interest. More specifically, one can calculate the expected frequency of CpGs as the ‘(frequency of C) × (frequency of G)’ assuming that random associations of C and G nucleotides determine the CpG frequency. CpG frequencies of sequences from species lacking DNA methylation, such as Drosophila, are well approximated by this estimate. By contrast, CpGs tend to be depleted in species where there is DNA methylation, due to mutations caused by deamination of cytosine bases.
It is convenient to use the observed frequency of CpGs normalized by the expected frequency of CpGs, often referred to as ‘CpG O/E’, to describe the depletion of CpGs. CpG O/E can be calculated as (observed frequency of CpG)/(expected frequency of CG), or [(Number of CG) × (sequence length)]/[(Number of C) × (Number of G)]. It should be noted that CpG O/E is calculated from genomic DNA and, therefore, reflects a historical appraisal of the effects of DNA methylation. Moreover, CpG O/E is only affected by DNA methylation in germline, which leads to inherited changes in DNA sequence between generations. Researchers have long noted that CpG O/Es from different species show variation consistent with the variation of DNA methylation [15]. For example, in humans, where DNA methylation is abundant, CpG O/E is around 0.2 for most genomic regions (figure 2a), indicating that a very high percentage of cytosine bases have mutated to thymine due to the effects of DNA methylation. CpG O/E has been a highly useful metric to infer within-genome variation of DNA methylation and served as an important aid to functional studies [17,18].
Figure 2.

Correspondence between DNA methylation and CpG O/E. (a) CpG O/E calculated in 500 bp sliding windows in the human chromosome 1 is inversely related to empirically measured DNA methylation in the human brain. Human chromosomes are generally hypermethylated and exhibit low CpG O/E. Regions devoid of DNA methylation maintain relatively high CpG O/E and are referred to as ‘CpG islands’. (b) CpG O/E and DNA methylation in honeybee. Honeybee chromosomes are generally devoid of DNA methylation, with the exception of peaks of DNA methylation observed in the so-called methylation islands [16]. CpG O/Es are generally high in the honeybee genome and show a reduction in regions corresponding to methylation islands. (Online version in colour.)
(b). CpG islands and lessons learned from DNA methylation of vertebrate genomes
A prime example of how analysis of CpG frequency has guided epigenetic studies is the investigation of so-called CpG islands in vertebrates. CpG islands are so named to indicate the presence of a relatively high frequency of CpGs relative to surrounding regions displaying low CpG frequencies [19,20]. CpG islands were initially identified as genomic regions that possessed low levels of DNA methylation [19]. Because these regions are ‘hypomethylated’, they are likely to be immune to the mutagenic effect of DNA methylation, and thus retain CpGs at frequencies higher than methylated regions (figure 2a). CpG islands stand out because most human and other vertebrate genomes are heavily methylated. Interestingly, CpG islands tend to be located near the 5′ end of genes and are associated with active transcription [20,21]. Numerous studies have revealed the functional importance of CpG islands. For example, genes harbouring CpG islands near their transcription start sites tend to be broadly expressed [21–23]. The use of CpG islands has been critical in functional annotation of the human genome in the pre-genomic era [24] and to this day; CpG islands continue to be among the most widely studied epigenetic regulatory markers.
CpG islands offer a rare opportunity to investigate the relationship between DNA methylation and sequence evolution in the human genome. Efforts to define CpG islands from genomic sequences rely heavily on the observation that CpG O/E and GC contents are often strongly correlated [25]. This pattern is in large part driven by the depletion of CpG dinucleotides affecting GC-poor regions more dramatically than GC-rich regions [26].
Another mechanism that can affect both CpG O/E and GC content is biased gene conversion. Briefly, gene conversion occurs when homologous recombination results in a mismatched base pair. In many genomes, when such a mismatch is corrected, the correction is biased to a G/C allele over an A/T allele, a process referred to as ‘biased gene conversion’ [27,28]. Biased gene conversion can increase GC content, and could directly generate new CpGs. For example, Cohen et al. [29] showed that biased gene conversion could maintain high GC and high CpG O/E regions during primate genome evolution. Therefore, CpG O/E, while being extremely useful to identify putatively hypomethylated regions of vertebrate genomes, can also increase due to biased gene conversion [29,30]. In addition, since biased gene conversion increases with recombination, it is important to consider recombination rates in comparative analyses of GC content and CpG O/E.
(c). DNA methylation and isochore evolution of vertebrate genomes
DNA methylation may have affected the evolution of large regions of DNA showing uniform GC content; such regions are known as ‘isochores’. GC content of genome sequences shows considerable variation in many species, particularly in warm-blooded vertebrates including mammals and birds. It was initially suggested that GC content homogeneity stretched thousands of base pairs and formed isochores [31]. Several hypotheses have been put forward to explain the evolutionary origin and maintenance of isochores. For example, a selective hypothesis [32] posits that GC-rich isochores confer selective advantages in warm-blooded species because they are thermally more stable than GC-poor isochores. By contrast, two other hypotheses focus on variation in mutation rates and recombination as causal mechanisms. First, GC content could vary across the genome according to variation in rates and patterns of mutations [33]. Second, isochores may originate through biased gene conversion associated with variation in recombination rate across the genome [27,34].
DNA methylation can influence isochores by impacting mutation and recombination. As discussed above, DNA methylation can cause C to T (or G to A on the complementary strand) mutations due to spontaneous deamination. Fryxell & Zuckerkandl [35] noted that deamination requires a temporary melting of double-stranded DNA, and that more thermodynamic energy is required to melt GC-rich DNA than to melt AT-rich DNA. Consequently, genomic regions already high in GC content may experience lower rates of mutations originating from deamination of methylated DNA. Low-GC regions, on the other hand, can more easily undergo deamination and subsequent GC to AT mutations. This positive feedback loop can drive the evolution of GC-poor regions and further reduce GC content. By contrast, GC-rich regions may gain more GC nucleotides, thus generating and maintaining isochores [35]. This hypothesis has been supported by empirical data demonstrating the melting effects on substitutions [36], prevalence of single nucleotide polymorphisms in human genomes [37], and germline DNA methylation and substitution patterns in avian genomes [38].
DNA methylation may also have a direct link to recombination. Experimental studies have demonstrated that recombination following double-strand breaks leads to the recruitment of DNA methyltransferases, which are the key enzymes involved in methylating DNA, and the generation of methylated regions of the double-strand break [39,40]. At the genome scale, germline methylation levels are positively correlated with inferred recombination rates [41]. Furthermore, recombination can directly cause mutation in some cases [42]. However, how these processes are causatively linked to each other on an evolutionary timescale requires further research.
(d). Identifying methylated regions using CpG contents
It was long recognized that CpG O/E values of different animal genomes showed substantial variation, and that many invertebrate genomes generally had much higher CpG O/E levels than other animal taxa [15,18]. The prevailing notion until the last decade was that this pattern arose because invertebrates generally lacked DNA methylation. However, several studies have since discovered that many invertebrate genomes contain all the genes necessary for a functional DNA methylation system and contain methylated CpGs [16,18,43–45]. Furthermore, taxonomically expanded sampling of invertebrate methylomes is revealing a great deal of diversity in genomic DNA methylation. Invertebrate genomes show nearly two orders of magnitude difference in DNA methylation. For example, honeybee genomes show less than 1% methylation [6]; by contrast, sponge genomes show approximately 80% methylation [46]. Some invertebrate genomes even exhibit DNA methylation of promoters and transposable elements (TEs) [46–48].
Studies of DNA methylation in invertebrates may benefit from the comparison of CpG O/E and experimentally measured DNA methylation, as the former reflects historical levels of DNA methylation in the germline, while the latter reveals current DNA methylation levels. These two measures are generally negatively correlated to each other across diverse plant and animal taxa [18,46,49,50] (although some exceptions warrant further study [48]). For example, in the human genome, most genomic regions are heavily methylated, and CpG O/E is reduced overall (figure 2a). Hypomethylated regions, represented by CpG islands (with high CpG O/E), are exceptions to this general pattern (figure 2a). By contrast, the honeybee shows little methylation throughout its genome. In terms of CpG O/E, methylated regions (methylated islands) appear as dips of CpG O/E in an otherwise high CpG O/E genome [51] (figure 2b). Overall, CpG O/E measures continue to play an important role in identifying putatively methylated regions. However, it should be noted that analysis of CpG O/E alone could miss regions that have recently acquired DNA methylation, as those regions may have not experienced sufficient mutational input to alter CpG contents.
3. Histone proteins as mediators of mutational effects in the genome
Most of the eukaryotic genome is compacted into nucleosomes. Nucleosomes consist of approximately 147 bp of DNA wrapped around an octamer of two copies of each of the four core histone proteins, H2A, H2B, H3 and H4 [52] (figure 3a). DNA bound into nucleosomes is less accessible to regulatory proteins than unbound DNA. Therefore, many regulatory processes in eukaryotes have been linked to interactions between histone proteins and DNA.
Figure 3.

(a) The eukaryotic nucleosome is composed of DNA (blue) wrapped around a core of eight histone proteins (red). The histone ‘tails’ projecting from the nucleosome are often chemically modified to contain epigenetic information. (b) The structure of the nucleosome leads to variation in mutation rate. For example, the minor groove of the DNA double helix which faces the histone core, shown by green arrows, experiences different mutation rates than other regions of the DNA double helix. Nucleosome structure [53] drawn in PyMOL [10].
Histone proteins undergo a variety of histone post-translation modifications (HPTMs) that affect gene function. These HPTMs are carried out by an array of ‘writer’ enzymes that modify histone proteins based on histone structural features and interactions with other regulatory proteins [54]. HPTMs affect gene regulation in two ways [55]. First, HPTMs may cause changes to the properties of the histone proteins themselves resulting in, for example, changes in the positions or stability of nucleosomes. Such changes in nucleosome properties may lead to differences in the compaction of the DNA and, consequently, to variation in transcription of genes in the region. Second, HPTMs may affect gene function by determining which transcription factors interact with DNA. The association of different transcription factors may increase or decrease transcription rates of genes. Thus, HPTMs hold important epigenetic information that has strong, heritable influences on the function of associated genes [55].
(a). Histone proteins affect DNA mutation
The packaging of DNA into nucleosomes has complex effects on the processes that generate and repair mutations [56,57]. DNA that is bound in nucleosomes is less prone to DNA ‘breathing’, whereby DNA opens into a single-stranded state. Single-stranded DNA mutates at a higher frequency than double-stranded DNA bound into nucleosomes. Consequently, genomic DNA that is bound by nucleosomes displays substantially lower mutation probabilities than DNA that resides in unoccupied locations [58]. The mechanistic basis for the effects of nucleosomes on mutation arises from changes to the three-dimensional structure of DNA when it is compacted [59,60]. DNA bases that are ‘outward’ facing from the nucleosome may experience higher mutational pressures. Moreover, DNA bases that reside in the minor or major grooves of the double helix also mutate at different rates [61] (figure 3b).
DNA compacted into heterochromatic and euchromatic regions of the genome possesses different epigenetic information [62] and can also be subject to different levels of DNA repair. Certain DNA repair mechanisms, such as the DNA mismatch or nucleotide excision repair pathways, operate differently in heterochromatic and euchromatic regions [63,64]. Moreover, bases near the edge of nucleosomes are more accessible to DNA repair machinery, thereby leading to nucleosome-related, nucleotide-position effects on DNA repair [59,60].
(b). Histone modifications as mediators of genome evolution
Certain HPTMs, such as histone methylation, ubiquitination and phosphorylation, are linked to variation in DNA mutation rate [56,57]. Such variation arises because HPTMs can lead to changes in chromatin structure that allow access to DNA by repair enzymes [65]. Modifications to histone proteins also affect the probability of DNA sequence mutation by altering the frequency of DNA break repair [66]. For instance, the acetylation of histone proteins promotes certain types of DNA repair, and the phosphorylation of a variant of histone H2A or the methylation of histones H4 and H3 stimulates DNA repair in some cases. Indeed, a variety of HPTMs or histone protein variants accompany DNA damage and may facilitate access to damaged DNA by repair proteins [67,68]. Finally, there are differences in mutational effects of epigenetically marked and unmarked histone proteins [69]. Consequently, HPTMs in germline chromatin can lead to evolutionary changes to genome sequences.
4. Transposable elements, genome structure and epigenetic information
TEs are DNA sequences that are able to move or be copied to new locations in the genome [70]. TEs comprise a very large fraction of eukaryotic genomes; they make upwards of 33% of the entire genome of many mammals and 75% of the genome of some plants [70,71]. Importantly, a high percentage of spontaneous mutations in plants and animals result directly from TE movement in the genome [70,72].
TEs can disrupt gene activity if they transpose into an existing, functional gene [73,74]. However, TEs can also introduce new exons into genes, thereby generating new substrate for evolution. TE movement may affect patterns of gene expression if a TE absorbs a host promoter or transcription factor binding sequence, and then transposes next to an existing gene [75]. TEs are also implicated in generating large deletions or duplications by facilitating ectopic recombination [70,71,76]. Moreover, insertion of TEs can change the sequence composition as well as the epigenetic landscape of the target region and thus affect the mutational spectrum. Thus, TEs cause a wide variety of mutagenic events and are among the most important factors affecting genome evolution [71,76].
(a). Epigenetic information represses mutations caused by transposable elements
Epigenetic mechanisms have important effects on TE activity and, therefore, on controlling the generation of mutations within eukaryotic genomes. Indeed, some epigenetic mechanisms are believed to have evolved specifically to control TE activity [74]. Therefore, epigenetic mechanisms are implicated in affecting genome mutation by suppressing or releasing TE movement within the genome [77].
DNA methylation is the best-documented epigenetic system associated with TE activity [78]. Methylation of TEs in mammals differs between sexes, age and tissues, and these differences correlate with the activity of TEs [78]. Animals and plants with experimentally deactivated DNA methylation systems experience an upregulation of TEs, thereby indicating a direct role of DNA methylation on suppressing TE activity [62,74]. Finally, a specific DNA methyltransferase gene, Dnmt3C, was recently discovered in some mammals. Dnmt3C apparently specializes in methylating TEs in the germline, therefore providing strong evidence that DNA methylation plays an important role in suppressing TE movement [79].
Other epigenetic systems also act to suppress TE activity and, consequently, genome mutations. For example, TE RNA can be processed through the small-interfering RNA (siRNA) pathway. siRNAs produced from TE RNA can be targeted by the RNA interference system (RNAi) [80]. The RNAi system can then degrade the target TE RNAs, thereby controlling the spread of TEs in the genome [81]. In addition, the siRNA pathway can lead to subsequent modifications to chromatin structure that ultimately act to silence TE genes [81].
PIWI-interacting RNAs (piRNAs) represent another epigenetic information system that affects the behaviour of TEs [74,82]. piRNAs are short, single-stranded, non-coding RNAs that are specifically defined as interacting with Piwi proteins. piRNAs are believed to have evolved in response to the effects of TEs in animals [83]. piRNAs silence TEs and, therefore, prevent harmful TE-induced mutations [82]. Specifically, the Piwi complex, which interacts with focal piRNAs (figure 4), can promote the generation of heterochromatic marks around TE DNA. The Piwi protein may lead to the silencing of TEs by allowing for the addition of repressive histone modification H3K9me3 or by preventing the deposition of the activating histone modification H3K4me2. piRNAs also facilitate the degradation of TE RNAs and prevent TE multiplication in animals. piRNAs complementary to sequences of TE mRNAs act as guides to protein complexes that either act to destroy the TE mRNA or assist in mRNA degradation [72]. Moreover, new types of piRNAs may be derived directly from invading TEs [85]. In this way, the piRNA system acts as a type of inherited immunity against TEs [82,83].
Figure 4.
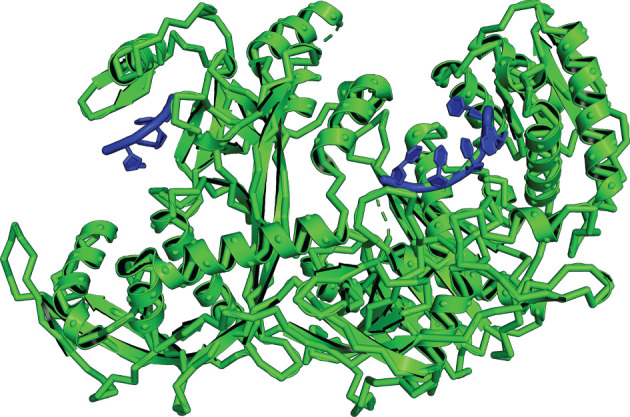
The silkworm PIWI-clade protein (green) bound to piRNA (blue). This protein family is involved in gene silencing through its interactions with piRNAs. piRNAs are non-coding RNAs involved in the suppression of transposable elements and, therefore, are epigenetic factors that indirectly prevent genome mutation and evolution. PIWI-piRNA structure [84] drawn in PyMOL [10].
Epigenetic systems interact in a variety of ways to suppress TEs. The epigenetic silencing of TEs by DNA methylation and non-coding RNAs is often linked to other epigenetic changes [62]. In plants and animals, suppression of TEs is associated with HPTMs that suppress TE activity [62,86–88]. Interestingly, the types of histone modifications associated with TEs are quite complex and depend on the TE sequence itself [86]. Thus, epigenetic systems may be highly tuned to dampening the effects of different TEs and preserving the integrity of the genome.
5. Evolution and epigenetics of duplicated genes
Epigenetic mechanism can affect genome evolution by influencing the fate of duplicate genes. Gene duplication is a fundamental source of genomic variation. Gene duplication can lead to the production of genes with new functions via neofunctionalization [89] or contribute to biological complexity by promoting separation of ancestral functions in a tissue, developmental stage or cell type via subfunctionalization [90]. A duplicated gene is likely to be functionally redundant to the original gene. Thus, loss-of-function mutations are expected to quickly accumulate, rendering the duplicate gene non-functional [91]. Therefore, the most critical step in the preservation of duplicated genes is the initial retention of both gene copies following duplication (e.g. [92]).
Epigenetic mechanisms can offer molecular pathways for duplicate gene retention and functional divergence. Epigenetic silencing of duplicate genes among tissues or developmental stages effectively ‘shields’ duplicate genes from natural selection and increases the probability of subfunctionalization and neofunctionalization [93]. For example, DNA methylation can reduce gene expression, thereby preventing a gene from producing a negative phenotype [94,95]. Indeed, human and yeast duplicate genes showed reduction of expression following duplication, which could facilitate the retention of gene copies [96].
Empirical data from diverse taxa demonstrate that epigenetic patterns of duplicate genes and singletons are distinct. Promoters of duplicate genes in mammals have higher DNA methylation compared to those of singletons [94,95]. Typically, the gene copy with a higher level of promoter DNA methylation tends to exhibit reduced expression compared to its sister copy, indicating that DNA methylation differences may contribute to expression differences [95]. Data from other species such as zebrafish [97] are consistent with this model.
Data from invertebrates and plants also support the role of DNA methylation in regulating duplicate genes [98,99]. Gene body methylation, which is the most common form of DNA methylation in invertebrates, is known to be positively associated with gene expression [4]. Interestingly, duplicated genes have lower levels of gene body methylation than singletons in honeybees [99], which is consistent with the idea that DNA methylation may affect duplicate gene function. Data from plants also support the idea that divergence of gene body methylation correlates with expression divergence [100]. Therefore, a model of DNA methylation and expression reduction may be applicable to divergent animal and plant taxa.
6. Conclusion
Epigenetic information is critical to organismal development, behaviour and function. In addition, many types of epigenetic information can affect genome evolution and structure. Epigenetic marks shape genome evolution through their direct and indirect interactions with chromatin. In particular, DNA methylation leads to a high frequency of single-base substitutions of cytosines in the genome. The packaging of eukaryotic DNA into nucleosomes also has strong effects on genome evolution, because the structure of the nucleosome affects mutation probabilities and DNA repair mechanisms. Epigenetic information in the form of DNA methylation, non-coding RNAs or HPTMs also strongly influences genome mutation by suppressing TE movement. Finally, the evolution of gene content within eukaryotic genomes is affected by epigenetic mechanisms that facilitate the process of gene duplication. Thus, overall, we suggest a greater appreciation of epigenetic information as a mediator of genome evolution. Epigenetic information is ultimately a cause of both organismal plasticity and molecular evolution in eukaryotes.
Acknowledgements
We thank Xin Wu for help with figure 2, and CJ Dyson and anonymous reviewers for comments on earlier versions of this manuscript.
Data accessibility
This article has no additional data.
Authors' contributions
S.V.Y. and M.A.D.G. developed and co-wrote the manuscript.
Competing interests
We declare we have no competing interests.
Funding
This research was partially supported by National Science Foundation grant nos IOS-2019799 and EF-2021635.
References
- 1.Waddington CH. 1957. The strategy of the genes; a discussion of some aspects of theoretical biology. London, UK: Allen & Unwin. [Google Scholar]
- 2.Bonasio R. 2015. The expanding epigenetic landscape of non-model organisms. J. Exp. Biol. 218, 114-122. ( 10.1242/Jeb.110809) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nicoglou A, Merlin F. 2017. Epigenetics: a way to bridge the gap between biological fields. Stud. Hist. Philos. Biol. Biomed. Sci. 66, 73-82. ( 10.1016/j.shpsc.2017.10.002) [DOI] [PubMed] [Google Scholar]
- 4.Glastad KM, Hunt BG, Goodisman MAD. 2019. Epigenetics in insects: genome regulation and the generation of phenotypic diversity. Annu. Rev. Entomol. 64, 185-203. ( 10.1146/annurev-ento-011118-111914) [DOI] [PubMed] [Google Scholar]
- 5.Bewick AJ, Hofmeister BT, Powers RA, Mondo SJ, Grigoriev IV, James TY, Stajich JE, Schmitz RJ. 2019. Diversity of cytosine methylation across the fungal tree of life. Nat. Ecol. Evol. 3, 479-490. ( 10.1038/s41559-019-0810-9) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zemach A, McDaniel IE, Silva P, Zilberman D. 2010. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science 328, 916-919. ( 10.1126/science.1186366) [DOI] [PubMed] [Google Scholar]
- 7.Yi SV. 2012. Birds do it, bees do it, worms and ciliates do it too: DNA methylation from unexpected corners of the tree of life. Genome Biol. 13, 174. ( 10.1186/Gb-2012-13-10-174) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis SH, et al. 2020. Widespread conservation and lineage-specific diversification of genome-wide DNA methylation patterns across arthropods. PLoS Genet. 16, e1008864. ( 10.1371/journal.pgen.1008864) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gruber DR, et al. 2018. Oxidative damage to epigenetically methylated sites affects DNA stability, dynamics and enzymatic demethylation. Nucleic Acids Res. 46, 10 827-10 839. ( 10.1093/nar/gky893) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DeLano WL. 2004. Use of PYMOL as a communications tool for molecular science. Abstr. Pap. Am. Chem. Soc. 228, U313-U314. [Google Scholar]
- 11.Blow MJ, et al. 2016. The epigenomic landscape of prokaryotes. PLoS Genet. 12, e1005854. ( 10.1371/journal.pgen.1005854) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iyer LM, Zhang DP, Aravind L. 2016. Adenine methylation in eukaryotes: apprehending the complex evolutionary history and functional potential of an epigenetic modification. Bioessays 38, 27-40. ( 10.1002/bies.201500104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coulondre C, Miller JH, Farabaugh PJ, Gilbert W. 1978. Molecular basis of base substitution hotspots in Escherichia coli. Nature 274, 775-780. ( 10.1038/274775a0) [DOI] [PubMed] [Google Scholar]
- 14.Walsh CP, Xu GL. 2006. Cytosine methylation and DNA repair. Curr. Top Microbiol. Immunol. 301, 283-315. ( 10.1007/3-540-31390-7_11) [DOI] [PubMed] [Google Scholar]
- 15.Bird AP. 1980. DNA methylation and the frequency of CpG in animal DNA. Nucleic Acids Res. 8, 1499-1504. ( 10.1093/nar/8.7.1499) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bewick AJ, Vogel KJ, Moore AJ, Schmitz RJ. 2017. Evolution of DNA methylation across insects. Mol. Biol. Evol. 34, 654-665. ( 10.1093/molbev/msw264) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yi SV, Goodisman MAD. 2009. Computational approaches for understanding the evolution of DNA methylation in animals. Epigenetics 4, 551-556. ( 10.4161/epi.4.8.10345) [DOI] [PubMed] [Google Scholar]
- 18.Thomas GWC, et al. 2020. Gene content evolution in the arthropods. Genome Biol. 21, 15. ( 10.1186/s13059-019-1925-7) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cooper DN, Taggart MH, Bird AP. 1983. Unmethylated domains in vertebrate DNA. Nucleic Acids Res. 11, 647-658. ( 10.1093/nar/11.3.647) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bird AP. 1986. CpG-rich islands and the function of DNA methylation. Nature 321, 209-213. ( 10.1038/321209a0) [DOI] [PubMed] [Google Scholar]
- 21.Saxonov S, Berg P, Brutlag DL. 2006. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA 103, 1412-1417. ( 10.1073/pnas.0510310103) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. 2007. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 39, 457-466. ( 10.1038/ng1990) [DOI] [PubMed] [Google Scholar]
- 23.Elango N, Yi SV. 2008. DNA methylation and structural and functional bimodality of vertebrate promoters. Mol. Biol. Evol. 25, 1602-1608. ( 10.1093/molbev/msn110) [DOI] [PubMed] [Google Scholar]
- 24.Deaton AM, Bird A. 2011. CpG islands and the regulation of transcription. Genes Dev. 25, 1010-1022. ( 10.1101/gad.2037511) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardiner-Garden M, Frommer M. 1987. CpG islands in vertebrate genomes. J. Mol. Biol. 196, 261-282. ( 10.1016/0022-2836(87)90689-9) [DOI] [PubMed] [Google Scholar]
- 26.Duret L, Galtier N. 2000. The covariation between TpA deficiency, CpG deficiency, and G + C content of human isochores is due to a mathematical artifact. Mol. Biol. Evol. 17, 1620-1625. ( 10.1093/oxfordjournals.molbev.a026261) [DOI] [PubMed] [Google Scholar]
- 27.Galtier N, Piganeau G, Mouchiroud D, Duret L. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159, 907-911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marais G. 2003. Biased gene conversion: implications for genome and sex evolution. Trends Genet. 19, 330-338. ( 10.1016/S0168-9525(03)00116-1) [DOI] [PubMed] [Google Scholar]
- 29.Cohen NM, Kenigsberg E, Tanay A. 2011. Primate CpG islands are maintained by heterogeneous evolutionary regimes involving minimal selection. Cell 145, 773-786. ( 10.1016/j.cell.2011.04.024) [DOI] [PubMed] [Google Scholar]
- 30.Yi SV. 2017. Insights into epigenome evolution from animal and plant methylomes. Genome Biol. Evol. 9, 3189-3201. ( 10.1093/gbe/evx203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Costantini M, Clay O, Auletta F, Bernardi G. 2006. An isochore map of human chromosomes. Genome Res. 16, 536-541. ( 10.1101/gr.4910606) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bernardi G. 2000. The compositional evolution of vertebrate genomes. Gene 259, 31-43. ( 10.1016/S0378-1119(00)00441-8) [DOI] [PubMed] [Google Scholar]
- 33.Wolfe KH, Sharp PM, Li WH. 1989. Mutation rates differ among regions of the mammalian genome. Nature 337, 283-285. ( 10.1038/337283a0) [DOI] [PubMed] [Google Scholar]
- 34.Duret L, Arndt PF. 2008. The impact of recombination on nucleotide substitutions in the human genome. PLoS Genet. 4, e1000071. ( 10.1371/journal.pgen.1000071) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fryxell KJ, Zuckerkandl E. 2000. Cytosine deamination plays a primary role in the evolution of mammalian isochores. Mol. Biol. Evol. 17, 1371-1383. ( 10.1093/oxfordjournals.molbev.a026420) [DOI] [PubMed] [Google Scholar]
- 36.Elango N, Kim SH, Program NCS, Yi S. 2008. Mutations of different molecular origins exhibit contrasting patterns of regional substitution rate variation. PLoS Comp. Biol. 4, e1000015. ( 10.1371/journal.pcbi.1000015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fryxell KJ, Moon WJ. 2005. CpG mutation rates in the human genome are highly dependent on local GC content. Mol. Biol. Evol. 22, 650-658. ( 10.1093/molbev/msi043) [DOI] [PubMed] [Google Scholar]
- 38.Mugal CF, Ellegren H. 2011. Substitution rate variation at human CpG sites correlates with non-CpG divergence, methylation level and GC content. Genome Biol. 12, R58. ( 10.1186/gb-2011-12-6-r58) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cuozzo C, et al. 2007. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 3, 1144-1162. ( 10.1371/journal.pgen.0030110) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morano A, et al. 2014. Targeted DNA methylation by homology-directed repair in mammalian cells. Transcription reshapes methylation on the repaired gene. Nucleic Acids Res. 42, 804-821. ( 10.1093/nar/gkt920) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zeng J, Yi SV. 2014. Specific modifications of histone tails, but not DNA methylation, mirror the temporal variation of mammalian recombination hotspots. Genome Biol. Evol. 6, 2918-2929. ( 10.1093/gbe/evu230) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Arbeithuber B, Betancourt AJ, Ebner T, Tiemann-Boege I. 2015. Crossovers are associated with mutation and biased gene conversion at recombination hotspots. Proc. Natl Acad. Sci. USA 112, 2109-2114. ( 10.1073/pnas.1416622112) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang Y, Jorda M, Jones PL, Maleszka R, Ling X, Robertson HM, Mizzen CA, Peinado MA, Robinson GE. 2006. Functional CpG methylation system in a social insect. Science 314, 645-647. ( 10.1126/science.1135213) [DOI] [PubMed] [Google Scholar]
- 44.Walsh TK, Brisson JA, Robertson HM, Gordon K, Jaubert-Possamai S, Tagu D, Edwards OR. 2010. A functional DNA methylation system in the pea aphid, Acyrthosiphon pisum. Insect Mol. Biol. 19, 215-228. ( 10.1111/j.1365-2583.2009.00974.x) [DOI] [PubMed] [Google Scholar]
- 45.Nasonia Genome Working Group, Werren JH, et al. 2010. Functional and evolutionary insights from the genomes of three parasitoid Nasonia species. Science 327, 343-348. ( 10.1126/science.1178028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Mendoza A, et al. 2019. Convergent evolution of a vertebrate-like methylome in a marine sponge. Nat. Ecol. Evol. 3, 1464-1473. ( 10.1038/s41559-019-0983-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Keller TE, Han P, Yi SV. 2016. Evolutionary transition of promoter and gene body DNA methylation across invertebrate-vertebrate boundary. Mol. Biol. Evol. 33, 1019-1028. ( 10.1093/molbev/msv345) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.de Mendoza A, Pflueger J, Lister R. 2019. Capture of a functionally active methyl-CpG binding domain by an arthropod retrotransposon family. Genome Res. 29, 1277-1286. ( 10.1101/gr.243774.118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S. 2007. Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat. Genet. 39, 61-69. ( 10.1038/Ng1929) [DOI] [PubMed] [Google Scholar]
- 50.Sarda S, Zeng J, Hunt BG, Yi SV. 2012. The evolution of invertebrate gene body methylation. Mol. Biol. Evol. 29, 1907-1916. ( 10.1093/molbev/mss062) [DOI] [PubMed] [Google Scholar]
- 51.Jeong H, Wu X, Smith B, Yi SV. 2018. Genomic landscape of methylation islands in hymenopteran insects. Genome Biol. Evol. 10, 2766-2776. ( 10.1093/gbe/evy203) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McGinty RK, Tan S. 2015. Nucleosome structure and function. Chem. Rev. 115, 2255-2273. ( 10.1021/cr500373h) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Davey CA, Sargent DF, Luger K, Maeder AW, Richmond TJ. 2002. Solvent mediated interactions in the structure of the nucleosome core particle at 1.9 angstrom resolution. J. Mol. Biol. 319, 1097-1113. ( 10.1016/S0022-2836(02)00386-8) [DOI] [PubMed] [Google Scholar]
- 54.Biswas S, Rao CM. 2018. Epigenetic tools (The Writers, The Readers and The Erasers) and their implications in cancer therapy. Eur. J. Pharmacol. 837, 8-24. ( 10.1016/j.ejphar.2018.08.021) [DOI] [PubMed] [Google Scholar]
- 55.Zentner GE, Henikoff S. 2013. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 20, 259-266. ( 10.1038/Nsmb.2470) [DOI] [PubMed] [Google Scholar]
- 56.Gonzalez-Perez A, Sabarinathan R, Lopez-Bigas N. 2019. Local determinants of the mutational landscape of the human genome. Cell 177, 101-114. ( 10.1016/j.cell.2019.02.051) [DOI] [PubMed] [Google Scholar]
- 57.Makova KD, Hardison RC. 2015. The effects of chromatin organization on variation in mutation rates in the genome. Nat. Rev. Genet. 16, 213-223. ( 10.1038/nrg3890) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen XS, Chen ZD, Chen H, Su ZJ, Yang JF, Lin FQ, Shi SH, He XL. 2012. Nucleosomes suppress spontaneous mutations base-specifically in eukaryotes. Science 335, 1235-1238. ( 10.1126/science.1217580) [DOI] [PubMed] [Google Scholar]
- 59.Brown AJ, Mao P, Smerdon MJ, Wyrick JJ, Roberts SA. 2018. Nucleosome positions establish an extended mutation signature in melanoma. PLoS Genet. 14, e1007823. ( 10.1371/journal.pgen.1007823) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pich O, Muinos F, Sabarinathan R, Reyes-Salazar I, Gonzalez-Perez A, Lopez-Bigas N. 2018. Somatic and germline mutation periodicity follow the orientation of the DNA minor groove around nucleosomes. Cell 175, 1074-1087. ( 10.1016/j.cell.2018.10.004) [DOI] [PubMed] [Google Scholar]
- 61.Hinz JM, Rodriguez Y, Smerdon MJ. 2010. Rotational dynamics of DNA on the nucleosome surface markedly impact accessibility to a DNA repair enzyme. Proc. Natl Acad. Sci. USA 107, 4646-4651. ( 10.1073/pnas.0914443107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Feng JX, Riddle NC. 2020. Epigenetics and genome stability. Mamm. Genome 31, 181-195. ( 10.1007/s00335-020-09836-2) [DOI] [PubMed] [Google Scholar]
- 63.Lim B, Mun J, Kim YS, Kim SY. 2017. Variability in chromatin architecture and associated DNA repair at genomic positions containing somatic mutations. Cancer Res. 77, 2822-2833. ( 10.1158/0008-5472.Can-16-3033) [DOI] [PubMed] [Google Scholar]
- 64.Janssen A, Colmenares SU, Karpen GH. 2018. Heterochromatin: guardian of the genome. Annu. Rev. Cell Dev. Biol. 34, 265-288. ( 10.1146/annurev-cellbio-100617-062653) [DOI] [PubMed] [Google Scholar]
- 65.Shen H, Laird PW. 2013. Interplay between the cancer genome and epigenome. Cell 153, 38-55. ( 10.1016/j.cell.2013.03.008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sawan C, Vaissiere T, Murr R, Herceg Z. 2008. Epigenetic drivers and genetic passengers on the road to cancer. Mutat. Res.-Fund. Mol. Mech. Mutagen. 642, 1-13. ( 10.1016/j.mrfmmm.2008.03.002) [DOI] [PubMed] [Google Scholar]
- 67.Meas R, Wyrick JJ, Smerdon MJ. 2019. Nucleosomes regulate base excision repair in chromatin. Mutat. Res.-Fund. Mol. Mech. Mutagen. 780, 29-36. ( 10.1016/j.mrrev.2017.10.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Banerjee DR, Deckard CE, Zeng Y, Sczepanski JT. 2019. Acetylation of the histone H3 tail domain regulates base excision repair on higher-order chromatin structures. Sci. Rep. 9, 15972. ( 10.1038/s41598-019-52340-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tolstorukov MY, Volfovsky N, Stephens RM, Park PJ. 2011. Impact of chromatin structure on sequence variability in the human genome. Nat. Struct. Mol. Biol. 18, 510-515. ( 10.1038/nsmb.2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schrader L, Schmitz J. 2019. The impact of transposable elements in adaptive evolution. Mol. Ecol. 28, 1537-1549. ( 10.1111/mec.14794) [DOI] [PubMed] [Google Scholar]
- 71.Bourgeois Y, Boissinot S. 2019. On the population dynamics of junk: a review on the population genomics of transposable elements. Genes 10, 419. ( 10.3390/genes10060419) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cosby RL, Chang NC, Feschotte C. 2019. Host-transposon interactions: conflict, cooperation, and cooption. Genes Dev. 33, 1098-1116. ( 10.1101/gad.327312.119) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Friedli M, Trono D. 2015. The developmental control of transposable elements and the evolution of higher species. Annu. Rev. Cell Dev. Biol. 31, 429-451. ( 10.1146/annurev-cellbio-100814-125514) [DOI] [PubMed] [Google Scholar]
- 74.Deniz O, Frost JM, Branco MR. 2019. Regulation of transposable elements by DNA modifications. Nat. Rev. Genet. 20, 417-432. ( 10.1038/s41576-019-0117-3) [DOI] [PubMed] [Google Scholar]
- 75.Percharde M, Sultana T, Ramalho-Santos M. 2020. What doesn't kill you makes you stronger: transposons as dual players in chromatin regulation and genomic variation. Bioessays 42, 1900232. ( 10.1002/bies.201900232) [DOI] [PubMed] [Google Scholar]
- 76.Rey O, Danchin E, Mirouze M, Loot C, Blanchet S. 2016. Adaptation to global change: a transposable element-epigenetics perspective. Trends Ecol. Evol. 31, 514-526. ( 10.1016/j.tree.2016.03.013) [DOI] [PubMed] [Google Scholar]
- 77.Danchin E, Pocheville A, Rey O, Pujol B, Blanchet S. 2019. Epigenetically facilitated mutational assimilation: epigenetics as a hub within the inclusive evolutionary synthesis. Biol. Rev. 94, 259-282. ( 10.1111/brv.12453) [DOI] [Google Scholar]
- 78.Jansz N. 2019. DNA methylation dynamics at transposable elements in mammals. DNA Methylation 63, 677-689. ( 10.1042/Ebc20190039) [DOI] [PubMed] [Google Scholar]
- 79.Barau J, Teissandier A, Zamudio N, Roy S, Nalesso V, Herault Y, Guillou F, Bourc'his D. 2016. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science 354, 909-912. ( 10.1126/science.aah5143) [DOI] [PubMed] [Google Scholar]
- 80.Castel SE, Martienssen RA. 2013. RNA interference in the nucleus: roles for small RNAs in transcription, epigenetics and beyond. Nat. Rev. Genet. 14, 100-112. ( 10.1038/nrg3355) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fultz D, Choudury SG, Slotkin RK. 2015. Silencing of active transposable elements in plants. Curr. Opin. Plant Biol. 27, 67-76. ( 10.1016/j.pbi.2015.05.027) [DOI] [PubMed] [Google Scholar]
- 82.Sarkar A, Volff JN, Vaury C. 2017. piRNAs and their diverse roles: a transposable element-driven tactic for gene regulation? FASEB J. 31, 436-446. ( 10.1096/fj.201600637RR) [DOI] [PubMed] [Google Scholar]
- 83.Ozata DM, Gainetdinov I, Zoch A, O'Carroll D, Zamore PD. 2019. PIWI-interacting RNAs: small RNAs with big functions. Nat. Rev. Genet. 20, 89-108. ( 10.1038/s41576-018-0073-3) [DOI] [PubMed] [Google Scholar]
- 84.Matsumoto N, Nishimasu H, Sakakibara K, Nishida KM, Hirano T, Ishitani R, Siomi H, Siomi MC, Nureki O. 2016. Crystal structure of silkworm PIWI-clade Argonaute Siwi bound to piRNA. Cell 167, 484-497. ( 10.1016/j.cell.2016.09.002) [DOI] [PubMed] [Google Scholar]
- 85.Hirakata S, Siomi MC. 2016. piRNA biogenesis in the germline: from transcription of piRNA genomic sources to piRNA maturation. BBA Gene Regul. Mech. 1859, 82-92. ( 10.1016/j.bbagrm.2015.09.002) [DOI] [PubMed] [Google Scholar]
- 86.He JP, et al. 2019. Transposable elements are regulated by context-specific patterns of chromatin marks in mouse embryonic stem cells. Nat. Commun. 10, 34. ( 10.1038/s41467-018-08006-y) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cui XK, Cao XF. 2014. Epigenetic regulation and functional exaptation of transposable elements in higher plants. Curr. Opin. Plant Biol. 21, 83-88. ( 10.1016/j.pbi.2014.07.001) [DOI] [PubMed] [Google Scholar]
- 88.Galindo-Gonzalez L, Sarmiento F, Quimbaya MA. 2018. Shaping plant adaptability, genome structure and gene expression through transposable element epigenetic control: focus on methylation. Agronomy 8, 180. ( 10.3390/agronomy8090180) [DOI] [Google Scholar]
- 89.Ohno S. 1970. Evolution by gene duplication. Berlin, Germany: Springer. [Google Scholar]
- 90.Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531-1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lynch M, Conery JS. 2000. The evolutionary fate and consequences of duplicate genes. Science 290, 1151-1155. [DOI] [PubMed] [Google Scholar]
- 92.Innan H, Kondrashov F. 2010. The evolution of gene duplications: classifying and distinguishing between models. Nat. Rev. Genet. 11, 97-108. ( 10.1038/nrg2689) [DOI] [PubMed] [Google Scholar]
- 93.Rodin SN, Riggs AD. 2003. Epigenetic silencing may aid evolution by gene duplication. J. Mol. Evol. 56, 718-729. ( 10.1007/s00239-002-2446-6) [DOI] [PubMed] [Google Scholar]
- 94.Chang AYF, Liao BY. 2012. DNA methylation rebalances gene dosage after mammalian gene duplications. Mol. Biol. Evol. 29, 133-144. ( 10.1093/molbev/msr174) [DOI] [PubMed] [Google Scholar]
- 95.Keller TE, Yi SV. 2014. DNA methylation and evolution of duplicate genes. Proc. Natl Acad. Sci. USA 111, 5932-5937. ( 10.1073/pnas.1321420111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Qian WF, Liao BY, Chang AYF, Zhang JZ. 2010. Maintenance of duplicate genes and their functional redundancy by reduced expression. Trends Genet. 26, 425-430. ( 10.1016/j.tig.2010.07.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhong ZX, Du K, Yu Q, Zhang YE, He SP.. 2016. Divergent DNA methylation provides insights into the evolution of duplicate genes in zebrafish. G3 Genes Genom. Genet. 6, 3581-3591. ( 10.1534/g3.116.032243) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kucharski R, Maleszka J, Maleszka R. 2016. A possible role of DNA methylation in functional divergence of a fast evolving duplicate gene encoding odorant binding protein 11 in the honeybee. Proc. R. Soc. B 283, 20160558. ( 10.1098/rspb.2016.0558) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dyson CJ, Goodisman MAD. 2020. Gene duplication in the honeybee: patterns of DNA methylation, gene expression, and genomic environment. Mol. Biol. Evol. 37, 2322-2331. ( 10.1093/molbev/msaa088) [DOI] [PubMed] [Google Scholar]
- 100.Wang J, Marowsky NC, Fan CZ. 2014. Divergence of gene body DNA methylation and evolution of plant duplicate genes. PLoS ONE 9, e110357. ( 10.1371/journal.pone.0110357) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This article has no additional data.